
Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties


Abstract
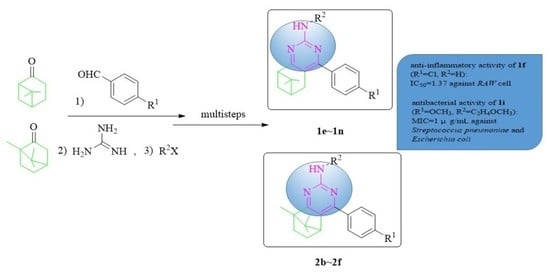
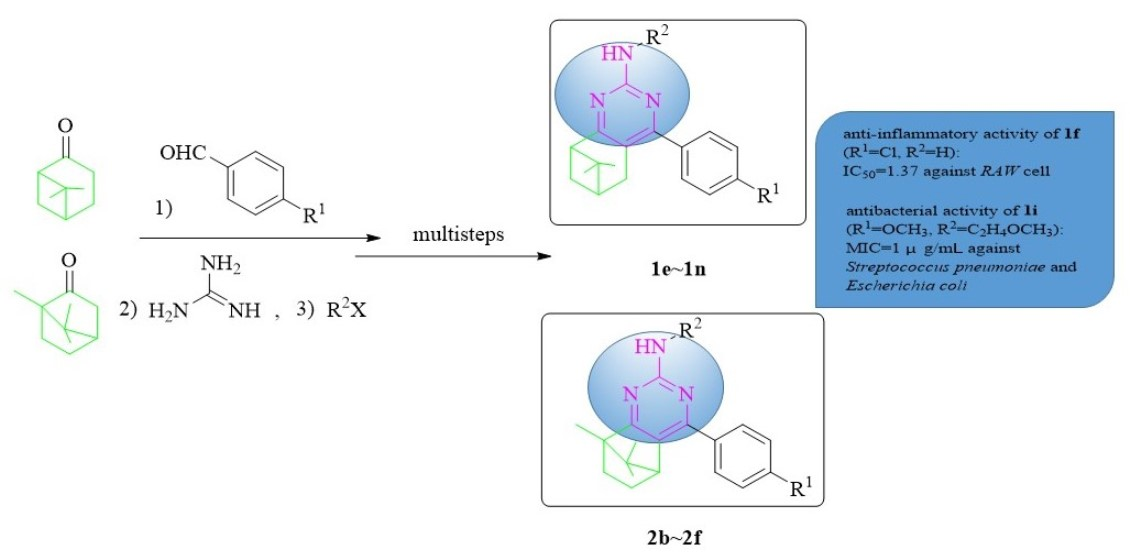
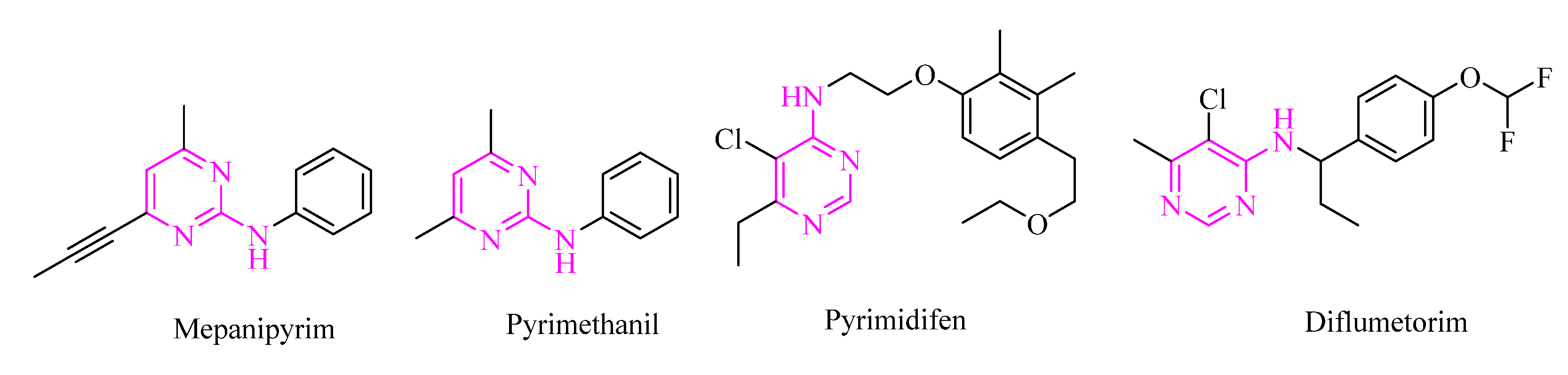
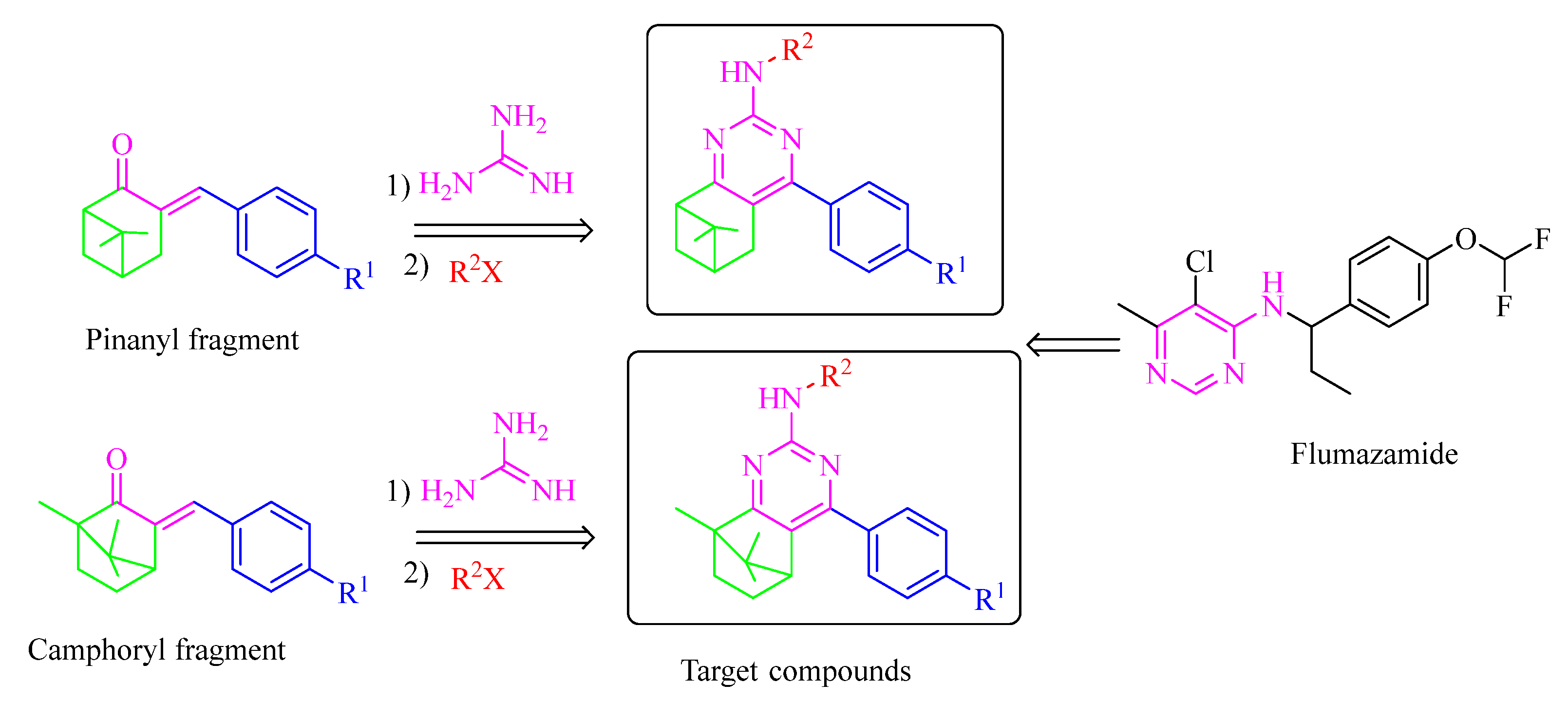
1. Introduction
2. Results and Discussion
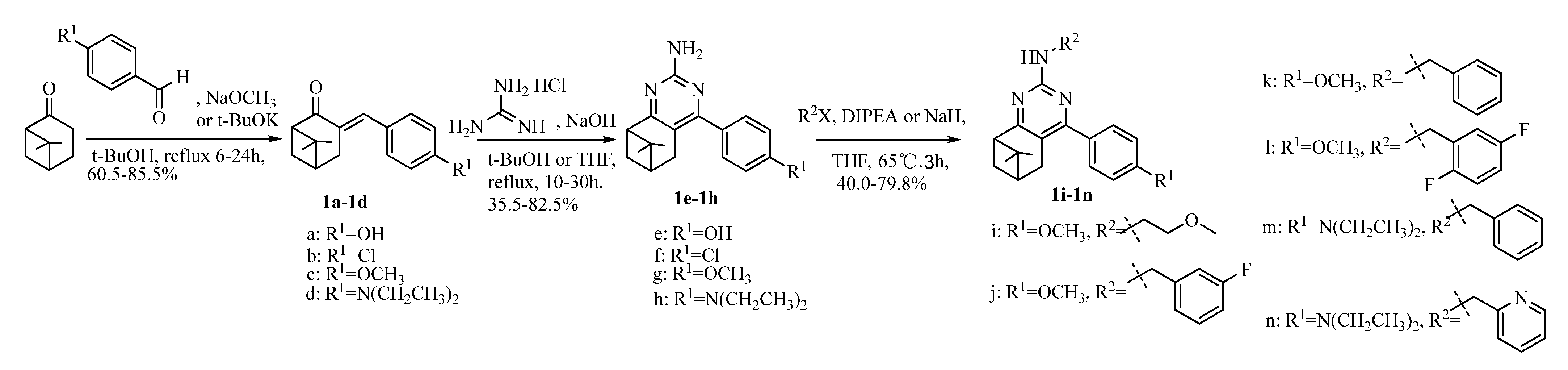
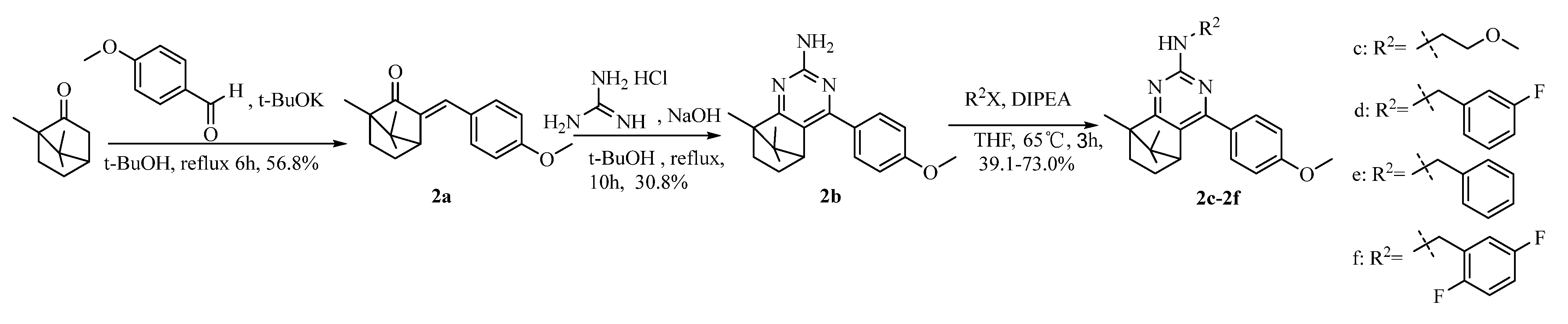
2.1. Chemistry
2.2. Structural Characterization of Compounds
2.3. Biological Evaluation
2.3.1. Antibacterial Activity Assay
2.3.2. Anti-Inflammatory Activity Assay
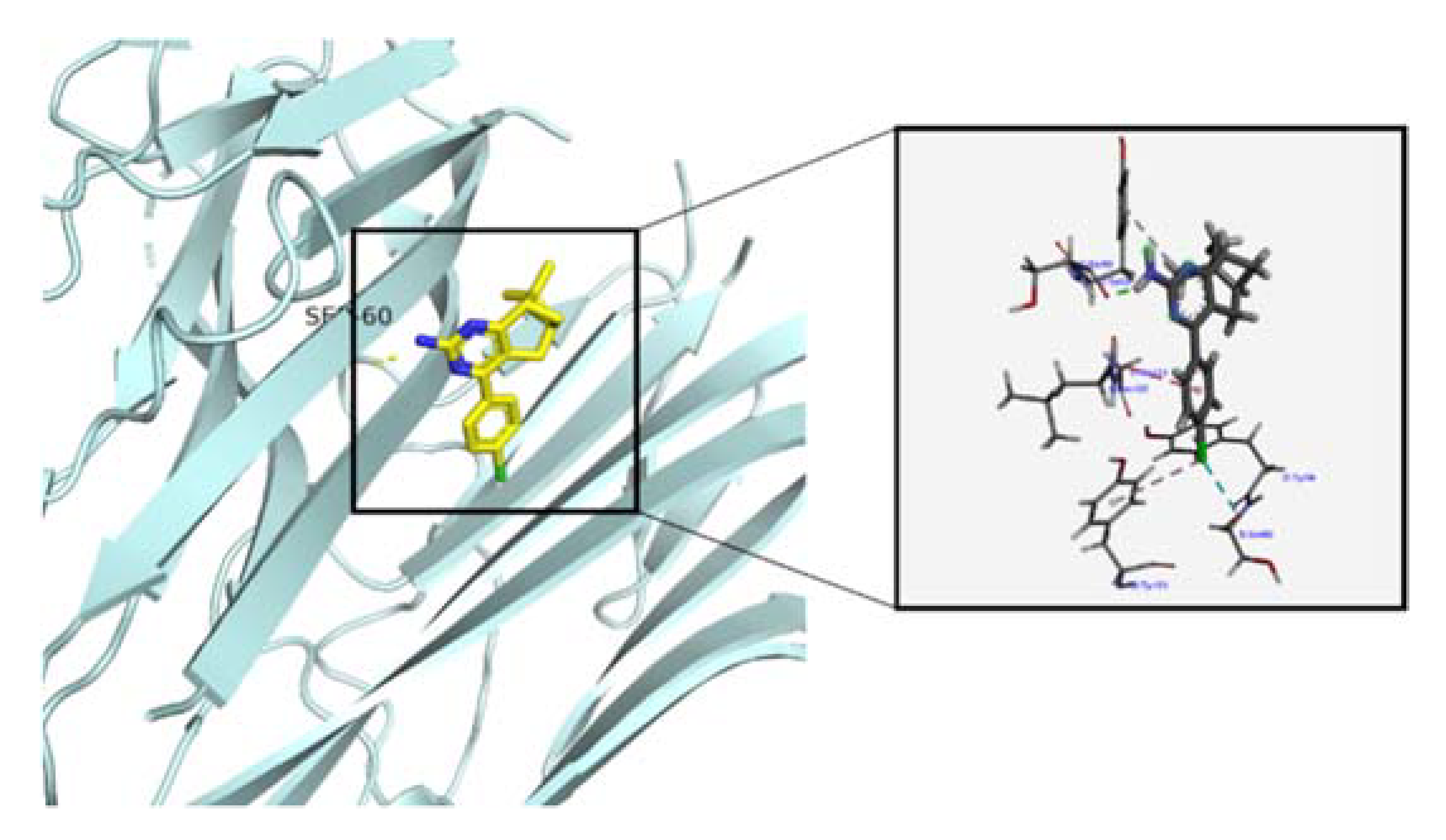
2.3.3. Molecular Docking Study
3. Conclusions
4. Experimental Section
4.1. Chemistry
4.1.1. Synthesis of Intermediate 3-Arylbenzylidene Nopinones 1a~1d
4.1.2. Synthesis of Pinanealkylpyrimidineamines 1e~1n
4.1.3. Synthesis of Pinanylpyrimidinamines 1i~1n
4.1.4. Synthesis of Intermediate 3-(4-Methoxybenzylidene)-1, 7, 7-Trimethylbicyclo [2.2.1] Heptan-2-One (2a)
4.1.5. Synthesis of Camphoryl Pyrimidine Amines 2b~2f
4.2. Antibacterial Activities Assay
4.3. Anti-Inflammatory Activities Assay
4.4. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giovanny, C.S.; Lucas, M.M.; Bruna, A.B.; Vitor, F.M.; Luiz, C.S.; Bruno, H.S. Heterocyclic compounds as antiviral drugs: Synthesis, structure–activity relationship and traditional applications. J. Heterocyclic. Chem. 2021, 58, 2226–2260. [Google Scholar]
- Liu, X.H.; Wang, Q.; Sun, Z.H.; David, E.W.; James, J.B.; Alden, S.E.; Tan, C.X.; Weng, J.Q. Synthesis and insecticidal activity of novel pyrimidine derivatives containing urea pharmacophore against Aedes aegypti. Pest Manag. Sci. 2017, 73, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Tenma, N.; Dind, B.P.; Kogaku, K.; Yukiko, Y.; Tomoya, U.; Takaya, M.; Ryosuke, K.; Shihori, H.; Yuki, E.; Shoya, Y.; et al. Total synthesis of cynaropicrin. Org. Biomol. Chem. 2021, 19, 6038–6044. [Google Scholar]
- Santosh, S.U.; Navanath, J.V.; Ajinkya, A.P.; Dattatraya, K.J.; Sunil, S.V.; Laxman, S.W.; Govind, B.K.; Deshmukh, M.B.; Prashant, V.A. Synthesis, anti-inflammatory, ulcerogenic and cyclooxygenase activities of indenopyrimidine derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 814–818. [Google Scholar]
- Scarim, C.B.; Pavan, F.R. Recent advancement in drug development of nitro(no2)-heterocyclic compounds as lead scaffolds for the treatment of mycobacterium tuberculosis. Drug Develop. Res. 2022, 83, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Türe, A.; Ergül, M.; Ergül, M.; Altun, A.; Küçükgüzel, I. Design, synthesis, and anticancer activity of novel 4-thiazolidinone-phenylaminopyrimidine hybrids. Mol. Divers. 2021, 25, 1025–1050. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.J.; Yang, Q. Development of a practical synthesis of a pyrimidine derivative with fungicidal activity. Org. Process. Res. Dev. 2019, 23, 2157–2165. [Google Scholar] [CrossRef]
- Sharga, B.M.; Krivovjaz, A.O.; Slivka, M.V.; Lambruch, L.M.; Cheypesh, A.V.; Lendel, V.G.; Nikolaychuk, V.I.; Markovich, V.P. Synthesis and antimicrobial activity of phenylselenyl tribromide and its fused thienopyrimidine derivatives. Farmacia 2016, 64, 512–520. [Google Scholar]
- Weig, A.W.; Barlock, S.L.; O’Connor, P.M.; Marciano, O.M.; Smith, R.; Ernst, R.K.; Melander, R.J.; Melander, C. A scaffold hopping strategy to generate new aryl-2-amino pyrimidine MRSA biofilm inhibitors. RSC Med. Chem. 2021, 12, 293–296. [Google Scholar] [CrossRef] [PubMed]
- El-Dean, A.M.K.; Abd-Ella, A.A.; Hassanien, R.; El-Sayed, M.E.A.; Zaki, R.M.; Abdel-Raheem, S.A.A. Chemical design and toxicity evaluation of new pyrimidothienotetrahydroisoquinolines as potential in-secticidal agent. Toxicol. Rep. 2019, 6, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.Z.; Liu, A.P.; Huang, M.Z.; Liu, M.H.; Pei, H.; Huang, L.; Yi, H.B.; Liu, W.D.; Hu, A.X. Design, synthesis, DFT study and antifungal activity of the derivatives of pyrazolecarboxamide containing thiazole or oxazole ring. Eur. J. Med. Chem. 2018, 149, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Hassaneen, H.M.; Saleh, F.M.; Abdallah, T.A.; Mohamed, Y.S.; Awad, E.M. Synthesis, reactions, and antimicrobial activity of some novel pyrazolo[3,4-d]pyrimidine, pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine, and pyrazolo[4,3-e][1,2,4]triazolo[3,4-c]pyrimidine derivatives. J. Heterocyclic. Chem. 2020, 57, 892–912. [Google Scholar] [CrossRef]
- Madhvi Utreja, D.; Sharma, S. Barbiturates: A review of synthesis and antimicrobial research progress. Curr. Org Synth. 2022, 19, 31–55. [Google Scholar] [CrossRef]
- Lewis, P.O.; PharmD, B. Risk factor evaluation for methicillin-resistant staphylococcus aureus and pseudomonas aeruginosain community-acquired pneumonia. Ann. Pharmacother. 2021, 55, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Tressler, A.B.; Markwei, M.; Fortin, C.; Yao, M.; Procop, G.E.; Soper, D.E.; Goje, O. Risks for recurrent vulvovaginal candidiasis caused by non-albicans Candida versus Candida Albicans. J. Womens Health 2021, 30, 1588–1596. [Google Scholar] [CrossRef]
- Riina, M.; Ulla, A.; Jenni, T.; Hannu, U.; Tytti, S. Antibacterial and oxidative stress-protective effects of five monoterpenes from softwood. Molecules 2022, 27, 1–15. [Google Scholar]
- Mahajan, A.; Singh, H.; Singh, A.; Agrawal, D.K.; Arora, A.; Chundawat, T.S. Trifluoromethylated quinolone-hydantoin hybrids: Synthesis and antibacterial evaluation. Science 2022, 4, 30. [Google Scholar] [CrossRef]
- Ana, C.M.; Pedro, L.R.; Irlan, A.F.; Luciana, S.; Marcus, T.S.; Sabrina, G.A.; Ricardo, D.C. Antifungal activity, mode of action, docking prediction and anti-biofilm effects of (+)-β-pinene enantiomers against candida spp. Curr. Top. Med. Chem. 2018, 18, 2481–2490. [Google Scholar]
- Rosa, T.; Mariarosaria, L.; Marco, B.; Simone, R.; Nicodemo, G.P. Salvia officinalis L. from Italy: A comparative chemical and biological study of its essential oil in the mediterranean context. Molecules 2021, 25, 5826. [Google Scholar]
- Chen, W.Y.; Vermaak, I.; Viljoen, A. Camphor-A fumigant during the black death and a coveted fragrant wood in ancient Egypt and Babylon-A Review. Molecules 2013, 18, 5434–5454. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Shernyukov, A.V.; Gatilov, Y.V.; Razumova, Y.V.; Zarubaevc, V.V.; Tretiak, D.S.; Pokrovsky, A.G.; Kiselev, O.L.; Salakhutdinov, N.F. Discovery of a new class of antiviral compounds: Camphorimine derivatives. Eur. J. Med. Chem. 2015, 105, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Q.; Liu, Y.; Li, M.; Mao, J.W.; Zhang, L.R.; Huang, R.B.; Jin, X.B.; Ye, L.B. Anti-tumor effect of α-pinene on human hepatoma cell lines through inducing G2/M cell cycle arrest. J. Pharmacol. Sci. 2015, 127, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.S.; Yu, H.; Xie, Y.F.; Guo, Y.H.; Fan, J.J.; Yao, W.R. The anti-inflammatory potential of Cinnamomum camphora (L.) J.Presl essential oil in vitro and in vivo. J. Ethnopharmacol. 2021, 267, 113516. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.Y. A preliminary study on the relation between camphor wood volatile matters and corrosion of museum metallic objects. IOP Conf. Ser. Earth Environ. Sci. 2020, 467, 012147. [Google Scholar] [CrossRef]
- Bryleva, Y.A.; Ustimenko, Y.P.; Plyusnin, V.F.; Mikheilis, A.V.; Shubin, A.A.; Glinskaya, L.A.; Komarov, V.Y.; Agafontsev, A.M.; Tkachev, A.V. Ln(III) complexes with a chiral 1H-pyrazolo[3,4-b]pyridine derivative fused with a -α-pinene moiety: Synthesis, crystal structure, and photophysical studies in solution and in the solid state. New. J. Chem. 2021, 45, 2276–2284. [Google Scholar] [CrossRef]
- Yang, J.L.; Fang, H.; Fang, X.Y.; Xu, X.; Yang, Y.Q.; Rui, J.; Wu, C.L.; Wang, S.F.; Xu, H.J. A novel tetrahydroquinazolin-2-amine-based high selective fluorescent sensor for Zn2+ from nopinone. Tetrahedron 2016, 72, 4503–4509. [Google Scholar] [CrossRef]
- Ponomarev, K.Y.; Morozova, E.A.; Suslov, E.V.; Korchagina, D.V.; Tolstikova, T.G.; Volcho, K.P.; Salakhutdinov, N.F. Synthesis and analgesic activity of 5,7- and 6-substituted diazaadamantanes containing monoterpene moieties. Chem. Nat. Comp. 2017, 53, 1131–1136. [Google Scholar] [CrossRef]
- Lin, G.S.; Bai, X.; Duan, W.G.; Cen, B.; Huang, M.; Lu, S.Z. High value-added application of sustainable natural forest product α pinene: Synthesis of myrtenal oxime esters as potential KARI inhibitors. ACS Sustain. Chem. Eng. 2019, 7, 7862–7868. [Google Scholar] [CrossRef]
- Rui, J.; Zhang, Q.; Wang, X.; Xu, X.; Xu, H.J.; Rao, W.D.; Wang, S.F. Synthesis and biological activity of novel pinanyl pyrazole acetamide derivatives. Chin. J. Org. Chem. 2017, 37, 218–225. [Google Scholar] [CrossRef][Green Version]
- Zhang, Q.J.; Wang, Y.Y.; Zhao, Y.X.; Ma, C.H.; Yang, Y.Q.; Xu, X.; Gu, W.; Wang, S.F. Synthesis and α-amylase inhibitory potential and molecular docking study of nopinone-based thiazolylhydrazone derivatives. Chin. J. Org. Chem. 2019, 39, 1372–1382. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Gu, W.; Shan, Y.; Liu, F.; Xu, X.; Yang, Y.Q.; Zhang, Q.J.; Zhang, Y.; Kuang, H.B.; Wang, Z.L.; et al. Design, synthesis and anticancer activity of novel nopinone-based thiosemicarbazone derivatives. Bioorg. Med. Chem. 2017, 27, 2360–2363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.Y.; Zhao, Y.X.; Gu, W.; Zhu, Y.Q.; Wang, S.F. Novel camphor-based pyrimidine derivatives induced cancer cell death through a ROS-mediated mitochondrial apoptosis pathway. RSC Adv. 2019, 9, 29711–29720. [Google Scholar] [CrossRef] [PubMed]
- Raji, M.; Le, T.M.; Csámpai, A.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Stereoselective synthesis and applications of pinane-based chiral 1,4-amino alcohol derivatives. Synthesis 2022, 54, 3831–3844. [Google Scholar]
- Deyno, S.; Mtewa, A.G.; Hope, D.; Bazira, J.; Makonnen, E.; Alete, P.E. Antibacterial activities of Echinops kebericho Mesfin Tuber extracts and isolation of the most active compound, dehydrocostus Lactone. Front. Pharmacol. 2021, 11, 1–13. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | R1 | R2 | Bacterial | Fungi | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| K. pneumoniae | S. pneumoniae | P. aeruginosa | S. aureus | E. coli | MRSA | B. cereus | C. albicans | |||
| 1e | OH | H | 32 | 32 | 16 | 32 | 8 | 64 | 8 | >1024 |
| 1f | Cl | H | 32 | 8 | 16 | 32 | 8 | 8 | 8 | >1024 |
| 1g | OCH3 | H | 64 | 32 | 64 | 1 | 8 | 8 | 32 | >1024 |
| 1h | N(C2H5)2 | H | 256 | >1024 | 128 | 16 | 128 | 8 | >1024 | >1024 |
| 1i | OCH3 |  | 128 | 1 | 32 | >1024 | 1 | 64 | 64 | 32 |
| 1j | OCH3 |  | 256 | 64 | 128 | 512 | 64 | 64 | >1024 | 32 |
| 1k | OCH3 |  | 512 | >1024 | 128 | 512 | 128 | 64 | >1024 | 64 |
| 1l | OCH3 |  | >1024 | >1024 | 128 | 16 | 512 | 8 | >1024 | 16 |
| 1m | N(C2H5)2 |  | >1024 | 32 | 128 | 512 | 64 | 64 | >1024 | >1024 |
| 1n | N(C2H5)2 |  | >1024 | >1024 | >1024 | >1024 | >1024 | >1024 | >1024 | >1024 |
| 2b | OCH3 | H | >1024 | 32 | 16 | >1024 | 8 | 64 | 32 | >1024 |
| 2c | OCH3 |  | 64 | >1024 | 256 | 16 | 32 | 128 | >1024 | 64 |
| 2d | OCH3 |  | >1024 | >1024 | 128 | 256 | 64 | 64 | >1024 | 64 |
| 2e | OCH3 |  | >1024 | >1024 | 128 | 512 | 128 | 32 | >1024 | 128 |
| 2f | OCH3 |  | 32 | 16 | 16 | 32 | 8 | 8 | 16 | 32 |
| amikacin b | - | - | 32 | 2 | 32 | 1 | 2 | 1 | 4 | -a |
| ketoconazole b | - | - | - | - | - | - | - | - | - | 16 |
| Compd. | MW a | CLogP b | pKa c | tPSA d |
|---|---|---|---|---|
| 1e | 281 | 3.73 | 8.81 | 70.97 |
| 1f | 300 | 4.86 | - | 50.74 |
| 1g | 295 | 4.18 | - | 59.97 |
| 1i | 353 | 5.01 | - | 55.21 |
| 2f | 435 | 7.26 | - | 45.98 |
| amikacin | 585 | −4.12 | 13.32 | 331.94 |
| 1l | 421 | 6.74 | - | 45.98 |
| ketoconazole | 531 | 3.64 | - | 66.84 |
| Compd. | (Anti-Inflammatory Activity)/μmol·L−1 (IC50 ± SD) |
|---|---|
| 1e | 2.32 ± 0.9 |
| 1f | 1.37 ± 0.41 |
| 1g | 3.0 ± 0.71 |
| 1i | 4.86 ± 0.84 |
| 2f | 1.87 ± 0.38 |
| Aspirin | 1.91 ± 0.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Wang, Y.; Wang, S.; Wu, H. Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties. Molecules 2022, 27, 8104. https://doi.org/10.3390/molecules27228104
Zhang M, Wang Y, Wang S, Wu H. Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties. Molecules. 2022; 27(22):8104. https://doi.org/10.3390/molecules27228104
Chicago/Turabian StyleZhang, Mingguang, Yunyun Wang, Shifa Wang, and Hongyan Wu. 2022. "Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties" Molecules 27, no. 22: 8104. https://doi.org/10.3390/molecules27228104
APA StyleZhang, M., Wang, Y., Wang, S., & Wu, H. (2022). Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties. Molecules, 27(22), 8104. https://doi.org/10.3390/molecules27228104