
An Efficient Approach to Large-Scale Ab Initio Conformational Energy Profiles of Small Molecules


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
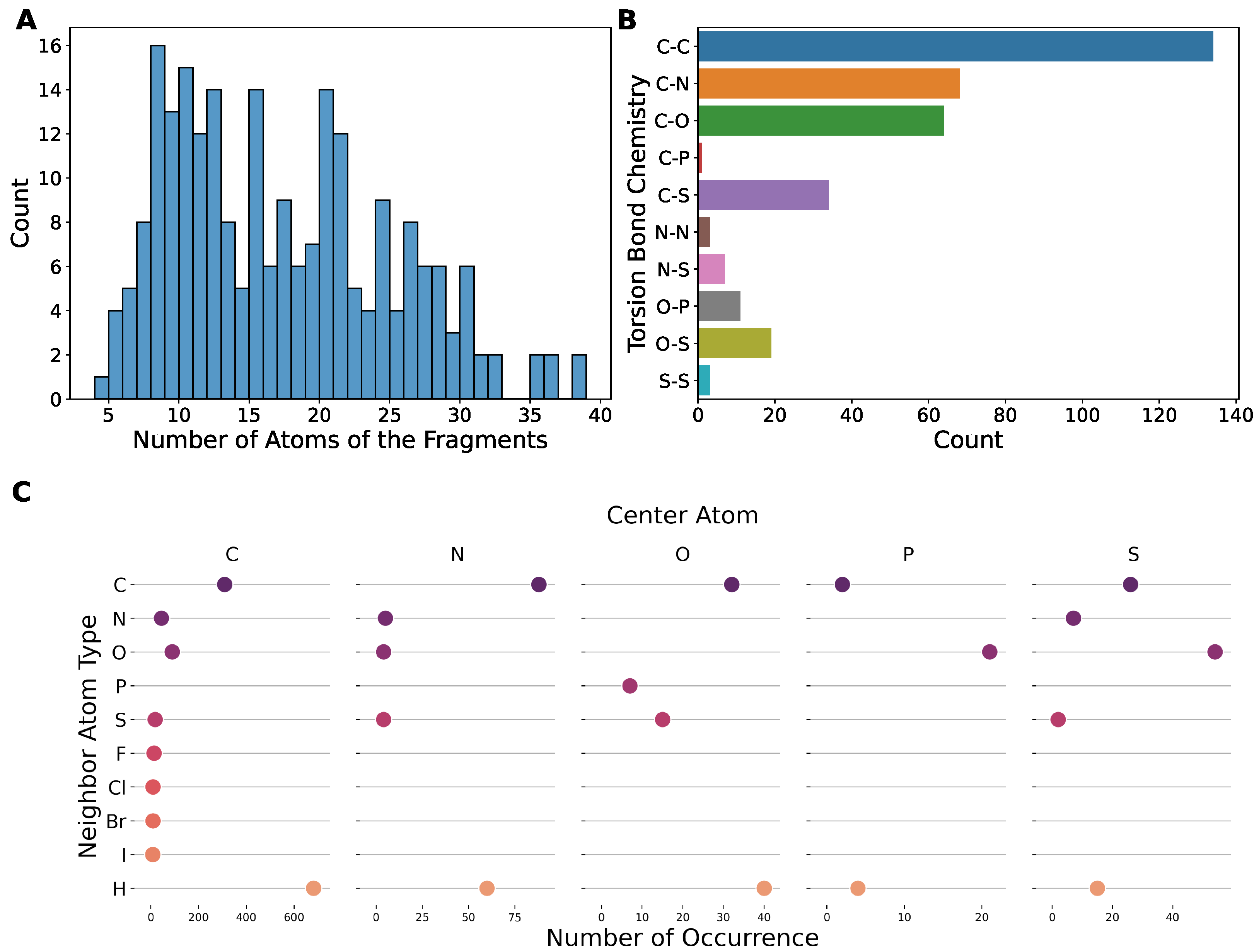
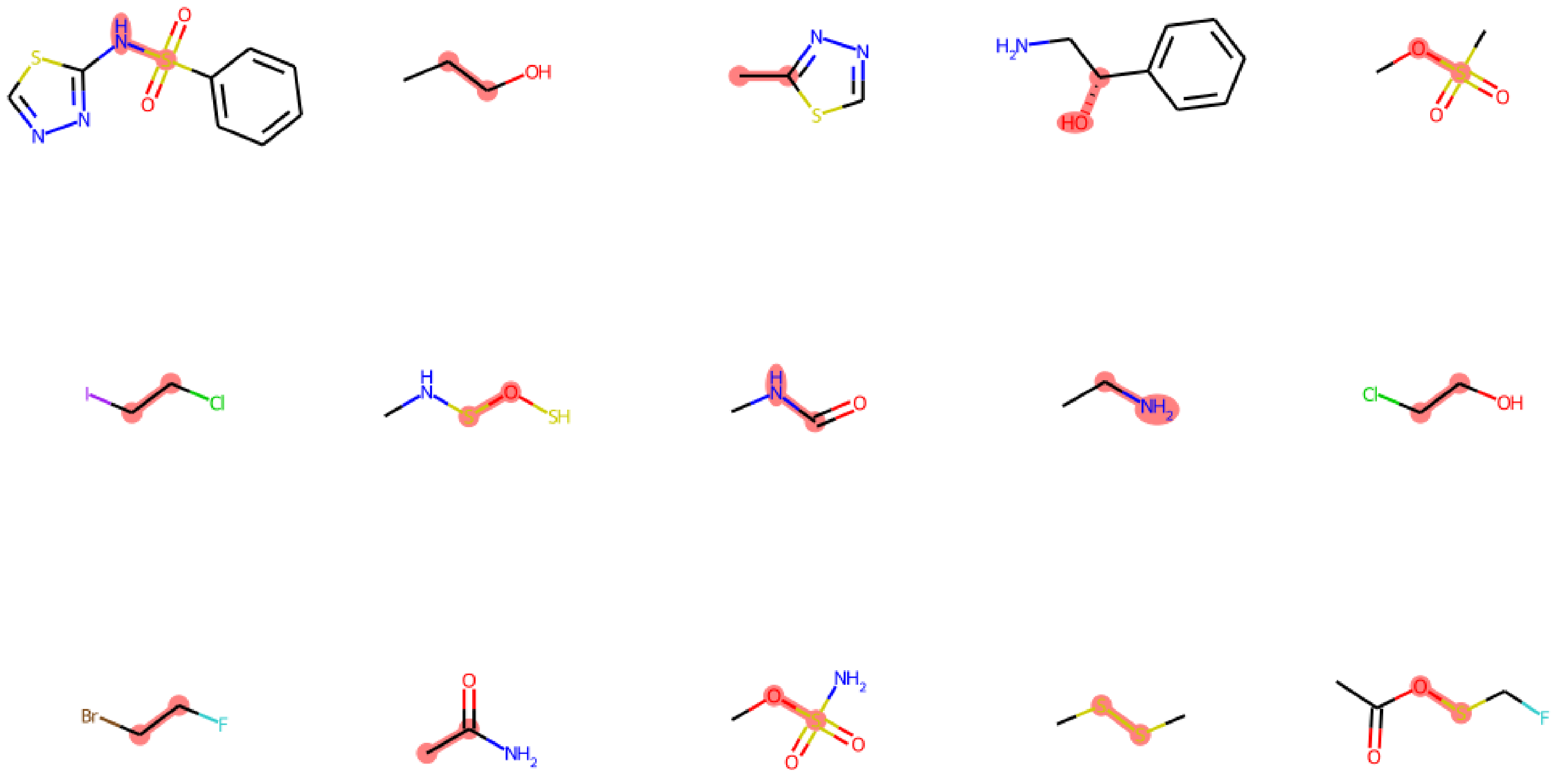
2.1. Our Dataset Has a Broad Coverage of Chemical Space
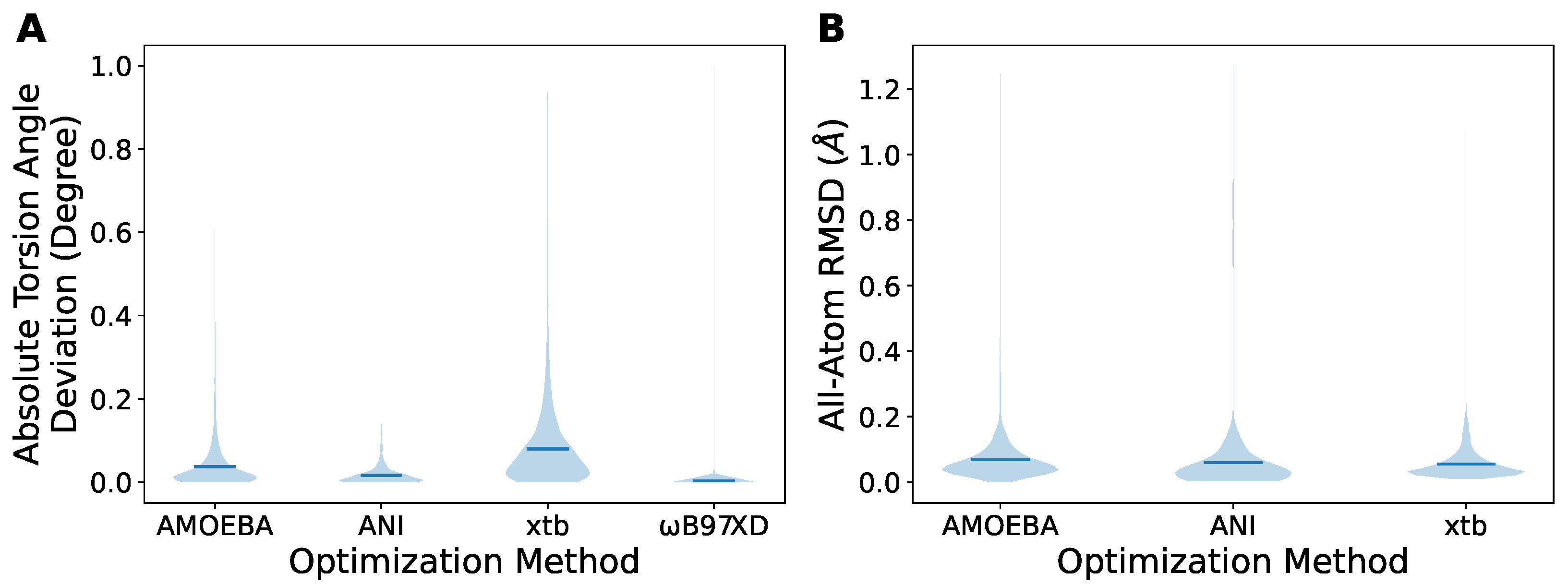
2.2. Cheaper Optimization Methods Can Also Provide Acceptable Geometry
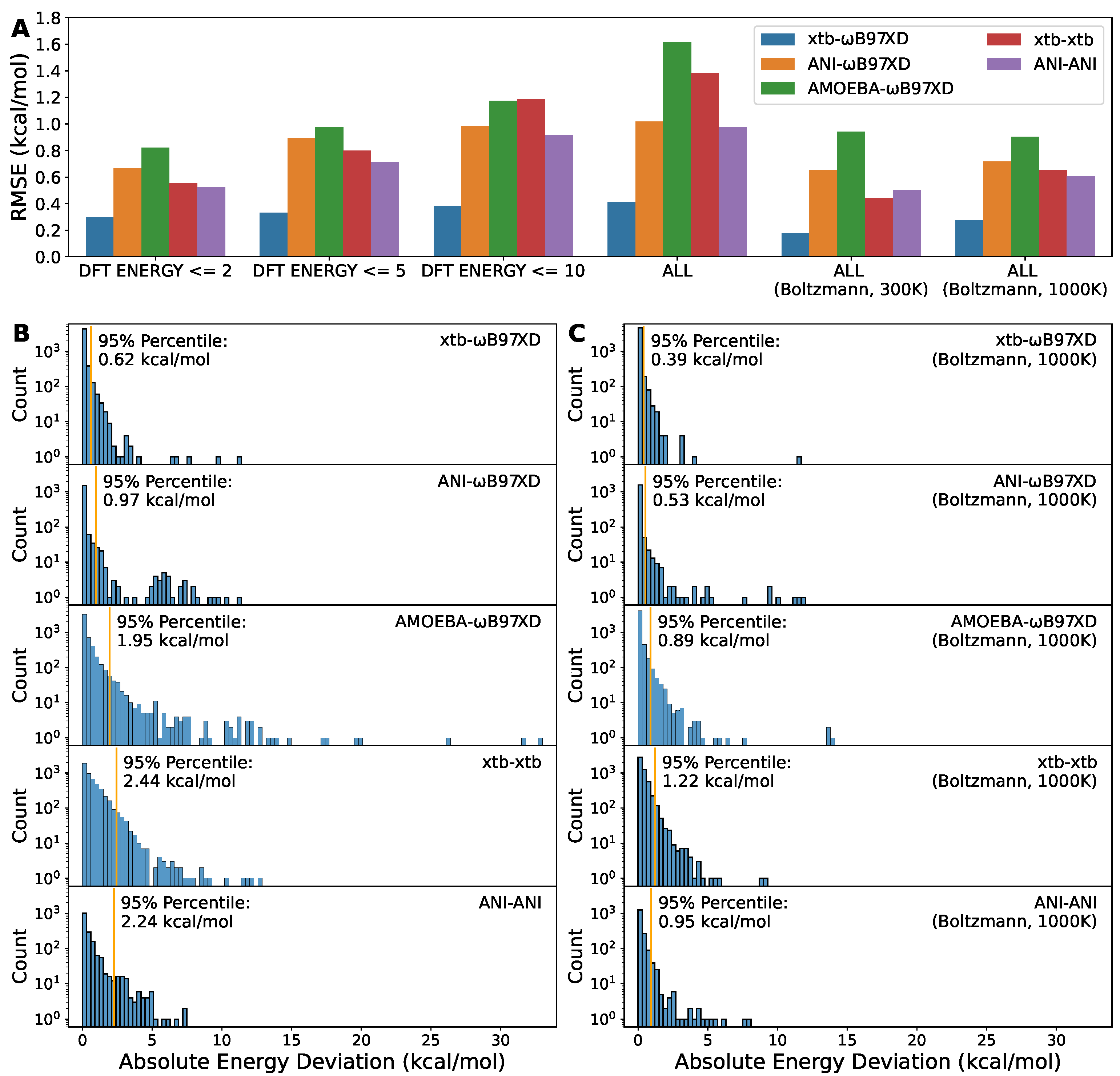
2.3. DFT Method Is Still Necessary to Obtain Satisfactory Torsion Energy Profile
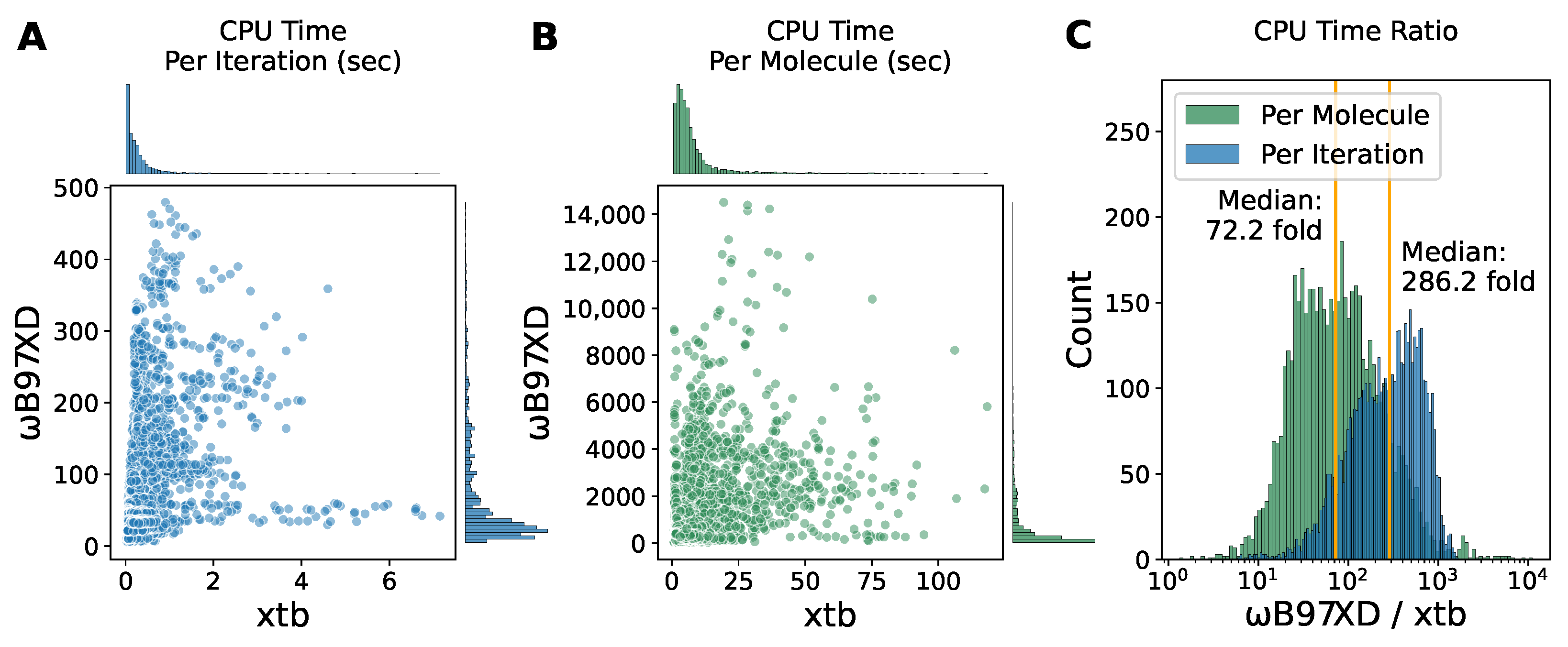
2.4. Inexpensive Computational Cost Highlights the Advantage of the Semi-Empirical Method xtb
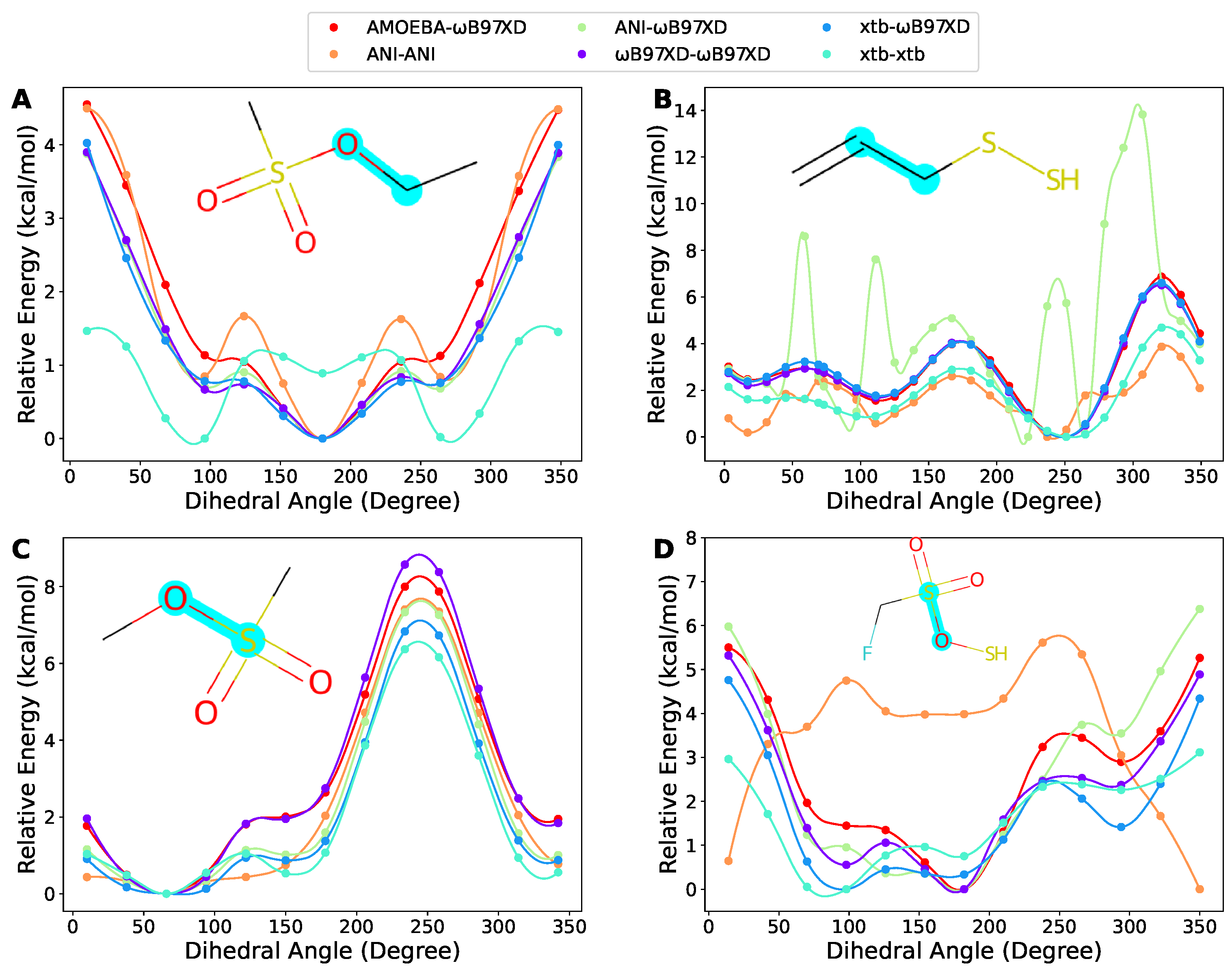
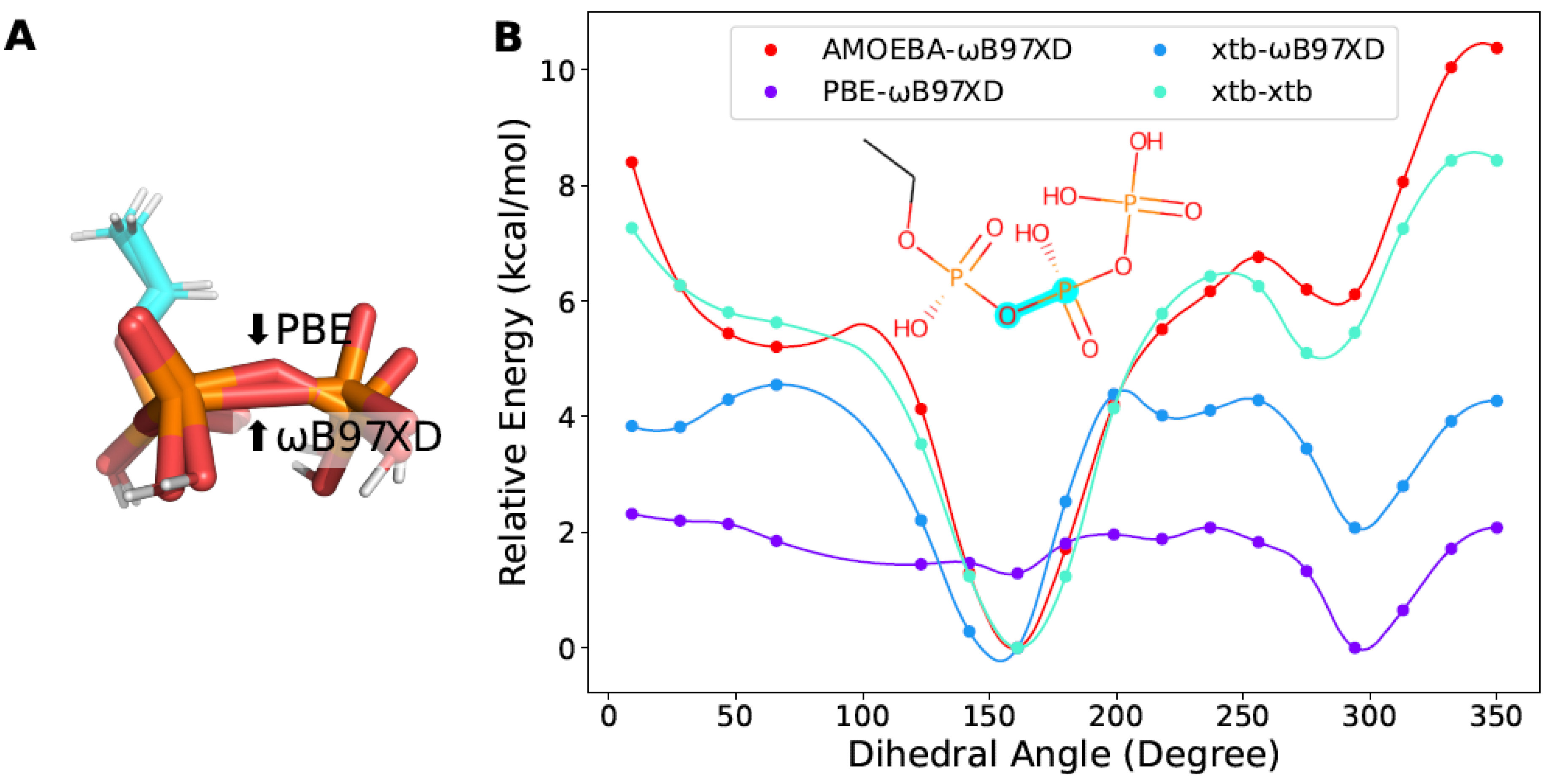
2.5. Case Discussion
3. Materials and Methods
3.1. Dataset
3.2. Computational Details
3.3. Metrics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wiberg, K.B. The concept of strain in organic chemistry. Angew. Chem. Int. Ed. Engl. 1986, 25, 312–322. [Google Scholar] [CrossRef]
- Gonthier, J.F.; Steinmann, S.N.; Wodrich, M.D.; Corminboeuf, C. Quantification of “fuzzy” chemical concepts: A computational perspective. Chem. Soc. Rev. 2012, 41, 4671–4687. [Google Scholar] [CrossRef] [PubMed]
- Peach, M.L.; Cachau, R.E.; Nicklaus, M.C. Conformational energy range of ligands in protein crystal structures: The difficult quest for accurate understanding. J. Mol. Recognit. 2017, 30, e2618. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Casarini, D. Recent trends in conformational analysis. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 613–641. [Google Scholar] [CrossRef]
- Boyd, D.B.; Lipkowitz, K.B. Molecular mechanics: The method and its underlying philosophy. J. Chem. Educ. 1982, 59, 269. [Google Scholar] [CrossRef]
- Engler, E.M.; Andose, J.D.; Schleyer, P.V. Critical evaluation of molecular mechanics. J. Am. Chem. Soc. 1973, 95, 8005–8025. [Google Scholar] [CrossRef]
- Hehre, W.J. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction: Irvine, CA, USA, 2003; Volume 2. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Zhang, C.; Lu, C.; Jing, Z.; Wu, C.; Piquemal, J.P.; Ponder, J.W.; Ren, P. AMOEBA polarizable atomic multipole force field for nucleic acids. J. Chem. Theory Comput. 2018, 14, 2084–2108. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xia, Z.; Zhang, J.; Best, R.; Wu, C.; Ponder, J.W.; Ren, P. Polarizable atomic multipole-based AMOEBA force field for proteins. J. Chem. Theory Comput. 2013, 9, 4046–4063. [Google Scholar] [CrossRef] [PubMed]
- Kania, A.; Sarapata, K.; Gucwa, M.; Wójcik-Augustyn, A. Optimal solution to the torsional coefficient fitting problem in force field parametrization. J. Phys. Chem. A 2021, 125, 2673–2681. [Google Scholar] [CrossRef]
- Zgarbová, M.; Rosnik, A.M.; Luque, F.J.; Curutchet, C.; Jurečka, P. Transferability and additivity of dihedral parameters in polarizable and nonpolarizable empirical force fields. J. Comput. Chem. 2015, 36, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Roux, B. Automated force field parameterization for nonpolarizable and polarizable atomic models based on ab initio target data. J. Chem. Theory Comput. 2013, 9, 3543–3556. [Google Scholar] [CrossRef] [PubMed]
- Morado, J.; Mortenson, P.N.; Verdonk, M.L.; Ward, R.A.; Essex, J.W.; Skylaris, C.K. Paramol: A package for automatic parameterization of molecular mechanics force fields. J. Chem. Inf. Model. 2021, 61, 2026–2047. [Google Scholar] [CrossRef]
- Wu, J.C.; Chattree, G.; Ren, P. Automation of AMOEBA polarizable force field parameterization for small molecules. Theor. Chem. Acc. 2012, 131, 1–11. [Google Scholar] [CrossRef]
- Walker, B.; Liu, C.; Wait, E.; Ren, P. Automation of AMOEBA polarizable force field for small molecules: Poltype 2. J. Comput. Chem. 2022, 43, 1530–1542. [Google Scholar] [CrossRef]
- Ponder, J.W.; Wu, C.; Ren, P.; Pande, V.S.; Chodera, J.D.; Schnieders, M.J.; Haque, I.; Mobley, D.L.; Lambrecht, D.S.; DiStasio, R.A., Jr.; et al. Current status of the AMOEBA polarizable force field. J. Phys. Chem. B 2010, 114, 2549–2564. [Google Scholar] [CrossRef]
- Schrödinger Release 2022-3: FEP+; Schrödinger, LLC: New York, NY, USA, 2021.
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Isayev, O.; Roitberg, A.E. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192–3203. [Google Scholar] [CrossRef] [PubMed]
- Devereux, C.; Smith, J.S.; Huddleston, K.K.; Barros, K.; Zubatyuk, R.; Isayev, O.; Roitberg, A.E. Extending the applicability of the ANI deep learning molecular potential to sulfur and halogens. J. Chem. Theory Comput. 2020, 16, 4192–4202. [Google Scholar] [CrossRef] [PubMed]
- Glick, Z.L.; Metcalf, D.P.; Koutsoukas, A.; Spronk, S.A.; Cheney, D.L.; Sherrill, C.D. AP-Net: An atomic-pairwise neural network for smooth and transferable interaction potentials. J. Chem. Phys. 2020, 153, 044112. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Zheng, H.; Yu, K. A Scalable Graph Neural Network Method for Developing an Accurate Force Field of Large Flexible Organic Molecules. J. Phys. Chem. Lett. 2021, 12, 7982–7987. [Google Scholar] [CrossRef]
- Glick, Z.L.; Koutsoukas, A.; Cheney, D.L.; Sherrill, C.D. Cartesian message passing neural networks for directional properties: Fast and transferable atomic multipoles. J. Chem. Phys. 2021, 154, 224103. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, P.; Chatterjee, P.; MacKerell, A.D., Jr. Deep Neural Network Model to Predict the Electrostatic Parameters in the Polarizable Classical Drude Oscillator Force Field. J. Chem. Theory Comput. 2022, 18, 1711–1725. [Google Scholar] [CrossRef]
- Park, C.W.; Kornbluth, M.; Vandermause, J.; Wolverton, C.; Kozinsky, B.; Mailoa, J.P. Accurate and scalable graph neural network force field and molecular dynamics with direct force architecture. NPJ Comput. Mater. 2021, 7, 1–9. [Google Scholar] [CrossRef]
- Smith, J.S.; Zubatyuk, R.; Nebgen, B.; Lubbers, N.; Barros, K.; Roitberg, A.E.; Isayev, O.; Tretiak, S. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 2020, 7, 1–10. [Google Scholar] [CrossRef]
- Donchev, A.G.; Taube, A.G.; Decolvenaere, E.; Hargus, C.; McGibbon, R.T.; Law, K.H.; Gregersen, B.A.; Li, J.L.; Palmo, K.; Siva, K.; et al. Quantum chemical benchmark databases of gold-standard dimer interaction energies. Sci. Data 2021, 8, 1–9. [Google Scholar] [CrossRef]
- Eastman, P.; Behara, P.K.; Dotson, D.L.; Galvelis, R.; Herr, J.E.; Horton, J.T.; Mao, Y.; Chodera, J.D.; Pritchard, B.P.; Wang, Y.; et al. SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials. arXiv 2022, arXiv:2209.10702. [Google Scholar]
- Axelrod, S.; Gomez-Bombarelli, R. GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Sci. Data 2022, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Nebgen, B.; Lubbers, N.; Isayev, O.; Roitberg, A.E. Less is more: Sampling chemical space with active learning. J. Chem. Phys. 2018, 148, 241733. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260. [Google Scholar]
- Gaus, M.; Cui, Q.; Elstner, M. DFTB3: Extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB). J. Chem. Theory Comput. 2011, 7, 931–948. [Google Scholar] [CrossRef] [PubMed]
- Sharapa, D.I.; Genaev, A.; Cavallo, L.; Minenkov, Y. A robust and cost-efficient scheme for accurate conformational energies of organic molecules. ChemPhysChem 2019, 20, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods II. Applications. J. Comput. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 2008, 120, 215–241. [Google Scholar]
- Kesharwani, M.K.; Karton, A.; Martin, J.M. Benchmark ab initio conformational energies for the proteinogenic amino acids through explicitly correlated methods. Assessment of density functional methods. J. Chem. Theory Comput. 2016, 12, 444–454. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. 2022, 134, e202205735. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Gordon, M.S.; Binkley, J.S.; Pople, J.A.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular-orbital methods. 22. Small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 2797–2803. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent molecular orbital methods. XII. Further extensions of Gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Curtiss, L.A.; McGrath, M.P.; Blaudeau, J.P.; Davis, N.E.; Binning Jr, R.C.; Radom, L. Extension of Gaussian-2 theory to molecules containing third-row atoms Ga–Kr. J. Chem. Phys. 1995, 103, 6104–6113. [Google Scholar] [CrossRef]
- Glukhovtsev, M.N.; Pross, A.; McGrath, M.P.; Radom, L. Extension of Gaussian-2 (G2) theory to bromine-and iodine-containing molecules: Use of effective core potentials. J. Chem. Phys. 1995, 103, 1878–1885. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+ G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- McLean, A.; Chandler, G. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z= 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Spitznagel, G.W.; Clark, T.; von Ragué Schleyer, P.; Hehre, W.J. An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na-Cl. J. Comput. Chem. 1987, 8, 1109–1116. [Google Scholar] [CrossRef]
- Smith, D.G.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Gao, X.; Ramezanghorbani, F.; Isayev, O.; Smith, J.S.; Roitberg, A.E. TorchANI: A free and open source PyTorch-based deep learning implementation of the ANI neural network potentials. J. Chem. Inf. Model. 2020, 60, 3408–3415. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Walker, B.D.; Liu, C.; Ren, P. An Efficient Approach to Large-Scale Ab Initio Conformational Energy Profiles of Small Molecules. Molecules 2022, 27, 8567. https://doi.org/10.3390/molecules27238567
Wang Y, Walker BD, Liu C, Ren P. An Efficient Approach to Large-Scale Ab Initio Conformational Energy Profiles of Small Molecules. Molecules. 2022; 27(23):8567. https://doi.org/10.3390/molecules27238567
Chicago/Turabian StyleWang, Yanxing, Brandon Duane Walker, Chengwen Liu, and Pengyu Ren. 2022. "An Efficient Approach to Large-Scale Ab Initio Conformational Energy Profiles of Small Molecules" Molecules 27, no. 23: 8567. https://doi.org/10.3390/molecules27238567
APA StyleWang, Y., Walker, B. D., Liu, C., & Ren, P. (2022). An Efficient Approach to Large-Scale Ab Initio Conformational Energy Profiles of Small Molecules. Molecules, 27(23), 8567. https://doi.org/10.3390/molecules27238567