Abstract
Pyrazole, an important pharmacophore and a privileged scaffold of immense significance, is a five-membered heterocyclic moiety with an extensive therapeutic profile, viz., anti-inflammatory, anti-microbial, anti-anxiety, anticancer, analgesic, antipyretic, etc. Due to the expansion of pyrazolecent red pharmacological molecules at a quicker pace, there is an urgent need to put emphasis on recent literature with hitherto available information to recognize the status of this scaffold for pharmaceutical research. The reported potential pyrazole-containing compounds are highlighted in the manuscript for the treatment of cancer and inflammation, and the results are mentioned in % inhibition of inflammation, % growth inhibition, IC50, etc. Pyrazole is an important heterocyclic moiety with a strong pharmacological profile, which may act as an important pharmacophore for the drug discovery process. In the struggle to cultivate suitable anti-inflammatory and anticancer agents, chemists have now focused on pyrazole biomolecules. This review conceals the recent expansion of pyrazole biomolecules as anti-inflammatory and anticancer agents with an aim to provide better correlation among different research going around the world.
1. Introduction
The pyrazole moiety is a heterocyclic ring system (five membered) with 3 C and 2 N in adjacent sites. In the year 1959, 1-pyrazolyl-alanine (natural pyrazole) was obtained from watermelons seeds [1,2]. Pyrazole compounds have an extensive past of applications, being used as herbicides, agrochemicals, and as active pharmaceutical agents. The current scenario of pyrazole moiety (Scheme 1) as a selective COX-2 inhibitor and anticancer agents has additionally emphasized their significance in pharmaceutical chemistry [3].
Scheme 1.
Five membered pyrazole moiety.
An organized observation of these groups of heterocyclic molecules exhibited that pyrazole-bearing therapeutic agents play an essential part in pharmaceutical sciences. The dominance of the pyrazole moiety in therapeutically potential lead compounds has encouraged the necessity for sophisticated and well-organized methods to prepare these heterocyclic compounds. The pyrazole moiety is shown to possess a broad range of biological profiles in the literature, mainly anticancer, anti-inflammatory, analgesic, antioxidant, anti-convulsant, anti-microbial, anti-mycobacterial, anti-amoebic, anti-depressant, hypotensive, and ACE inhibitors [4]. Several pyrazole-containing moieties have previously found their therapeutic application clinically, as NSAIDs such as celecoxib, lonazolac, tepoxaline, antipyrine (analgesic and antipyretic), phenylbutazone (in rheumatoid arthritis, osteoarthritis), novalgine, deracoxib, difenamizole, mepirizole, SC-560, crizotinib, pyrazofurin, and many more are already in the market [5]. Such drugs are listed in Table 1. In this updated review, our chief intention is to accentuate the anti-inflammatory and anticancer profiles displayed by the pyrazole moiety (Figure 1). The mechanism of action of anticancer activity are given in Figure 2. The different kinds of pharmacological activities of various compounds containing the pyrazole moiety are summarized in Figure 3.

Table 1.
Representative drugs containing the pyrazole scaffold.
Figure 1.
Pharmacological profile of pyrazole.
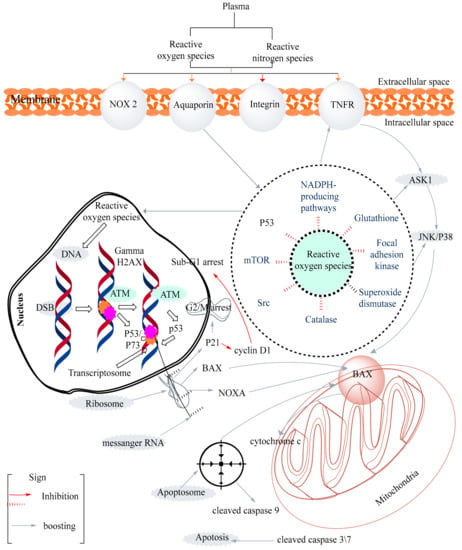
Figure 2.
The mechanism of action of the anticancer activity pathway.
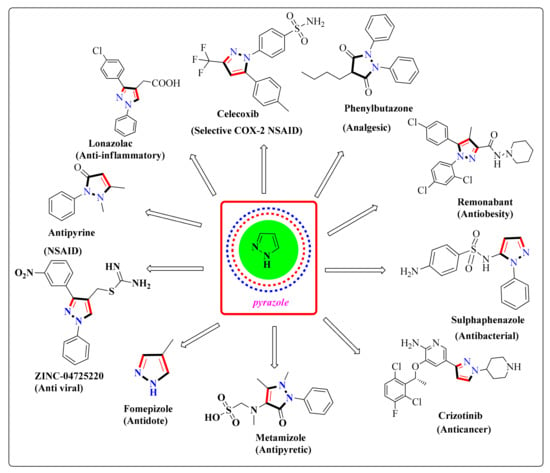
Figure 3.
The pyrazole scaffold, exhibiting various pharmacological activities.
2. Pyrazole Biomolecules as Cancer Therapeutics
Cancer is an uncontrolled abnormal cell growth [20] and a severe life-threatening condition globally [21,22]. It is a class of several illnesses [23] that are initiated to be deadly, followed by cardiovascular disorders in mortality and morbidity [24,25]. Although huge innovative and operative elucidations to cancer have been made, and quite clear margins in the management of cancer are exhibited, hereafter, it is estimated as the principle cause of mortality in the upcoming generation [26]. Presently, pharmaceutical companies are putting large amounts of money into the innovation and advancement of potential and safe anticancer drugs with fewer side effects [27,28]. Therefore, 1,3,4-trisubstituted pyrazole derivatives were prepared and evaluated for cytotoxic potential by Abadi et al., N-(1-{1-[4-nitrophen]-3-phephenyl-1H-pyrazol-4-yl}methylene)-2-chlorobenzohydrazide (2) showed satisfactory potential for MCF7, SF-268, and NCI-H460 cell lines with GI50, TGI50, and LC50 values of 3.79, 12.50, and 42.30 µM [29] (Refer to Table 2 for the structure of the compound). Bouabdallah et al., reported concerning N,N-bis[(3, 5-dimethylpyrazol-1-yl) methyl]aniline (3) and screened against Hep-2 and P815. These derivatives exhibited significant cytotoxic potential (IC50 = 3.25 mg/mL and 17.82 mg/mL) against Hep-2 and P815 cancer cell lines [30] (Refer to Table 2 for the structure of the compound). Wei et al., described ethyl-1-(2-hydroxy-3-aroxypropyl)-3-aryl-1H-pyrazole-5-carboxylate derivatives, among which, compound 4 was found to be most potent in countering A549 growth (IC50 = 26 µM) [31] (Refer to Table 2 for the structure of the compound). Xia et al., prepared 1-arylmethyl-3-aryl-1H-pyrazole-5-carbohydrazide and screened for antitumor activity. Compound 5 displayed significant cell apoptosis and potent antitumor activity with growth inhibitory properties (IC50 = 49.85 μM) [32]. Fan et al., carried out synthesis of 1-(2′-hydroxy-3′-aroxypropyl)-3-aryl-1H-pyrazole-5-carbohydrazide derivatives and evaluated cytotoxicity against A549 cell lines. Compounds 6 and 7 brought about A549 cell line autophagy without causing apoptosis and 1-(3-(4-chlorophenoxy)-2-hydroxypropyl)-3-(4-chlorophenyl)-1H-pyrazole-5-carbohydrazide showed maximum autophagy in NCIH460 cells lines with IC50 of 48 μM and 32 μM at 48 h [33]. Abdel-Aziz et al., synthesized and screened compound 8, which exhibited significant potential for Hep-2 cancer cell lines with IC50 = 0.74 mg/mL [34]. Zheng et al., described a new class of (E)-1-(4-tert-butylbenzyl-N′-(1-(5-chloro-2-hydroxyphenyl)ethylidene)-3-(4-chlorophenyl)-1H-pyrazole-5-carbohydrazides among which compound 9 offered IC50 = 0.28 µM and caused A549 cell line apoptosis [35]. Zheng et al., described some Oxime-linked pyrazole derivatives. Compound 10 indicated the highest potential for the A549 cancer cell line with in vitro cytotoxicity (IC50 = 14.5 µM) [36] (Refer to Table 2 for the structure of the compound). Newer derivatives of 3-(Biphenyl-2-yl)-5-(4-methoxyphenyl)-1H-prazole were investigated by Shaw et al., against NCI-H460, SW620, OVCA, and AGS cell lines, in which compound 11 revealed GI50 of 0.67, 0.89, 0.73, and 0.79 µM [37] (Refer to Table 2 for the structure of the compound). Poly-substituted pyrazole derivatives having antitumor potential were prepared by Rostom et al. The amino cyano pyrazole 12 showed GI50 0.36 µM, TGI 8.78 µM, and LC50 MG-MID values of 69.3 µM, whereas tricyclic-napthopyrazole-containing derivative 13 showed GI50 = 0.08 µM, TGI 30.9 µM, and MG-MID values of 93.3 µM [38] (Refer to Table 2 for the structure of the compound). Newer pyrazolic compounds were developed and assessed for antitumor activity by Insuasty et al., Compounds 14 and 15 appeared most active with GI50 values ranging between 0.04 to 11.4 µM against K-562, UO-31, SR, and HOP-92 cancer cell lines [39]. Nitulescu et al., prepared different substituted pyrazole derivatives in which compound 16 appeared potent with a mean log of GI50 = −5.75 [40]. Zhang et al., reported 3-(1H-indole-3-yl)-1H-pyrazole-5-carbohydrazide derivatives; where compound 17 and 18 exhibited important antiproliferative potential for HepG-2 with an IC50 of 0.71 μM, BT474 with an IC50 value of 1.39 μM, and BGC823 cell lines with an IC50 value of 0.71 μM compared to the standard 5-fluorouracil (5-FU). The results validated that both derivatives arrested cell cycle at S phase [41] (Refer to Table 2 for the structure of the compound).
Table 2.
Representative compound structures 2 to 18 and their anticancer activity.
El-Gamal et al., prepared 3-(3-chloro-5-hydroxyphenyl)-N-(2-(4-methylpiperazin-1-yl)ethyl)-4-(pyridine-4-yl)-1H-pyrazole-1-carboxamide derivatives, in which compound 19 was the most powerful anticancer agent for A375 cell lines with IC50 = 4.2 μM [42] (Refer to Table 3 for the structure of the compound). N-((1,3-diphenyl-1H-pyrazol-4-yl)methyl) aniline-containing derivatives were explored by Huang et al., who assessed them for anticancer potential and cyclin-dependent kinase-2 (CDK2) inhibitory properties in vitro, with an IC50 of 0.98 ± 0.06μM. Compound 20 reflected excellent anticancer potential against MCF-7 with IC50 values of 1.88 ± 0.11 and B16-F10 cancer cell lines with IC50 = 2.12 ± 0.15 μM [43]. Compound 21, displaying significant anticancer efficacy for HCT116 (IC50 = 0.39 ± 0.06 μM) and MCF-7 (IC50 = 0.46 ± 0.04 μM) cell lines, was reported by Li et al., and displayed inhibition of Aurora-A kinase with IC50 = 0.16 ± 0.03 µM [44]. Novel derivatives of the pyrazole moiety were prepared by Mohareb and Kumar et al., in which compounds 22 and 23 were screened against the MCF7 cancer cell line with IC50 = 0.01 µM, NCI-H460 with IC50 = 0.03 µM and SF-268 with IC50 = 31.5 µM against doxorubicin as the standard drug [27,45] (Refer to Table 3 for the structure of the compound). Sun et al., prepared 1,3-diphenyl-N-(phenylcarbamothioyl)-1H-pyrazole-4-carboxamide derivatives and assessed for CDK inhibition potential. Compound 24 caused inhibition of CDK2 (IC50 = 25 nM) and arrest of Go/G1 phase in A549 cancer cell lines in a dose reliant fashion [46]. Inceler et al., evaluated 1, 3-diarylpyrazole containing derivatives for anticancer potential among which compound 25 displayed GI50 values of 25.2 ± 3.2 and 28.3 ± 1.53 µM, against Raji and HL60 cancer cell lines [47] (Refer to Table 3 for the structure of the compound). Gamal et al., prepared newer pyrazole derivatives of which compound 26 displayed potential anticancer results against A375 cell line [48]. Xu et al., evaluated N-arylpyrazoles on Bel-7402, HL-60, BGC-823 and KB among which compound 27 caused significant inhibition of Bel-7402 cells (1.5 times) as compared to established drug cisplatin [49]. Koca et al., prepared of 4-benzoyl-1,5-diphenyl-N-(substitutedphenylcarbamothioyl)-1H-pyrazole-3-carboxamide derivatives through one pot synthesis and assessed for anticancer potential on HepG2, Jurkat, DLD-1, human T cell lymphoblast cancer cell lines. Compound 28 exhibited significant potential to be considered for inhibition of kinase [21] (Refer to Table 3 for the structure of the compound). Zheng et al., developed pyrazole linked benzimidazole derivative 29 and evaluated on U937, K562, HT29 and A549 cancer cell lines, LoVo and in vitro inhibition of Aurora A/B kinase [50]. 5-phenyl-1H-pyrazol derivatives were prepared and assessed for BRAF (V600E) inhibition. Compound 30 displayed significant inhibition with an IC50 = 0.19 µM as reported by Dong et al.; anti-cancer assays showed many derivatives displaying maximum anticancer potential against WM266.4 and A375 with IC50 = 1.50 to 1.32 µM, i.e., equivalent to vemurafenib [51] (Refer to Table 3 for the structure of the compound).
Table 3.
Representative compound structure 19 to 30 and their anticancer activity.
Mohareb et al., assessed the antitumor potential accompanied with pyrazole-containing derivative 31 for NCI-H460, MCF-7, and SF268 cell lines [52] (Refer to Table 4 for the structure of the compound). Nakao et al., prepared newer 1-(1H)-5-methylbenzimidazol-2-yl)-4-benzyl-3-methyl-1H-pyrazol-5-ol derivatives, among which compound 32 exhibited potential PCA-1/ALKBH3 inhibitory activity as an anti-prostate-cancer molecule [53]. Xing et al., developed novel pyrazole hydrazide derivative 33 that exposed significant activity on B16-F10 and MCF-7 cancer cell lines with IC50 = 0.49 ± 0.07 µM and 0.57 ± 0.03 µM [54]. In 2014, Aydın et al., prepared pyrazole-biphenyl derivatives in which compound 34 reflected 69.95% inhibition and apoptosis of K-562 cells. [55] (Refer to Table 4 for the structure of the compound). Cankara et al., prepared pyrazole containing amide derivatives and determined the anticancer potential against Huh-7, HCT-116, and MCF-7 cell lines through a sulforhodamine B assay [56]. Compound 35 showed promising cytotoxicity against HCT-116, Huh-7, and MCF-7 cell lines with IC50 = 1.1 µM, 1.6 µM, and 3.3 µM with arresting cell cycle at the SubG1/G1 phase. Kumar et al. [57] synthesized pyrazole-arylethanone derivatives under green conditions and evaluated them for cytotoxicity on HepG2, HCT116, and CDK2 and EGFR cell lines. Compound 36 showed cytotoxicity to all cell lines except SKOV3. On CDK2 and EGFR cell lines, compound 36 and 37 presented a similar potential in comparison to carboplatin. Pyrazole carboxamide derivatives were prepared by Lu et al., under moderate reaction conditions and assessed for anticancer activity. DNA binding interaction and kinase inhibition were examined to understand the mechanisms [58] (Refer to Table 4 for the structure of the compound). The anticancer assessment displayed good inhibition against tHCT116 and HepG2 and decreased inhibition of kinase by compound 38 with a supreme binding affinity towards DNA (Kpym-5) = 1.06 × 105 M−1). Zhang et al., reported pyrazole-5-carboxamide-containing derivatives and assessed them for anticancer potential against A549 cell lines [59]. Compound 39 yielded significant inhibition at a concentration of 10 µM. Yao et al., prepared pyrazole derivatives showing disubstitution at 1 and 3 and evaluated their anticancer potential against BGC823, Hela, MCF7, A549, Molt-4, U937, K562, and HT1080 [60] (Refer to Table 4 for the structure of the compound). Compound 40 presented acceptable selectivity and anticancer potential against class I and IIb HDAC isoforms. Wen et al., prepared pyrazole-containing derivatives and evaluated for histone deacetylase inhibition. Compound 41 confirmed noteworthy antitumor potential in a xenografted model of HCT-116 [61]. Zhao et al., developed the excellent pyrazole derivative 42, which showed its anticancer potential with IC50 = 0.12 µM and 0.16 µM on WM 266.4 and MCF-7 cell lines [62] (Refer to Table 4 for the structure of the compound). Kamal et al., reported a novel series of pyrazole derivatives, among which compound 43 displayed noteworthy cytotoxicity [63]. Hu et al., synthesized some novel compounds, in which compound 44 showed considerable inhibition for BCR-Bal kinase (IC50 = 14.2 nM) [64] (Refer to Table 4 for the structure of the compound). Nitulescu et al., prepared novel pyrazole-containing derivatives and investigated them for anticancer potency. Compound 45 appeared as a potential anticancer agent [65] (Refer to Table 4 for the structure of the compound). Newer substituted pyrazole derivatives were prepared by Rai et al., and assessed for anticancer activity. Compound 46 exhibited equivalent anticancer potential compared to rug doxorubicin [24] (Refer to Table 4 for the structure of the compound). Nitulescu et al., performed ultrasound-assisted synthesis of pyrazole derivatives and evaluated them for anticancer potential. Compound 47 appeared as a strong and potential anticancer agent [66] (Refer to Table 4 for the structure of the compound).
Table 4.
Representative compound structures 31 to 47 and their anticancer activity.
Khloya et al., developed sulfonamide derivative 48, which was found to cause better inhibition of the carbonic anhydrase enzyme against acetazolamide compared to the standard drug [67]. Tao et al., synthesized pyrazole derivatives in which compound 49 appeared to be the most significant anticancer compound against EGFR and HER-2 tyrosine kinase with IC50 values of 0.26 µM and 0.20 µM [68] (Refer to Table 5 for the structure of the compound). Reddy et al., designed some pyrazole-containing derivatives and evaluated them for anticancer activity. Compound 50 prompted significant inhibition against MCF-7, A549, and HeLa with IC50 values between 0.83–1.81 µM [69] (Refer to Table 5 for the structure of the compound). Pyrazole–oxindole conjugates were developed and evaluated for anticancer potential and tubulin polymerization inhibition by Kamal et al., among which compound 51 significantly inhibited tubulin assembly [70] (Refer to Table 5 for the structure of the compound). Shi et al., prepared novel pyrazole derivatives of which compound 52 appeared as a potential anticancer agent [71]. Ali et al., prepared imidazo[2,1-b] thiazoles-linked pyrazole derivatives and evaluated them for antitumor potency. In vitro results revealed the potential of compound 53 against renal UO-31 and CNS SNB-75 cell lines [72] (Refer to Table 5 for the structure of the compound). Reddy et al., designed and synthesized 1H-pyrazolo-[4,3-d]pyrimidin-7(6H)-ones and assessed it for anticancer potential. Compound 54 showed potential anticancer results against HeLa, CAKI-I, PC-3, MiaPaca-2, and A549 cancer cell lines [73] (Refer to Table 5 for the structure of the compound). Zhang et al., reported on pyrazolyl hydroxamic acid derivatives and assessed their potential against A549 cancer cell lines to determine their anti-proliferative activity, in which compound 55 appeared as the most potent compound [59]. Imidazo[1,2-b] pyrazole derivatives were prepared by Grosse et al., and evaluated through MTT assay. Compound 56 showed significant inhibition with IC50 below 5 µM against human and murine cancer cell lines [74]. Pirol et al., designed and prepared 5-(p-tolyl)-1-(quinolin-2-yl)pyrazole-3-carboxylic-acid-based amide derivatives and assessed them for anticancer potential. Compound 57 exhibited the most potential [56]. Jin et al., designed pyrazole-based 4-([1, 2, 4]triazolo[1,5-a]pyridine-6-yl)-5(3)-(6-methylpyridin-2-yl)imidazole derivatives and evaluated them for ALK5 inhibition, of which compound 58 yielded the most significant results [75]. Park et al., prepared trisubstituted pyrazole-containing derivatives and assessed them for ROS1 kinase inhibition. Compound 59 exhibited five times greater potential against crizotinib [76] (Refer to Table 5 for the structure of the compound). Wang et al., prepared 5-phenyl-1H-pyrazole analogues encompassing a niacinamide ring and assessed them for antiproliferative potential. Compound 60 reflected potential cytotoxicity with IC50 = 2.63 µM and 3.16 µM against WM 266.64 and A375 [77]. Kamal et al., developed 4b-amidopodophyllotoxin heteroaromatic conjugates and assessed them for anticancer potential. Compound 61 revealed excellent antitumor potential against A549 cell lines [78]. Novel pyrazole-linked carbothioamide derivatives were prepared by Zhu et al., and assessed for anticancer potential. Compound 62 exhibited the most cytotoxic potential with IC50 = 6.51 µM against Raji cell lines [79]. Zheng et al., developed pyrazole-linked benzimidazole conjugates and assessed them for Aurora A/B kinase inhibition potential and anticancer activity. Compound 63 displayed significant inhibition [50]. Compound 64 was prepared by Miyamoto et al., through hybridization and optimization of different imidazo[1,2-b]pyridazines, which caused the inhibition of VEGFR2 kinase with IC50 = 0.95 nM. Furthermore, it inhibited VEGF-induced proliferation of human umbilical vein endothelial cells with IC50 = 0.30 nM [80] (Refer to Table 5 for the structure of the compound).
Table 5.
Representative compound structures 48 to 64 and their anticancer activity.
According to the molecular modeling methodology, Bavetsias et al., reported on imidazo[4,5-b] pyridine linked pyrazole derivatives and assessed them for selective Aurora-A kinase inhibition. Compound 65 caused inhibition of Aurora-A kinase with IC50 = 0.067 µM [81] (Refer to Table 6 for the structure of the compound). Sun et al., synthesized pyrazole-linked thiourea derivatives and explored them for CDK2 inhibition. Compound 66 appeared to have the most potential as a CDK2 inhibitor with IC50 = 25 nM, as well as inhibited proliferation of H460, MCF-F, and A549 in the range of 0.75–4.21 µM [47]. Li et al., prepared N-1,3-triphenyl-1H-pyrazole-4-carboxamide derivatives and assessed them for anticancer potential and binding against Aurora-A kinase. Compound 67 showed significant inhibition for HCT116 (IC50 = 0.39 ± 0.06 µM) and MCF-7 (0.46 ± 0.04 µM) cell lines and IC50 = 00.16 ± 0.03 µM against Aurora-A kinase [44] (Refer to Table 6 for the structure of the compound). Liu et al., prepared pyrazole-linked peptidomimetics, in which compound 68 reflected the potential inhibition of A549 cell lines with IC50 = 36.12 µM [82]. Lv et al., synthesized 5-benzyl-2-phenylpyrazolo[1,5-a]pyrazin-4,6(5H, 7H)-dione derivatives, in which compound 69 selectively inhibited H322 cell lines, along with the induction of apoptosis [83] (Refer to Table 6 for the structure of the compound). Bai et al., developed novel 1-acyl-3-amino-1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole derivatives and assessed them for antitumor potential against the HCT-116 cancer cell line, in which compound 70 showed most the potent results and was further designated for future cytotoxicity estimation against many other cancer cell lines. This compound transcended (R)-roscovitine by 4–28 times in terms of its potency. Moreover, this compound was also screened against twelve kinases and its interaction with CDK5 and glycogen synthase kinase-3b [84] (Refer to Table 6 for the structure of the compound). Newer pyrazolo[3,4-d]pyrimide-containing derivatives were prepared by Huang et al., where compound 71 displayed cytotoxicity against NCI-H226 and NPC-TW01 cell lines with GI50 = 18–30 µM [85] (Refer to Table 6 for the structure of the compound). Strocchi et al., developed pyrazole analogues and assessed them against HCC and SNU449 cancer cell lines. Compound 72 appeared most potent with IC50 = 50–100 µM and effectively blocked cell cycle progression and induced apoptosis [86]. Metwally et al., reported pyrazole and pyrazolotriazine derivatives and tested them for anticancer potential, in which compound 73 exhibited significant results [87]. Novel farnesyltransferase inhibitors containing phenothiazine were developed by Baciu-Atudosie et al., and determined against NCI-60 cancer cell lines. Compound 74 revealed significant cytostatic activity against HCT-116, SKMEL-5, and LOX IMVI cancer cell lines [88]. Bondock et al., prepared hybrid 1,3,4-oxadiazole derivatives and assessed them for anticancer potential. Compound 75 appeared to be the most potent against four cell lines [89]. Deslandes et al., prepared garnulatimide- and isogranulatimide-containing pyrazole derivatives and evaluated their anticancer activity, in which compound 76 displayed the highest potential results with an IC50 value of 11 ± 4 µM [90] (Refer to Table 6 for the structure of the compound). Puthiyapurayil et al., prepared novel S-substituted-1,3,4-oxadiazole linked N-methyl-4-(trifluoromethyl)phenylpyrazole derivatives combinatorial library and evaluated them for cytotoxic activity through an MTT assay. Compound 77 showed potential anti-proliferative results for MCF-7 cell lines with IC50 = 15.54 µM [91] (Refer to Table 6 for the structure of the compound). El-borai et al., prepared pyrazolopyridine derivatives through microwave irradiation and evaluated them for antitumor activity, in which compound 78 showed the maximum potency [92] (Refer to Table 6 for the structure of the compound). Ferrocenyl pyrazole-linked chiral aminoethanol derivatives were reported by Shen et al., and assessed for H322 lung and A549 cancer cell line inhibition, in which compound 79 displayed the maximum anticancer potential [93]. Abd El Hamid et al., prepared novel pyrazolopyrimidine derivatives, where compound 80 yielded the most significant anticancer activity with an IC50 value of 7.5 nM [94]. Vujasinovic et al., prepared pyrazole derivatives from different routes, among which compound 81 revealed maximum potency with a significant safety profile [95] (Refer to Table 6 for the structure of the compound).
Table 6.
Representative compound structures 65 to 81 and their anticancer activity.
Yamamoto et al., synthesized newer pyrazole containing derivatives and investigated them for anticancer potential. Compound 82 showed most the potential antiproliferative results against a CRPC model of LNCap-hr cell lines [96] (Refer to Table 7 for the structure of the compound). Newly synthesized pyrazole-based imidazopyridine derivatives were determined for antitumor potential by Bavetsias et al., in which compound 83 caused the most significant Aurora kinases inhibition [97]. Hanan et al., prepared newer pyrazolopyrimidine derivatives and assessed them for inhibition of Janus Kinase. Compound 84 appeared to be the derivative with the most potential with an IC50 = 7.4 nM [98]. Newhouse et al., prepared several non-Oxime containing pyrazole derivatives and assessed their anticancer activity. Compound 85 demonstrated appreciable B-Raf kinase inhibition [99] (Refer to Table 7 for the structure of the compound). Newer pyrazole derivatives were prepared by Zheng et al., and evaluated for anticancer activity. Compound 86 potentially inhibited H322 carcinoma and A549 cancer cell lines [100]. Jin et al., prepared 1-substituted-3-(6-methylpyridin-2-yl)-4-([1,2,4]triazolo[1,5-a]pyridin-6-yl)pyrazole derivatives and assessed them for cytotoxic potential. Compound 87 appeared to be the derivative with the most potential with IC50 = 0.57 nM [101]. Pyrazole linked molecules were prepared by Maggio et al., and investigated for antiproliferative activity. Compound 88 appeared to be the most significant anticancer agent with growth inhibition (GI50) ranging from 14–18 µM against HL60 and K562 [102]. Christodoulou et al., prepared trisubstituted pyrazole derivatives and assessed them for anti-angiogenic potential. Compound 89 appeared to be the most potent with IC50 = 12 μM for 89a and 25 ± 1.4 μM for 89b and inhibited MCF-7 with IC50 = 26 ± 2.2 μM and Hela with IC50 = 37 ± 2.6 μM carcinoma cells in vitro [103]. Lv et al., prepared pyrazole derivatives and assessed them for EGFR kinase inhibition. Compound 90 appeared most potential with IC50 of 0.07 μM, in comparison to erlotinib [104] (Refer to Table 7 for the structure of the compound). Niculescu-Duvaz et al., reported novel 1-arylmethyl-3-aryl-1H-pyrazole-5-carbohydrazide derivatives and assessed them for A549 cell line inhibition and induction of apoptosis. Compound 91 was found to be the most potent with significant logP (3.12–4.94) values [105] (Refer to Table 7 for the structure of the compound). Zheng et al., synthesized 3-aryl-1-(4-tert-butylbenzyl)-1H-pyrazole-5-carbohydrazide hydrazone derivatives and assessed them for A549 cell inhibition. Compound 92 exhibited the maximum inhibition of lung cancer cell lines [35]. Newer 3, 4-disubstituted pyrazole derivatives were prepared by Lin et al., and assessed for cyclin-dependent kinase inhibitory and anticancer activity on human cancerous cells [106]. Compound 93 was found to be the most effective. A novel series of 7-phenyl-7H-pyrazolo[4,3-e]-[1,2,4,]triazolo[1,5-c]pyrimidine-2-thione derivatives was prepared by Ghorab et. al. and assessed for Ehrlich Ascites Carcinoma cell lines against doxorubicin. Compound 94 yielded the most potent results [107]. Radek et al., reported pyrazolo[4,3-d]pyrimidine derivatives and assessed them for CDK2 inhibition, in which compound 95 appeared to be the most potent [108]. Dawood et al., prepared 2-(4-(pyrazol-4-yl)thiazol-2-ylimino)-1,3,4-thiadiazole derivatives, where compound 96 appeared as the best potential antiproliferative agent [109]. Singla et al., prepared newer pyrazolo[3,4-d]pyrimidine linked 4-(1H-benzimidazol-2-yl)-phenylamine derivatives and assessed their antitumor potency against NCI-60 cancer cell lines. Compound 97 exhibited the best potential inhibition at 10 µM concentration with average GI50 values of 1.30 μM [110]. Krystof et al., prepared 4-arylazo-3,5-diamino-1H-pyrazole derivatives and investigated them for anticancer activity and CDK inhibition. Compound 98 appeared as the best potential derivative with significant selectivity and cellular effects [111] (Refer to Table 7 for the structure of the compound). Balbi et al., developed pyrazole scaffolds and assessed them against A2780 cells, A549 cells, and P388 cell lines. Compound 99 significantly bound to α- and β-tubulin and caused molecular distortion, leading to disassembly of the microtubules [112] (Refer to Table 7 for the structure of the compound).
Table 7.
Representative compound structures 82 to 99 and their anticancer activity.
Raquib et al., synthesized ethyl-4-(3-(aryl)-1-phenyl-1H-pyrazol-4-yl)-2-Oxo-6-(pyridin-3-yl)cyclohex-3-enecarboxylate derivatives and evaluated them for topoisomerase-IIa inhibition and anticancer potential against MCF-7, NCI-H460, HeLa, and HEK-293T. Compound 100 showed 70.82% topoisomerase-IIa inhibition (100 µM) and significant anticancer potential against cancer cell lines with IC50 values of 7.01 ± 0.60 µM (HeLa), 8.55 ± 0.35 µM (NCIH460), and 14.31 ± 0.90 µM (MCF-7) [113] (Refer to Table 8 for the structure of the compound). Wang et al., prepared indolyl-substituted 1,4,6,7-tetrahydropyrano [4,3-c] pyrazole derivatives and assessed them for anticancer activity by MTT assay on MCF-7, HGC-27, PC-3, and EC-109 cell lines. Compound 101 displayed three times more activity than 5-fluorouracil [114]. Minu et al., reported mono- and di-substituted 2,3,3a,4,5,6-hexahydrocyclopenta[c]pyrazole derivatives and assessed them for anticancer potential on MCF-7 and A589 cell lines. Compound 102 displayed the best potential results with IC50 = 15.6 µM and 19.8 µM against MCF-7 and A589 cell lines [115]. Hafez et al., synthesized [4-amino-3-(4-chlorophenyl)-1H-pyrazol-5-yl](3,5-dimethyl-1H-pyrazol-1-yl)-methanone derivatives and assessed them for anticancer potential. Compounds 103 showed greater anticancer potential activity than doxorubicin as the standard drug [116] (Refer to Table 8 for the structure of the compound). Lv et al., synthesized newer (E)-1,3-diphenyl-1H-pyrazole-containing derivatives bearing the O-benzyl Oxime group and evaluated them for immunosuppression. Compound 104 displayed the best potential inhibition with IC50 = 1.18 µM for lymph node cells and 0.28 µM for PI3K [117] (Refer to Table 8 for the structure of the compound). Reddy et al., developed (N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1,3-diphenyl-1H-pyrazole-4-carboxamide derivatives and investigated them for CDK (I/II) inhibition. Compound 105 exhibited significant inhibition on MIAPaCa-2, MCF-7, and HeLa cell lines in the range of 0.13–0.7 µM (GI50) against nocodazole (0.81–0.95 µM) [118]. Yoon et al., reported a new and specific RET inhibitor as 5-aminopyrazole-4-carboxamide lead molecules to improve the metabolic stability of the pyrazolopyrimidines. Compound 106 revealed potent results against gatekeeper mutant (IC50 = 252 nM) and wild-type RET (IC50 = 44 nM) [119] (Refer to Table 8 for the structure of the compound). Thaher et al., reported 3-(4-fluorophenyl)-4-(pyridin-4-yl)-1-(aryl)-1H-pyrazol-5-amine as p38α inhibitors, of which compound 107 showed the maximum potential with IC50 in nanomolar range on B-Raf V600E, Src, B-Raf wt, VEGFR-2, and EGFRs, establishing it as an upright lead for novel anticancer potential [120]. Al-Suwaidan et al., prepared 1-(4-chlorophenyl)-4,4,4-trifluoro-2-(2-(4-methoxyphenyl)hydrazone)butane-1,3-dione derivatives and assessed them for in vitro antitumor activity. Compound 108 showed potential results with a GI50 value of 6.61 µM, TGI value of 42.66 µM, and LC50 value of 93.33 µM in comparison to 5-fluorouracil [121] (Refer to Table 8 for the structure of the compound). Pyrazoline-based hydroxamic acid derivatives were synthesized and assessed for anticancer potential. Compound 109 was recognized from a library of derivatives and showed a significant inhibitory effect against cancer cell types. When compared to side chains in the meta position, hydroxamate side chains in the ortho position exhibited greater biological activity. When compared to the reference drug Bestatin (IC50 > 500 μM against all cancer cell lines), compound 109 substituted with formamide demonstrated potent anti-proliferative activity (IC50 = 54.2 ± 4.9 μM, 116.5 ± 8.2 μM, 18.7 ± 1.6 μM, 156.0 ± 7.5 μM, 65.0 ± 2.8 μM, and 65.0 ± 2.8 μM against U93, PLC/PRF/5, K562, ES-2, PC-3, and HepG2). K562 was the most susceptible to pyrazoline-based compounds among all these cell lines. Additionally, compound 109 inhibited aminopeptidase N (APN) with IC50 = 0.16 ± 0.02 μM, which was higher than one order of scale lower than bestatin (IC50 = 9.4 ± 0.5 μM) [122] (Refer to Table 8 for the structure of the compound). Furthermore, 1,2,4-oxadiazole merged 1,2,3-triazole-pyrazole derivatives were prepared and tested by MTT assay against DU-145, PC3, MCF-7, and A549 cancer lines using etoposide as the standard drug. The anticancer activity of compound 110 (with the 3,5-dinitro group on the phenyl ring) was found to be the strongest, with IC50 = 0.01 ± 0.008 µM, 0.45 ± 0.023 µM, 0.081 ± 0.0012 µM, and 1.77 ± 0.33 µM against PC3, A549, MCF-7, and DU-145 cell lines. Compared to etoposide (IC50 = 1.36 ± 0.22 µM, 0.11 ± 0.066 µM, 0.13 ± 0.072 µM, and 1.50 ± 0.19 µM against A549, MCF-7, PC-3, and DU-145), the compound substituted with 4-nitro substituent on the phenyl ring connected to the oxadiazole skeleton showed decent anticancer activity (IC50 = 3.08 ± 0.135 µM, 1.97 ± 0.45 µM, 2.39 ± 1.56 µM, 2.11 ± 0.024 µM against A549, DU-145, PC-3, and MCF-7, respectively). Substitution of the 3,5-dinitro group with 4-chloro, 3,5-dimethoxy, or 4-methoxy group resulted in decreased activity; however, the replacement of the 3,5-dinitro group with a 4-Bromo, electron-rich group (3,4,5-trimethoxy) resulted in reasonable anticancer activity [123] (Refer to Table 8 for the structure of the compound). Pyrazoles incorporating thiazole derivatives were synthesized, and their bioactivity as a target for leukemia cancer was investigated. The most active compounds were pyrazole derivatives, with rising activity seen for pyrazole derivatives including a thiazole ring. Compound 111 with N-(3-methoxy-2-hydroxybenzal)-3-substituted(p-chlorophenyl)-4-cyano-5-oxopyrazol-1-thiocarboxamide against HL-60 was the most effective, with an IC50 = 1.35 µM, in comparison to Dox’s with IC50 = 2.02 µM. When compared with the untreated control, compound 111 enhanced the G2/M phase from 11.05% to 39.22%. Furthermore, in comparison to the untreated control, compound 111 can enhance the proportion of late apoptosis from 0.31 to 4.31%. With an IC50 value of 56.04 nM, compound 111 inhibited Topo II activity more effectively than etoposide (IC50 = 41.11 nM). The cytotoxic action of compound 111 is mostly attributable to its high DNA Topo II inhibition activity, according to this finding [124] (Refer to Table 8 for the structure of the compound). Novel pyrazole–pyrazoline-hybrid-containing derivatives were prepared and assessed as cancer-associated selective COX-2 inhibitors. Compound 112 indeed had a greater COX-2 selectivity index, as well as strong anticancer potential towards A549, SiHa, HepG2, and COLO205, (IC50 = 4.94 ± 0.05 µM, 4.94 ± 0.05 µM, 2.09 ± 0.01 µM, and 4.86 ± 0.01 µM) compared to 5-fluorouracil as a standard drug (IC50 = 2.08 ± 0.01 µM, 5.78 ± 0.04 µM, 4.35 ± 0.01 µM, 4.00 ± 0.01 µM, 19.01 ± 0.21 µM, respectively, against MCF-7, SiHa, A549, and COLO205, HepG2). The compound with no substitutions on ring B was revealed as being the most active (compound 112). The activity reduced as the electronegativity of ring B increased. Increases in electronegativity on ring B and electro positivity on ring C were observed to have a positive influence on activity. The study found that methoxy-containing compounds were more suited for in vitro anticancer activities (compound 112). The activity reduced as the electronegativity of ring-C rose. Because the compounds were more specific for the COX2 enzyme, it was possible to conclude that they showed anticancer effects via COX2 inhibition [125] (Refer to Table 8 for the structure of the compound).
Table 8.
Representative compound structures 100 to 112 and their anticancer activity.
3. Pyrazole Biomolecules as Inflammation Therapeutics
Inflammation is a physiological response to destructive stimuli encompassing blood vessels, immune cells, and chemical mediators (TNF-α, NO, IL-6) [126,127]. Pain is stressful and a horrible response due to injury in tissues or organs [128,129]. NSAIDs are a significant group of drug molecules recommended to cope with inflammatory conditions and to mitigate pain [130,131]. Nevertheless, excessive usage of NSAIDs frequently results in numerous adverse conditions such as gastrointestinal mucosal damage, renal toxicity, intolerance, and hepatotoxicity [132,133]. Future advancement/improvement of the therapeutic potential of anti-inflammatory and analgesic drugs (NSAIDs) lacking unwanted effects is still to be revealed by pharmaceutical scientists [134,135]. That is why several pyrazole bearing agents have also been established by chemists and druggists. Balbi et al., synthesized 5-(2,6,6-trimethyl-2-cyclohexen-1-yl)ethenyl-1H-pyrazolein derivatives, among them, compound 113 appeared to be a potent inhibitor of neutrophil chemotactic responsiveness in comparison to diclofenac [136] (Refer to Table 9 for the structure of the compound). Hall et al., developed methylene linked with pyrazoles, in which compound 114 appeared as a potent EP1 receptor antagonist [137]. Bandgar et al., prepared diaryl pyrazole derivatives and assessed them for anti-inflammatory potential against TNF-α and IL-6. Compound 115 showed IL-6 inhibition (42% at 10 μM) compared to flavopiridol (72–87% inhibition at 0.5 μM) and dexamethasone (85% inhibition at 1 μM) [138] (Refer to Table 9 for the structure of the compound). Abdel-Hafez et al., reported pyrazole-NO hybrid derivatives and assessed them for release of NO and their anti-inflammatory potential. Compound 116 displayed the maximum NO release and significant anti-inflammatory potential in comparison to indomethacin. [139] (Refer to Table 9 for the structure of the compound). Furthermore, 3-(4-methoxyphenyl)-N-propyl-5-(1H-pyrrol-2-yl)-1H-pyrazole-1-carbothioamide derivatives were developed by Gokhan-Kelekci et al., Compound 117 exhibited anti-inflammatory and analgesic potential [140]. Bis(3-aryl-4,5-dihydro-1H-pyrazole-1-thiocarboxamides) derivatives were prepared by Barsoum et al., and assessed for anti-inflammatory activity. Compound 118 appeared to be the best potential derivative in comparison to indomethacin [141]. Chandra et al., reported newer pyrazole derivatives and determined their anti-inflammatory and analgesic potential. Compound 119 showed significant results at three graded doses (25 mg/kg, 50 mg/kg, and 100 mg/kg p.o.) [142]. Ruiz et al., prepared (Z)-3-(2-(argiodiazenyl)phenyl)-N-((4-(5-(p-tolyl)-3-(trifluoromethyl)-1H-pyrazol-1yl)phenyl)sulfonyl)propanamide derivatives (120) and assessed them for inhibition of COX-2 compared to celecoxib [143]. Burguete et al., synthesized newer pyrazole derivatives and assessed them for anti-inflammatory potential, in which compound 121 showed significant anti-inflammatory potential [144] (Refer to Table 9 for the structure of the compound). Sandeep et al., prepared 3-substituted-1-aryl-5-phenyl-6-anilino-pyrazolo[3,4-d]pyrimidin-4-one derivatives and tested them for anti-inflammatory potential. Compound 122 revealed significant edema inhibition of 35–39 after 3 h of administering synthesized derivatives compared to diclofenac sodium and celecoxib (25 mg/kg). [145]. Hamdy et al., prepared pyrazolopyridine derivatives, among which compound 123 appeared to be a potent anti-inflammatory inhibitor [146]. Sharath et al., carried out the synthesis of newer indole-linked pyrazole derivatives and evaluated them for anti-inflammatory potential, in which compound 124 appeared as the most active derivative [147]. Thore et al., reported ethyl-5-amino-3-methylthio-1H-pyrazole-4-carboxylate derivatives and evaluated them for analgesic and anti-inflammatory properties via the writhing test. Compound 125 showed good analgesic and anti-inflammatory potential (25 mg/kg) with 0.9–1.12 ulcerogenic index in comparison to diclofenac sodium, which showed ulcerogenic index values of 3.10 [148]. Bandgar et al., prepared 1-methyl-5-(2,4,6-trimethoxyphenyl)-1H-pyrazole derivatives and assessed them for anti-inflammatory potential. Compound 126 appeared to be the most potent. [149]. Keche et al., prepared 1-acetyl-3-(3,4-dimethoxyphenyl)-5-(4-(3-(arylureido/arylthioureido/arylsulfonamid o)phenyl)-4,5-dihydropyrazole derivatives, in which compound 127 reflected the highest potential inhibition of inflammation in comparison to diclofenac sodium [150] (Refer to Table 9 for the structure of the compound). Selvam et al., prepared 1-(4-substitutedphenyl)-3-phenyl-1H-pyrazole-4-carbaldehyde derivatives and evaluated them for anti-inflammatory potential, among which 128 yielded excellent results [151]. El-Sayed et al., prepared pyrazole derivatives, among which compound 129 showed optimum COX-2 inhibition (IC50 = 0.26 µM and SI = 192.3) compared to celecoxib (IC50 = 0.28 µM and SI = 178.57) [127] (Refer to Table 9 for the structure of the compound).
Table 9.
Representative compound structures 113 to 129 and their anti-inflammatory activity.
Nagarapu et al., reported 3-phenyl-N-[3-(4-phenylpiperazin-1yl)propyl]-1H-pyrazole-5-carboxamide derivatives, among which compound 130 appeared to be the best potential anti-inflammatory derivative [152] (Refer to Table 10 for the structure of the compound). Bandgar et al., developed 3,5-diaryl pyrazole derivatives, in which compound 131 revealed excellent inhibition of inflammation [138] (Refer to Table 10 for the structure of the compound). Barsoum et al., synthesized bis(3-aryl-4,5-dihydro-1H-pyrazole-1-carboxamide) derivatives and evaluated them for inhibition of inflammation, in which compound 132 appeared as the best potential derivative compared to indomethacin [141] (Refer to Table 10 for the structure of the compound). Burguete et al., reported newer pyrazole derivatives and assessed them for anti-inflammatory activity. Compound 133 displayed good inhibition of inflammation [144]. Bekhit et al., prepared 1H-pyrazole scaffolds and assessed them for anti-inflammatory and analgesic potential. Many derivatives displayed inhibition of inflammation with reduced ulcerogenic potential. Compound 134 showed significant and selective COX-2 inhibition in comparison to indomethacin [153] (Refer to Table 10 for the structure of the compound). Amir et al., reported anti-inflammatory potential of 3,5-dimethylpyrazole derivatives and 3-methylpyrazole-5-one derivatives. Compounds 135a and 135b showed significant anti-inflammatory, analgesic, lipid peroxidation, and ulcerogenic activities. [154] (Refer to Table 10 for the structure of the compound). Furthermore, 4-[((1-(2-hydroxyethyl)-3,5-dimethylpyrazole-4-yl)azo)phenyl]-4-(2-phenethyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione derivatives were prepared and assessed for analgesic effect by Oruc et al using morphine as the standard drug. Compound 136 yielded the most potent results [155]. Bekhit et al., prepared derivatives containing the pyrazolyl benzene sulfonamide moiety and assessed them for anti-inflammatory activity. Compound 137 displayed significant results in comparison to indomethacin [156] (Refer to Table 10 for the structure of the compound). Chowdhury et al., prepared newer 4-[2-(4-methyl(amino)sulfonylphenyl)-trifluoromethyl-2H-pyrazol-3-yl]-1,2,3,6-tetrahydropyri -dine derivatives and inspected them for inhibition of inflammation. The outcomes showed that compound 138 exhibited the maximum inhibition of inflammation in comparison to celecoxib and aspirin [157] (Refer to Table 10 for the structure of the compound). Abdel-Hafez et al., prepared pyrazole-NO hybrids and assessed them for potential inhibition of inflammation. Compound 139 appeared potent in comparison to indomethacin [139]. El-Din et al., developed newer pyrazole derivatives and assessed them for anti-inflammatory potential [158]. Compound 140 presented the best potential outcomes with minimal ulcerogenicity without affecting renal function. Bekhit et al., reported 4-thiazolylpyrazolyl derivatives and tested them for inhibition of inflammation, among which compound 141 displayed significant inhibition and was considered as a potential COX-2 inhibitor [159]. Youssef et al., reported dipyrazoleethandiamides and cyano-1-phenyl-1H-pyrazol-5-yl]acetamide derivatives, of which compound 142 showed significant anti-inflammatory activities [160] (Refer to Table 10 for the structure of the compound). A novel class of 3-phenyl-N-[3-(4-phenylpiperazine-1-yl)propyl]-1H-pyrazole-5-carboxamide derivatives were prepared by Nagarapu et al., in which compound 143 yielded excellent results [152]. Shrivastava et al., prepared pyrazole containing 1,5-diaryl derivatives and evaluated them for anti-inflammatory potential. Compound 144 demonstrated the significant inhibition of inflammation in comparison to control and standard [161]. N-(substitutedaryl/alkylcarbamothioyl)-4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide compounds were prepared and investigated by Kucukguzel et al., for anti-inflammatory potential. The outcomes suggested that compound 145 revealed potential activity without causing any cell damage to the liver, kidney, and brain in comparison to celecoxib [162]. Calis et al., reported pyrazole-3(5)-carboxylic acid derivatives and tested them for anti-inflammatory potential. Compound 146 was designated with the highest anti-inflammatory potential compared to indomethacin and aspirin as standard drugs [163] (Refer to Table 10 for the structure of the compound).
Table 10.
Representative compound structures 130 to 146 and their anti-inflammatory activity.
Chavan et al., described novel pyrazole amalgamated flavones and assessed them for inhibition of inflammation. Compound 147, the 1H-pyrazole derivative with the 2,3,6-trimethoxyphenyl group, showed the best potency [164] (Refer to Table 11 for the structure of the compound). Baytas et al., prepared (E)-3-[3-(pyridin-4-yl)-1-phenyl-1H-pyrazole-4-yl]acrylamides and tested them for anti-platelet and anti-inflammatory activity [165]. Compound 148 revealed significant activity. Ragab et al., reported 1,3,4-trisubstitutedpyrazole containing derivatives and evaluated them for their anti-inflammatory and analgesic potential and ulcerogenicity [166] (Refer to Table 11 for the structure of the compound). Compounds 149, 150, and 151 showed good inhibition of inflammation and analgesic potential. They showed less selectivity for COX (I/II) but displayed good tolerance to GIT in comparison to phenylbutazone. Derivatives of pyrazole carboxamides bearing naproxen were developed by El-Sehemi et al., and assessed for anti-inflammatory potential. Compound 152 appeared to be the best potential derivative [167] (Refer to Table 11 for the structure of the compound). Hassan et al., prepared celecoxib analogues with a benzofuran ring and assessed them for COX (I/II) inhibitory bioassay. Among all these compounds, compound 153 displayed noteworthy inhibition of inflammation. The outcome exposed the vital role of derivatives containing C-3-pyridine-3-yl to exhibit better anti-inflammatory potential [6]. Kumar et al., reported pyrazolyl pyrazoline-containing derivatives and tested them for anti-inflammatory potential. Compound 154 appeared to have the most potential as an anti-inflammatory compound [168]. Mohammed et al., prepared hydrazone linked pyrazole derivatives. These compounds were evaluated for anti-inflammatory activity, blocking PGE2 production in blood serum of adult wistar rats, COX-1/2 inhibition, and ulcerogenicity. Compound 155 displayed significant anti-inflammatory efficacy compared to diclofenac, with no or minimal ulcerogenicity observed in comparison with indomethacin as a standard drug. The pyrazole derivatives demonstrated SI of 11.1 for COX (I/II) inhibition [169] (Refer to Table 11 for the structure of the compound). Pyrazole derivatives were prepared by Tewari et al., and investigated for anti-inflammatory potential and COX-2 inhibition [170]. Among these, compound 156 with IC50 = 0.44 µM and 157 with IC50 = 0.51 µM showed the most significant results compared to celecoxib with IC50 = 0.002 µM for COX-2 selectivity and paw volume of 1.46 and 1.11 compared to the reference drug nimesulide, which showed a paw volume of 1.26. Alegaon et al., prepared 1,3,4-trisubstituted pyrazole derivatives and assessed them for anti-inflammatory potential [171] (Refer to Table 11 for the structure of the compound). Compound 158 presented 64.6% inhibition of inflammation in comparison to diclofenac with 86.72% inhibition. Bansal et al. [172] prepared 1,3,4-oxadiazole-linked diarylpyrazole derivatives, in which compound 159 exhibited potential COX-2 inhibition with maximum selectivity compared to aspirin. Wang et al., synthesized pyrazole-linked benzamide derivatives and evaluated them for inverse agonistic activity on retinoic acid receptor Y. Compound 160 was identified as a potential lead molecule [173]. Newer pyrazole derivatives were prepared and evaluated for anti-inflammatory activity by Li et al.; among these, compound 161 exhibited better potency in comparison to indomethacin and ibuprofen [174] (Refer to Table 11 for the structure of the compound).
Table 11.
Representative compound structures 147 to 161 and their anti-inflammatory activity.
Wang et al., synthesized benzamide-containing pyrazole derivatives and investigated them for anti-inflammatory potential. Compound 162 showed potential anti-inflammatory results [173]. El-Fekyet al., carried out a series of quinoline-integrated pyrazole scaffolds and assessed them for anti-inflammatory property. Compound 163 revealed the maximum inhibition and more binding affinity for the COX-2 binding position [175] (Refer to Table 12 for the structure of the compound). Newer pyrazole-containing heterocycles were prepared by Kendre et al., and tested for analgesic potential. Scaffold 164 displayed good analgesic activity [176]. Kumar et al., synthesized novel pyrazole derivatives and identified anti-inflammatory activity. Compound 165 appeared to be the best potential inhibitor of inflammation [177]. Pelcman et al., prepared pyrazole carboxamide derivatives and screened them for inhibitory activity against 15-lipoxygenase. Scaffold 166 appeared to show significant anti-inflammatory potential with good % inhibition and analgesic activity [178] (Refer to Table 12 for the structure of the compound). Compound 167 was identified as the most potent and selective inhibitor of COX-2 with IC50 = 1.33 µM. Newer pyrazole-linked analogues were prepared and tested for analgesic potential by Abdellatif et al.; among these synthesized compounds, compound 168 demonstrated the highest anti-inflammatory action post carrageenan after 3 h of inflammation initiation (89% inhibition) as compared to the standard drug celecoxib (80% inhibition) [179] (Refer to Table 12 for the structure of the compound). Abdel-Aziz et al., prepared newer pyrazole derivatives, of which compound 169 exhibited remarkable anti-inflammatory potential with ED50 = 68 ± 2.2 mg/kg and 51 ± 0.7 mg/kg [180]. Doma et al., reported novel pyrazole analogues and evaluated them for anti-inflammatory activity using 0.1% carrageenan and injecting it into the subplantar region of the rat’s right paw. Derivative 170 was found to be the most significant member of the class [181]. Newer benzophenones were reported by Bandgar et al., and assessed for analgesic potential. Compound 171 showed significant inhibition of inflammation and analgesic activity [149] (Refer to Table 12 for the structure of the compound). Jadhav et al., prepared pyrazole derivatives and tested them for inhibition of inflammation. Compound 172 demonstrated the highest anti-inflammatory results when compared to diclofenac [182]. Vijesh et al., synthesized newer pyrazole-bearing 1,2,4-triazole and benzoxazole derivatives and assessed their anti-inflammatory potential. Compound 173, containing dichlorothiophene and triazole, was confirmed to have good analgesic and anti-inflammatory potential [183]. Aggarwal et al., reported many pyrazole-containing derivatives and investigated their analgesic efficacy. Compound 174 revealed noteworthy inhibition of inflammation (62–76%) in comparison to indomethacin (78%) [184] (Refer to Table 12 for the structure of the compound). Among the leads established by Yewale et al., compound 175 appeared as the most potent molecule with superior anti-inflammatory potential compared to diclofenac sodium and equivalent results against the standard drug celecoxib (dose 25 mg/kg) [145] (Refer to Table 12 for the structure of the compound).
Table 12.
Representative compound structures 162 to 175 and their anti-inflammatory activity.
Kamble et al., reported thiazole-bearing pyrazole derivatives and assessed them for anti-inflammatory potential. Compound 176 yielded the most effective results [185] (Refer to Table 13 for the structure of the compound). Chougala et al., prepared coumarin-bearing pyrano[2,3-c]pyrazole derivatives and screened them via an anti-inflammatory bioassay against protein denaturation and HRBC membrane stabilization model. Compound 177 exhibited the most potent anti-inflammatory results [186]. Somakala et al., reported newer pyrazolylurea derivatives and evaluated them for their p38α MAPK (mitogen-activated protein kinase) inhibition. Among these compounds, compound 178 showed p38α MAPK inhibition with IC50 values ranging 0.069 ± 0.07 mmol/L against SB203580 (IC50 = 0.043 ± 3.62 mmol/L). Compound 178 also indicated promising in vivo anti-inflammatory potency (80.93% inhibition) compared to diclofenac sodium (81.62% inhibition) [187]. Maddila et al., prepared 1,3,4-thiadiazole linked pyrazole-3-carboxamide derivatives and evaluated them for anti-inflammatory potential. Compound 179 showed significant inhibition of inflammation (77.27% and 81.00% at 3 h and 5 h) in comparison to indomethacin (74.82% and 80.32% at 3 h and 5 h) [188]. Abdellatif et al., synthesized 1,3,5-triaryl-4,5-dihydro-1H-pyrazole derivatives and assessed their inhibition of COX enzymes, anti-inflammatory potential, analgesic potential and ulcerogenicity. Compound 180 displayed significant anti-inflammatory potential with ED50 = 53.99 µmol/kg in comparison to celecoxib with ED50 = 82.15 µmol/kg, with lessen ulcer index (1.20 for compound 180 and 2.90 for celecoxib) [189]. Elshemya et al., prepared triazine-linked pyrazole derivatives and assessed them for inhibition of COX-2. Compound 181 displayed the best potential results with IC50 = 0.74 μM compared to celecoxib with IC50 = 0.78 μM [190] (Refer to Table 13 for the structure of the compound). Levent et al., reported diarylpyrazole-bearing carboxamide derivatives as newer antiplatelet drugs that interfere with platelet aggregation and inflammation (in cardiovascular diseases). Compound 182 revealed potential results with IC50 = 5.7–8.3 nM [191]. Unlu et al., prepared novel regioisomeric 1-(3-pyridazinyl)-5-arylpyrazole derivatives and evaluated them for anti-inflammatory and analgesic potential. Compound 183 displayed inhibitory potency for COX-I/II and can be a lead for future investigations [192] (Refer to Table 13 for the structure of the compound). A novel class of 1-phenylpyrazolo [3,4-d]pyrimidine derivatives were prepared by Bakr et al., and assessed for inhibition of COX, anti-inflammatory activity, and ulcerogenicity. Compound 184 displayed a higher edema inhibition of 34–68%; however, the in vivo active compound showed flexible ulceration (ulcer index = 0.33–4.0) compared to celecoxib (ulcer index = 0.33) [193]. Chandak et al., synthesized new 2-amino-substituted 4-coumarinylthiazoles-bearing benzenesulphonamide moieties and determined their anti-inflammatory potential. Compound 185 revealed promising anti-inflammatory activity (0.11 ± 0.16% IA) equivalent to the standard drug indomethacin (0.17 ± 0.03% IA) [194]. Faour et al., synthesized di-pyrazole comprising N-phenylpyrazole nucleus containing polysubstituted pyrazole moieties through different amide linkages and investigated them for inhibition of COX-2, LPS induced iNOS, and anti-inflammatory and analgesic activities. Compound 186 inhibited COX-2 with IC50 = 11.2 µM with 78% inhibition of inflammation, compared to 53% inhibition by diclofenac, and 54% protection against pain compared to 52% protection by diclofenac [195]. Alam et al., prepared hybrid pyrazole analogues, among which compound 187 exhibited anti-inflammatory potential with 80.63% inhibition after 4 h in comparison to ibuprofen (with 81.32% inhibition after 4 h and inhibition against COX-1/2 and TNF-α). The same compound showed high COX-2 inhibition, with half maximal inhibitory concentration of 1.79 μM compared to celecoxib [196] (Refer to Table 13 for the structure of the compound).
Table 13.
Representative compound structures 176 to 187 and their anti-inflammatory activity.
Compound 188 (AD732) was designed as a pyrazole derivative with anti-inflammatory and analgesic properties. Compound 188 (AD732) showed a strong inhibitory effect against COX-2 (IC50 = 0.57 ± 0.04 µM) rather than COX-1. Compound 188 (AD732) was compared to the standard celecoxib (IC50 = 0.26 ± 0.32 μM target on COX-1, IC50 = 2.62 ± 0.02 μM target on COX-2) and indomethacin medications (IC50 = >100.00 μM target on COX-1, IC50 = <0.30 μM target on COX-2) for anti-inflammatory action. In comparison to celecoxib, the COX-2 inhibitory activity of compound 188 (AD732) was less effective in vitro, suggesting that it may have less cardiovascular toxic effects. To summarize, compound 188 (AD732) appears to be a better and more efficient compound with intriguing promise for pain and inflammatory control. When treated rats were assessed 24 h following a single higher dose treatment, neither celecoxib nor compound 188 (AD732) caused stomach ulcers. The superior gastrointestinal safety of AD732 is based on its favorable inhibitory efficacy against COX-2 over COX-1 [197] (Refer to Table 14 for the structure of the compound). A novel pyrazole derivative was prepared and evaluated for inhibition of COX-1/2. The pyrazole ring’s bridging amide N generates a reasonably stable H-bond with Ser 530. Compounds 189(a), 189(b), 189(c), and 189(d) showed high activity and selectivity against COX-2 (IC50 = 39.43, 61.24, 38.73, and 39.14 nM). All compounds reported selectivity indices of 22.21, 14.35, 17.47, and 13.10, for 189(a), 189(b), 189(c), and 189(d), correspondingly. With IC50 = 34.21 nM, compound 189a replaced with acetamide morpholine appeared to be a potential COX-2 inhibitor. The IC50 values for compounds 189c and 189d substituted with fluoro and methoxy groups were 38.73 and 39.14 nM, correspondingly. In vivo, these compounds were superior to or equivalent to celecoxib as anti-inflammatory drugs. In silico, the same molecules exhibited comparable interactions to SC-588. Surprisingly, these drugs preferentially reduced the synthesis of the triggered microsomal PGE2 synthase [198] (Refer to Table 14 for the structure of the compound). Pyrazole and triazole compounds were synthesized, docking studies were performed, and their biological activity towards selective COX-2 inhibitors was investigated. Compound 190a, when substituted with sulfonamide on one N-aromatic ring, exhibited the highest inhibitory action against COX-2 with IC50 = 0.017 ± 0.001 μM and COX-1 with IC50 = 0.263 ± 0.016 μM. Compound 190b, which was replaced with nitro on the N-aromatic ring, inhibited COX-1 more effectively, with COX-1 (IC50 = 0.012 ± 0.001 μM). All compounds were compared with the other existing drug celecoxib, which demonstrated anti-inflammatory action towards COX-1 and COX-2 with IC50 = 1.479 ± 0.089 μM and 0.004 ± 0.000 μM, correspondingly. Because of its lower volume and far more optimal arrangement of the NO2 in same area, 190b was the most effective pyrazole-containing COX-1 inhibitor. The presence of a spacer in the inhibitor structure is important because it places the aromatic ring in a favorable location for hydrophobic interaction in COX-2. There are 2 aryl groups on the heterocyclic cores (pyrazole or triazole) that are not the same as those on most COX-2 inhibitors [199] (Refer to Table 14 for the structure of the compound).
Table 14.
Representative compound structures 188 to 191 and their anti-inflammatory activity.
Novel pyrazolo[3,4-d]pyridazinone-containing derivatives were developed and assessed against the alterations of discoidin domain receptor1 (DDR1) as anti-inflammatory agents. Compound 191 demonstrated strong inhibition of discoidin domain receptor1 (DDR1) with an IC50 value of 10.6 ± 1.9 nM and significant selectivity against 430 kinases. In a dextran sulphate sodium prompted animal colitis model, compound 191 potently reduced the production of pro-inflammatory cytokines and DDR1 auto phosphorylation in cells. The removal or relocation of the trifluoromethyl group in Compound 191 resulted in a significant loss of efficacy against the DDR1 outcome. The activity was significantly reduced when trifluoromethyl-substituted phenyl was replaced with n-butyl or cyclohexyl. To retain robust DDR1 inhibitory activity levels ranging from 27.4 to 60.4 nM, the acetylaminophenyl group at the R1 position might be substituted with phenyl or substituted phenyl. In mice’s bone-marrow-derived macrophages and human THP-11 generated macrophages, compound 191 effectively reduced LPS-induced production of these pro-inflammatory cytokines at 10 µM. Compound 191 suppressed basal autophosphorylation of discoidin domain receptor1 (DDR1) with an EC50 of 34.4 nM, which was more powerful than the positive control DDR1-IN-1, which had an EC50 of 114.5 nM [200].
4. Authors Perspective
The main goal of this revised study is to highlight the anticancer and anti-inflammatory profiles of the pyrazole molecule. Cancer is an uncontrollable abnormal cell proliferation that is a severe life-threatening disorder worldwide. Pharmaceutical firms are now investing heavily in developing and enhancing viable and safe anticancer treatments with minimal side effects. Pyrazole derivatives exhibit anti-inflammatory properties and have been shown to target several cancer cell lines, such as COX (I/II). The description includes a library of powerful compounds against cancer cell lines and inflammation. Among them, certain effective compounds and their SAR structures are strongly associated with biological activity and predicted some additional derivatives that approach structural alterations. We addressed the anticancer therapeutic action of compounds; all derivatives showed strong inhibitory activity, but certain powerful compounds demonstrated good inhibitory activity against cancer cell lines.
4.1. Pyrazole Biomolecules as Cancer Therapeutics
Compound 21 exhibited the most strong biological activity against HCT116, MCF-7, and Aurora-A kinase inhibitory activity cell lines, with IC50 values of 0.39 ± 0.06 µM, 0.46 ± 0.04 µM, and 0.16 ± 0.03 µM, which were equivalent to the positive control. The scaffolds of the known inhibitors of Aurora-A kinase can be categorized into four main groups, A–D. The scaffold in A contains 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole; the scaffold in B contains pyrrolo[2,3-b]pyrimidine; the scaffold in C contains quinoline; and the scaffold in D contains 2-anilino-diaminopyrimidine. Benzamide is a significant pharmacophore of natural compounds as well as a synthetic precursor for a variety of medications. Benzamide has been shown to have several pharmacological effects, including anti-inflammatory, anticancer, and anti-fungal properties. To create a more potent reaction with the Aurora-A kinase domains, a benzamide moiety was transferred to the pyrazole moiety to create the N phenyl-1H-pyrazole-4-carboxamide template. The highest activity was found for 21 compounds with a para-NO2 group on the A-rings and a para-OEt group on the B-rings [44] (refer to Figure 4 for Structure and SAR). In a separate investigation, compound 30 demonstrated a significant inhibitory effect against WM266.4 and A375 with IC50 values ranging from 1.50 to 1.32 µM. It makes use of a common scaffold comprised of a terminal aromatic group (phenyl or pyrazole) that occupies the allosteric pocket generated by the displacement of the DFG loop and an amide linker that links an aryl group in the hydrophobic pocket to a hinge-binding heterocycle. The use of an amide bond would allow for the quick and efficient screening of several candidate hinge-binding groups. The original molecule was modified to see how the substituents affected the BRAFV600E inhibitory effects. Urea derivatives have higher inhibitory effects than Schiff base compounds and related intermediate amines. Changes in substituents on the benzene rings had a minor impact on the activity of the compounds. In contrast, differences in the skeleton had a greater impact, which may explain why substituents on the benzene rings had a minor impact on the ability for molecular bonding between BRAFV600E and the compounds. According to the findings, the pyrazolyl core and the phenyl urea motif are critical in the inhibition of BRAFV600E [51] (refer to Figure 4 for Structure and SAR).
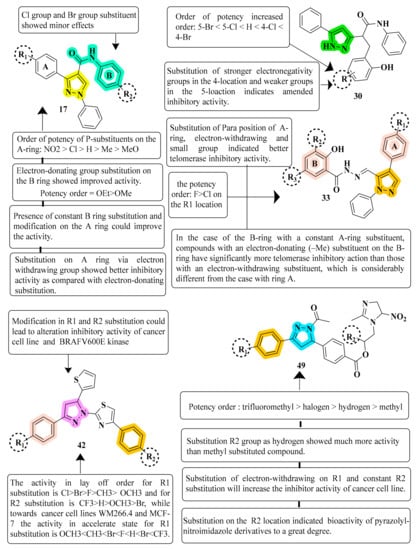
Figure 4.
Structure and SAR of potent anticancer activity compounds 17, 30, 33, 42, and 49.
Compounds with the OH and Me groups on the B-ring, compound 33, resulted in notable activity. Compound 33 displayed the most powerful effect, inhibiting the growth of MCF-7 and B16-F10 cells with IC50 values of 0.57 ± 0.03 and 0.49 ± 0.07 µM, respectively, and inhibiting the activity of telomerase with an IC50 of 1.9 ± 0.43 µM. More than one way to achieve powerful coordination may be achieved by using hydrazone derivatives and, specifically, acyl hydrazones. Acylhydrazone derivatives have the potential to form complexes with metal ions, such as Fe3+ and Ni2+, resulting in the inhibition of a physiologic reaction catalyzed by ions or the promotion of the absorption, transportation, distribution, and metabolism of drugs in the body, respectively. The F group outperformed the Cl group in antiproliferative action on the A ring, which might be attributable to the fluorine substituent’s greater lipophilicity and metabolic stability. The presence of fluorine often increases the solubility of lipids, which increases the rates of absorption and transport of drugs. Compound 33, based on its data structure, may be useful in designing and manufacturing stronger telomerase inhibitors [54] (refer to Figure 4 for Structure and SAR).
A series of novel BRAF-targeting pyrazole-based derivatives developed recently may prove useful against cancer. Some of them proved to have potential antitumor activity, particularly compound 42, which had an IC50 value of 0.12 µM against the cell line WM266.4 and 0.16 µM against the cell line MCF-7 and which also showed the most potential antiproliferative activity of apoptosis by flow cytometry experiments. Common scaffolds consist of a terminal aromatic group (phenyl or pyrazole) that fills the allosteric pocket. Thriophene and thiazole groups were demonstrated to have antibacterial, anti-inflammatory, and antiviral properties according to generalized research. The relevant compounds were formed by displacing the DFG loop and an amide linker and linking an aryl group in the hydrophobic pocket to a hinge-binding heterocycle. The introduction of an amide bond would enable the rapid and effective screening of different hinge-binding groups. Changes in substituents on the benzene thiazole rings had only a small effect on the compounds’ activity. As a consequence, compound 42 and the other pyrazole derivatives with effective thiazole and thiophene pharmacophores are promising candidates for further research as anticancer medicines [62](refer to Figure 4 for Structure and SAR).As shown in the figure below, compound 49 acts as a potent inhibitor of HER-2/EGFR, possessing a half-life of 0.26 m/0.51 m compared to the positive controls erlotinib (IC50 = 0.41 µM for HER-2 and IC50 = 0.20 µM for EGFR) and lapatinib (IC50 = 0.54 µM for HER-2 and IC50 = 0.28 µM for EGFR). On the mechanistic level, nitroimidazole derivatives drew much attention because of their ability to enter and accumulate in tumor areas. To boost the antiproliferation activity of pyrazolyl-nitroimidazole derivatives against Hela or HepG2 cell lines, electron-withdrawing groups were shown to be preferred to electron-donating groups. This indicates that the synthesized chemicals’ significant inhibitory effects on cell growth were directly connected to their kinase inhibitory actions [68] (refer to Figure 4 for Structure and SAR).
These molecules are intriguing novel frameworks for the case of cancer therapies due to their simplicity of synthesis and extraordinary biological activity. Compound 50 inhibited MCF-7, A549, and HeLa cells significantly, with IC50 values ranging from 0.83–1.81 µM. Furthermore, the compounds induced G1 phase cell cycle arrest in MCF-7 cells by inhibiting cyclin D1 and CDK2. It was observed that structural alterations and modifications on the B ring of pyrazolobenzimidazole derivatives resulted in distinct cytotoxic effects by studying the difference in selectivity of the three cell lines to the compounds. As a result, pyrazole-benzimidazole hybrids may lower cell cycle regulators, including cyclin D1 and CDK2 in MCF-7 cells, causing cell cycle arrest and growth suppression [69] (refer to Figure 5 for Structure and SAR). Because the pyrazolo[3,4-d]pyrimidine nucleus is an isostere of the purine nucleus, it has an anticancer effect via serving as an ATP competitive inhibitor for several kinase enzymes. It has been reported that hydrazinyl derivatives have anticancer properties, particularly in breast cancer cell lines. There is evidence that a methyl sulphonyl ring at position 3 enhances the antitumor activity of pyrazolo[3,4-d]pyrimidine nucleic. This study showed that compound 6d (X = H, R = 4-F) showed the highest IC50 at 7.5 nM. More research is needed to understand the precise mechanism of antitumor activity and investigate the SAR of various nucleus locations [94] (refer to Figure 5 for Structure and SAR). A high-throughput screening method identified the pyrazolo[1,5-a]pyrimidine scaffold as a potent Jak2 inhibitor. Increased polarity and the elimination or inhibition of metabolic hotspots such as N-methyl groups are two often used approaches for increasing stability against CYP-mediated metabolism. The H-bond accepting properties of oxygen atoms in heteroaromatic ring systems are weak; on the other hand, sp2 nitrogens of heteroaromatics tend to be strong H-bond acceptors. Compound 84 was found to be the most promising derivative, with an IC50 of 7.4 nM. Compound 84 offers a good combination of cell potency, oral exposure, and in vivo pSTAT5 knockdown, making it a feasible tool for initial Jak2-dependent efficacy studies. Any alteration at the para-position of the N-aryl group reduces the inhibitory potency of all Jak family members considerably. The pyrazolo[1,5-a]pyrimidine scaffold was shown to be a strong inhibitor of Jak2 in a high-throughput screening study [98] (refer to Figure 5 for Structure and SAR).
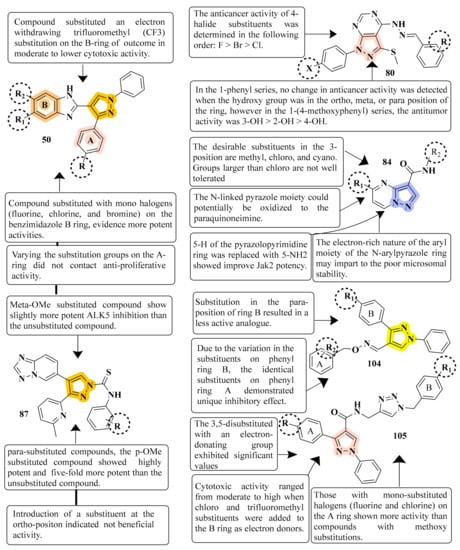
Figure 5.
Structure and SAR of potent anticancer activity compounds 50, 80, 87, 104, and 105.
Several of the pyridyl-substituted five-membered heterocyclic rings were boosted in their inhibitory activity and selectivity by adding methylene, methylene amino, or amino methylene linkages between their core rings and their phenyl rings. The most active compound 87 inhibited ALK5 phosphorylation with a IC50 value of 0.57 nM and demonstrated 94 percent inhibition at 100 nM in a luciferase reporter test using HaCaT cells irreversibly transfected with a p3TP-Luc construct. There was a significant increase in both the inhibitory activity and selectivity of compound 87 against p38a MAP kinase upon the incorporation of the [1,2,4]triazolo[1,5-a]pyridin-6-yl and phenycarbothioamide moiety at positions 4 and 1 of the pyrazole ring, respectively [101] (refer to Figure 5 for Structure and SAR).This work demonstrated the strong immunosuppressive actions of a variety of new pyrazole derivatives having an O-benzyl oxime moiety. Compound 104 had the greatest inhibitory efficacy (IC50 = 1.18 µM for lymph node cells and IC50 = 0.28 µM for PI3K). In particular, a variety of synthesized pyrazole oxime ether analogs were shown to have significant immunosuppressive effects in the low micromolar range. A study of the A-ring para substituents revealed that an electron-withdrawing group had increased immunosuppressive action, and the potency order is F > Cl > OMe > H > Me. The molecules with a potent inhibition effect towards IL-6 produced in ConA-stimulated murine lymph node cells were investigated [117] (refer to Figure 5 for Structure and SAR). The synthesis of N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1,3-diphenyl-1H-pyrazole-4-carboxamides was reported well as their biological evaluation. Nocodazole (8.01–0.95 µM) exhibited significant inhibition against MIAPaCa-2, MCF-7, and HeLa cell lines with compound 105 (GI50) in the range of 0.13–0.7 µM (GI50). Using the pyrazole moiety with two aryl ring systems, A and B, this study investigated the compounds with electron-withdrawing and electron-donating groups, i.e., chloro, fluoro, and fluoro trifluoromethyl groups on the ring systems (A and B). This compound 105 resulted in cell cycle arrest in MCF-7 cells and downregulation of CDK1 expression in MCF-7 cells when it was treated with it. As a result, they can be regarded as promising lead molecules for creating more effective anticancer medicines against breast cancer cells [118] (refer to Figure 5 for Structure and SAR).
APN inhibition was best with compound 109, which had an IC50 value of 0.16 ± 0.02 μM, a value over one order of magnitude less than that of bestatin (IC50 = 9.4 ± 0.5 μM). Depending on where R was substituted on the pyrazoline ring, these compounds could have significantly different APN inhibitory potencies. B series compounds have higher inhibitory activity when compared to APN equivalents. As determined by the structure–activity relationships (SARs) of A series compounds, only ortho- or meta-substituted hydroxamates can be synthesized from B series compounds, while para-substituted hydroxamates are inactive. Compound 109 might be a starting point for developing more powerful APN inhibitors. To test human APN against the ES-2 and PLC/PRF/5 cell surfaces, compounds with significant porcine kidney APN inhibitory properties were selected. APN is abundantly expressed on both cell lines’ membranes [122] (refer to Figure 6 for Structure and SAR). The strongest inhibitory effect was achieved by compound 111 with N-(3-methoxy-2-hydroxybenzal)-3-substituted(p-chlorophenyl)-4-cyano-5-oxopyrazol-1-thiocarboxamide against HL-60, with an IC50 of 1.35 μM compared to Dox’s IC50 of 2.02 μM. Compound 111 improved the G2/M phase from 11.05 percent to 39.22 percent compared to the untreated control. The highest intense cytotoxic action was demonstrated by compound 111. In a study that included activity assays of apoptosis detection and Topoisomerase II inhibition, compound 111 was found to be an effective inhibitor of Topo II [124] (refer to Figure 6 for Structure and SAR).
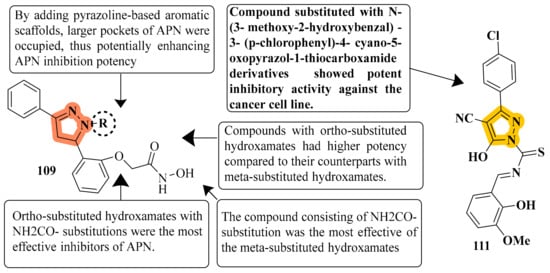
Figure 6.
Structure and SAR of potent anticancer activity compounds 109 and 111.
4.2. Pyrazole Biomolecules as Inflammation Therapeutics
The efficacy of new pyrazole and pyrazoline compounds to inhibit ovine COX-1 and COX-2 isozymes was assessed utilizing an in vitro cyclooxygenase (COX) inhibition test. Among the molecules examined, 129 exhibited excellent COX-2 inhibitory efficacy (IC50 = 0.26 µM) and selectivity (SI) = >192.3, which is equivalent to the reference medication celecoxib (IC50 = 0.28 µM and selectivity index = 178.57). Compound 129 is overly lipophilic due to the presence of trifluoromethyl fragments; therefore, it should serve as a lead molecule for further optimization of certain biological properties and ADME characteristics [127] (refer to Figure 7 for Structure and SAR). Novel 1-(4-methane(amino)sulfonylphenyl) derivatives 5th (4-substituted-aminomethyl-phenyl)-3-trifluoromethyl-1H-pyrazoles were developed, synthesized, and tested for biological activity. Compound 168, derived from the library, exhibited significant anti-inflammatory action. Compared with the standard celecoxib drug (80% inhibition), compound 168 demonstrated the greatest anti-inflammatory effect post carrageenan, 3 h after inflammation onset (89% inhibition) [179]. A total of fifteen pyrazolyl urea derivatives were produced and tested for their ability to inhibit p38 MAPK and act as antioxidants. Among the tested compounds, derivative 178 was discovered to be the most powerful, and its binding mechanism inside the p38 MAPK was also documented. Compound 178, which has a 4- chloro group and has significant p38 MAPK activity, also had the greatest anti-inflammatory efficacy (80.93 percent inhibition). The substitution of 2-chloro and 3-chloro groups for the 4-chloro groups resulted in a modest reduction in inactivity (76.19 percent and 78.06 percent inhibition, respectively). In both the in vitro and in vivo models, the monosubstituted electron-withdrawing group in the phenyl ring linked to the pyrazole nucleus showed strong anti-inflammatory efficacy. Compound 178 was found to be a strong anti-inflammatory agent, equivalent to the standard reference medication diclofenac sodium. In addition, compared to conventional medicine, this molecule had a lower ulcerogenic potential and the least activity in causing oxidative stress in tissues. In contrast, pyrazole compounds with a urea pharmacophore exhibited comparable anti-inflammatory effects and better TNF-inhibitory characteristics in the investigations. Moreover, pyrazolyl urea derivatives were investigated for p38 MAPK inhibition and demonstrated substantial efficacy [187] (refer to Figure 7 for Structure and SAR).
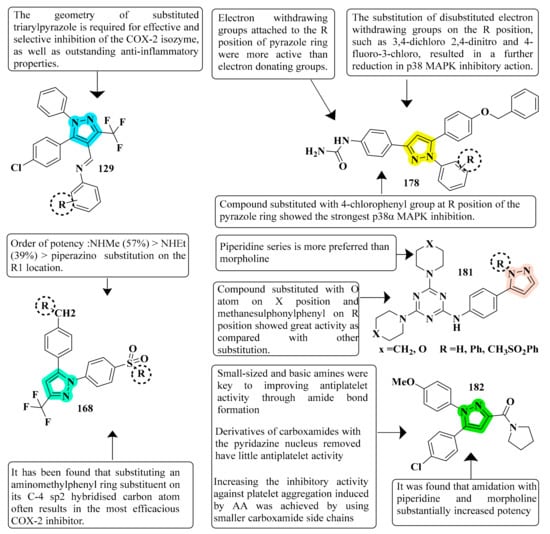
Figure 7.
Structure and SAR of potent anti-inflammatory activity compounds 129, 168, 178, 181, and 182.
An inhibitor of COX-2 with a five-membered pharmacophore, such as pyrazole, and isoxazole, which is polysubstituted 1,3,5-triazines. Compound 181 from synthesized derivatives showed promising findings with an IC50 of 0.74 μM vs. celecoxib’s IC50 of 0.78 μM. When a methanesulphonylphenyl pyrazole moiety was added, the selectivity increased significantly. To summarize, when the triazine core was replaced with pyrazoline, isoxazole, or unsubstituted pyrazole moieties, a considerable COX-2 selectivity was observed. Thus, scaffolds derived from Compound 181 might be further developed, and their biological activity tested [190] (refer to Figure 7 for Structure and SAR). The pyridazine 6-position was substituted with phenyl, and the carboxyl arm in the central pyrazole was extended by adding polar substituents. In addition, pyrazole compounds without either the 1-pyridazine or the 5-phenyl groups were synthesized. These derivatives were designed to explore how a side chain of different sizes and basicity at three positions of the pyrazole could affect the antiplatelet activity and to learn about the effects of exposure to vicinal diaries about the central heterocycle, as well as the necessary presence of central pyrazoles on their biological profiles. Various compounds of this class had different inhibitory effects on platelet aggregation induced by AA and collagen, mainly depending on the shape and size of the carboxamide molecules at the 3-position of the pyrazole ring. However, only small carboxamide molecules specifically inhibited collagen-induced platelet aggregation. Finally, we describe compound 182 as a new potent antiplatelet agent with an IC50 in the two- to one-digit nanomolar range (IC50 values of 5.7 nM). Because of their great efficacy in the cellular milieu, these simple pyrazole derivatives may hold promise for developing more effective molecules for cardiovascular disease intervention [191] (refer to Figure 7 for Structure and SAR).
The primary pyrazole core of selective COX2 inhibitors (celecoxib and SC558) was engaged, and structural alterations led to the identification of additional pyrazole analogs. In comparison to ibuprofen (which inhibited COX-1/2 and TNF- after 4 h and exhibited 81.32 percent inhibition), compound 187 exhibited 80.63 percent inhibition at 4 h. We, therefore, found that methylamine pyrazole derivatives provided the requisite geometry to effectively and selectively inhibit the COX2 enzyme and provide good anti-inflammatory properties when substituted for sulfonamide pyrazole derivatives. As a result, the hydrophobic benzyloxyphenyl group was introduced in the same way as the celecoxib trifluoromethyl group to boost the affinity for COX2 [196] (refer to Figure 8 for Structure and SAR). A novel pyrazole derivative was created and evaluated for its potential to inhibit COX-1/2 activity. There was good selectivity towards COX-2 inhibition in many pyrazoles bearing the benzenesulfonamide moiety, and consequently, these compounds had powerful anti-inflammatory activity. Compounds 189(a), 189(b), 189(c), and 189(d) showed high activity and selectivity against COX-2 (IC50 = 39.43, 61.24, 38.73, and 39.14 nM). All compounds reported selectivity indices of 22.21, 14.35, 17.47, and 13.10 for 189(a), 189(b), 189(c), and 189(d), correspondingly. In vivo, these compounds were superior to or equivalent to celecoxib as anti-inflammatory drugs. Interestingly, these molecules specifically reduced the induced microsomal PGE2 synthase synthesis, indicating that additional research into these derivatives is needed [198] (refer to Figure 8 for Structure and SAR).
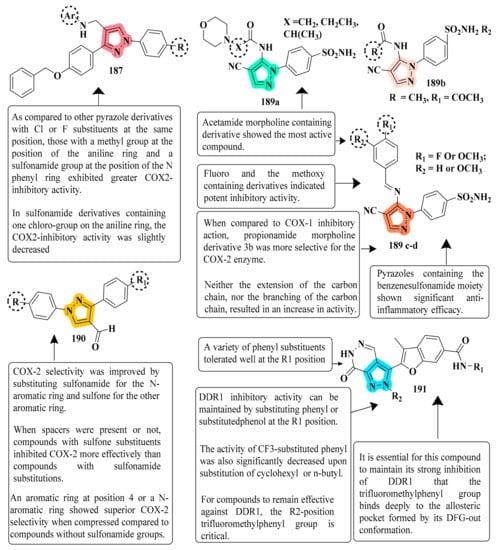
Figure 8.
Structure and SAR of potent anti-inflammatory activity compounds 187, 189, 190 and 191.
In an easy synthetic procedure, a series of diaryl pyrazole and triazole derivatives to produce selective COX-2 inhibitors were designed and synthesized. Among the synthesized derivatives, compound 190a with a sulfonamide substitution on one N-aromatic ring had the strongest inhibitory action against COX-2 with IC50 = 0.017 ± 0.001 μM and COX-1 with IC50 = 0.263 ± 0.016 μM. In contrast, compound 190b, which has nitro on its N-aromatic ring, inhibited COX-1 more effectively (IC50 = 0.012 ± 0.001 μM). The best COX-2 selectivity was obtained with sulfonamide and sulfone substituted on the N-aromatic ring. More interestingly, the heterocyclic cores (pyrazole or triazole) contain two not vicinal aryls in contrast with the most commonly known COX-2 inhibitors. Additional work is needed to synthesize diverse pyrazoles and triazoles with different spacer lengths and substitutions in order to assess their role in COX-2 inhibitory activity and selectivity [199] (refer to Figure 8 for Structure and SAR). Researchers developed and tested new compounds known as pyrazolo[3,4-d]pyridazinones as anti-inflammatory drugs against discoidin domain receptor 1 (DDR1) modifications. Mouse bone-marrow-derived and human THP-11 macrophages were effectively decreased by compound 191 at ten micromolar concentrations when exposed to LPS. Compound 191 inhibited pro-inflammatory cytokines and autophosphorylation of DDR1 in cells in a mouse colitis model induced by dextran sulfate sodium (DSS). DDR1-IN-1, a positive control compound, had an EC50 of 114.5 nM, whereas compound 191 suppressed basal autophosphorylation at 34.4 nM, more powerful than the positive control compound. As the N-phenyl group of DC-1 was exposed above the solvent-exposed area of DDR1, further modifications can be made to this group in DDR1 [200] (refer to Figure 8 for Structure and SAR).
5. Conclusions
This literature survey focused on merging different pharmacophoric subunits of a large number of pyrazolic analogues to create potential lead compounds showing anticancer and anti-inflammatory activities. It also highlighted the prestige of these biomolecules in the design, discovery, and advancement of novel drug molecules.
Author Contributions
Conceptualization, M.J.A. and O.A.; methodology, M.J.N., F.N. and A.M.; software, M.M.G. and P.A.; validation, S.A., M.I. and F.S.; formal analysis, H.K.T. and M.I.; investigation, M.I., F.S. and S.A.; resources, M.I.; data curation, P.A.; writing—original draft preparation, M.J.A.; writing—review and editing, S.A., M.I. and F.S.; visualization, O.A.; supervision, O.A.; project administration, O.A.; funding acquisition, M.I. and H.K.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are thankful to AlMaarefa University for their financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles; Wiley-VCH: Hoboken, NJ, USA, 2003; ISBN 3527307206. [Google Scholar]
- Naim, M.J.; Alam, O.; Nawaz, F.; Alam, M.J.; Alam, P. Current status of pyrazole and its biological activities. J. Pharm. Bioallied Sci. 2016, 8, 2–17. [Google Scholar] [PubMed]
- Alam, J.; Alam, O.; Ali, R.; Naim, M.J.; Khan, S.A. Synthesis and anti-inflammatory activity of some new thiadiazole linked pyrazole benzene sulphonamides as cyclooxygenase inhibitors. Orient. J. Chem. 2015, 31, 1873–1885. [Google Scholar] [CrossRef]
- Katz, A.M.; Pearsom, C.M.; Kannedy, J.H. A clinical trial of indomethacin in rheumatoid arthritis. Clin. Pharmacol. Ther. 1965, 6, 25–30. [Google Scholar] [CrossRef]
- Ilango, K.; Valentina, P. Textbook of Medicinal Chemistry, 1st ed.; Keerthi Publishers: Coimbatore, India, 2007; pp. 327–333. [Google Scholar]
- Hassan, G.S.; Abou-Seri, S.M.; Kamel, G.; Ali, M.M. Celecoxib analogues bearing benzofuran moiety as cyclooxygenase-2 inhibitors: Design, synthesis and evaluation as potential anti-inflammatory agents. Eur. J. Med. Chem. 2014, 76, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, G.; Rose, T.; Rissler, K. Determination of lonazolac and its hydroxyl and o-sulfated metabolites on-line sample preparation liquid chromatography with fluorescence detection. J. Chromatogr. B 2022, 786, 295–305. [Google Scholar]
- Giorgi, M.; Mengozzi, G.; Rafaelli, A.; Saba, A. Characterization of in vivo plasma metabolites of tepoxalin in horses using LC-MS-MS. J. Pharm. Anal. Biomed. Anal. 2011, 56, 45–53. [Google Scholar] [CrossRef]
- Brune, K. The early history of non-opioid analgesics. Acute Pain 1997, 1, 33–40. [Google Scholar] [CrossRef]
- De-Grauw, J.C.; Van Loon, J.P.; Van de Lest, C.H.; Brunott, A.; van Weeren, P.R. In vivo effects of phenylbutazone on inflammation and cartilage-derived biomarkers in equine joints with acute synovitis. Vet. J. 2014, 201, 51–56. [Google Scholar] [CrossRef]
- Song, Z. In vitro detecting ultra-trace novalgin in medicine and human urine by chemiluminescence. Talanta 2003, 60, 161–170. [Google Scholar] [CrossRef]
- Dowling, G.; Malone, E. Analytical strategy for the confirmatory analysis of the non-steroidal anti-inflammatory drugs firocoxib, propyphenazone, ramifenazone and piroxicam in bovine plasma by liquid chromatography tandem mass spectrometry. J. Pharm. Biomed. 2011, 56, 359–365. [Google Scholar] [CrossRef]
- Cox, S.K.; Roark, J.; Gassel, A.; Tobias, K. Determination of deracoxib in feline plasma samples using high performance liquid chromatography. J. Chromatogr. B 2005, 819, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Tsutomu, K.; Toshitaka, N. Effects of 1,3-diphenyl-5-(2-dimethylaminopropionamide)-pyrazole[difenamizole] on a conditioned avoidance response. Neuropharmacology 1978, 17, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lozano, J.; Server-Carrioa, J.; Escrivaa, E.; Folgadoa, J.V.; Mollaa, C.; Lezamab, L. X-ray crystal structure and electronic properties of chlorobis(mepirizole)copper(II) tetrafluoroborate (mepirizole = 4-methoxy-2-(5- methoxy-3-methyl-1H-pyrazol-1-yl)-6-methylpyrimidine). Polyhedron 1997, 16, 939–944. [Google Scholar] [CrossRef]
- Calvello, R.; Panaro, M.A.; Carbone, M.L.; Cianciulli, A.; Perrone, M.G.; Vitale, P.; Malerba, P.; Scilimati, A. Novel selective COX-1 inhibitors suppress neuroinflammatory mediators in LPS-stimulated N13 microglial cells. Pharmacol. Res. 2015, 65, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Y.; Ji, F.Q. A molecular dynamics investigation on crizotinib resistance mechanism of C1156Y mutation in ALK. Biochem. Biophys. Res. Commun. 2012, 423, 319–324. [Google Scholar] [CrossRef]
- Wyde, P.R.; Gilbert, B.E.; Ambrose, M.W. Comparison of the anti-respiratory syncytial virus activity and toxicity of papaverine hydrochloride and pyrazofurin in vitro and in vivo. Antivir. Res. 1989, 11, 15–26. [Google Scholar] [CrossRef]
- Kapadia, G.J.; Tokuda, H.; Sridhar, R.; Balasubramanian, V.; Takayasu, J.; Bu, P.; Enjo, F.; Takasaki, M.; Konoshima, T.; Nishino, H. Cancer chemopreventive activity of synthetic colorants used in foods, pharmaceuticals and cosmetic preparations. Cancer Lett. 1998, 129, 87–95. [Google Scholar] [CrossRef]
- Frank, A.O.; Feldkamp, M.D.; Kennedy, J.P.; Waterson, A.G.; Pelz, N.F.; Patrone, J.D.; Vangamudi, B.; Camper, D.V.; Rossanese, O.W.; Chazin, W.J.; et al. Discovery of a potent inhibitor of replication protein a protein-protein interactions using a fragment-linking approach. J. Med. Chem. 2013, 56, 9242–9250. [Google Scholar] [CrossRef]
- Koca, I.; Ozgur, A.; Coskun, K.A.; Tutar, Y. Synthesis and anticancer activity of acyl thioureas bearing pyrazole moiety. Bioorg. Med. Chem. 2013, 21, 3859–3865. [Google Scholar] [CrossRef]
- Cui, J.J. Targeting receptor tyrosine kinase MET in Cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef]
- Desai, B.; Dixon, K.; Farrant, E.; Feng, Q.; Gibson, K.R.; van Hoorn, W.P.; Mills, J.; Morgan, T.; Parry, D.M.; Ramjee, M.K.; et al. Rapid discovery of a novel series of Abl kinase inhibitors by application of an integrated microfluidic synthesis and screening platform. J. Med. Chem. 2013, 56, 3033–3047. [Google Scholar] [CrossRef] [PubMed]
- Rai, U.S.; Isloor, A.M.; Shetty, P.; Pai, K.S.R.; Fun, H.K. Synthesis and in vitro biological evaluation of new pyrazole chalcones and heterocyclic diamides as potential anticancer agents. Arab. J. Chem. 2015, 8, 317–321. [Google Scholar]
- Su, Q.; Ioannidis, S.; Chuaqui, C.; Almeida, L.; Alimzhanov, M.; Bebernitz, G.; Bell, K.; Block, M.; Howard, T.; Huang, S.; et al. Discovery of 1-methyl-1H-imidazole derivatives as potent JAK2 inhibitors. J. Med. Chem. 2014, 57, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in copper complexes as anticancer agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.A.; Saidachary, G.; Mallesham, G.; Sridhar, B.; Jain, N.; Kalivendi, S.V.; Rao, V.J.; Raju, B.C. Synthesis, anticancer activity and photophysical properties of novel substituted 2-oxo-2H-chromenylpyrazolecarboxylates. Eur. J. Med. Chem. 2013, 65, 389–402. [Google Scholar] [CrossRef]
- Aguilar, A.; Zhou, H.; Chen, J.; Liu, L.; Bai, L.; McEachern, D.; Yang, C.; Meagher, J.; Stuckey, J.; Wang, S. A Potent, Highly Efficacious, Bcl-2/Bcl-xL inhibitor. J. Med. Chem. 2013, 56, 3048–3067. [Google Scholar] [CrossRef]
- Abadi, A.H.; Eissa, A.A.H.; Hassan, G.S. Synthesis of novel 1, 3, 4-trisubstituted pyrazole derivatives and their evaluation as antitumor and antiangiogenic agents. Chem. Pharm. Bull. 2003, 51, 838–844. [Google Scholar] [CrossRef]
- Bouabdallah, I.; M’Barek, L.A.; Zyad, A.; Ramdani, A.; Zidane, I.; Melhaoui, A. Anticancer effect of three pyrazole derivatives. Nat. Prod. Res. 2006, 20, 1024–1030. [Google Scholar] [CrossRef]
- Wei, F.; Zhao, B.X.; Huang, B.; Zhang, L.; Sun, C.H.; Dong, W.L.; Shin, D.S.; Miao, J.Y. Design, synthesis, and preliminary biological evaluation of novel ethyl 1-(2′-hydroxy-3′-aroxypropyl)-3-aryl-1H-pyrazole-5-carboxylate. Bioorg. Med. Chem. Lett. 2006, 16, 6342–6347. [Google Scholar] [CrossRef]
- Xia, Y.; Dong, Z.W.; Zhao, B.X.; Ge, X.; Meng, N.; Shin, D.S.; Miao, J.Y. Synthesis and structure–activity relationships of novel 1-arylmethyl-3-aryl-1H-pyrazole-5-carbohydrazide derivatives as potential agents against A549 lung cancer cells. Bioorg. Med. Chem. 2007, 15, 6893–6899. [Google Scholar] [CrossRef]
- Fan, C.D.; Zhao, B.X.; Wei, F.; Zhang, G.H.; Dong, W.L.; Miao, J.Y. Synthesis and discovery of autophagy inducers for A549 and H460 lung cancer cells, novel 1-(2′-hydroxy-3′-aroxypropyl)-3-aryl-1H-pyrazole-5-carbohydrazide derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 3860–3864. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, H.A.; El-Zahabi, H.S.A.; Dawood, K.M. Microwave-assisted synthesis and in-vitro anti-tumor activity of 1, 3, 4-triaryl-5-N-arylpyrazole-carboxamides. Eur. J. Med. Chem. 2010, 45, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.W.; Wu, L.L.; Zhao, B.X.; Dong, W.L.; Miao, J.Y. Synthesis of novel substituted pyrazole-5-carbohydrazide hydrazone derivatives and discovery of a potent apoptosis inducer in A549 lung cancer cells. Bioorg. Med. Chem. 2009, 17, 1957–1962. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.W.; Li, Y.; Geb, D.; Zhao, B.-X.; Liu, Y.-R.; Lv, H.-S.; Ding, J.; Miao, J.-Y. Synthesis of novel oxime-containing pyrazole derivatives and discovery of regulators for apoptosis and autophagy in A549 lung cancer cells. Bioorg. Med. Chem. Lett. 2010, 20, 4766–4770. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.Y.; Liau, H.H.; Lu, P.J.; Yang, C.N.; Lee, C.H.; Chen, J.Y.; Xu, Z.; Flynn, G. 3, 5-Diaryl-1H-pyrazole as a molecular scaffold for the synthesis of apoptosis-inducing agents. Bioorg. Med. Chem. 2010, 18, 3270–3278. [Google Scholar] [CrossRef]
- Rostom, S.A. Polysubstituted pyrazoles, part 6. Synthesis of some 1-(4-chlorophenyl)-4-hydroxy-1H-pyrazol-3-carbonyl derivatives linked to nitrogenous heterocyclic ring systems as potential antitumor agents. Bioorg. Med. Chem. 2010, 18, 2767–2776. [Google Scholar] [CrossRef]
- Insuasty, B.; Tigreros, A.; Orozco, F.; Quiroga, J.; Abonia, R.; Nogueras, M.; Sanchez, A.; Cobo, J. Synthesis of novel pyrazolic analogues of chalcones and their 3-aryl-4-(3-aryl-4,5-dihydro-1H-pyrazol-5-yl)-1-phenyl-1H-pyrazole derivatives as potential antitumor agents. Bioorg. Med. Chem. 2010, 18, 4965–4974. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Draghici, C.; Missir, A.V. Synthesis of new pyrazole derivatives and their anticancer evaluation. Eur. J. Med. Chem. 2010, 45, 4914–4919. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, G.; Zhao, G.; Xu, W.; Huo, L. Synthesis and cytotoxic activity of novel 3-(1H-indol-3-yl)-1H-pyrazole-5-carbohydrazide derivatives. Eur. J. Med. Chem. 2011, 46, 5868–5877. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Sim, T.B.; Hong, J.H.; Cho, J.H.; Yoo, K.H.; Oh, C.H. Synthesis of 1H-Pyrazole-1-carboxamide Derivatives and Their Antiproliferative Activity against Melanoma Cell Line. Arch. Pharm. Chem. Life Sci. 2011, 344, 197–204. [Google Scholar] [CrossRef]
- Huang, X.F.; Lu, X.; Zhang, Y.; Song, G.Q.; He, Q.L.; Li, Q.S.; Yang, X.H.; Wei, Y.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of N-((1, 3-diphenyl-1H-pyrazol-4-yl) methyl) aniline derivatives as novel anticancer agents. Bioorg. Med. Chem. 2012, 20, 4895–4900. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, X.; Xing, M.; Yang, X.H.; Zhao, T.T.; Gong, H.B.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of N, 1, 3-triphenyl-1H-pyrazole-4-carboxamide derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 3589–3593. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; El-Sayed, N.N.; Abdelaziz, M.A. Uses of cyanoacetylhydrazine in heterocyclic synthesis: Novel synthesis of pyrazole derivatives with anti-tumor activities. Molecules 2012, 17, 8449–8463. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lv, X.-H.; Qiu, H.-Y.; Wang, Y.-T.; Du, Q.-R.; Li, D.-D.; Yang, Y.-H.; Zhu, H.-L. Synthesis, biological evaluation and molecular docking studies of pyrazole derivatives coupling with a thiourea moiety as novel CDKs inhibitors. Eur. J. Med. Chem. 2013, 68, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Inceler, N.; Yılmaz, A.; Baytas, S.N. Synthesis, characterization, anticancer, and antioxidant activity of some new thiazolidin-4-ones in MCF-7 cells. Med. Chem. Res. 2012, 22, 3109–3118. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Park, Y.S.; Chi, D.Y.; Yoo, K.H.; Oh, C.H. New triarylpyrazoles as broad-spectrum anticancer agents: Design, synthesis, and biological evaluation. Eur. J. Med. Chem. 2013, 65, 315–322. [Google Scholar] [CrossRef]
- Xu, S.; Li, S.; Tang, Y.; Zhang, J.; Wang, S.; Zhou, C.; Li, X. Design, synthesis, and biologic evaluation of some novel N-arylpyrazole derivatives as cytotoxic agents. Med. Chem. Res. 2013, 22, 5610–5616. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, M.; Ling, X.; Liu, Y.; Xue, Y.; An, L.; Gu, N.; Jin, M. Design, synthesis, quantum chemical studies and biological activity evaluation of pyrazole–benzimidazole derivatives as potent Aurora A/B kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3523–3530. [Google Scholar] [CrossRef]
- Dong, J.J.; Li, Q.S.; Wang, S.F.; Li, C.Y.; Zhao, X.; Qiu, H.Y.; Zhao, M.Y.; Zhu, H.L. Synthesis, biological evaluation and molecular docking of novel 5-phenyl-1H-pyrazol derivatives as potential BRAFV600E inhibitors. Organic & Biomolecular Chemistry. Org. Biomol. Chem. 2013, 11, 6328–6337. [Google Scholar]
- Mohareb, R.M.; Abdallah, A.E.M.; Abdelaziz, M.A. New approaches for the synthesis of pyrazole, thiophene, thieno [2, 3-b] pyridine, and thiazole derivatives together with their anti-tumor evaluations. Med. Chem. Res. 2014, 23, 564–579. [Google Scholar] [CrossRef]
- Nakao, S.; Mabuchi, M.; Shimizu, T.; Itoh, Y.; Takeuchi, Y.; Ueda, M.; Mizuno, H.; Shigi, N.; Ohshio, I.; Jinguji, K.; et al. Design and synthesis of prostate cancer antigen-1 (PCA-1/ALKBH3) inhibitors as anti-prostate cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Zhao, T.-T.; Ren, Y.-J.; Peng, N.-N.; Yang, X.-H.; Li, X.; Zhang, H.; Liu, G.-Q.; Zhang, L.-R.; Zhu, H.-L. Synthesis, biological evaluation, and molecular docking studies of pyrazolyl-acylhydrazone derivatives as novel anticancer agents. Med. Chem. Res. 2014, 23, 3274–3286. [Google Scholar] [CrossRef]
- Aydın, S.; Kaushik-Basu, N.; Ozbas¸-Turan, S.; Akbuga, J.; Mega Tiber, P.; Orun, O.; Gurukumar, K.R.; Basu, A.; Küçükgüzel, S.G. Synthesis of 1-aroyl-3, 5-dimethyl-1H-pyrazoles as anti-HCV and anticancer agents. Lett. Drug Des. Discov. 2014, 11, 121–131. [Google Scholar] [CrossRef]
- Cankara Pirol, S.; Caliskan, B.; Durmaz, I.; Atalay, R.; Banoglu, E. Synthesis and preliminary mechanistic evaluation of 5-(p-tolyl)-1-(quinolin-2-yl)pyrazole-3-carboxylic acid amides with potent antiproliferative activity on human cancer cell lines. Eur. J. Med. Chem. 2014, 20, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kaur, K.; Karelia, D.N.; Beniwal, V.; Gupta, G.K.; Sharma, A.K.; Gupta, A.K. Synthesis and biological evaluation of some 2-(3, 5-dimethyl-1H-pyrazol-1-yl)-1-arylethanones: Antibacterial, DNA photocleavage, and anticancer activities. Eur. J. Med. Chem. 2014, 81, 267–276. [Google Scholar] [CrossRef]
- Lu, Y.; Ran, T.; Lin, G.; Jin, Q.; Jin, J.; Li, H.; Guo, H.; Lu, T.; Wang, Y. Novel 1H-pyrazole-3-carboxamide derivatives: Synthesis, anticancer evaluation and identification of their DNA-binding interaction. Chem. Pharm. Bull. 2014, 62, 238–246. [Google Scholar] [CrossRef]
- Zhang, J.F.; Li, M.; Miao, J.Y.; Zhao, B.X. Biological activities of novel pyrazolyl hydroxamic acid derivatives against human lung cancer cell line A549. Eur. J. Med. Chem. 2014, 83, 516–525. [Google Scholar] [CrossRef]
- Yao, Y.; Liao, C.; Li, Z.; Wang, Z.; Sun, Q.; Liu, C.; Yang, Y.; Tu, Z.; Jiang, S. Design, synthesis, and biological evaluation of 1, 3-disubstituted-pyrazole derivatives as new class I and IIb histone deacetylase inhibitors. Eur. J. Med. Chem. 2014, 86, 639–652. [Google Scholar] [CrossRef]
- Wen, J.; Bao, Y.; Niu, Q.; Yang, J.; Fan, Y.; Li, J.; Jing, Y.; Zhao, L.; Liu, D. Identification of N-(6-mercaptohexyl)-3-(4-pyridyl)-1H-pyrazole-5-carboxamide and its disulfide prodrug as potent histone deacetylase inhibitors with in vitro and in vivo anti-tumor efficacy. Eur. J. Med. Chem. 2016, 109, 350–359. [Google Scholar] [CrossRef]
- Zhao, M.Y.; Yin, Y.; Yu, X.W.; Sangani, C.B.; Wang, S.F.; Lu, A.M.; Yang, L.F.; Lv, P.C.; Jiang, M.G.; Zhu, H.L. Synthesis, biological evaluation and 3D-QSAR study of novel 4,5-dihydro-1H-pyrazole thiazole derivatives as BRAF (V600E) inhibitors. Bioorg. Med. Chem. 2015, 23, 46–54. [Google Scholar] [CrossRef]
- Kamal, A.; Shaik, A.B.; Polepalli, S.; Kumar, G.B.; Reddy, V.S.; Mahesh, R.; Garimella, S.; Jain, N. Synthesis of arylpyrazole linked benzimidazole conjugates as potential microtubule disruptors. Bioorg. Med. Chem. 2015, 23, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zheng, Y.; Li, Z.; Wang, Y.; Lv, Y.; Qin, X.; Zeng, C. Design, synthesis, and biological activity of phenyl-pyrazole derivatives as BCR-ABL kinase inhibitors. Bioorg. Med. Chem. 2015, 23, 3147–3152. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Draghici, C.; Olaru, O.T.; Matei, L.; Ioana, A.; Dragu, L.D.; Bleotu, C. Synthesis and apoptotic activity of new pyrazole derivatives in cancer cell lines. Bioorg. Med. Chem. 2015, 23, 5799–5808. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Matei, L.; Aldea, I.M.; Draghici, C.; Olaru, O.T.; Bleotu, C. Ultrasound-assisted synthesis and anticancer evaluation of new pyrazole derivatives as cell cycle inhibitors. Arab. J. Chem. 2019, 12, 816–824. [Google Scholar] [CrossRef]
- Khloya, P.; Ceruso, M.; Ram, S.; Supuran, C.T.; Sharma, P.K. Sulfonamide bearing pyrazolylpyrazolines as potent inhibitors of carbonic anhydrase isoforms I, II, IX and XII. Bioorg. Med. Chem. Lett. 2015, 25, 3208–3212. [Google Scholar] [CrossRef]
- Tao, X.X.; Duan, Y.T.; Chen, L.W.; Tang, D.J.; Yang, M.R.; Wang, P.F.; Xu, C.; Zhu, H.L. Design, synthesis and biological evaluation of pyrazolyl-nitroimidazole derivatives as potential EGFR/HER-2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 677–683. [Google Scholar] [CrossRef]
- Reddy, T.S.; Kulhari, H.; Reddy, V.G.; Bansal, V.; Kamal, A.; Shukla, R. Design, synthesis and biological evaluation of 1,3-diphenyl-1H-pyrazole derivatives containing benzimidazole skeleton as potential anticancer and apoptosis inducing agents. Eur. J. Med. Chem. 2015, 101, 790–805. [Google Scholar] [CrossRef]
- Kamal, A.; Shaik, A.B.; Jain, N.; Kishor, C.; Nagabhushana, A.; Supriya, B.; Kumar, G.B.; Chourasiya, S.S.; Suresh, Y.; Mishra, R.K.; et al. Design and synthesis of pyrazole-oxindole conjugates targeting tubulin polymerization as new anticancer agents. Eur. J. Med. Chem. 2015, 92, 501–513. [Google Scholar] [CrossRef]
- Shi, J.B.; Tang, W.J.; Qi, X.B.; Li, R.; Liu, X.H. Novel pyrazole-5-carboxamide and pyrazole-pyrimidine derivatives: Synthesis and anticancer activity. Eur. J. Med. Chem. 2015, 90, 889–896. [Google Scholar] [CrossRef]
- Ali, A.R.; El-Bendary, E.R.; Ghaly, M.A.; Shehata, I.A. Synthesis, in vitro anticancer evaluation and in silico studies of novel imidazo [2,1-b]thiazole derivatives bearing pyrazole moieties. Eur. J. Med. Chem. 2014, 75, 492–500. [Google Scholar] [CrossRef]
- Reddy, G.L.; Guru, S.K.; Srinivas, M.; Pathania, A.S.; Mahajan, P.; Nargotra, A.; Bhushan, S.; Vishwakarma, R.A.; Sawant, S.D. Synthesis of 5-substituted-1H-pyrazolo[ 4,3-d]pyrimidin-7(6H)-one analogues and their biological evaluation as anticancer agents: mTOR inhibitors. Eur. J. Med. Chem. 2014, 80, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Grosse, S.; Mathieu, V.; Pillard, C.; Massip, S.; Marchivie, M.; Jarry, C.; Bernard, P.; Kiss, R.; Guillaumet, G. New imidazo [1,2-b]pyrazoles as anticancer agents: Synthesis, biological evaluation and structure activity relationship analysis. Eur. J. Med. Chem. 2014, 84, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Park, H.J.; Park, S.J.; Sheen, Y.Y.; Kim, D.K. 4-([1,2,4]Triazolo [1,5-a]pyridin-6-yl)-5(3)-(6-methylpyridin-2-yl)imidazole and pyrazole derivatives as potent and selective inhibitors of transforming growth factor-beta type I receptor kinase. Bioorg. Med. Chem. 2014, 22, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Al-Sanea, M.M.; Abdelazem, A.Z.; Park, H.M.; Roh, E.J.; Park, H.M.; Yoo, K.H.; Sim, T.; Tae, J.S.; Lee, S.H. Structure-based optimization and biological evaluation of trisubstituted pyrazole as a core structure of potent ROS1 kinase inhibitors. Bioorg. Med. Chem. 2014, 22, 3871–3878. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Zhu, Y.L.; Zhu, P.T.; Makawana, J.A.; Zhang, Y.L.; Zhao, M.Y.; Lv, P.C.; Zhu, H.L. Design, synthesis and biological evaluation of novel 5-phenyl-1H-pyrazole derivatives as potential BRAF V600E inhibitors. Bioorg. Med. Chem. 2014, 22, 6201–6208. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Tamboli, J.R.; Vishnuvardhan, M.V.; Adil, S.F.; Nayak, V.L.; Ramakrishna, S. Synthesis and anticancer activity of heteroaromatic linked 4β-amido podophyllotoxins as apoptotic inducing agents. Bioorg. Med. Chem. Lett. 2013, 23, 273–280. [Google Scholar] [CrossRef]
- Zhu, S.L.; Wu, Y.; Liu, C.J.; Wei, C.Y.; Tao, J.C.; Liu, H.M. Synthesis and in vitro cytotoxic activity evaluation of novel heterocycle bridged carbothioamide type isosteviol derivatives as antitumor agents. Bioorg. Med. Chem. Lett. 2013, 23, 1343–1346. [Google Scholar] [CrossRef]
- Miyamoto, N.; Sakai, N.; Hirayama, T.; Miwa, K.; Oguro, Y.; Oki, H.; Okada, K.; Takagi, T.; Iwata, H.; Awazu, Y.; et al. Discovery of N-[5-({2-[(cyclopropylcarbonyl)amino]imidazo [1,2-b]pyridazin-6-yl}oxy)-2-methylphenyl]-1,3-dimethyl-1H-pyrazole-5-carboxamide (TAK-593), a highly potent VEGFR2 kinase inhibitor. Bioorg. Med. Chem. 2013, 21, 2333–2345. [Google Scholar] [CrossRef]
- Bavetsias, V.; Faisal, A.; Crumpler, S.; Brown, N.; Kosmopoulou, M.; Joshi, A.; Atrash, B.; Perez-Fuertes, Y.; Schmitt, J.A.; Boxall, K.J.; et al. Aurora isoform selectivity: Design and synthesis of imidazo [4,5-b] pyridine derivatives as highly selective inhibitors of Aurora-A kinase in cells. J. Med. Chem. 2013, 56, 9122–9135. [Google Scholar] [CrossRef]
- Liu, Y.-R.; Luo, J.-Z.; Duan, P.-P.; Shao, J.; Zhao, B.-X.; Miao, J.-Y. Synthesis of pyrazole peptidomimetics and their inhibition against A549 lung cancer cells. Bioorg. Med. Chem. Lett. 2012, 22, 6882–6887. [Google Scholar] [CrossRef]
- Lv, H.-S.; Kong, X.-Q.; Ming, Q.-Q.; Jin, X.; Miao, J.-Y.; Zhao, B.-X. Synthesis of 5- benzyl- phenylpyrazolo[1,5-a]pyrazin-4,6(5H,7H)-dione derivatives and discovery of an apoptosis inducer for H322 lung cancer cells. Bioorg. Med. Chem. Lett. 2012, 22, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.-G.; Yu, D.-K.; Wang, J.-X.; Zhang, H.; He, H.-W.; Shao, R.-G.; Li, X.-M.; Wang, Y.-C. Design, synthesis and anticancer activity of 1-acyl-3-amino-1, 4, 5, 6-tetrahydropyrrolo[3,4-c]pyrazole derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 6947–6951. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Wang, L.-Y.; Chang, C.-H.; Kuo, Y.-H.; Kaneko, K.; Takayama, H.; Kimura, M.; Juang, S.-H.; Wong, F.F. One-pot synthesis and anti-proliferative evaluation of pyrazolo[3,4- d]pyrimidine derivatives. Tetrahedron 2012, 68, 9658–9664. [Google Scholar] [CrossRef]
- Strocchi, E.; Fornari, F.; Minguzzi, M.; Gramantieri, L.; Milazzo, M.; Rebuttini, V.; Breviglieri, S.; Camaggi, C.M.; Locatelli, E.; Bolondi, L.; et al. Design, synthesis and biological evaluation of pyrazole derivatives as potential multi-kinase inhibitors in hepatocellular carcinoma. Eur. J. Med. Chem. 2012, 48, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Metwally, M.A.; Gouda, M.A.; Harmal, A.N.; Khalil, A.M. Synthesis, antitumor, cytotoxic and antioxidant evaluation of some new pyrazolotriazines attached to antipyrine moiety. Eur. J. Med. Chem. 2012, 56, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Baciu-Atudosie, L.; Ghinet, A.; Farce, A.; Dubois, J.; Belei, D.; Bîcu, E. Synthesis and biological evaluation of new phenothiazine derivateives bearing a pyrazole unit as protein farnesyltransferase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 6896–6902. [Google Scholar] [CrossRef]
- Bondock, S.; Adel, S.; Etman, H.A.; Badria, F.A. Synthesis and antitumor evaluation of some new 1,3,4-oxadiazole-based heterocycles. Eur. J. Med. Chem. 2012, 48, 192–199. [Google Scholar] [CrossRef]
- Deslandes, S.; Lamoral-Theys, D.; Frongia, C.; Chassaing, S.; Bruyere, C.; Lozach, O.; Meijer, L.; Ducommun, B.; Kiss, R.; Delfourne, E. Synthesis and biological evaluation of analogues of the marine alkaloids granulatimide and isogranulatimide. Eur. J. Med. Chem. 2012, 54, 626–636. [Google Scholar] [CrossRef]
- Puthiyapurayil, P.; Poojary, B.; Chikkanna, C.; Buridipad, S.K. Design, synthesis and biological evaluation of a novel series of 1,3,4-oxadiazole bearing N-methyl-4 (trifluoromethyl)phenyl pyrazole moiety as cytotoxic agents. Eur. J. Med. Chem. 2012, 53, 203–210. [Google Scholar] [CrossRef]
- El-borai, M.A.; Rizk, H.F.; Abd-Aal, M.F.; El-Deeb, I.Y. Synthesis of pyrazolo[3,4- b]pyridines under microwave irradiation in multi-component reactions and their antitumor and antimicrobial activities—Part 1. Eur. J. Med. Chem. 2012, 48, 92–96. [Google Scholar] [CrossRef]
- Shen, S.L.; Zhu, J.; Li, M.; Zhao, B.-X.; Miao, J.-Y. Synthesis of ferrocenyl pyrazole-containing chiral aminoethanol derivatives and their inhibition against A549 and H322 lung cancer cells. Eur. J. Med. Chem. 2012, 54, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Abd El Hamid, M.K.; Mihovilovic, M.D.; El-Nassan, H.B. Synthesis of novel pyrazolo[3,4-d]pyrimidine derivatives as potential anti-breast cancer agents. Eur. J. Med. Chem. 2012, 57, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Vujasinovic, I.; Paravic-Radicevic, A.; Mlinaric-Majerski, K.; Brajsa, K.; Bertosa, B. Synthesis and biological validation of novel pyrazole derivatives with anticancer activity guided by 3D-QSAR analysis. Bioorg. Med. Chem. 2012, 20, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tomita, N.; Suzuki, Y.; Suzaki, T.; Kaku, T.; Hara, T.; Yamaoka, M.; Kanzaki, N.; Hasuoka, A.; Baba, A.; et al. Design, synthesis, and biological evaluation of 4-arylmethyl-1-phenylpyrazole and 4-aryloxy-1-phenylpyrazole derivatives as novel androgen receptor antagonists. Bioorg. Med. Chem. 2012, 20, 2338–2352. [Google Scholar] [CrossRef] [PubMed]
- Bavetsias, V.; Crumpler, S.; Sun, C.; Avery, S.; Atrash, B.; Faisal, A.; Moore, A.S.; Kosmopoulou, M.; Brown, N.; Sheldrake, P.W.; et al. Optimization of imidazo[4,5-b]pyridine based kinase inhibitors: Identification of a dual FLT3/Aurora kinase inhibitor as an orally bioavailable preclinical development candidate for the treatment of acute myeloid leukemia. J. Med. Chem. 2012, 55, 8721–8734. [Google Scholar] [CrossRef] [PubMed]
- Hanan, E.J.; van Abbema, A.; Barrett, K.; Blair, W.S.; Blaney, J.; Chang, C.; Eigenbrot, C.; Flynn, S.; Gibbons, P.; Hurley, C.A.; et al. Discovery of potent and selective pyrazolopyrimidine janus kinase 2 inhibitors. J. Med. Chem. 2012, 55, 10090–10107. [Google Scholar] [CrossRef]
- Newhouse, B.J.; Hansen, J.D.; Grina, J.; Welch, M.; Topalov, G.; Littman, N.; Callejo, M.; Martinson, M.; Galbraith, S.; Laird, E.R.; et al. Non-oxime pyrazole based inhibitors of B-Raf kinase. Bioorg. Med. Chem. Lett. 2011, 21, 3488–3492. [Google Scholar] [CrossRef]
- Zheng, L.-W.; Shao, J.-H.; Zhao, B.-X.; Miao, J.-Y. Synthesis of novel pyrazolo[1,5-a]pyrazin-4(5H)-one derivatives and their inhibition against growth of A549 and H322 lung cancer cells. Bioorg. Med. Chem. Lett. 2011, 21, 3909–3913. [Google Scholar] [CrossRef]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Rao, K.S.; Mohan, A.V.N.; Park, C.-Y.; Son, J.-Y.; Sheen, Y.Y.; Kim, D.-K. Synthesis and biological evaluation of 1-substituted-3-(6-methylpyridin-2-yl)-4-([1,2,4] triazolo[1,5-a]pyridin-6-yl)pyrazoles as transforming growth factor-b type-1 receptor kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 6049–6053. [Google Scholar] [CrossRef]
- Maggio, B.; Raimondi, M.V.; Raffa, D.; Plescia, F.; Cascioferro, S.; Cancemi, G.; Tolomeo, M.; Grimaudo, S.; Daidone, G. Synthesis and antiproliferative activity of 3-(2-chloroethyl)-5-methyl-6-phenyl-8-(trifluoromethyl)-5,6-dihydropyrazolo[3,4-f][1,2,3,5]tetrazepin-4-(3H)-one. Eur. J. Med. Chem. 2015, 96, 98–104. [Google Scholar] [CrossRef]
- Christodoulou, M.S.; Liekens, S.; Kasiotis, K.M.; Haroutounian, S.A. Novel pyrazole derivatives: Synthesis and evaluation of anti-angiogenic activity. Bioorg. Med. Chem. 2010, 18, 4338–4350. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.; Li, H.; Sun, J.; Zhou, Y.; Zhu, H. Synthesis and biological evaluation of pyrazole derivatives containing thiourea skeleton as anticancer agents. Bioorg. Med. Chem. 2010, 18, 4606–4614. [Google Scholar] [CrossRef]
- Niculescu-Duvaz, D.; Niculescu-Duvaz, I.; Suijkerbuijk, B.M.J.M.; Menard, D.; Zambon, A.; Nourry, A.; Davies, L.; Manne, H.A.; Friedlos, F.; Ogilvie, L.; et al. Novel tricyclic pyrazole BRAF inhibitors with imidazole or furan central scaffolds. Bioorg. Med. Chem. 2010, 18, 6934–6952. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Chiu, G.; Yu, Y.; Connolly, P.J.; Li, S.; Lu, Y.; Mary, A.; Angel, R.; Pesquera, F.; Stuart, L.; et al. Design, synthesis, and evaluation of 3, 4-disubstituted pyrazole analogues as anti-tumor CDK inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4557–4561. [Google Scholar] [CrossRef] [PubMed]
- Ghorab, M.M.; Ismail, Z.H.; Abdel-Gawad, S.M.; Abdel Aziem, A. Antimicrobial activity of amino acid, imidazole, and sulfonamide derivatives of pyrazolo[3, 4-d] pyrimidine. Heteroat. Chem. 2004, 15, 57. [Google Scholar] [CrossRef]
- Radek, J.; Havli, L.; McNae, I.W.; Walkinshaw, M.D.; Voller, J.; Sturc, A.; Navratilov, J.; Kuzma, M.; Mistrik, M.; Bartek, J.; et al. Pyrazolo[4, 3-d] pyrimidine bioisostere of roscovitine: Evaluation of a novel selective inhibitor of cyclin-dependent kinases with antiproliferative activity. J. Med. Chem. 2011, 54, 2980–2993. [Google Scholar]
- Dawood, K.M.; Eldebss, T.M.A.; El-Zahabi, H.S.A.; Yousef, M.H.; Metz, P. Synthesis of some new pyrazole-based 1,3-thiazoles and 1,3,4-thiadiazoles as anticancer agents. Eur. J. Med. Chem. 2013, 70, 740–749. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Singh, R.; Tandon, V.; Paul, K. Novel pyrazolo[3, 4-d] pyrimidine with 4-(1H-benzimidazol-2-yl)-phenylamine as broad spectrum anticancer agents: Synthesis, cell based assay, topoisomerase inhibition, DNA intercalation and bovine serum albumin studies. Eur. J. Med. Chem. 2017, 126, 24–35. [Google Scholar] [CrossRef]
- Krystof, V.; Cankar, P.; Frysová, I.; Slouka, J.; Kontopidis, G.; Dzubák, P.; Hajdúch, M.; Srovnal, J.; Azevedo, W.F., Jr.; Orság, M.; et al. 4-Arylazo-3, 5-diamino-1 H-pyrazole CDK inhibitors: SAR study, crystal structure in complex with CDK2, selectivity, and cellular effects. J. Med. Chem. 2006, 49, 6500. [Google Scholar] [CrossRef]
- Balbi, A.; Anzaldi, M.; Macciò, C.; Aiello, C.; Mazzei, M.; Gangemi, R.; Castagnola, P.; Miele, M.; Rosano, C.; Viale, M. Synthesis and biological evaluation of novel pyrazole derivatives with anticancer activity. Eur. J. Med. Chem. 2011, 46, 5293–5309. [Google Scholar] [CrossRef]
- Alam, R.; Wahi, D.; Singh, R.; Sinha, D.; Tandon, V.; Grover, A. Design, synthesis, cytotoxicity, HuTopoIIα inhibitory activity and molecular docking studies of pyrazole derivatives as potential anticancer agents. Bioorg. Chem. 2016, 69, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Q.; Yang, H.; He, B.; Jia, Y.K.; Meng, S.Y.; Zhang, C.; Liu, H.M.; Liu, F.W. A novel domino approach for synthesis of indolyl tetrahydropyrano[4, 3-c] pyrazole derivatives as anticancer agents. Tetrahedron 2016, 72, 5769–5775. [Google Scholar] [CrossRef]
- Minu, M.; Singh, D.; Mahaddalkar, T.; Lopus, M.; Winter, P.; Ayoub, A.T.; Missiaen, K.; Tilli, T.M.; Pasdar, M.; Tuszynski, J. Chemical synthesis, pharmacological evaluation and in silico analysis of new 2, 3, 3a, 4, 5, 6-hexahydrocyclopenta [c] pyrazole derivatives as potential anti-mitotic agents. Bioorg. Med. Chem. Lett. 2016, 26, 3855–3861. [Google Scholar] [CrossRef] [PubMed]
- Hafez, H.N.; El-Gazzar, A.R.; Al-Hussain, S.A. Novel pyrazole derivatives with oxa/thiadiazolyl, pyrazolyl moieties and pyrazolo[4, 3-d]-pyrimidine derivatives as potential antimicrobial and anticancer agents. Bioorg. Med. Chem. Lett. 2016, 26, 2428–2433. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.H.; Li, Q.S.; Ren, Z.L.; Chu, M.J.; Sun, J.; Zhang, X.; Xing, M.; Zhu, H.L.; Cao, H.Q. (E)-1, 3-diphenyl-1H-pyrazole derivatives containing O-benzyl oxime moiety as potential immunosuppressive agents: Design, synthesis, molecular docking and biological evaluation. Eur. J. Med. Chem. 2016, 108, 586–593. [Google Scholar] [CrossRef]
- Reddy, V.G.; Reddy, T.S.; Nayak, V.L.; Prasad, B.; Reddy, A.P.; Ravikumar, A.; Taj, S.; Kamal, A. Design, synthesis and biological evaluation of N-((1-benzyl-1H-1, 2, 3-triazol-4-yl) methyl)-1, 3-diphenyl-1H-pyrazole-4-carboxamides as CDK1/Cdc2 inhibitors. Eur. J. Med. Chem. 2016, 122, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Shin, I.; Nam, Y.; Kim, N.D.; Lee, K.B.; Sim, T. Identification of a novel 5-amino-3-(5-cyclopropylisoxazol-3-yl)-1-isopropyl-1H-pyrazole-4-carboxamide as a specific RET kinase inhibitor. Eur. J. Med. Chem. 2017, 125, 1145–1155. [Google Scholar] [CrossRef]
- Thaher, B.A.; Arnsmann, M.; Totzke, F.; Ehlert, J.E.; Kubbutat, M.H.; Schachtele, C.; Zimmermann, M.O.; Koch, P.; Boeckler, F.M.; Laufer, S.A. Tri-and Tetrasubstituted Pyrazole Derivates: Regioisomerism switches activity from p38MAP kinase to important cancer Kinases. J. Med. Chem. 2012, 55, 961–965. [Google Scholar] [CrossRef]
- Al-Suwaidan, I.A.; Abdel-Aziz, N.I.; El-Azab, A.S.; El-Sayed, M.A.; Alanazi, A.M.; El-Ashmawy, M.B.; Abdel-Aziz, A.A. Antitumor evaluation and molecular docking study of substituted 2-benzylidenebutane-1, 3-dione, 2-hydrazonobutane-1, 3-dione and trifluoromethyl-1H-pyrazole analogues. J. Enzym. Inhib. Med. Chem. 2015, 30, 679–687. [Google Scholar] [CrossRef]
- Cao, J.; Zang, J.; Ma, C.; Li, X.; Hou, J.; Li, J.; Huang, Y.; Xu, W.; Wang, B.; Zhang, Y. Design, Synthesis, And Biological Evaluation Of Pyrazoline-Based Hydroxamic Acid Derivatives As Aminopeptidase N (APN) Inhibitors. Chem. Med. Chem. 2018, 13, 431–436. [Google Scholar] [CrossRef]
- Mohan, G.; Sridhar, G.; Laxminarayana, E.; Chary, M. Synthesis and Biological Evaluation of 1,2,4-Oxadiazole Incorporated 1,2,3-Triazole-Pyrazole Derivatives As Anticancer Agents. Chem. Data Collect. 2021, 34, 100735. [Google Scholar]
- Mahmoud Youssef Moustafa, A. In vitro Anti Leukemia Cancer Activity of Some Novel Pyrazole Derivatives and Pyrazoles Containing Thiazole Moiety. Am. J. Heterocycl. Chem. 2019, 5, 55. [Google Scholar] [CrossRef][Green Version]
- Akhtar, W.; Marella, A.; Alam, M.; Khan, M.; Akhtar, M.; Anwer, T.; Khan, F.; Naematullah, M.; Azam, F.; Rizvi, M.; et al. Design and Synthesis Of Pyrazole–Pyrazoline Hybrids As Cancer-Associated Selective COX-2 Inhibitors. Arch. Pharm. 2020, 354, 2000116. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Knerr, L.; Akerud, T.; Hallberg, K.; Oster, L.; Rohman, M.; Osterlund, K.; Beisel, H.G.; Olsson, T.; Brengdhal, J.; et al. Discovery of a novel pyrazole series of group X secreted phospholipase A2 inhibitor (sPLA2X) via fragment based virtual screening. Bioorg. Med. Chem. Lett. 2014, 24, 5251–5255. [Google Scholar] [CrossRef] [PubMed]
- Shaveta, A.; Singh, M.; Kaur, S.; Sharma, R.; Bhatti, P. Rational design, synthesis and evaluation of chromone-indole and chromone-pyrazole based conjugates: Identification of a lead for anti-inflammatory drug. Eur. J. Med. Chem. 2014, 77, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.M. New pyrimido[5,4-e]pyrrolo[1,2-c]pyrimidines: Synthesis, 2D-QSAR, anti-inflammatory, analgesic and ulcerogenicity studies. Eur. J. Med. Chem. 2012, 55, 12–22. [Google Scholar] [CrossRef]
- Pandya, K.M.; Battula, S.; Patel, N.B. Design & Synthesis of InCl3 Catalyzed Novel Pyrazole Conjugates with 2°-Amines; Discover Their in Vitro Antimicrobial & DFT Studies. Polycycl. Aromat. Compd. 2022. [Google Scholar] [CrossRef]
- Oliveira, S.M.; Silva, C.R.; Wentz, A.P.; Paim, G.R.; Correa, M.S.; Bonacorso, H.G.; Prudente, A.S.; Otuki, M.F.; Ferreira, J. Antinociceptive effect of 3-(4-fluorophenyl)-5-trifluoromethyl-1H-1-tosylpyrazole. A celecoxib structural analogue in models of pathological pain. Pharmacol. Biochem. Behav. 2014, 124, 396–404. [Google Scholar] [CrossRef]
- El-Sayed, M.A.; Abdel-Aziz, N.I.; Abdel-Aziz, A.A.; El-Azab, A.S.; El-Tahir, K.E. Synthesis, biological evaluation and molecular modelling study of pyrazole and pyrazoline derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Part 2. Bioorg. Med. Chem. 2012, 20, 3306–3316. [Google Scholar] [CrossRef]
- Qiu, K.M.; Yan, R.; Xing, M.; Wang, H.H.; Cui, H.E.; Gong, H.B.; Zhu, H.L. Synthesis, biological evaluation and molecular modelling of dihydro-pyrazolylthiazolinone derivatives as potential COX-2 inhibitors. Bioorg. Med. Chem. 2012, 20, 6648–6654. [Google Scholar] [CrossRef]
- Hussain, S.; Kaushik, D. Novel 1-substituted-3, 5-dimethyl-4-[(substitute phenyl) diazenyl] pyrazole derivatives: Synthesis and pharmacological activity. J. Saudi Chem. Soc. 2015, 19, 274–281. [Google Scholar] [CrossRef]
- Brullo, C.; Spisani, S.; Selvatici, R.; Bruno, O. N-Aryl-2-phenyl-2, 3-dihydroimidazo[1,2-b]pyrazole-1-carboxamides 7-substituted strongly inhibiting both fMLP-OMe- and IL-8-induced human neutrophil chemotaxis. Eur. J. Med. Chem. 2012, 47, 573–579. [Google Scholar] [CrossRef]
- Naour, M.L.; Akgun, E.; Yekkirala, A.; Lunzer, M.M.; Powers, M.D.; Kalyuzhny, A.E.; Portoghese, P.S. Bivalent ligands that target mu opioid (MOP) and cannabinoid1 (CB1) receptors are potent analgesics devoid of tolerance. J. Med. Chem. 2013, 56, 5505–5513. [Google Scholar] [CrossRef] [PubMed]
- Balbi, A.; Anzaldi, M.; Mazzei, M.; Mariangela, M.; Dallegri, F.; Ottenello, L. Synthesized a compound 5-(2, 6, 6-Trimethyl-2-cyclohexen-1-yl) ethenyl-1H-pyrazole. Bioorg. Med. Chem. 2007, 14, 5152–5160. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Billinton, A.; Brown, S.H.; Clayton, N.M.; Chowdhury, A.; Giblin, G.M.P.; Goldsmith, P.; Hayhow, T.G.; Hurst, D.N.; Kilford, I.R.; et al. Non-acidic pyrazole EP 1 receptor antagonists with in vivo analgesic efficacy. Bioorg. Med. Chem. Lett. 2008, 18, 3392–3399. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Totre, J.V.; Gawande, S.S.; Khobragade, C.N.; Warangkar, S.C.; Kadam, P.D. Synthesis of novel 3,5-diaryl pyrazole derivatives using combinatorial chemistry as inhibitors of tyrosinase as well as potent anticancer, anti-inflammatory agents. Bioorg. Med. Chem. 2010, 18, 6149–6155. [Google Scholar] [CrossRef]
- Abdel-Hafez, E.M.N.; Abuo-Rahma, G.E.A.A.; Abdel-Aziz, M.; Radwan, M.F.; Hassan, H.F. Design, synthesis and biological investigation of certain pyrazole-3-carboxylic acid derivatives as novel carriers for nitric oxide. Bioorg. Med. Chem. 2009, 17, 3829–3837. [Google Scholar] [CrossRef]
- Gokhan-Kelekci, N.; Yabanoglu, S.; Kupeli, E.; Salgın, U.; Ozgen, O.; Ucar, G.; Yesilada, E.; Kendi, E.; Yesiladaf, A.; Bilgina, A.A. A new therapeutic approach in Alzheimer disease: Some novel pyrazole derivatives as dual MAO-B inhibitors and anti-inflammatory analgesics. Bioorg. Med. Chem. 2007, 15, 5775–5786. [Google Scholar] [CrossRef]
- Barsoum, F.F.; Girgis, A.S. Facile synthesis of bis (4, 5-dihydro-1H-pyrazole-1-carboxamides) and their thio-analogues of potential PGE2 inhibitory properties. Eur. J. Med. Chem. 2009, 31, 2172–2177. [Google Scholar] [CrossRef]
- Chandra, T.; Garg, N.; Lata, S.; Saxena, K.K.; Kumar, A. Synthesis of substituted acridinyl pyrazoline derivatives and their evaluation for anti-inflammatory activity. Eur. J. Med. Chem. 2010, 45, 1772–1776. [Google Scholar] [CrossRef]
- Ruiz, J.F.M.; Kedziora, K.; Keogh, B.; Maguire, J.; Reilly, M.; Windle, H.; Kelleher, D.P.; Gilmer, J.F. A double prodrug system for colon targeting of benzenesulfonamide COX-2 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 6636–6640. [Google Scholar] [CrossRef]
- Burguete, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Villar, R.; Vicente, E.; Solano, B.; Ancizu, S.; Perez-Silanes, S.; Aldanaa, I.; Monge, A. Synthesis and anti-inflammatory/antioxidant activities of some new ring substituted 3-phenyl-1-(1,4-di-N-oxide quinoxalin-2-yl)-2-propen-1-one derivatives and of their 4,5-dihydro-(1H)-pyrazole analogues. Bioorg. Med. Chem. Lett. 2007, 17, 6439–6443. [Google Scholar] [CrossRef]
- Sandeep, B.Y.; Saurabh, B.G.; Kamalkishor, G.B.; Rupesh, U.S. Novel 3-substituted-1-aryl-5-phenyl-6-anilinopyrazolo[3,4-d]pyrimidin-4-ones:Docking, synthesis and pharmacological evaluation as a potential anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2012, 22, 6616–6620. [Google Scholar]
- Hamdy, N.A.; Gamal-Eldeen, A.M. New pyridone, thioxopyridine, pyrazolopyridine and pyridine derivatives that modulate inflammatory mediators in stimulated RAW 264.7 murine macrophage. Eur. J. Med. Chem. 2009, 30, 4547–4556. [Google Scholar] [CrossRef] [PubMed]
- Sharath, V.; Kumar, H.V.; Naik, N. Synthesis of novel indole based scaffolds holding pyrazole ring as anti-inflammatory and antioxidant agents. J. Pharm. Res. 2013, 6, 785–790. [Google Scholar] [CrossRef]
- Thore, S.N.; Gupta, S.V.; Baheti, K.G. Novel ethyl-5-amino-3-methylthio-1H-pyrazole- 4-carboxylates: Synthesis and pharmacological activity. J. Chem. Soc. 2016, 20, 259–264. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Chavan, H.V.; Adsul, L.K.; Thakare, V.N.; Shringare, S.N.; Shaikh, R.; Gacche, R.N. Design, synthesis, characterization and biological evaluation of novel pyrazole integrated benzophenones. Bioorg. Med. Chem. Lett. 2013, 23, 912–916. [Google Scholar] [CrossRef]
- Keche, A.P.; Hatnapure, G.D.; Tale, R.H.; Rodgec, A.H.; Kamble, V.M. Synthesis, anti-inflammatory and antimicrobial evaluation of novel 1-acetyl-3,5-diaryl-4,5-dihydro (1H) pyrazole derivatives bearing urea, thiourea and sulfonamide moieties. Bioorg. Med. Chem. Lett. 2012, 22, 6611–6615. [Google Scholar] [CrossRef]
- Selvam, T.P.; Kumar, P.V.; Saravanan, G.; Prakash, C.R. Microwave-assisted synthesis, characterization and biological activity of novel pyrazole derivatives. J. Saudi Chem. Soc. 2014, 18, 1015–1021. [Google Scholar] [CrossRef]
- Nagarapu, L.; Mateti, J.; Gaikwad, H.K.; Bantu, R.; Rani, M.S.; Subhashini, N.J.P. Synthesis and anti-inflammatory activity of some novel 3-phenyl-N-[3-(4-phenylpiperazin-1yl) propyl]-1H-pyrazole-5-carboxamide derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 4138–4140. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Abdel-Aziem, T. Design, synthesis and biological evaluation of some pyrazole derivatives as anti-inflammatory-antimicrobial agents. Bioorg. Med. Chem. 2004, 12, 1935–1945. [Google Scholar] [CrossRef]
- Amir, M.; Kumar, S. Synthesis and anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation activities of 3,5-dimethyl pyrazoles, 3-methyl pyrazol-5-ones and 3,5-disubstituted pyrazolines. Indian J. Chem. 2005, 44, 2532–2537. [Google Scholar]
- Oruc, E.E.; Kocyigit-Kaymakcioglu, B.; Oral, B.; Altunbas-Toklu, H.Z.; Kabasakal, L.; Rollas, S. Synthesis of some novel azo derivatives of 3, 5-dimethly-1-(2-hydroxyethyl) pyrazole as potent analgesic agents. Arch. Pharm. Chem. Life Sci. 2006, 339, 267–272. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Ashour, H.M.A.; Abdel Ghany, Y.S.; El-Din, A.; Bekhit, A.; Baraka, A. Synthesis and biological evaluation of some thiazolyl and thiadiazolyl derivatives of 1H-pyrazole as anti-inflammatory antimicrobial agents. Eur. J. Med. Chem. 2008, 43, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.A.; Khaled Abdellatif, R.A.; Dong, Y.; Knaus, E.E. Synthesis of new4-[2-(4-methyl(amino)sulfonylphenyl)-5-trifluoromethyl-2H-pyrazol-3-yl]-1,2,3, 6-tetrahydropyridines:A search for novel nitric oxide donor anti-inflammatory agents. Bioorg. Med. Chem. 2008, 16, 8882–8888. [Google Scholar] [CrossRef] [PubMed]
- El-Din, M.M.; Senbel, A.M.; Bistawroos, A.A.; El-Mallah, A.; El-Din, N.A.N.; Bekhit, A.A.; Abd El Razik, H.A. A Novel COX-2 Inhibitor Pyrazole Derivative Proven Effective as an Anti-inflammatory and Analgesic Drug. Basic Clin. Pharmacol. Toxicol. 2010, 108, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Bekhit, A.A.; Fahmy, H.T.Y.; Rostom, S.A.F.; Bekhit, A.E.D.A. Synthesis and biological evaluation of some thiazolylpyrazole derivatives as dual anti-inflammatory antimicrobial agents. Eur. J. Med. Chem. 2010, 45, 6027–6038. [Google Scholar] [CrossRef]
- Youssef, A.M.; Neeland, E.G.; Villanueva, E.B.; White, M.S.; El-Ashmawy, I.M.; Patrick, B.; Klegeris, A.; Abd-El-Aziz, A.S. Synthesis and biological evaluation of novel pyrazole compounds. Bioorg. Med. Chem. 2010, 18, 5685–5696. [Google Scholar] [CrossRef]
- Shrivastava, P.; Singh, P.; Tewari, A.K. Synthesis of pyrazole-based 1, 5-diaryl compounds as potent anti-inflammatory agents. Med. Chem. Res. 2012, 21, 2465–2475. [Google Scholar] [CrossRef]
- Kucukguzel, S.G.; Coşkun, I.; Aydın, G.; Aktay, S.; Gursoy, O.; Cevik Ozakpınar, O.B.; Ozsavc, D.; Sener, A.; Kaushik-Basu, N.; Basu, A.; et al. Synthesis and characterization of celecoxib derivatives as possible anti-inflammatory, analgesic, antioxidant, anticancer and anti-HCV agents. Molecules 2013, 18, 3595–3614. [Google Scholar] [CrossRef]
- Çalışkan, B.; Yılmaz, A.; Evren, İ.; Menevşe, S.; Uludag, O.; Banoglu, E. Synthesis and evaluation of analgesic, anti-inflammatory, and anticancer activities of new pyrazole-3 (5)-carboxylic acid derivatives. Med. Chem. Res. 2013, 22, 782–793. [Google Scholar] [CrossRef]
- Chavan, H.V.; Bandgar, B.P.; Adsul, L.K.; Dhakane, V.D.; Bhale, P.S.; Thakare, V.N.; Masand, V. Design, synthesis, characterization and anti-inflammatory evaluation of novel pyrazole amalgamated flavones. Bioorg. Med. Chem. Lett. 2013, 23, 1315–1321. [Google Scholar] [CrossRef]
- Baytas, S.N.; Inceler, N.; Ozkan, Y.; Unlu, S.; Sahin, M.F. Novel 1, 3-diarylpyrazole acrylamides: Synthesis, antiplatelet activity screening, and in silico evaluation studies. Med. Chem. Res. 2013, 22, 5922–5933. [Google Scholar] [CrossRef]
- Ragab, F.A.; Abdel Gawad, N.M.; Georgey, H.H.; Said, M.F. Synthesis of novel 1, 3, 4-trisubstituted pyrazoles as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2013, 63, 645–654. [Google Scholar] [CrossRef]
- El-Sehemi, A.G.; Bondock, S.; Ammar, Y.A. Transformations of naproxen into pyrazolecarboxamides: Search for potent anti-inflammatory, analgesic and ulcerogenic agents. Med. Chem. Res. 2014, 23, 827–838. [Google Scholar] [CrossRef]
- Kumar, P.; Chandak, N.; Kaushik, P.; Sharma, C.; Kaushik, D.; Aneja, K.R.; Sharma, P.K. Benzenesulfonamide bearing pyrazolylpyrazolines: Synthesis and evaluation as anti-inflammatory-antimicrobial agents. Med. Chem. Res. 2014, 23, 882–895. [Google Scholar] [CrossRef]
- Mohammed, K.O.; Nissan, Y.M. Synthesis, Molecular Docking, and Biological Evaluation of Some Novel Hydrazones and Pyrazole Derivatives as Anti-inflammatory Agents. Chem. Biol. Drug Des. 2014, 84, 473–488. [Google Scholar] [CrossRef]
- Tewari, A.K.; Singh, V.P.; Yadav, P.; Gupta, G.; Singh, A.; Goel, R.K.; Shinde, P.; Mohan, C.G. Synthesis, biological evaluation and molecular modeling study of pyrazole derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Bioorg. Chem. 2014, 56, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Alegaon, S.G.; Alagawadi, K.R.; Garg, M.K.; Dushyant, K.; Vinod, D. 1, 3, 4-Trisubstituted pyrazole analogues as promising anti-inflammatory agents. Bioorg. Chem. 2014, 54, 51–59. [Google Scholar] [CrossRef]
- Bansal, S.; Bala, M.; Suthar, S.K.; Choudhary, S.; Bhattacharya, S.; Bhardwaj, V.; Singla, S.; Joseph, A. Design and synthesis of novel 2-phenyl-5-(1,3-diphenyl-1H-pyrazol-4-yl)-1,3,4-oxadiazoles as selective COX-2 inhibitors with potent anti-inflammatory activity. Eur. J. Med. Chem. 2014, 80, 167–174. [Google Scholar] [CrossRef]
- Wang, T.; Banerjee, D.; Bohnert, T.; Chao, J.; Enyedy, I.; Fontenot, J.; Guertin, K.; Jones, H.; Lin, E.Y.; Marcotte, D.; et al. Discovery of novel pyrazole-containing benzamides as potent RORγ inverse agonists. Bioorg. Med. Chem. Lett. 2015, 25, 2985–2990. [Google Scholar] [CrossRef]
- Li, Y.R.; Li, C.; Liu, J.C.; Guo, M.; Zhang, T.Y.; Sun, L.P.; Zheng, C.J.; Piao, H.R. Synthesis and biological evaluation of 1,3-diaryl pyrazole derivatives as potential antibacterial and anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2015, 25, 5052–5057. [Google Scholar] [CrossRef] [PubMed]
- El-Feky, S.A.; Abd El-Samii, Z.K.; Osman, N.A.; Lashine, J.; Kamel, M.A.; Thabet, H. Synthesis, molecular docking and anti-inflammatory screening of novel quinoline incorporated pyrazole derivatives using the Pfitzinger reaction II. Bioorg. Chem. 2015, 58, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Kendre, B.V.; Landge, M.G.; Bhusare, S.R. Synthesis and biological evaluation of some novel pyrazole, isoxazole, benzoxazepine, benzothiazepine and benzodiazepine derivatives bearing an aryl sulfonate moiety as antimicrobial and anti-inflammatory agents. Arab. J. Chem. 2019, 12, 2091–2097. [Google Scholar] [CrossRef]
- Kumar, R.S.; Arif, I.A.; Ahamed, A.; Idhayadhulla, A. Anti-inflammatory and antimicrobial activities of novel pyrazole analogues. Saudi J. Biol. Sci. 2016, 23, 614–620. [Google Scholar] [CrossRef]
- Pelcman, B.; Sanin, A.; Nilsson, P.; Schaal, W.; Olofsson, K.; Krog-Jensen, C.; Forsell, P.; Hallberg, A.; Larhed, M.; Boesen, T.; et al. N- Substituted pyrazole-3-carboxamides as inhibitors of human 15-lipoxygenase. Bioorg. Med. Chem. Lett. 2015, 25, 3017–3023. [Google Scholar] [CrossRef]
- Abdellatif, K.R.; Moawad, A.; Knaus, E.E. Synthesis of new 1-(4-methane(amino)sulfonylphenyl)-5-(4-substituted-aminomethylphenyl)-3-trifluoro methyl-1H-pyrazoles: A search for novel nitric oxide donor anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2014, 24, 5015–5021. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.A.; Al-Rashood, K.A.; El-Tahir, K.E.; Suddek, G.M. Synthesis of N-benzenesulfonamide-1H-pyrazoles bearing arylsulfonyl moiety: Novel celecoxib analogues as potent anti-inflammatory agents. Eur. J. Med. Chem. 2014, 80, 416–422. [Google Scholar] [CrossRef]
- Doma, A.; Kulkarni, R.; Palakodety, R.; Sastry, G.N.; Sridhara, J.; Garlapati, A. N-Pyrazole derivatives as potent inhibitors of c-Jun N-terminal kinase: Synthesis and SAR studies. Bioorg. Med. Chem. 2014, 22, 6209–6219. [Google Scholar] [CrossRef]
- Jadhav, S.Y.; Shirame, S.P.; Kulkarni, S.D.; Patil, S.B.; Pasale, S.K.; Bhosale, R.B. PEG mediated synthesis and pharmacological evaluation of some fluoro substituted pyrazoline derivatives as anti-inflammatory and analgesic agents. Bioorg. Med. Chem. Lett. 2013, 23, 2575–2578. [Google Scholar] [CrossRef]
- Vijesh, A.M.; Isloor, A.M.; Shetty, P.; Sundershan, S.; Fun, H.K. New pyrazole derivatives containing 1, 2, 4-triazoles and benzoxazoles as potent antimicrobial and analgesic agents. Eur. J. Med. Chem. 2013, 62, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Bansal, A.; Rozas, I.; Kelly, B.; Kaushik, P.; Kaushik, D. Synthesis, biological evaluation and molecular modeling study of 5-trifluoromethyl- Delta(2)-pyrazoline and isomeric 5/3-trifluoromethylpyrazole derivatives as anti-inflammatory agents. Eur. J. Med. Chem. 2013, 70, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Kamble, R.D.; Meshram, R.J.; Hese, S.V.; More, R.A.; Kamble, S.S.; Gacche, R.N.; Dawane, B.S. Synthesis and in silico investigation of thiazoles bearing pyrazoles derivatives as anti-inflammatory agents. Comput. Biol. Chem. 2016, 61, 86–96. [Google Scholar] [CrossRef]
- Chougala, B.M.; Samundeeswari, S.; Holiyachi, M.; Shastri, L.A.; Dodamani, S.; Jalalpure, S.; Dixit, S.R.; Joshi, S.D.; Sunagar, V.A. Synthesis, characterization and molecular docking studies of substituted 4-coumarinylpyrano[2, 3-c] pyrazole derivatives as potent antibacterial and anti-inflammatory agents. Eur. J. Med. Chem. 2017, 125, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Somakala, K.; Amir, M. Synthesis, characterization and pharmacological evaluation of pyrazolyl urea derivatives as potential anti-inflammatory agents. Acta Pharm. Sin. B 2017, 7, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Maddila, S.; Gorle, S.; Sampath, C.; Lavanya, P. Synthesis and anti-inflammatory activity of some new 1, 3, 4-thiadiazoles containing pyrazole and pyrrole nucleus. J. Saudi Chem. Soc. 2012, 20, S306–S312. [Google Scholar] [CrossRef]
- Abdellatif, K.R.; Fadaly, W.A. Design, synthesis, cyclooxygenase inhibition and biological evaluation of new 1, 3, 5-triaryl-4,5-dihydro-1H-pyrazole derivatives possessing amino/methanesulfonyl pharmacophore. Bioorg. Chem. 2017, 70, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Elshemy, H.A.H.; Abdelall, E.K.A.; Azouz, A.A.A.; Moawad, W.A.M.; Ali Safwat, N.M. Synthesis, anti-inflammatory, cyclooxygenases inhibitions assays and histopathological study of poly-substituted 1,3,5-triazines: Confirmation of regiospecific pyrazole cyclization by HMBC. Eur. J. Med. Chem. 2017, 127, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Levent, S.; Çalışkan, B.; Çiftçi, M.; Özkan, Y.; Yenicesu, İ.; Ünver, H.; Banoglu, E. Pyrazole derivatives as inhibitors of arachidonic acid-induced platelet aggregation. Eur. J. Med. Chem. 2013, 64, 42–53. [Google Scholar] [CrossRef]
- Unlu, S.; Banoglu, E.; Ito, S.; Niiya, T.; Eren, G.; Kçelik, B.; Şahin, M.F. Synthesis, characterization and preliminary screening of regioisomeric 1-(3-pyridazinyl)-3-arylpyrazole and 1-(3-pyridazinyl)-5-arylpyrazole derivatives towards cyclooxygenase inhibition. J. Enzym. Inhib. Med. Chem. 2007, 22, 351–361. [Google Scholar] [CrossRef]
- Bakr, R.B.; Azouz, A.A.; Abdellatif, K.R. Synthesis, cyclooxygenase inhibition, anti-inflammatory evaluation and ulcerogenic liability of new 1-phenylpyrazolo[3, 4-d] pyrimidine derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Chandak, N.; Kumar, P.; Kaushik, P.; Varshney, P.; Sharma, C.; Kaushik, D.; Jain, S.; Aneja, K.R.; Sharma, P.K. Dual evaluation of some novel 2-amino-substituted coumarinyl thiazoles as anti-inflammatory–antimicrobial agents and their docking studies with COX-1/COX-2 active sites. J. Enzym. Inhib. Med. Chem. 2014, 29, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Faour, W.H.; Mroueh, M.; Daher, C.F.; Elbayaa, R.Y.; Ragab, H.M.; Ghoneim, A.I.; El-mallah, A.I.; Ashour, H.M. Synthesis of some new amide-linked bipyrazoles and their evaluation as anti-inflammatory and analgesic agents. J. Enzym. Inhib. Med. Chem. 2016, 31, 1079–1094. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.J.; Alam, O.; Khan, S.A.; Naim, M.J.; Islamuddin, M.; Deora, G.S. Synthesis, anti-inflammatory, analgesic, COX-1/2-inhibitory activity, and molecular docking studies of hybrid pyrazole analogues. Drug Des. Devel. Ther. 2016, 10, 3529–3543. [Google Scholar] [CrossRef] [PubMed]
- Bekhit, A.; Nasralla, S.; El-Agroudy, E.; Hamouda, N.; El-Fattah, A.; Bekhit, S.; Amagase, K.; Ibrahim, T. Investigation of The Anti-Inflammatory And Analgesic Activities Of Promising Pyrazole Derivative. Eur. J. Pharm. Sci. 2021, 168, 106080. [Google Scholar] [CrossRef]
- Hassan, G.; Abdel Rahman, D.; Abdelmajeed, E.; Refaey, R.; Alaraby Salem, M.; Nissan, Y. New Pyrazole Derivatives: Synthesis, Anti-Inflammatory Activity, Cycloxygenase Inhibition Assay and Evaluation of Mpges. Eur. J. Med. Chem. 2019, 171, 332–342. [Google Scholar] [CrossRef]
- Assali, M.; Abualhasan, M.; Sawaftah, H.; Hawash, M.; Mousa, A. Synthesis, Biological Activity, And Molecular Modeling Studies Of Pyrazole And Triazole Derivatives As Selective COX-2 Inhibitors. J. Chem. 2020, 2020, 6393428. [Google Scholar] [CrossRef]
- Tan, X.; Li, C.; Yang, R.; Zhao, S.; Li, F.; Li, X.; Chen, L.; Wan, X.; Liu, X.; Yang, T.; et al. Discovery of Pyrazolo[3,4-D]Pyridazinone Derivatives As Selective DDR1 Inhibitors Via Deep Learning Based Design, Synthesis, And Biological Evaluation. J. Med. Chem. 2022, 65, 103–119. [Google Scholar] [CrossRef]



















































































































































































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).