
Dioxygen Reactivity of Copper(I)/Manganese(II)-Porphyrin Assemblies: Mechanistic Studies and Cooperative Activation of O2




Abstract
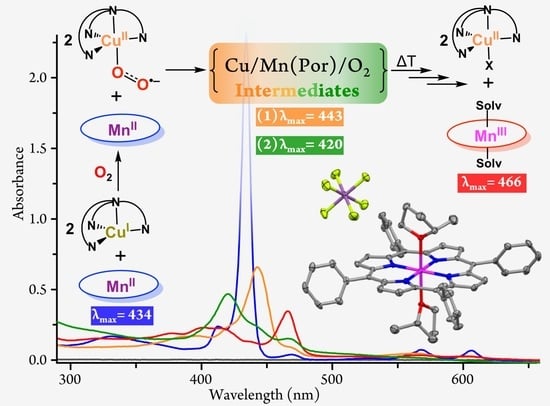
:
1. Introduction
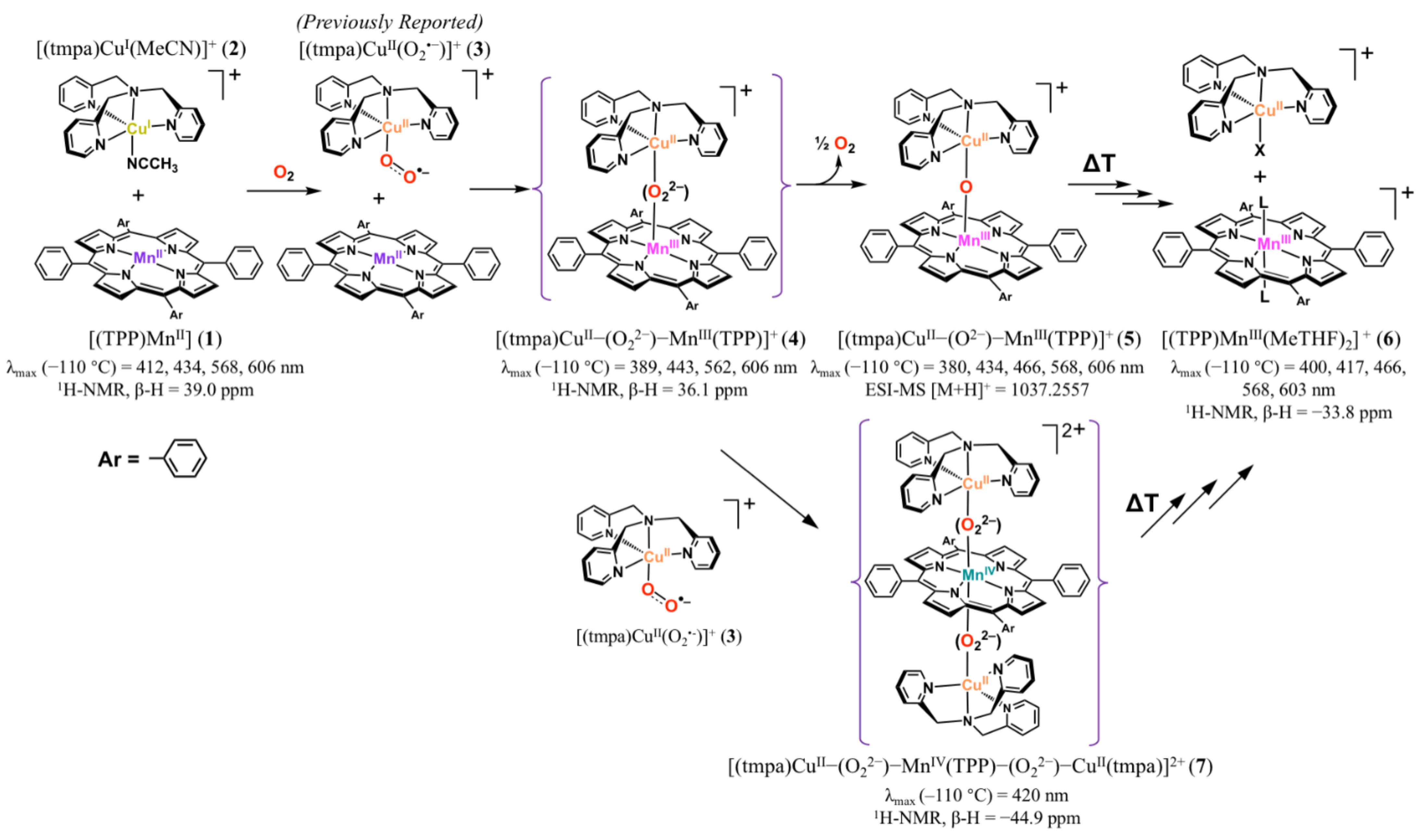
2. Results and Discussion
2.1. UV-vis Absorption Spectroscopy
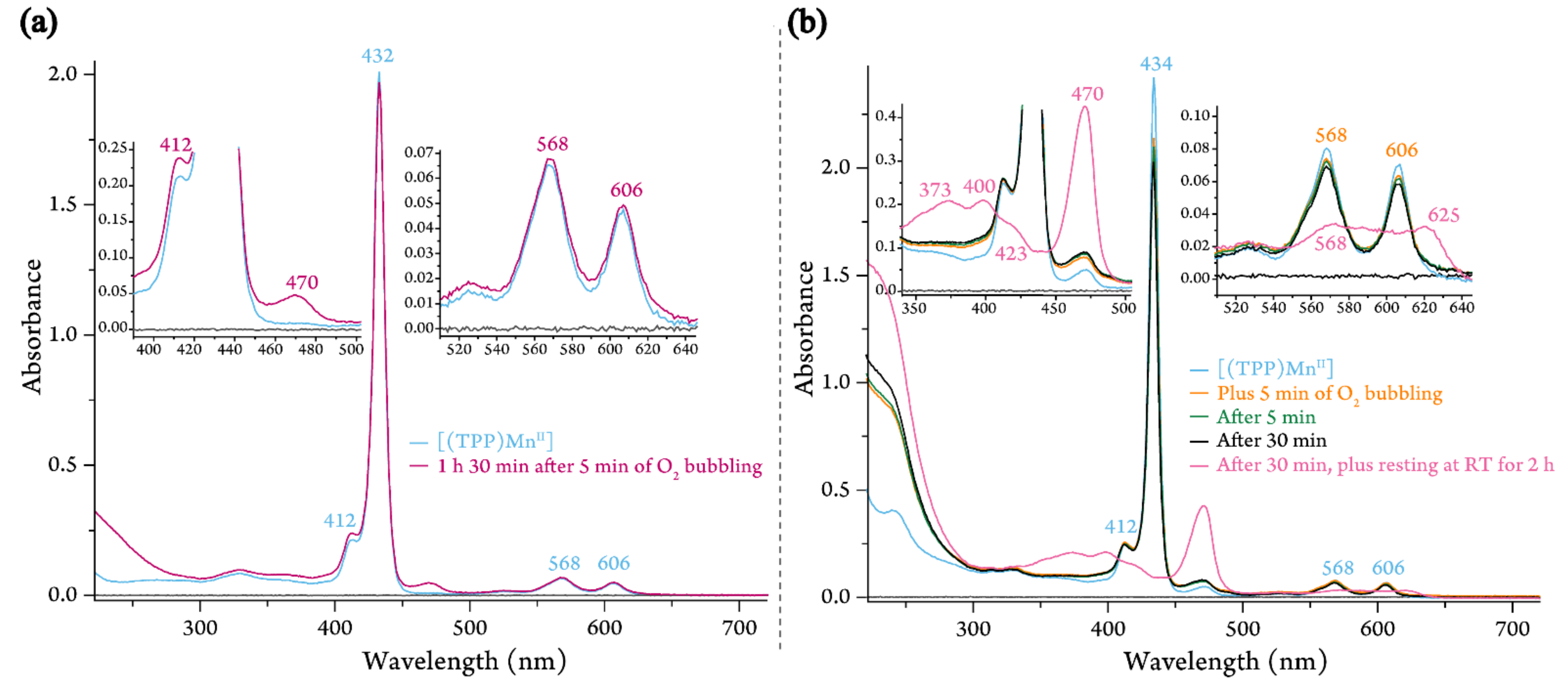
2.1.1. Dioxygen Chemistry of [(TPP)MnII]
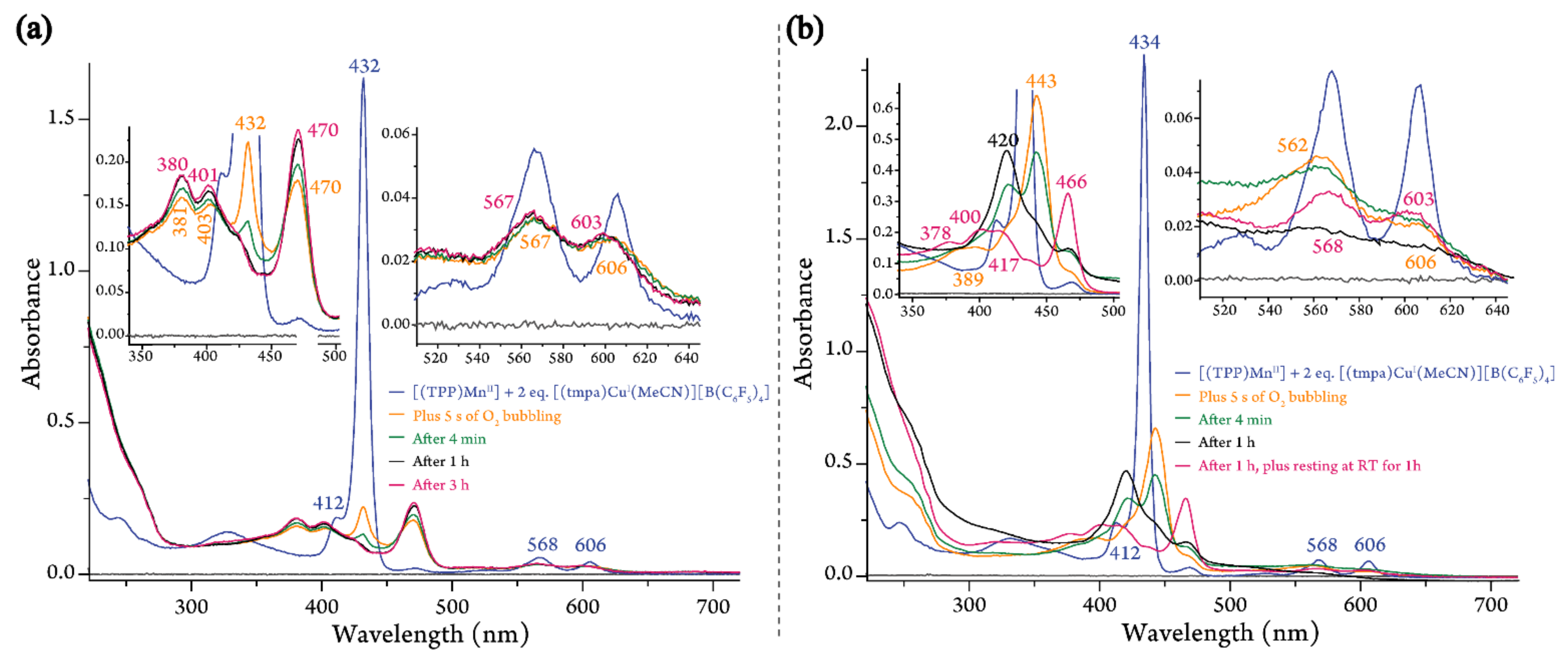
2.1.2. Dioxygen Chemistry of [(TPP)MnII]/[(tmpa)CuI(MeCN)][B(C6F5)4]
2.1.3. Dioxygen Chemistry of [(TPP)MnII]/2 [(tmpa)CuI(MeCN)][B(C6F5)4]
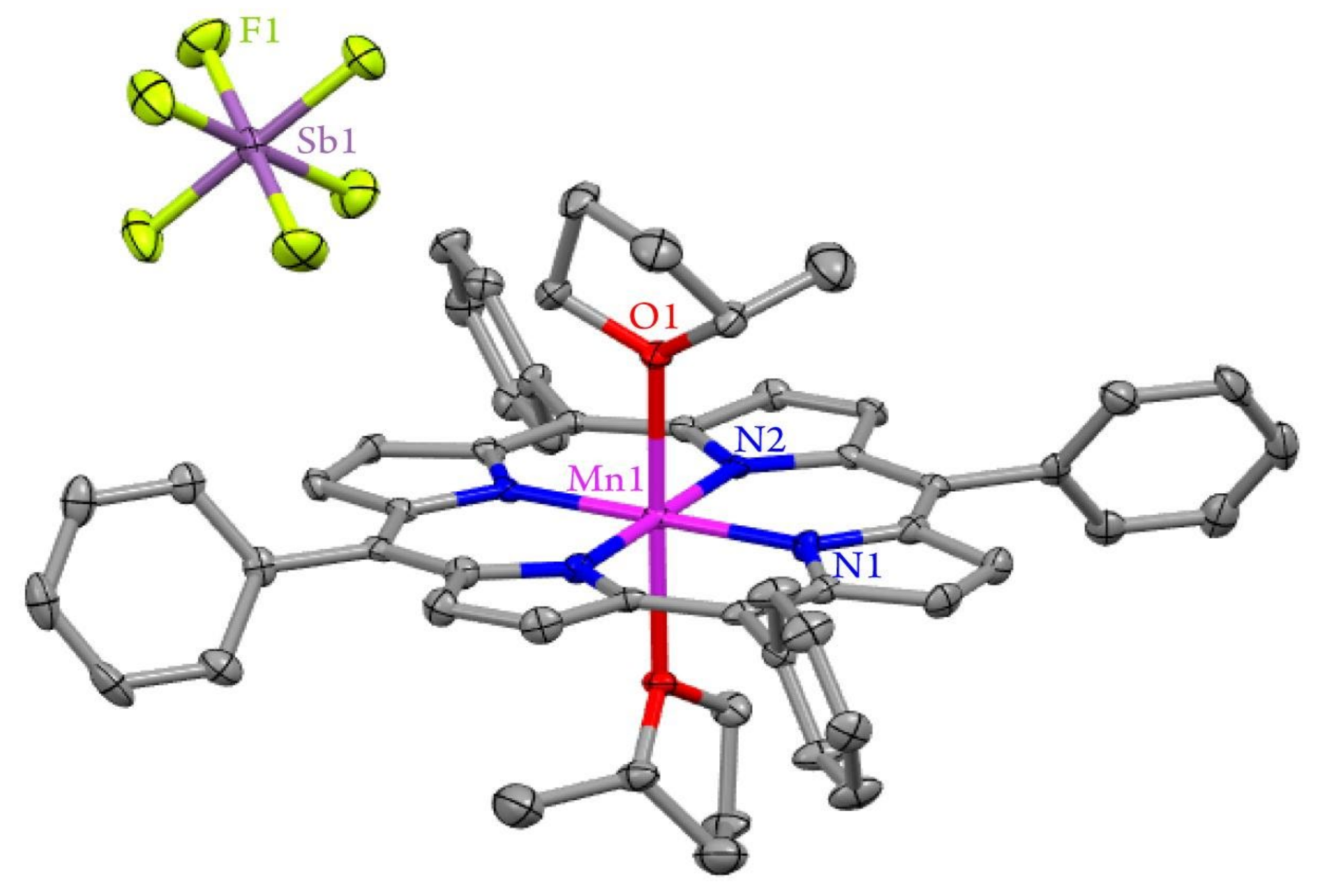
2.2. X-ray Structure of [(TPP)MnIII(MeTHF)2]SbF6
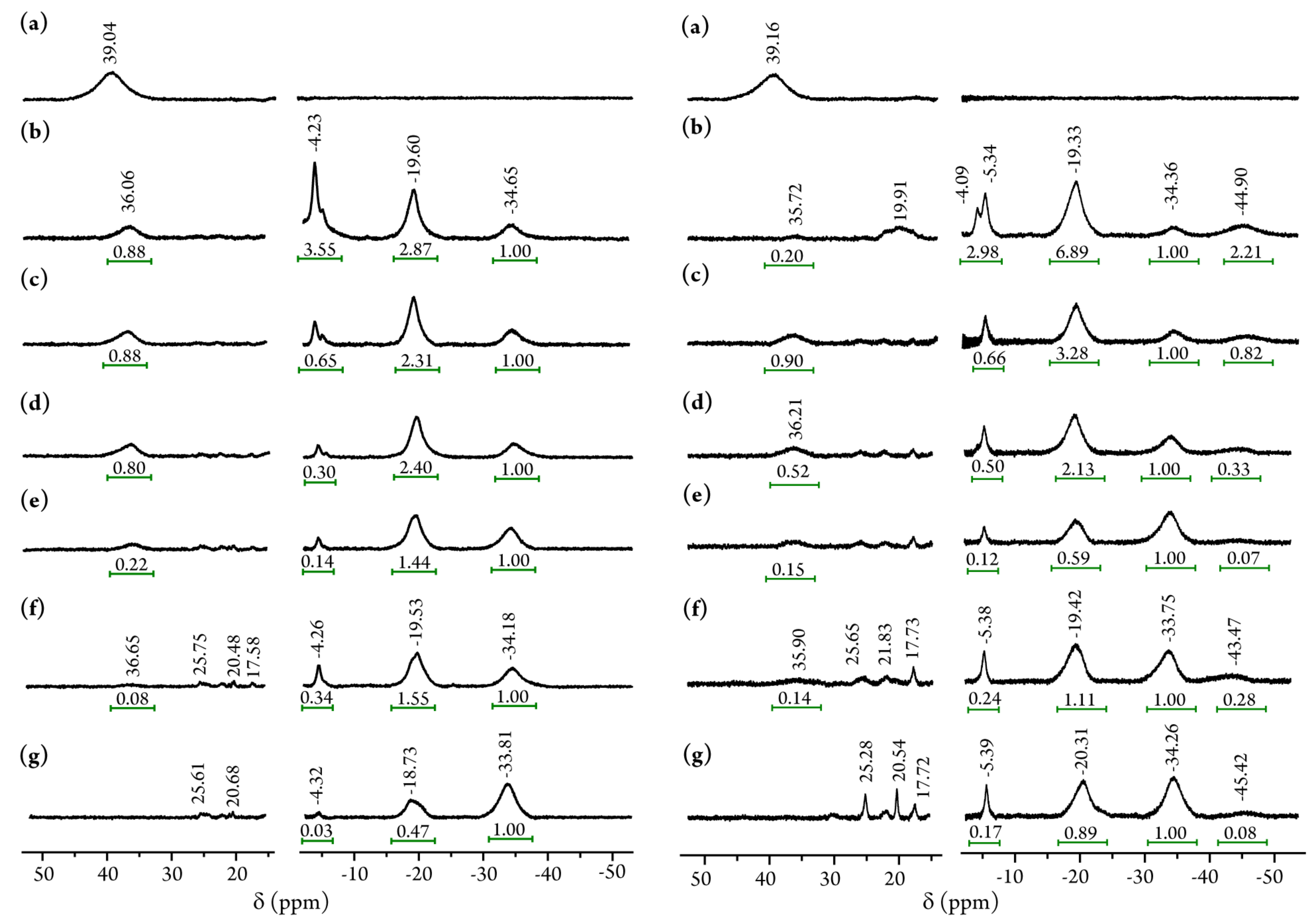
2.3. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.4. Electrospray Ionization Mass Spectrometry (ESI-MS)
2.5. Fourier-Transform Infrared Spectroscopy (FT-IR)
3. Conclusions
4. Materials and Methods
4.1. General Methods
4.2. Synthesis and Characterization
4.2.1. Synthesis of [(TPP)MnII]
4.2.2. Synthesis of [(TPP)MnIII(THF)2]SbF6
4.3. Dioxygen Reactivity Studies
4.3.1. Room-Temperature UV-vis Measurements for [(TPP)MnII]
4.3.2. Low-Temperature UV-vis Measurements for [(TPP)MnII]
4.3.3. Room-Temperature UV-vis Measurements for 1:1 and 1:2 Mixtures of [(TPP)MnII] and [(tmpa)CuI(MeCN)][B(C6F5)4]
4.3.4. Low-Temperature UV-vis Measurements for 1:1 and 1:2 Mixtures of [(TPP)MnII] and [(tmpa)CuI(MeCN)][B(C6F5)4]
4.3.5. 1H-Nuclear Magnetic Resonance (1H-NMR) Measurements
4.4. Crystallographic Studies
4.5. Electrospray Ionization Mass Spectrometry (ESI-MS) Measurements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pecoraro, V.L.; Baldwin, M.J.; Gelasco, A. Interaction of Manganese with Dioxygen and Its Reduced Derivatives. Chem. Rev. 1994, 94, 807–826. [Google Scholar] [CrossRef]
- Pecoraro, V.L.; Gelasco, A.; Baldwin, M.J. Reactivity and Mechanism of Manganese Enzymes. In Mechanistic Bioinorganic Chemistry; Advances in Chemistry; American Chemical Society: Washington, DC, USA, 1996; Volume 246, pp. 265–301. [Google Scholar]
- Lingappa, U.F.; Monteverde, D.R.; Magyar, J.S.; Valentine, J.S.; Fischer, W.W. How Manganese Empowered Life with Dioxygen (and vice versa). Free Radic. Biol. Med. 2019, 140, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Emsley, J. Manganese. In Nature’s Building Bocks: An A–Z Guide to the Elements; Oxford University Press: Oxford, UK, 2001; pp. 249–253. [Google Scholar]
- Law, N.A.; Caudle, M.T.; Pecoraro, V.L. Manganese Redox Enzymes and Model Systems: Properties, Structures, and Reactivity. In Advances in Inorganic Chemistry; Sykes, A.G., Ed.; Academic Press: Cambridge, MA, USA, 1998; Volume 46, pp. 305–440. [Google Scholar]
- Dismukes, G.C. Manganese Enzymes with Binuclear Active Sites. Chem. Rev. 1996, 96, 2909–2926. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Richards, N.G.J. Biological Functions Controlled by Manganese Redox Changes in Mononuclear Mn-Dependent Enzymes. Essays Biochem. 2017, 61, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groves, J.T.; Watanabe, Y.; McMurry, T.J. Oxygen Activation by Metalloporphyrins. Formation and Decomposition of an Acylperoxymanganese(III) Complex. J. Am. Chem. Soc. 1983, 105, 4489–4490. [Google Scholar] [CrossRef]
- Liu, W.; Groves, J.T. Manganese Porphyrins Catalyze Selective C−H Bond Halogenations. J. Am. Chem. Soc. 2010, 132, 12847–12849. [Google Scholar] [CrossRef]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St Clair, D.; Batinic-Haberle, I. Manganese Superoxide Dismutase, MnSOD and Its Mimics. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 794–814. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Dong, H.; Li, J.; Cheng, B.; Huang, Y.; Feng, Y.; Lei, A. Spectroscopic Observation of Iodosylarene Metalloporphyrin Adducts and Manganese(V)-oxo Porphyrin Species in a Cytochrome P450 Analogue. Nat. Commun. 2012, 3, 1190. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Groves, J.T. Manganese Catalyzed C–H Halogenation. Acc. Chem. Res. 2015, 48, 1727–1735. [Google Scholar] [CrossRef]
- Huang, X.; Zhuang, T.; Kates, P.A.; Gao, H.; Chen, X.; Groves, J.T. Alkyl Isocyanates via Manganese-Catalyzed C–H Activation for the Preparation of Substituted Ureas. J. Am. Chem. Soc. 2017, 139, 15407–15413. [Google Scholar] [CrossRef]
- Passard, G.; Dogutan, D.K.; Qiu, M.; Costentin, C.; Nocera, D.G. Oxygen Reduction Reaction Promoted by Manganese Porphyrins. ACS Catal. 2018, 8, 8671–8679. [Google Scholar] [CrossRef]
- Guo, M.; Seo, M.S.; Lee, Y.-M.; Fukuzumi, S.; Nam, W. Highly Reactive Manganese(IV)-Oxo Porphyrins Showing Temperature-Dependent Reversed Electronic Effect in C–H Bond Activation Reactions. J. Am. Chem. Soc. 2019, 141, 12187–12191. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kates, P.A.; Dilger, A.K.; Cheng, P.T.; Ewing, W.R.; Groves, J.T. Manganese-Catalyzed Desaturation of N-Acyl Amines and Ethers. ACS Catal. 2019, 9, 9513–9517. [Google Scholar] [CrossRef]
- Klaine, S.; Bratcher, F.; Winchester, C.M.; Zhang, R. Formation and Kinetic Studies of Manganese(IV)-oxo Porphyrins: Oxygen Atom Transfer Mechanism of Sulfide Oxidations. J. Inorg. Biochem. 2020, 204, 110986. [Google Scholar] [CrossRef]
- Zhang, L.; Lee, Y.-M.; Guo, M.; Fukuzumi, S.; Nam, W. Unprecedented Reactivities of Highly Reactive Manganese(III)–Iodosylarene Porphyrins in Oxidation Reactions. J. Am. Chem. Soc. 2020, 142, 19879–19884. [Google Scholar] [CrossRef]
- Mann, S.I.; Nayak, A.; Gassner, G.T.; Therien, M.J.; DeGrado, W.F. De Novo Design, Solution Characterization, and Crystallographic Structure of an Abiological Mn–Porphyrin-Binding Protein Capable of Stabilizing a Mn(V) Species. J. Am. Chem. Soc. 2021, 143, 252–259. [Google Scholar] [CrossRef]
- Kostopoulos, N.; Banse, F.; Fave, C.; Anxolabéhère-Mallart, E. Modulating Alkene Reactivity from Oxygenation to Halogenation via Electrochemical O2 Activation by Mn Porphyrin. Chem. Commun. 2021, 57, 1198–1201. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, J.; Zhang, L.; Lee, Y.-M.; Fukuzumi, S.; Nam, W. Enthalpy–Entropy Compensation Effect in Oxidation Reactions by Manganese(IV)-Oxo Porphyrins and Nonheme Iron(IV)-Oxo Models. J. Am. Chem. Soc. 2021, 143, 18559–18570. [Google Scholar] [CrossRef]
- Weschler, C.J.; Hoffman, B.M.; Basolo, F. Synthetic Oxygen Carrier. Dioxygen Adduct of a Manganese Porphyrin. J. Am. Chem. Soc. 1975, 97, 5278–5280. [Google Scholar] [CrossRef]
- Hoffman, B.M.; Szymanski, T.; Brown, T.G.; Basolo, F. The Dioxygen Adducts of Several Manganese(II) Porphyrins. Electron Paramagnetic Resonance Studies. J. Am. Chem. Soc. 1978, 100, 7253–7259. [Google Scholar] [CrossRef]
- VanAtta, R.B.; Strouse, C.E.; Hanson, L.K.; Valentine, J.S. Peroxo(tetraphenylporphinato)manganese(III) and Chloro(tetraphenylporphinato)manganese(II) Anions. Synthesis, Crystal Structures, and Electronic Structures. J. Am. Chem. Soc. 1987, 109, 1425–1434. [Google Scholar] [CrossRef]
- Phung, Q.M.; Pierloot, K. The Dioxygen Adducts of Iron and Manganese Porphyrins: Electronic Structure and Binding Energy. Phys. Chem. Chem. Phys. 2018, 20, 17009–17019. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.S.; Quinn, A.E. Reaction of Superoxide with the Manganese(III) Tetraphenylporphine Cation. Inorg. Chem. 1976, 15, 1997–1999. [Google Scholar] [CrossRef]
- Adam, S.M.; Wijeratne, G.B.; Rogler, P.J.; Diaz, D.E.; Quist, D.A.; Liu, J.J.; Karlin, K.D. Synthetic Fe/Cu Complexes: Toward Understanding Heme-Copper Oxidase Structure and Function. Chem. Rev. 2018, 118, 10840–11022. [Google Scholar] [CrossRef] [PubMed]
- Karlin, K.D.; Wei, N.; Jung, B.; Kaderli, S.; Niklaus, P.; Zuberbuehler, A.D. Kinetics and Thermodynamics of Formation of Copper-Dioxygen Adducts: Oxygenation of Mononuclear Copper(I) Complexes Containing Tripodal Tetradentate Ligands. J. Am. Chem. Soc. 1993, 115, 9506–9514. [Google Scholar] [CrossRef]
- Karlin, K.D.; Kaderli, S.; Zuberbühler, A.D. Kinetics and Thermodynamics of Copper(I)/Dioxygen Interaction. Acc. Chem. Res. 1997, 30, 139–147. [Google Scholar] [CrossRef]
- Zhang, C.X.; Kaderli, S.; Costas, M.; Kim, E.; Neuhold, Y.-M.; Karlin, K.D.; Zuberbühler, A.D. Copper(I)−Dioxygen Reactivity of [(L)CuI]+ (L = Tris(2-pyridylmethyl)amine): Kinetic/Thermodynamic and Spectroscopic Studies Concerning the Formation of Cu−O2 and Cu2−O2 Adducts as a Function of Solvent Medium and 4-Pyridyl Ligand Substituent Variations. Inorg. Chem. 2003, 42, 1807–1824. [Google Scholar] [CrossRef]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [Green Version]
- Williamson, M.M.; Hill, C.L. Isolation and Characterization of a Five-Coordinate Manganese(III) Porphyrin Cation. Crystal and Molecular Structure of Aquo(tetraphenylporphinato)manganese(III) Triflate. Inorg. Chem. 1986, 25, 4668–4671. [Google Scholar] [CrossRef]
- Galinato, M.G.I.; Brocious, E.P.; Paulat, F.; Martin, S.; Skodack, J.; Harland, J.B.; Lehnert, N. Elucidating the Electronic Structure of High-Spin [MnIII(TPP)Cl] Using Magnetic Circular Dichroism Spectroscopy. Inorg. Chem. 2020, 59, 2144–2162. [Google Scholar] [CrossRef]
- Carrasco, M.C.; Hematian, S. (Hydr)oxo-Bridged Heme Complexes: From Structure to Reactivity. J. Porphyr. Phthalocyanines 2019, 23, 1286–1307. [Google Scholar] [CrossRef] [Green Version]
- Citek, C.; Herres-Pawlis, S.; Stack, T.D.P. Low Temperature Syntheses and Reactivity of Cu2O2 Active-Site Models. Acc. Chem. Res. 2015, 48, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; Stern, M.K. Synthesis, Characterization, and Reactivity of Oxomanganese(IV) Porphyrin Complexes. J. Am. Chem. Soc. 1988, 110, 8628–8638. [Google Scholar] [CrossRef]
- Groves, J.T.; Lee, J.; Marla, S.S. Detection and Characterization of an Oxomanganese(V) Porphyrin Complex by Rapid-Mixing Stopped-Flow Spectrophotometry. J. Am. Chem. Soc. 1997, 119, 6269–6273. [Google Scholar] [CrossRef]
- Low, D.W.; Abedin, S.; Yang, G.; Winkler, J.R.; Gray, H.B. Manganese Microperoxidase-8. Inorg. Chem. 1998, 37, 1841–1843. [Google Scholar] [CrossRef] [Green Version]
- Camenzind, M.J.; Hollander, F.J.; Hill, C.L. Syntheses, Ground Electronic State, and Crystal and Molecular Structure of the Monomeric Manganese(IV) Porphyrin Complex Dimethoxy(5,10,15,20-tetraphenylporphinato)manganese(IV). Inorg. Chem. 1982, 21, 4301–4308. [Google Scholar] [CrossRef]
- Smegal, J.A.; Schardt, B.C.; Hill, C.L. Isolation, Purification, and Characterization of Intermediate (Iodosylbenzene)metalloporphyrin Complexes from the (Tetraphenylporphinato)manganese(III)-iodosylbenzene Catalytic Hydrocarbon Functionalization System. J. Am. Chem. Soc. 1983, 105, 3510–3515. [Google Scholar] [CrossRef]
- Kaustov, L.; Tal, M.E.; Shames, A.I.; Gross, Z. Spin Transition in a Manganese(III) Porphyrin Cation Radical, Its Transformation to a Dichloromanganese(IV) Porphyrin, and Chlorination of Hydrocarbons by the Latter. Inorg. Chem. 1997, 36, 3503–3511. [Google Scholar] [CrossRef]
- Hatano, K.; Anzai, K.; Iitaka, Y. The Crystal and Molecular Structure of Bis(methanol)-α,β,γ,δ-tetraphenylporphinatomanganese(III) Perchlorate-Methanol. A Molecular Structure Relevant to the Intermediate-spin Six Coordinate Iron(III) Porphyrin. Bull. Chem. Soc. Jpn. 1983, 56, 422–427. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.L.; Williamson, M.M. Structural and Electronic Properties of Six-Coordinate Manganese(III) Porphyrin Cations. Crystal and Molecular Structure of Bis(N,N-dimethylformamide)(Tetraphenylporphinato)manganese(III) Perchlorate, [MnIII TPP(DMF)2]+ClO4. Inorg. Chem. 1985, 24, 2836–2841. [Google Scholar] [CrossRef]
- Hill, C.L.; Williamson, M.W. Electronic and Structural Properties of a Reactive Metalloporphyrin with N-oxide Axial Ligands. Crystal and Molecular Structure of Bis(2,6-lutidine N-oxide)(Tetraphenylporphinato)manganese(III) Perchlorate. Inorg. Chem. 1985, 24, 3024–3030. [Google Scholar] [CrossRef]
- Scheidt, W.R.; Pearson, W.B.; Gosal, N. Structure of Bis(methanol)(meso-tetraphenylporphinato)manganese(III) Hexachloroantimonate Bis(tetrachloroethane) Solvate. Acta Cryst. C 1988, 44, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Bhyrappa, P.; Wilson, S.R.; Suslick, K.S. Hydrogen-Bonded Porphyrinic Solids: Supramolecular Networks of Octahydroxy Porphyrins. J. Am. Chem. Soc. 1997, 119, 8492–8502. [Google Scholar] [CrossRef]
- Tong, S.-l.; Zhang, J.; Yan, Y.; Hu, S.; Yu, J.; Yu, L. Self-Assembled Supramolecular Architecture with Alternating Porphyrin and Phthalocyanine, Bonded by Hydrogen Bonding and π–π Stacking. Solid State Sci. 2011, 13, 1967–1971. [Google Scholar] [CrossRef]
- Williamson, M.N.; Hill, C.L. Molecular Stereochemistry of Aquamanganese(III) Porphyrins. Demonstrable effect of π-Arene-Porphyrin Interaction in the metal Coordination Environment in a Metalloporphyrin. Inorg. Chem. 1987, 26, 4155–4160. [Google Scholar] [CrossRef]
- Salomon, R.G.; Ghosh, S.; Raychaudhuri, S. Homogeneous Metal-Catalyzed Photochemistry in Organic Synthesis. In Photosensitive Metal—Organic Systems; Advances in Chemistry; American Chemical Society: Washington, DC, USA, 1993; Volume 238, pp. 315–333. [Google Scholar]
- Fox, S.J.; Chen, L.; Khan, M.A.; Richter-Addo, G.B. Nitrosoarene Complexes of Manganese Porphyrins. Inorg. Chem. 1997, 36, 6465–6467. [Google Scholar] [CrossRef]
- Shirazi, A.; Goff, H.M. Characterization of Superoxide-Metalloporphyrin Reaction Products: Effective Use of Deuterium NMR Spectroscopy. J. Am. Chem. Soc. 1982, 104, 6318–6322. [Google Scholar] [CrossRef]
- Carrasco, M.C.; Dezarn, K.J.; Khan, F.S.T.; Hematian, S. Protonation of the Oxo-Bridged Heme/Copper Assemblies: Modeling the Oxidized State of the Cytochrome c Oxidase Active Site. J. Inorg. Biochem. 2021, 225, 111593. [Google Scholar] [CrossRef]
- Chufán, E.E.; Verani, C.N.; Puiu, S.C.; Rentschler, E.; Schatzschneider, U.; Incarvito, C.; Rheingold, A.L.; Karlin, K.D. Generation and Characterization of [(P)M−(X)−Co(TMPA)]n+ Assemblies; P = Porphyrinate, M = FeIII and CoIII, X = O2−, OH−, O22−, and TMPA = Tris(2-pyridylmethyl)amine. Inorg. Chem. 2007, 46, 3017–3026. [Google Scholar] [CrossRef]
- Wasser, I.M.; Martens, C.F.; Verani, C.N.; Rentschler, E.; Huang, H.-w.; Moënne-Loccoz, P.; Zakharov, L.N.; Rheingold, A.L.; Karlin, K.D. Synthesis and Spectroscopy of μ-Oxo (O2−)-Bridged Heme/Non-heme Diiron Complexes: Models for the Active Site of Nitric Oxide Reductase. Inorg. Chem. 2004, 43, 651–662. [Google Scholar] [CrossRef]
- Tyeklar, Z.; Jacobson, R.R.; Wei, N.; Murthy, N.N.; Zubieta, J.; Karlin, K.D. Reversible Reaction of Dioxygen (and Carbon Monoxide) with a Copper(I) Complex. X-ray Structures of Relevant Mononuclear Cu(I) Precursor Adducts and the Trans-(μ-1,2-peroxo)dicopper(II) Product. J. Am. Chem. Soc. 1993, 115, 2677–2689. [Google Scholar] [CrossRef]
- Kincaid, J.; Nakamoto, K. Vibrational Spectra of Transition Metal Complexes of Tetraphenylporphine. J. Inorg. Nucl. Chem. 1975, 37, 85–89. [Google Scholar] [CrossRef]
- Hematian, S.; Siegler, M.A.; Karlin, K.D. Heme/Copper Assembly Mediated Nitrite and Nitric Oxide Interconversion. J. Am. Chem. Soc. 2012, 134, 18912–18915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, A.D.; Longo, F.R.; Kampas, F.; Kim, J. On the Preparation of Metalloporphyrins. J. Inorg. Nucl. Chem. 1970, 32, 2443–2445. [Google Scholar] [CrossRef]
- Ghiladi, R.A.; Kretzer, R.M.; Guzei, I.; Rheingold, A.L.; Neuhold, Y.-M.; Hatwell, K.R.; Zuberbühler, A.D.; Karlin, K.D. (F8TPP)FeII/O2 Reactivity Studies {F8TPP = Tetrakis(2,6-difluorophenyl)porphyrinate(2−)}: Spectroscopic (UV−Visible and NMR) and Kinetic Study of Solvent-Dependent (Fe/O2 = 1:1 or 2:1) Reversible O2-Reduction and Ferryl Formation. Inorg. Chem. 2001, 40, 5754–5767. [Google Scholar] [CrossRef]
- Hematian, S.; Siegler, M.A.; Karlin, K.D. Nitric Oxide Generation from Heme/Copper Assembly Mediated Nitrite Reductase Activity. J. Biol. Inorg. Chem. 2014, 19, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXL-2018: Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 2018. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex * | Solvent | λmax (nm) | Reference |
|---|---|---|---|
| {(TPP)MnIV[(O22−)CuII(tmpa)]2}2+ (7) | MeTHF | 420 | This work |
| [(TPP)MnIV(OCH3)2] | methanol | 360, 420, 520 | [39] |
| [Cl(TPP)MnIV(OIPh)]2O | chlorobenzene | 421, 502 | [40] |
| [(TMP)MnIV(O)(OH)] | DCM | 422 | [8] |
| [(TMP)MnIVCl2] | DCM | 420, 530, 620, 720 | [41] |
| benzene | 420, 535, 720 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Khan, F.S.T.; Hematian, S. Dioxygen Reactivity of Copper(I)/Manganese(II)-Porphyrin Assemblies: Mechanistic Studies and Cooperative Activation of O2. Molecules 2022, 27, 1000. https://doi.org/10.3390/molecules27031000
Li R, Khan FST, Hematian S. Dioxygen Reactivity of Copper(I)/Manganese(II)-Porphyrin Assemblies: Mechanistic Studies and Cooperative Activation of O2. Molecules. 2022; 27(3):1000. https://doi.org/10.3390/molecules27031000
Chicago/Turabian StyleLi, Runzi, Firoz Shah Tuglak Khan, and Shabnam Hematian. 2022. "Dioxygen Reactivity of Copper(I)/Manganese(II)-Porphyrin Assemblies: Mechanistic Studies and Cooperative Activation of O2" Molecules 27, no. 3: 1000. https://doi.org/10.3390/molecules27031000
APA StyleLi, R., Khan, F. S. T., & Hematian, S. (2022). Dioxygen Reactivity of Copper(I)/Manganese(II)-Porphyrin Assemblies: Mechanistic Studies and Cooperative Activation of O2. Molecules, 27(3), 1000. https://doi.org/10.3390/molecules27031000