
Phytocannabinoid Compositions from Cannabis Act Synergistically with PARP1 Inhibitor against Ovarian Cancer Cells In Vitro and Affect the Wnt Signaling Pathway


 , ,
, , 
Abstract
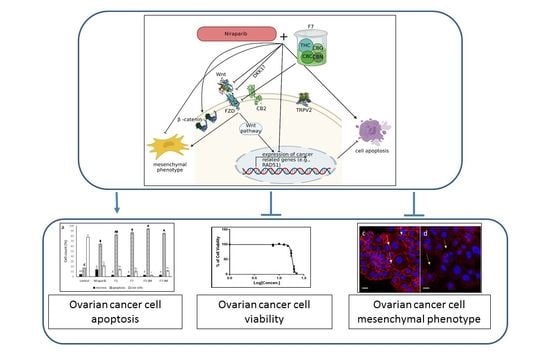
1. Introduction
2. Results
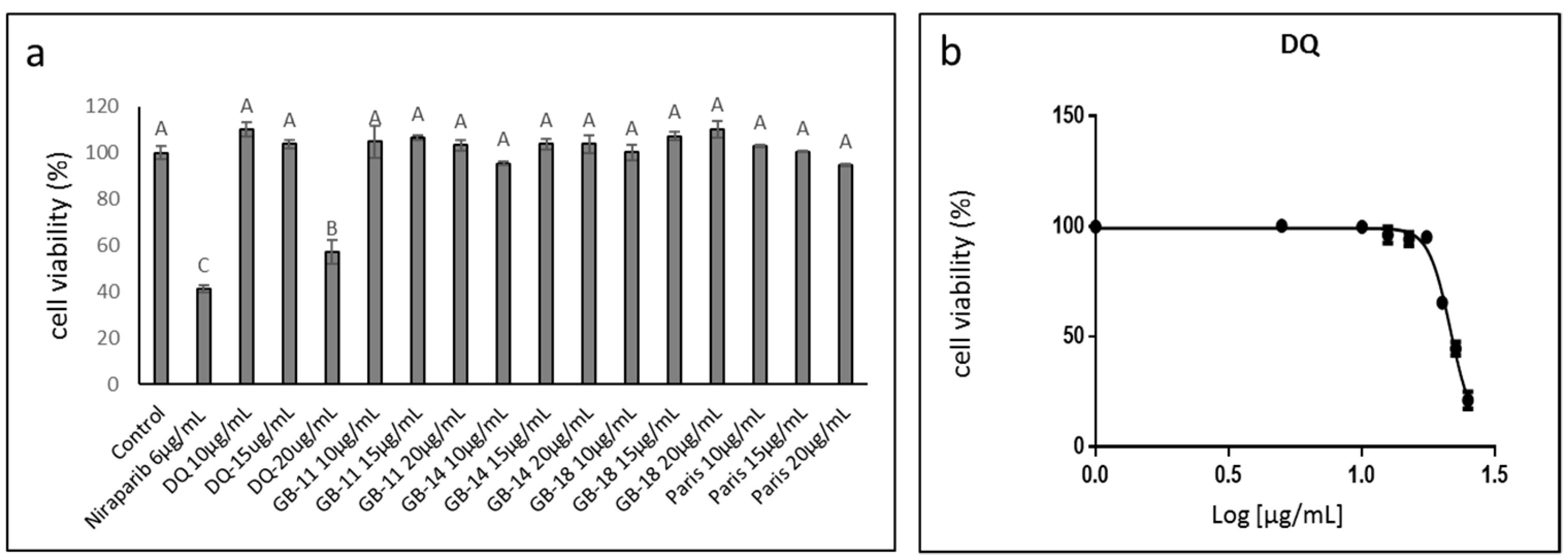
2.1. Determining Cannabis Strains with Cytotoxic Activity against Ovarian Cancer Cell Lines
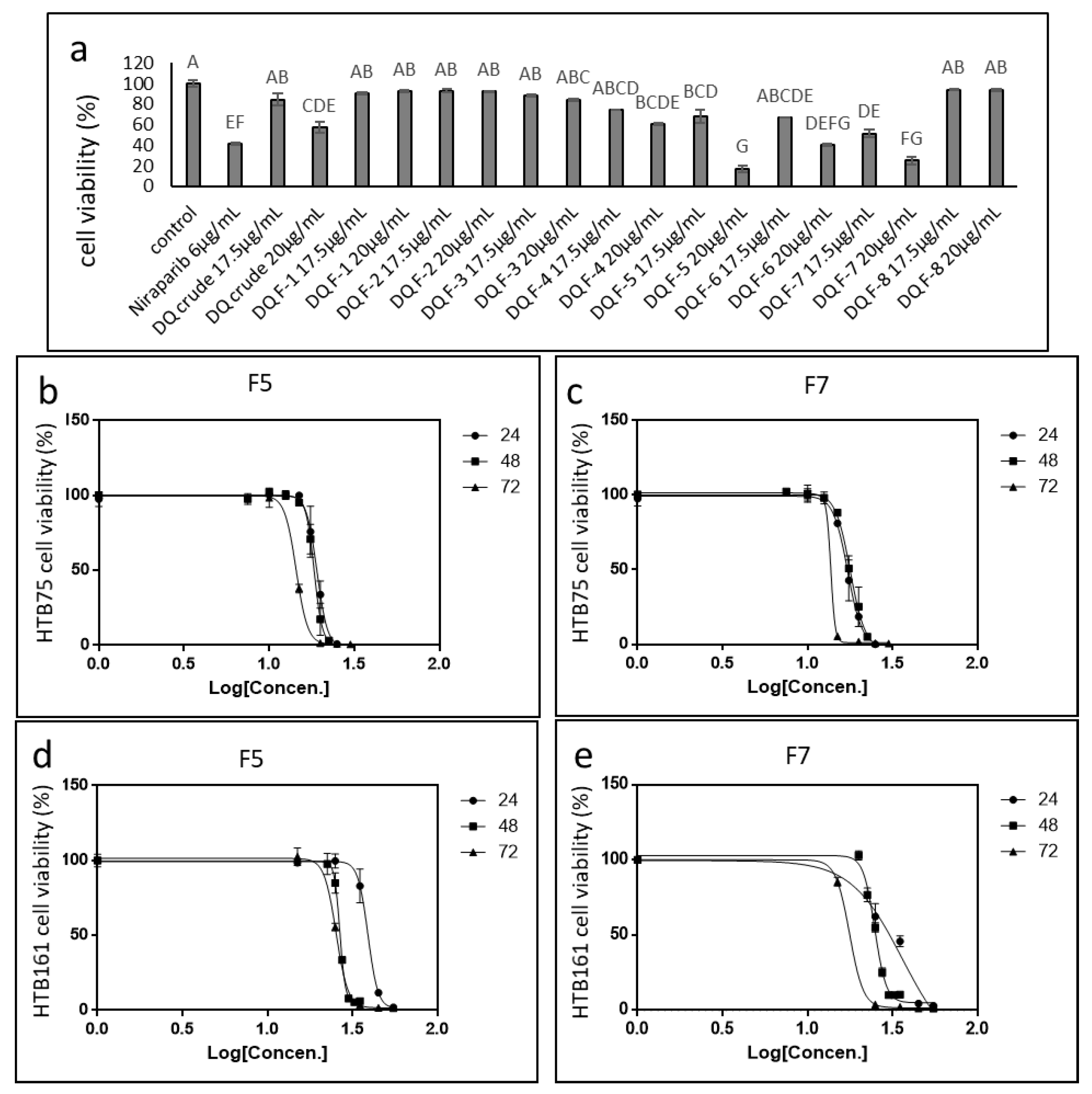
2.2. Identifying the Active Fractions of the DQ Cannabis Strain
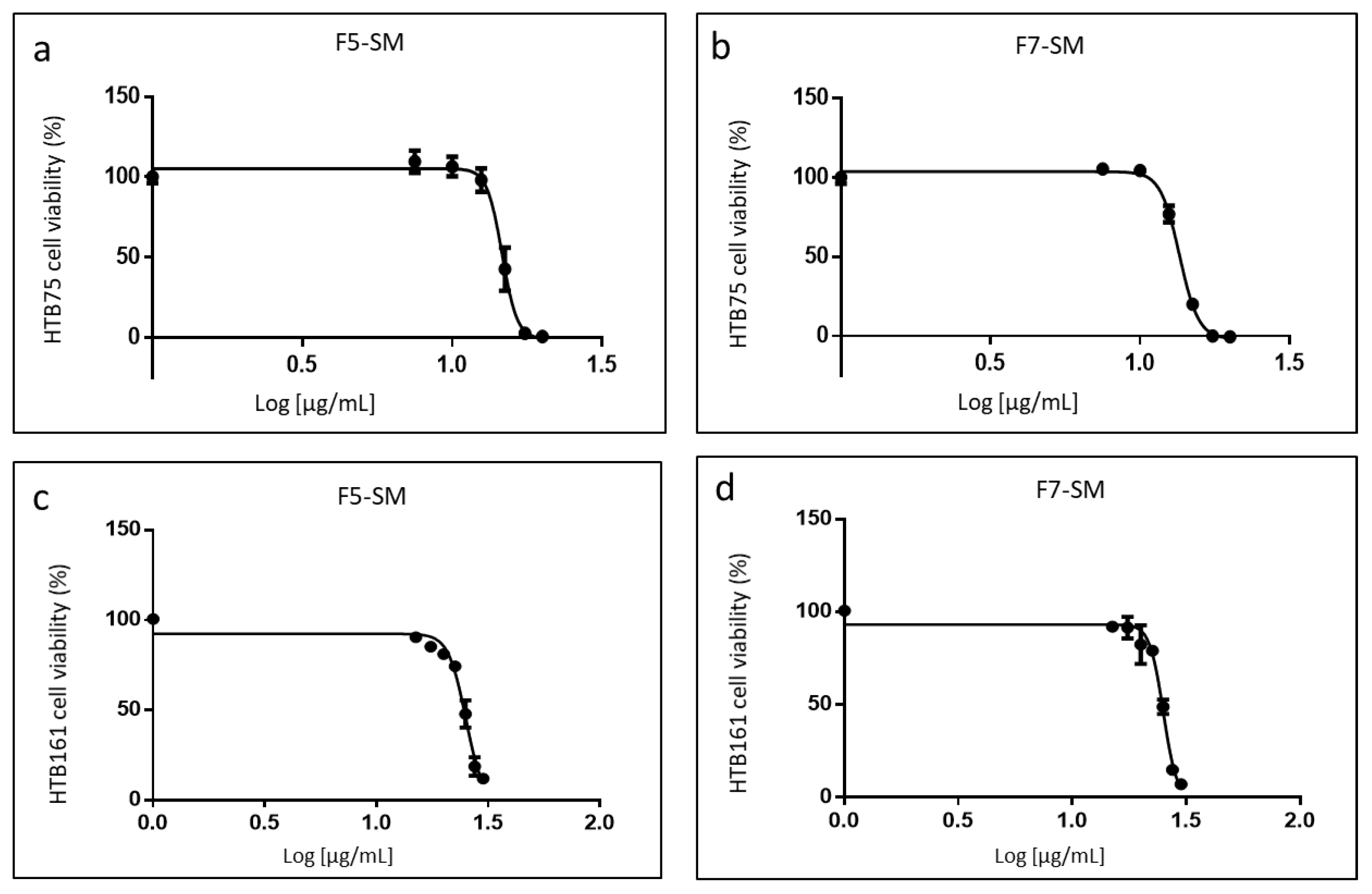
2.3. Determining the Activity of the Phytocannabinoid F5 and F7 Standard Mixes
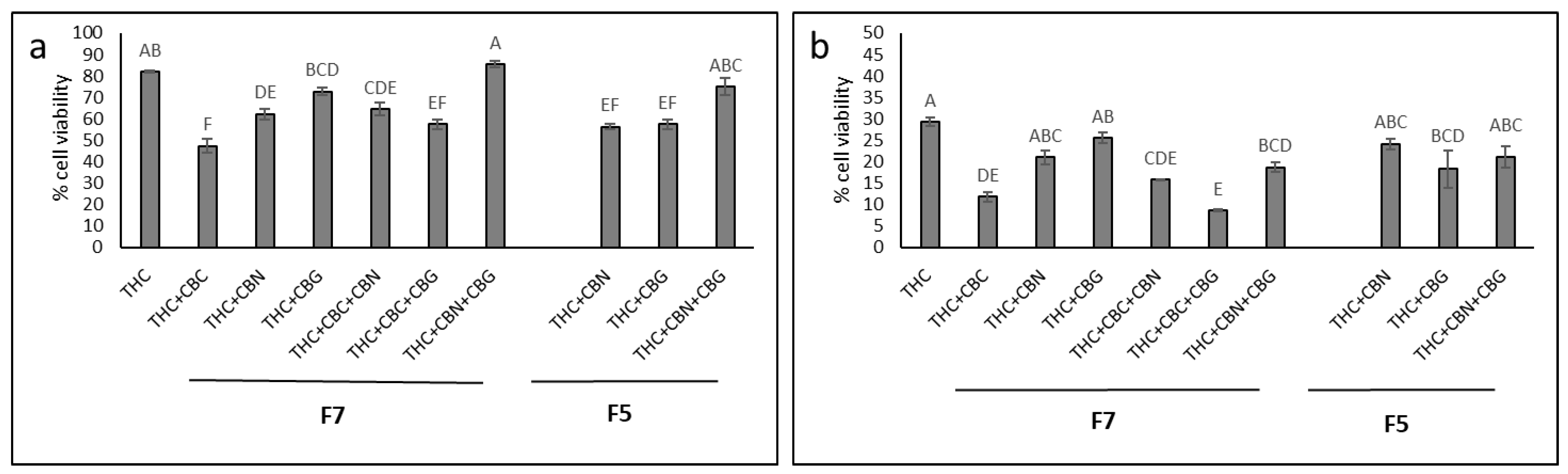
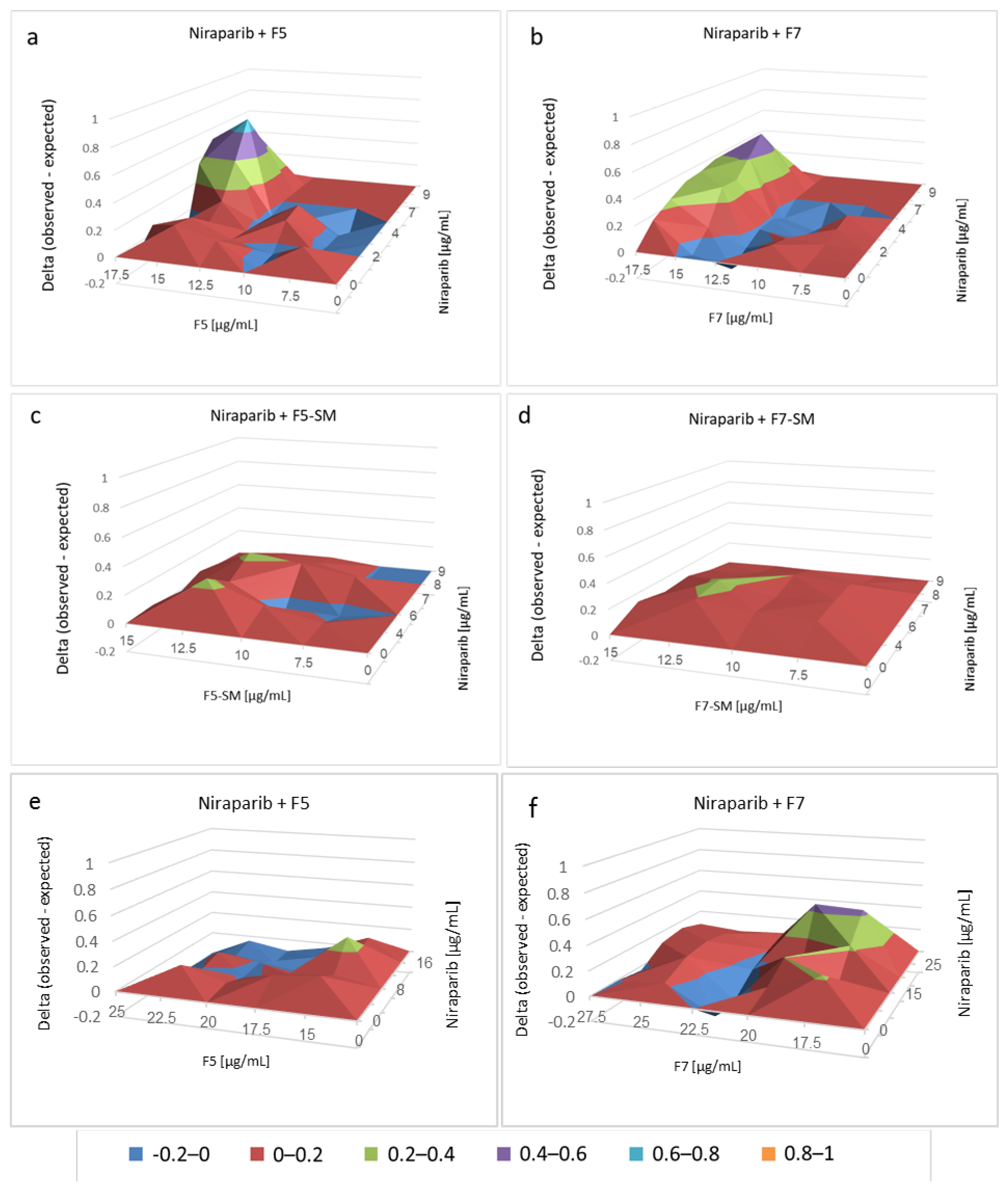
2.4. Determining the Most Effective Combinations of Phytocannabinoids
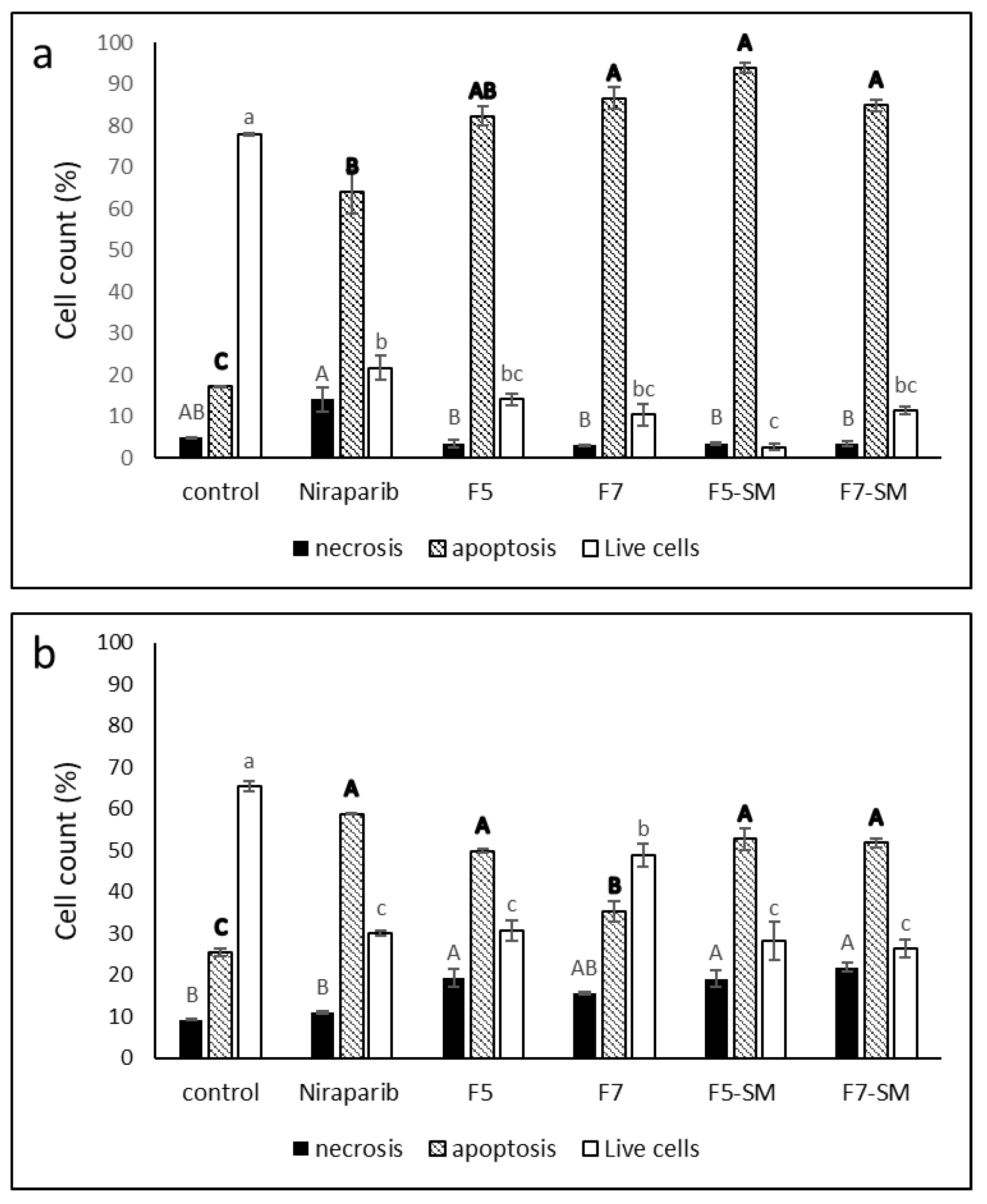
2.5. Determining the Effects of Treatment on Cell Apoptosis
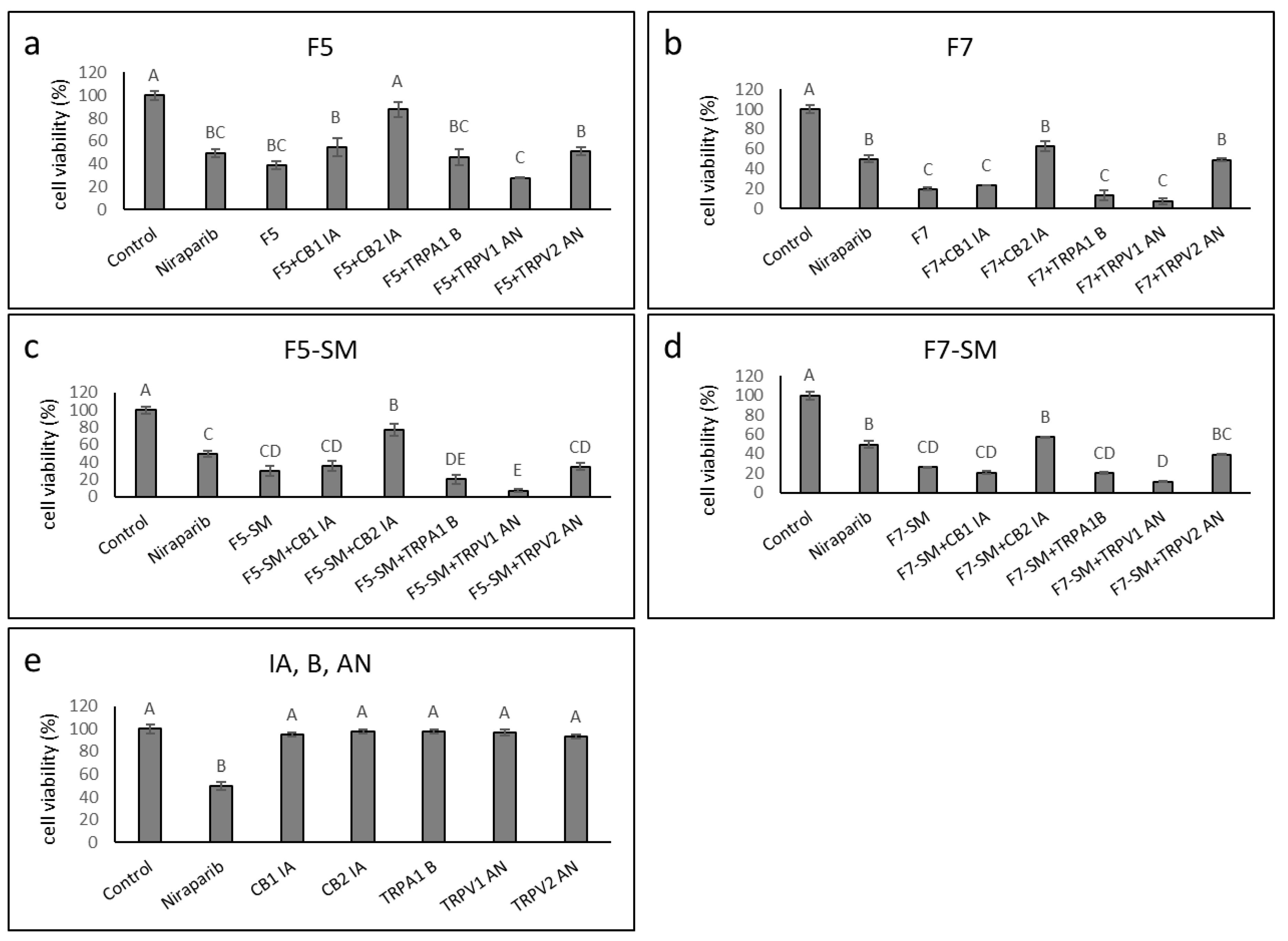
2.6. Determining the Synergistic Effects of CB1 and CB2 Receptor Inverse Agonists, a TRPA1 Receptor Blocker, and TRPV1 or TRPV2 Receptor Antagonists on Cytotoxic Activity
2.7. Determining the Effects of Combined Treatments of Cannabis Fractions or Compounds with Chemotherapy Drugs
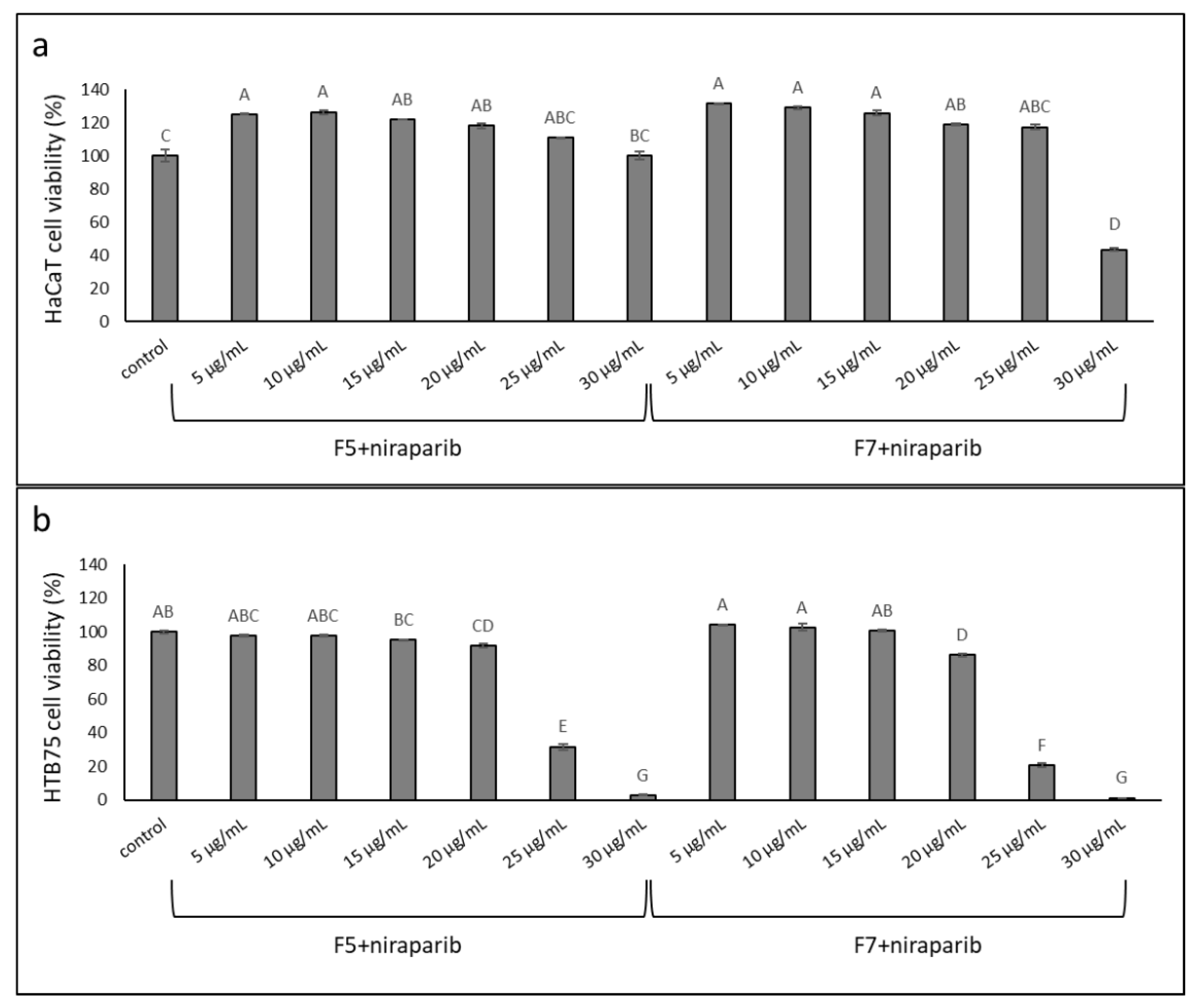
2.8. Determining the Effects of Treatment on Normal Cells
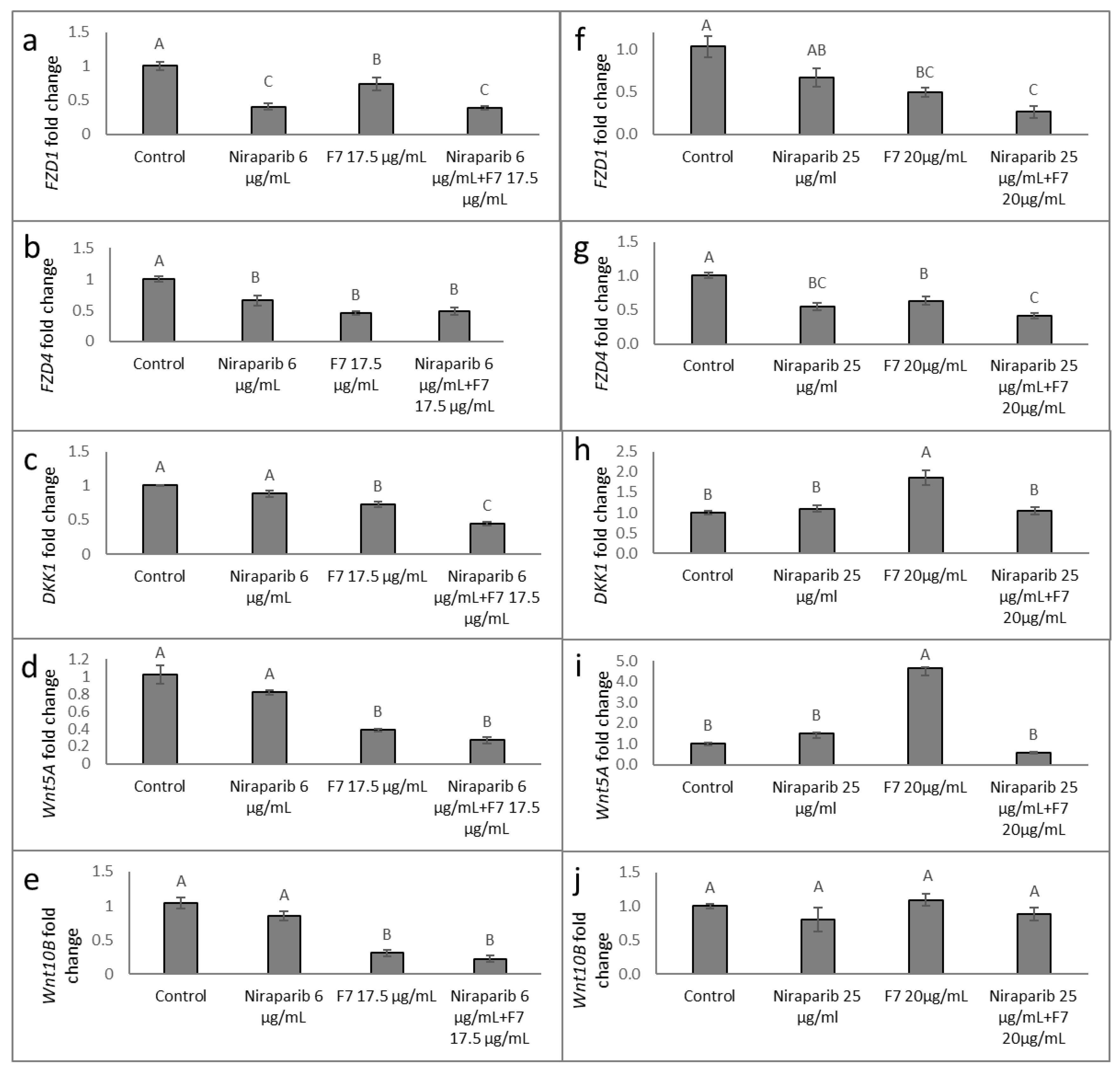
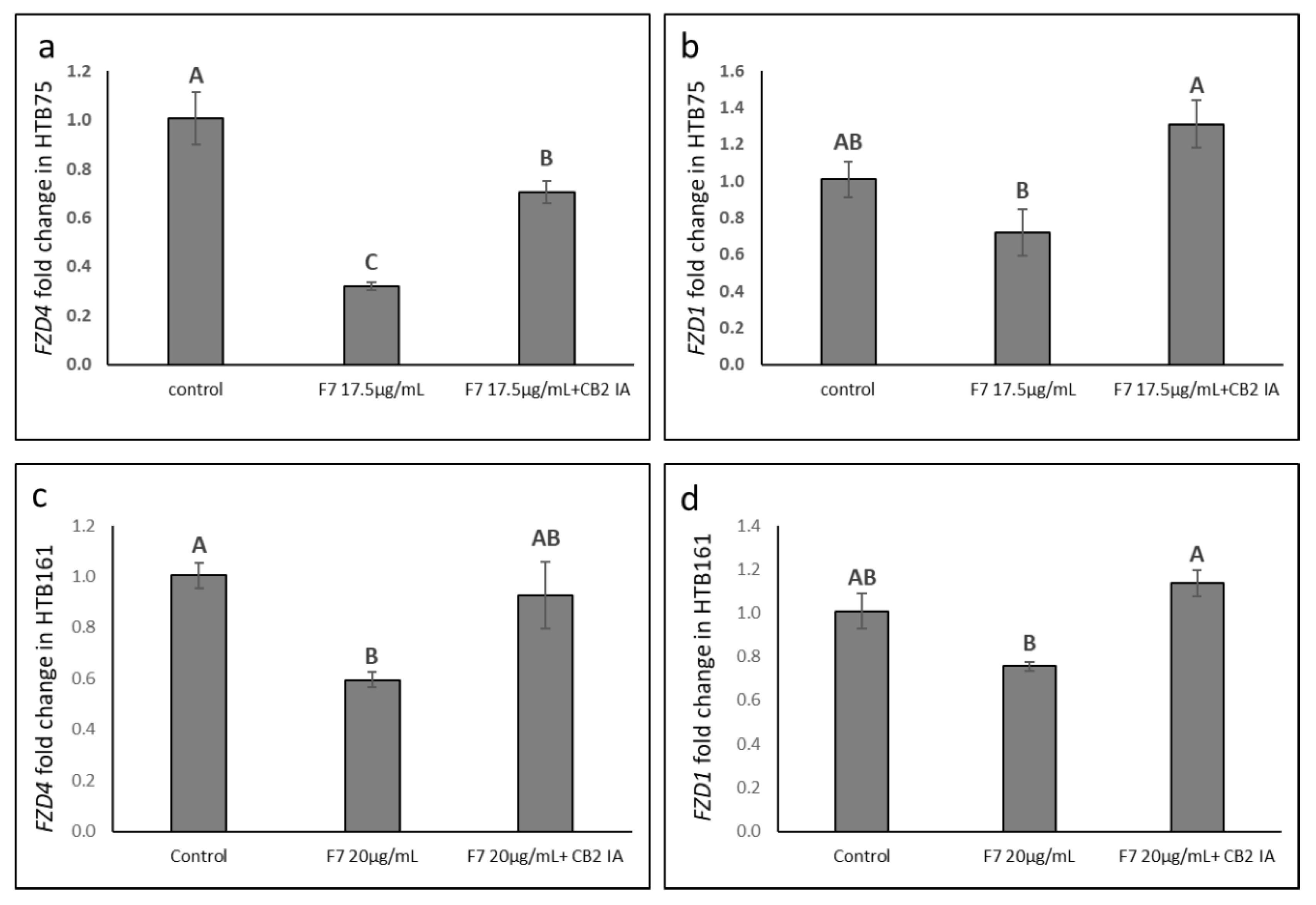
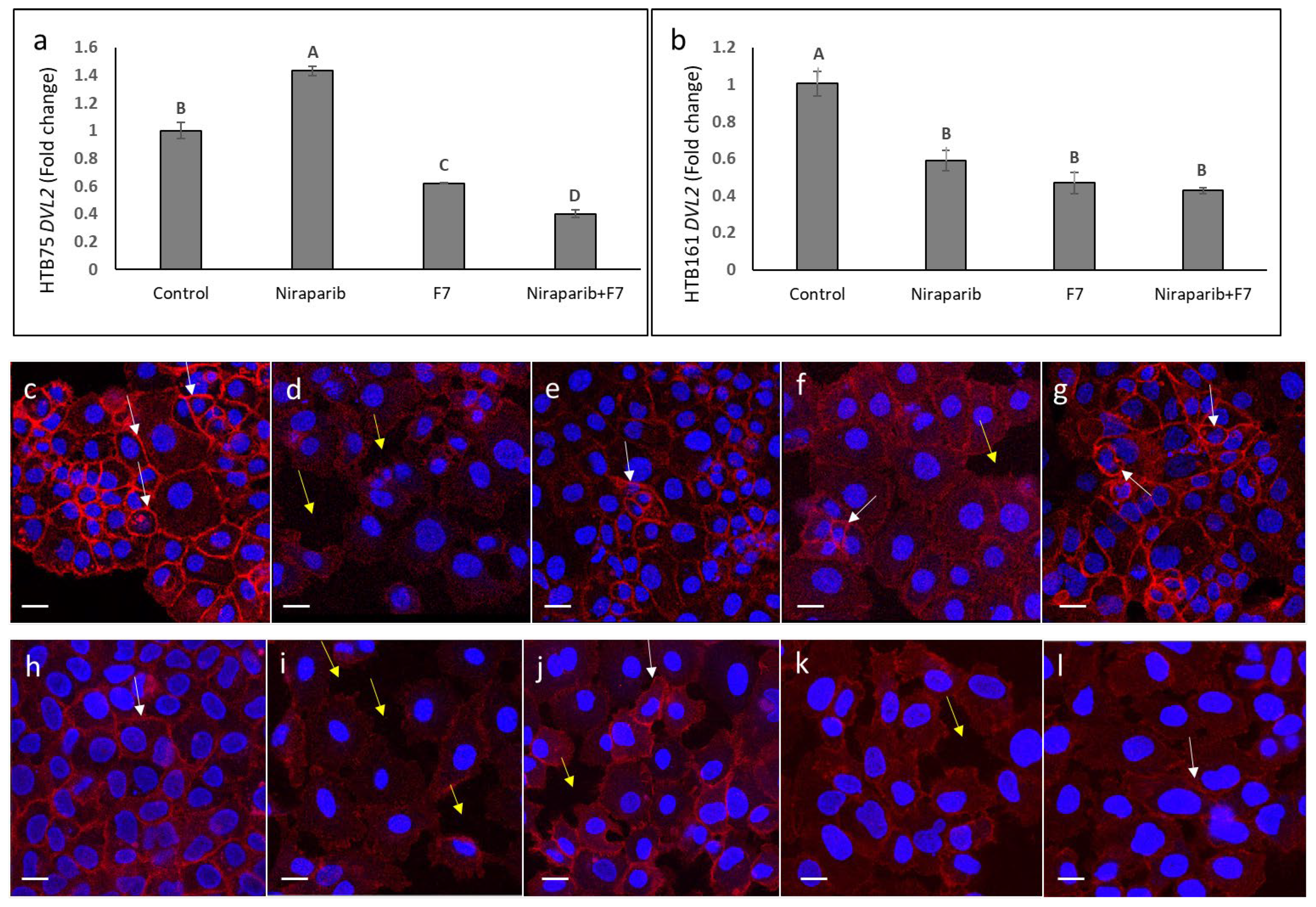
2.9. Determining the Effects of Treatment on Wnt Pathway-Related Gene Expression
2.10. Determining the Effects of Treatment on Epithelial-Mesenchymal Transition Phenotype
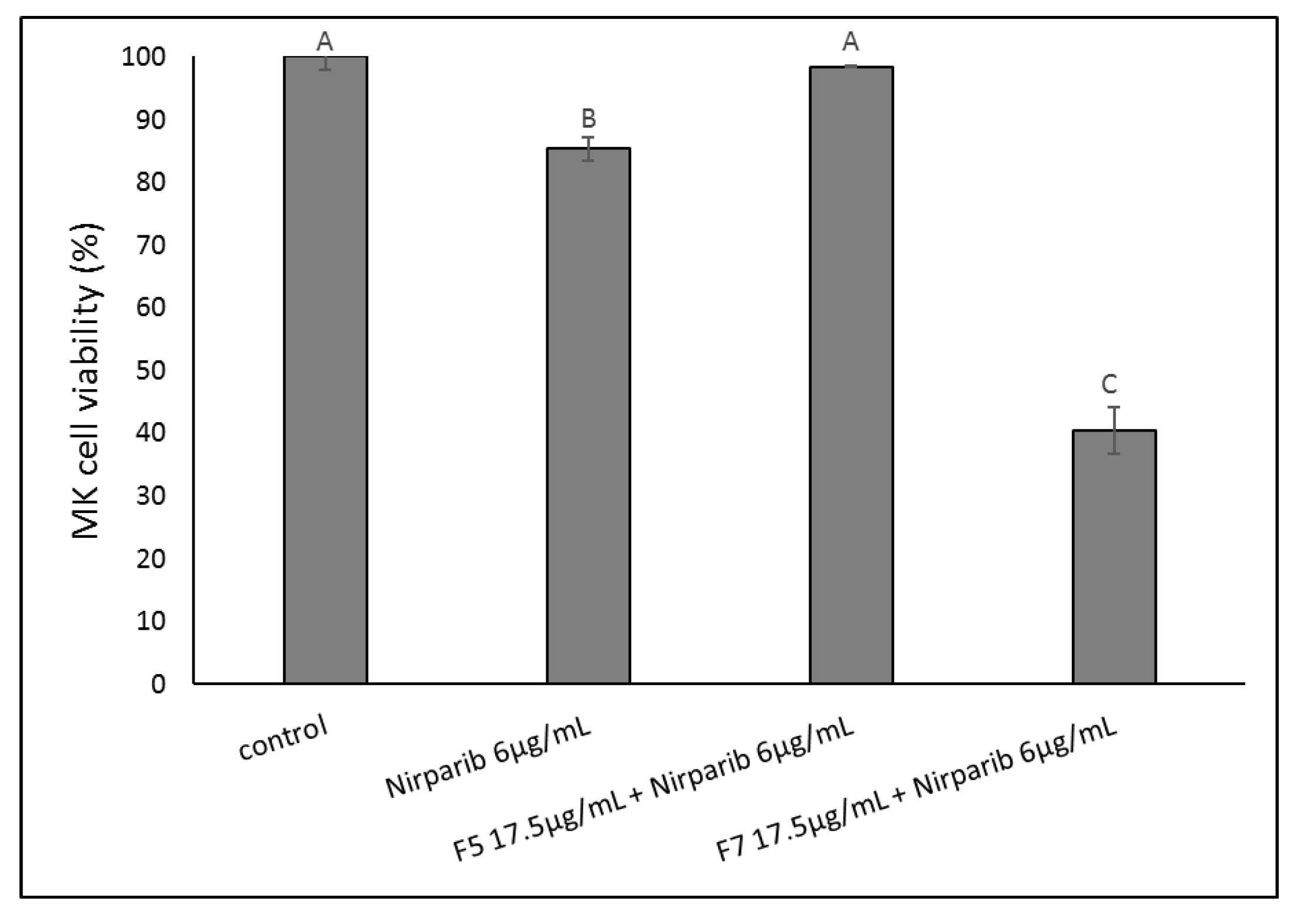
2.11. Determining the Effects of Treatment on an OC Patient’s Cells
3. Discussion
4. Materials and Methods
4.1. Plant Extraction
4.2. Extract Fractionation
4.3. Chemical Analysis
4.4. Standard/Material Preparation and Use
4.5. Cell Culture
4.6. Isolation of OC Cells from Patient MK
4.7. Cell Viability Assay
4.8. Flow Cytometry Analysis
4.9. Staining and Confocal Microscopy
4.10. Analysis of Combined Drug Effects
4.11. Quantitative Real-Time PCR
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Menon, U.; Gentry-Maharaj, A.; Burnell, M.; Singh, N.; Ryan, A.; Karpinskyj, C.; Carlino, G.; Taylor, J.; Massingham, S.K.; Raikou, M. Ovarian cancer population screening and mortality after long-term follow-up in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2021, 397, 2182–2193. [Google Scholar] [CrossRef]
- Barnett, R. Ovarian cancer. Lancet 2016, 387, 1265. [Google Scholar] [CrossRef]
- Corroon, J.; Sexton, M.; Bradley, R. Indications and administration practices amongst medical cannabis healthcare providers: A cross-sectional survey. BMC Fam. Pract. 2019, 20, 174. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Meyer, S.M.; Muñoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef]
- Gülck, T.; Møller, B.L. Phytocannabinoids: Origins and biosynthesis. Trends Plant Sci. 2020, 25, 985–1004. [Google Scholar] [CrossRef]
- Aizpurua-Olaizola, O.; Soydaner, U.; Öztürk, E.; Schibano, D.; Simsir, Y.; Navarro, P.; Etxebarria, N.; Usobiaga, A. Evolution of the cannabinoid and terpene content during the growth of Cannabis sativa plants from different chemotypes. J. Nat. Prod. 2016, 79, 324–331. [Google Scholar] [CrossRef]
- Ramer, R.; Hinz, B. Cannabinoids as anticancer drugs. Adv. Pharmacol. 2017, 80, 397–436. [Google Scholar]
- Baram, L.; Peled, E.; Berman, P.; Yellin, B.; Besser, E.; Benami, M.; Louria-Hayon, I.; Lewitus, G.M.; Meiri, D. The heterogeneity and complexity of Cannabis extracts as antitumor agents. Oncotarget 2019, 10, 4091. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sánchez, A.I.; Torres-Suárez, A.I.; Cohen, M.; Delie, F.; Bastida-Ruiz, D.; Yart, L.; Martin-Sabroso, C.; Fernández-Carballido, A. PLGA nanoparticles for the intraperitoneal administration of CBD in the treatment of ovarian cancer: In Vitro and In Ovo assessment. Pharmaceutics 2020, 12, 439. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sánchez, A.; Fernández-Carballido, A.; Delie, F.; Cohen, M.; Martin-Sabroso, C.; Mezzanzanica, D.; Figini, M.; Satta, A.; Torres-Suárez, A. Enhancing ovarian cancer conventional chemotherapy through the combination with cannabidiol loaded microparticles. Eur. J. Pharm. Biopharm. 2020, 154, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Barrie, A.M.; Gushue, A.C.; Eskander, R.N. Dramatic response to Laetrile and cannabidiol (CBD) oil in a patient with metastatic low grade serous ovarian carcinoma. Gynecol. Oncol. Rep. 2019, 29, 10. [Google Scholar] [CrossRef]
- Nguyen, V.H.L.; Hough, R.; Bernaudo, S.; Peng, C. Wnt/β-catenin signalling in ovarian cancer: Insights into its hyperactivation and function in tumorigenesis. J. Ovarian Res. 2019, 12, 122. [Google Scholar] [CrossRef]
- Kaur, A.; Lim, J.Y.S.; Sepramaniam, S.; Patnaik, S.; Harmston, N.; Lee, M.A.; Petretto, E.; Virshup, D.M.; Madan, B. WNT inhibition creates a BRCA-like state in Wnt-addicted cancer. EMBO Mol. Med. 2021, 13, e13349. [Google Scholar] [CrossRef]
- Patel, S.; Alam, A.; Pant, R.; Chattopadhyay, S. Wnt signaling and its significance within the tumor microenvironment: Novel therapeutic insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef]
- Arend, R.C.; Londoño-Joshi, A.I.; Straughn, J.M., Jr.; Buchsbaum, D.J. The Wnt/β-catenin pathway in ovarian cancer: A review. Gynecol. Oncol. 2013, 131, 772–779. [Google Scholar] [CrossRef]
- Peeri, H.; Shalev, N.; Vinayaka, A.C.; Nizar, R.; Kazimirsky, G.; Namdar, D.; Anil, S.M.; Belausov, E.; Brodie, C.; Koltai, H. Specific Compositions of Cannabis sativa Compounds Have Cytotoxic Activity and Inhibit Motility and Colony Formation of Human Glioblastoma Cells In Vitro. Cancers 2021, 13, 1720. [Google Scholar] [CrossRef]
- Mazuz, M.; Moyal, L.; Hodak, E.; Nadarajan, S.; Vinayaka, A.C.; Gorovitz-Haris, B.; Lubin, I.; Drori, A.; Drori, G.; Van Cauwenberghe, O. Synergistic cytotoxic activity of cannabinoids from cannabis sativa against cutaneous T-cell lymphoma (CTCL) in-vitro and ex-vivo. Oncotarget 2020, 11, 1141. [Google Scholar] [CrossRef]
- García-Morales, L.; Castillo, A.M.; Tapia Ramírez, J.; Zamudio-Meza, H.; Domínguez-Robles, M.D.C.; Meza, I. CBD reverts the mesenchymal invasive phenotype of breast cancer cells induced by the inflammatory cytokine IL-1β. Int. J. Mol. Sci. 2020, 21, 2429. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Davis, D.A.; Tomar, S.; Roy, L.; Gurler, H.; Xie, J.; Lantvit, D.D.; Cardenas, H.; Fang, F.; Liu, Y. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol. Oncol. 2015, 138, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Tomko, A.M.; Whynot, E.G.; Ellis, L.D.; Dupré, D.J. Anti-cancer potential of cannabinoids, terpenes, and flavonoids present in cannabis. Cancers 2020, 12, 1985. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Schiano Moriello, A.; Iappelli, M.; Verde, R.; Stott, C.G.; Cristino, L.; Orlando, P.; Di Marzo, V. Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: Pro-apoptotic effects and underlying mechanisms. Br. J. Pharmacol. 2013, 168, 79–102. [Google Scholar] [CrossRef]
- Anis, O.; Vinayaka, A.C.; Shalev, N.; Namdar, D.; Nadarajan, S.; Anil, S.M.; Cohen, O.; Belausov, E.; Ramon, J.; Mayzlish Gati, E. Cannabis-derived compounds cannabichromene and Δ9-tetrahydrocannabinol interact and exhibit cytotoxic activity against urothelial cell carcinoma correlated with inhibition of cell migration and cytoskeleton organization. Molecules 2021, 26, 465. [Google Scholar] [CrossRef] [PubMed]
- Guida, M.; Ligresti, A.; De Filippis, D.; D’Amico, A.; Petrosino, S.; Cipriano, M.; Bifulco, G.; Simonetti, S.; Orlando, P.; Insabato, L. The levels of the endocannabinoid receptor CB2 and its ligand 2-arachidonoylglycerol are elevated in endometrial carcinoma. Endocrinology 2010, 151, 921–928. [Google Scholar] [CrossRef]
- Ren, X.; Hao, W.; Liu, J.; Li, Y.; Wang, B.; Zu, X.; Xue, H. Study on the clinical significance of TRPV2 and MMP2 expressions in ovarian cancer. Biocell 2021, 45, 521. [Google Scholar] [CrossRef]
- Caruso, D.; Papa, A.; Tomao, S.; Vici, P.; Panici, P.B.; Tomao, F. Niraparib in ovarian cancer: Results to date and clinical potential. Ther. Adv. Med. Oncol. 2017, 9, 579–588. [Google Scholar] [CrossRef]
- Mirza, M.R.; Bergmann, T.K.; Mau-Sørensen, M.; Christensen, R.d.; Åvall-Lundqvist, E.; Birrer, M.J.; Jørgensen, M.; Roed, H.; Malander, S.; Nielsen, F. A phase I study of the PARP inhibitor niraparib in combination with bevacizumab in platinum-sensitive epithelial ovarian cancer: NSGO AVANOVA1/ENGOT-OV24. Cancer Chemother. Pharmacol. 2019, 84, 791–798. [Google Scholar] [CrossRef]
- Available online: https://www.cancernetwork.com/view/eu-approves-olaparib-in-combination-with-bevacizumab-to-treat-advanced-ovarian-cancer (accessed on 6 June 2022).
- Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-olaparib-plus-bevacizumab-maintenance-treatment-ovarian-fallopian-tube-or-primary (accessed on 6 June 2022).
- Nalli, Y.; Dar, M.S.; Bano, N.; Rasool, J.U.; Sarkar, A.R.; Banday, J.; Bhat, A.Q.; Rafia, B.; Vishwakarma, R.A.; Dar, M.J. Analyzing the role of cannabinoids as modulators of Wnt/β-catenin signaling pathway for their use in the management of neuropathic pain. Bioorg. Med. Chem. Lett. 2019, 29, 1043–1046. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Zhang, S.; Lu, X.; Wu, J.; Yu, K.; Ji, A.; Lu, W.; Wang, Z.; Wu, J. IQGAP1 promotes pancreatic cancer progression and epithelial-mesenchymal transition (EMT) through Wnt/β-catenin signaling. Sci. Rep. 2019, 9, 7539. [Google Scholar] [CrossRef] [PubMed]
- Bernaudo, S.; Salem, M.; Qi, X.; Zhou, W.; Zhang, C.; Yang, W.; Rosman, D.; Deng, Z.; Ye, G.; Yang, B. Cyclin G2 inhibits epithelial-to-mesenchymal transition by disrupting Wnt/β-catenin signaling. Oncogene 2016, 35, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Cheng, J.; Chen, Y.; Wang, W.; Chen, J.; Mao, G. Potential role and prognostic importance of dishevelled-2 in epithelial ovarian cancer. Int. J. Gynecol. Obstet. 2017, 138, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solis, C.; Escamilla-Ramirez, A.; Jimenez-Farfan, D.; Castillo-Rodriguez, R.A.; Flores-Najera, A.; Cruz-Salgado, A. Crosstalk of the Wnt/β-catenin signaling pathway in the induction of apoptosis on cancer cells. Pharmaceuticals 2021, 14, 871. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kumar, S.; Sharma, A.; Chauhan, S.S.; Bhatla, N.; Kumar, S.; Bakhshi, S.; Gupta, R.; Kumar, L. PARP-1 inhibitor modulate β-catenin signaling to enhance cisplatin sensitivity in cancer cervix. Oncotarget 2019, 10, 4262. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, V.; Wright, J.A.; Churchill, M.; Wang, T.; Rosati, R.; Lannagan, T.R.; Vrbanac, L.; Richardson, A.B.; Kobayashi, H.; Price, T. Medium-throughput drug screening of patient-derived organoids from colorectal peritoneal metastases to direct personalized therapy. Clin. Cancer Res. 2020, 26, 3662–3670. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef]
- Wei, C.; Zhang, X.; He, S.; Liu, B.; Han, H.; Sun, X. MicroRNA-219-5p inhibits the proliferation, migration, and invasion of epithelial ovarian cancer cells by targeting the Twist/Wnt/β-catenin signaling pathway. Gene 2017, 637, 25–32. [Google Scholar] [CrossRef]
- Li, B.; Cai, S.; Zhao, Y.; He, Q.; Yu, X.; Cheng, L.; Zhang, Y.; Hu, X.; Ke, M.; Chen, S. Nerve growth factor modulates the tumor cells migration in ovarian cancer through the WNT/β-catenin pathway. Oncotarget 2016, 7, 81026. [Google Scholar] [CrossRef]
- Nutt, D.J.; Phillips, L.D.; Barnes, M.P.; Brander, B.; Curran, H.V.; Fayaz, A.; Finn, D.P.; Horsted, T.; Moltke, J.; Sakal, C. A multicriteria decision analysis comparing pharmacotherapy for chronic neuropathic pain, including cannabinoids and cannabis-based medical products. Cannabis Cannabinoid Res. 2021, 7, 482–500. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | CB2 Expression (Mean ± SE) | TRPV2 Expression (Mean ± SE) |
|---|---|---|
| Control | 1.00 ± 0.12 A | 1.00 ± 0.09 a |
| F5 17.5 μg/mL | 1.06 ± 0.11 A | 1.15 ± 0.11 a |
| F7 17.5 μg/mL | 1.11 ± 0.10 A | 1.15 ± 0.13 a |
| RAD51 Expression in HTB161 (Mean ± SE) | RAD51 Expression in HTB75 (Mean ± SE) | |||
|---|---|---|---|---|
| Duration of Treatment | 6 h | 9 h | 6 h | 9 h |
| Control | 1.00 ± 0.04 A | 1.00 ± 0.00 a | 1.00 ± 0.10 A | 1.01 ± 0.12 a |
| F7 | 0.54 ± 0.01 B | 0.73 ± 0.05 b | 0.67 ± 0.04 A | 0.89 ± 0.05 a |
| Niraparib | 0.63 ± 0.02 B | 0.59 ± 0.02 bc | 1.30 ± 0.24 A | 1.02 ± 0.16 a |
| Niraparib+F7 | 0.65 ± 0.08 B | 0.55 ± 0.03 c | 0.83 ± 0.09 A | 1.15 ± 0.04 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalev, N.; Kendall, M.; Anil, S.M.; Tiwari, S.; Peeri, H.; Kumar, N.; Belausov, E.; Vinayaka, A.C.; Koltai, H. Phytocannabinoid Compositions from Cannabis Act Synergistically with PARP1 Inhibitor against Ovarian Cancer Cells In Vitro and Affect the Wnt Signaling Pathway. Molecules 2022, 27, 7523. https://doi.org/10.3390/molecules27217523
Shalev N, Kendall M, Anil SM, Tiwari S, Peeri H, Kumar N, Belausov E, Vinayaka AC, Koltai H. Phytocannabinoid Compositions from Cannabis Act Synergistically with PARP1 Inhibitor against Ovarian Cancer Cells In Vitro and Affect the Wnt Signaling Pathway. Molecules. 2022; 27(21):7523. https://doi.org/10.3390/molecules27217523
Chicago/Turabian StyleShalev, Nurit, Michelle Kendall, Seegehalli M. Anil, Sudeep Tiwari, Hadar Peeri, Navin Kumar, Eduard Belausov, Ajjampura C. Vinayaka, and Hinanit Koltai. 2022. "Phytocannabinoid Compositions from Cannabis Act Synergistically with PARP1 Inhibitor against Ovarian Cancer Cells In Vitro and Affect the Wnt Signaling Pathway" Molecules 27, no. 21: 7523. https://doi.org/10.3390/molecules27217523
APA StyleShalev, N., Kendall, M., Anil, S. M., Tiwari, S., Peeri, H., Kumar, N., Belausov, E., Vinayaka, A. C., & Koltai, H. (2022). Phytocannabinoid Compositions from Cannabis Act Synergistically with PARP1 Inhibitor against Ovarian Cancer Cells In Vitro and Affect the Wnt Signaling Pathway. Molecules, 27(21), 7523. https://doi.org/10.3390/molecules27217523