
Transition Metal–(μ-Cl)–Aluminum Bonding in α-Olefin and Diene Chemistry



Abstract
1. Introduction
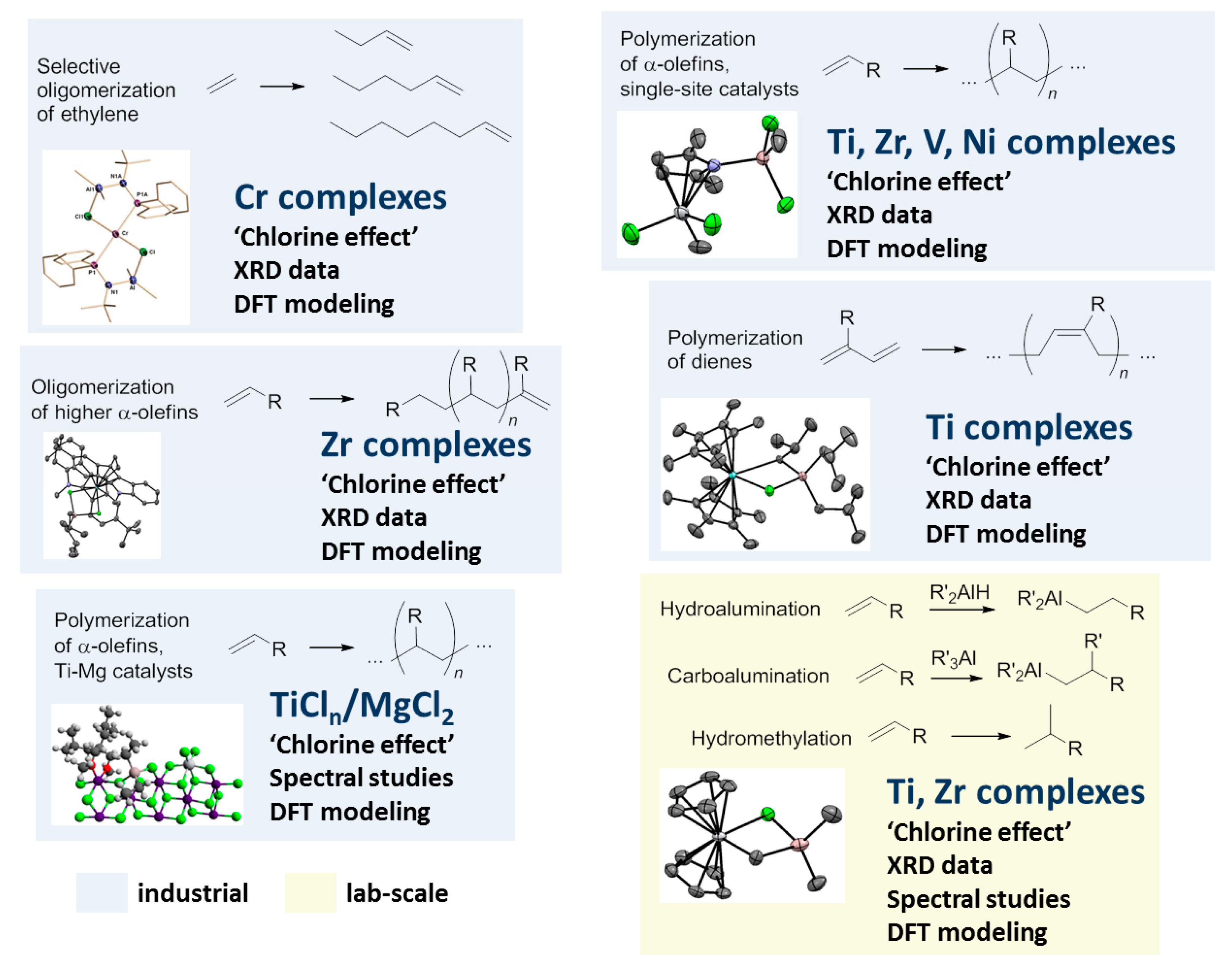
2. Complexes of Ti
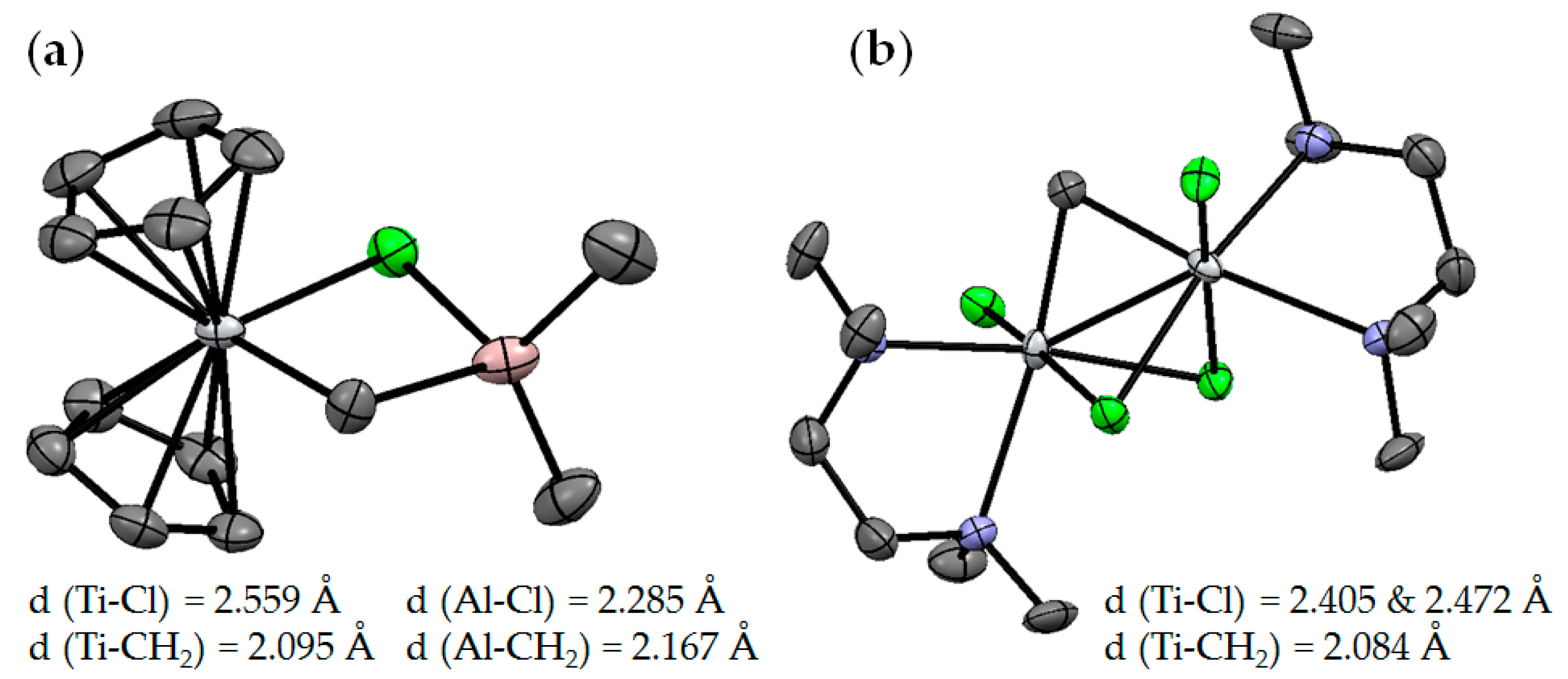
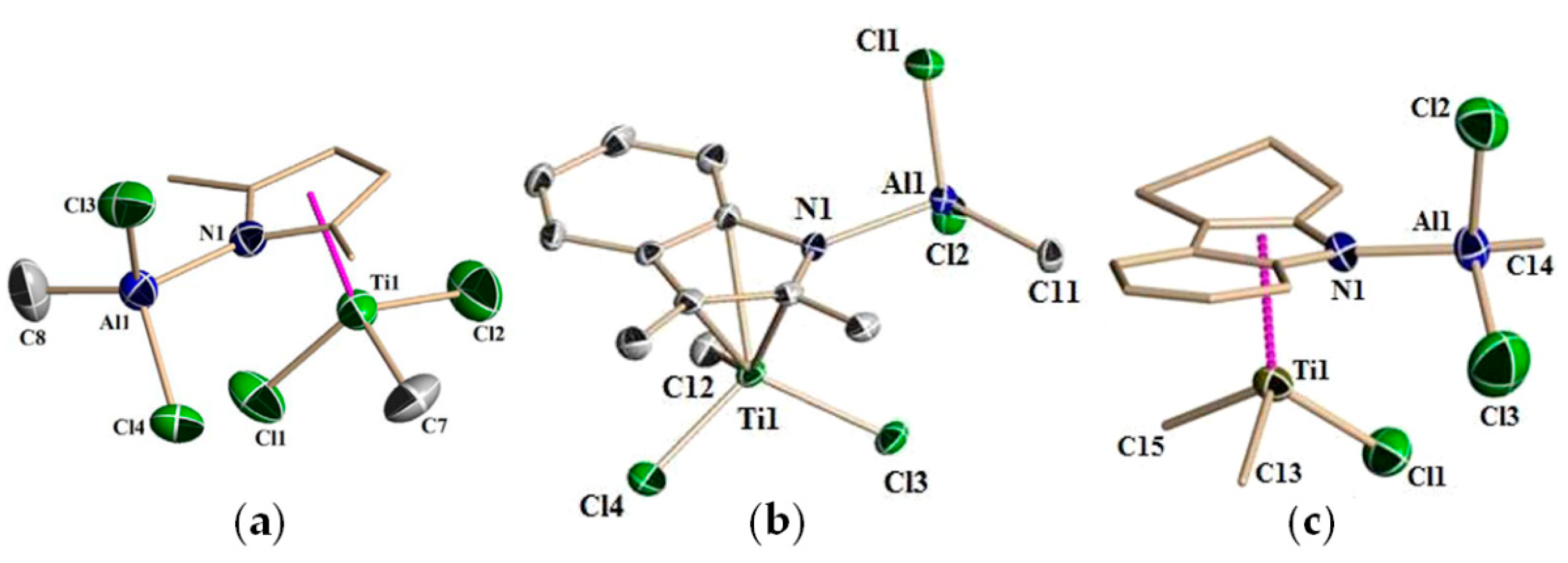
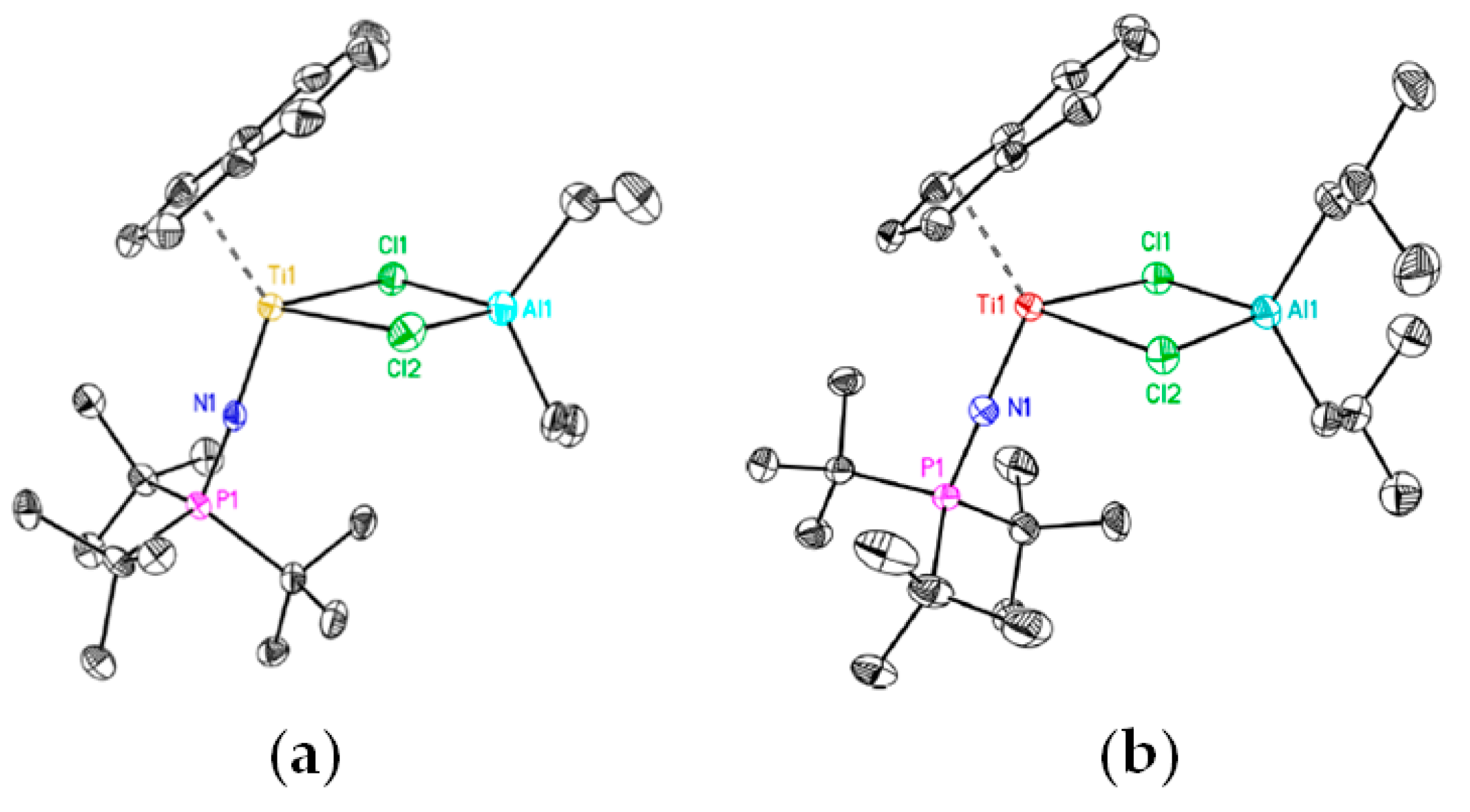
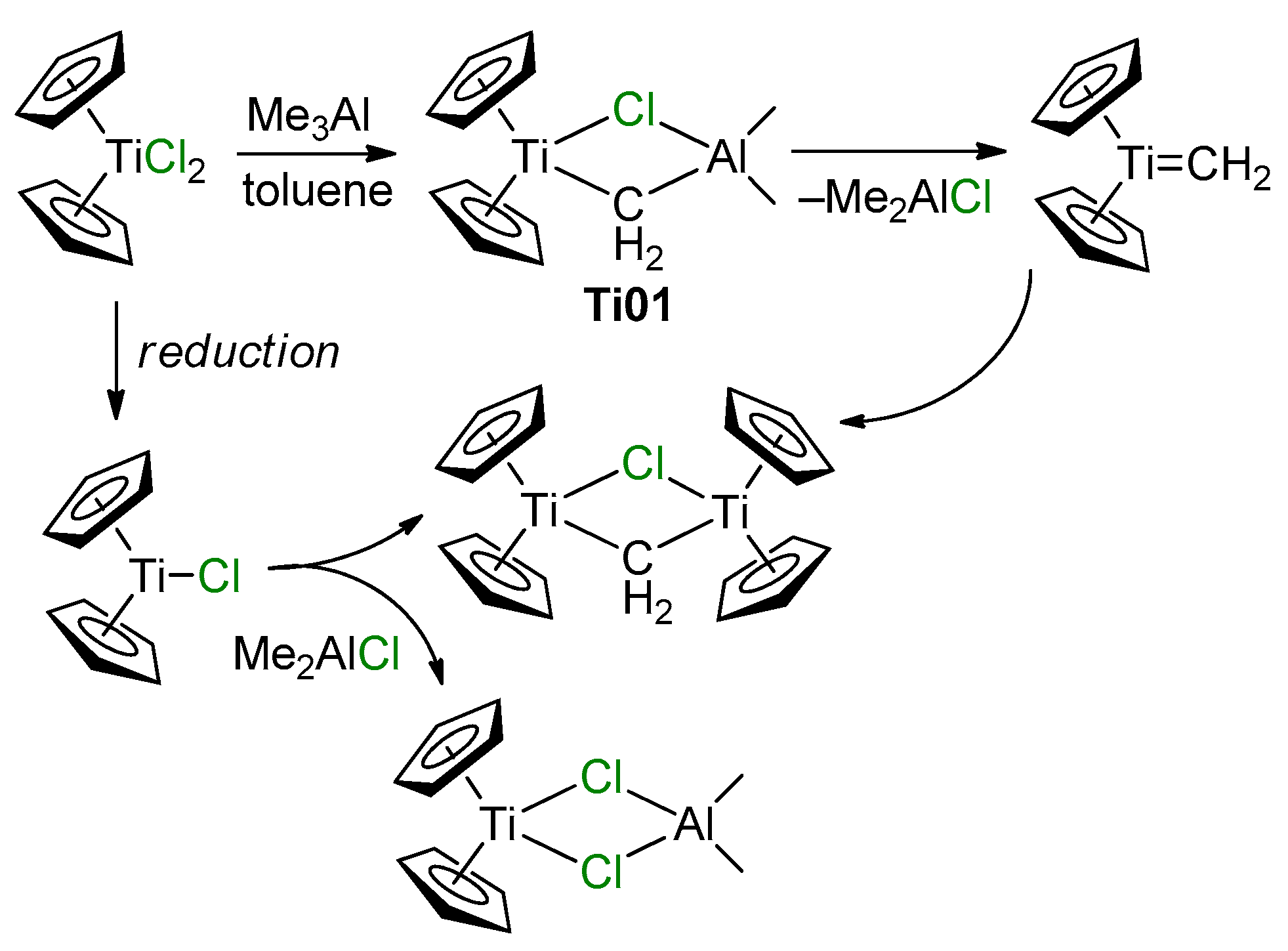
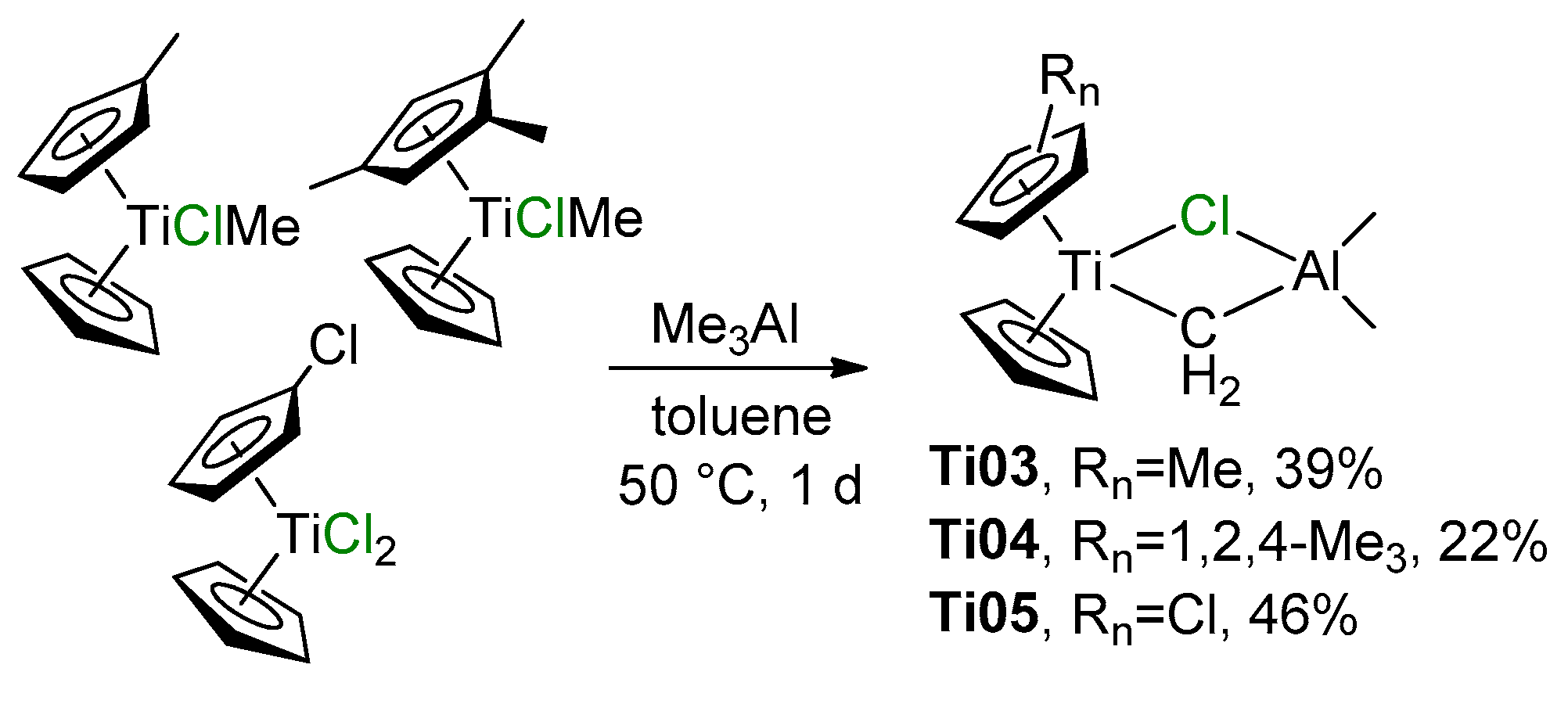
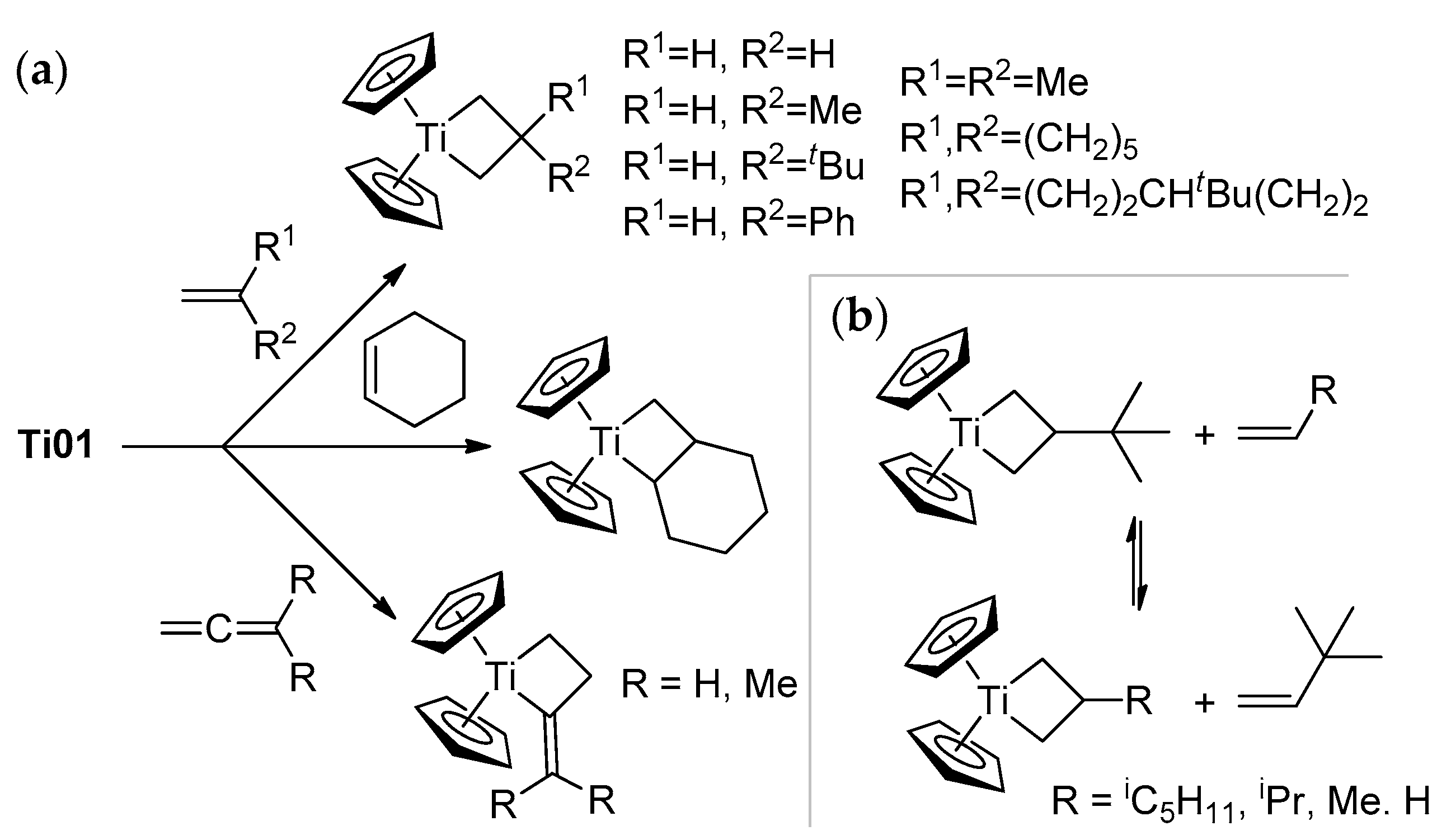
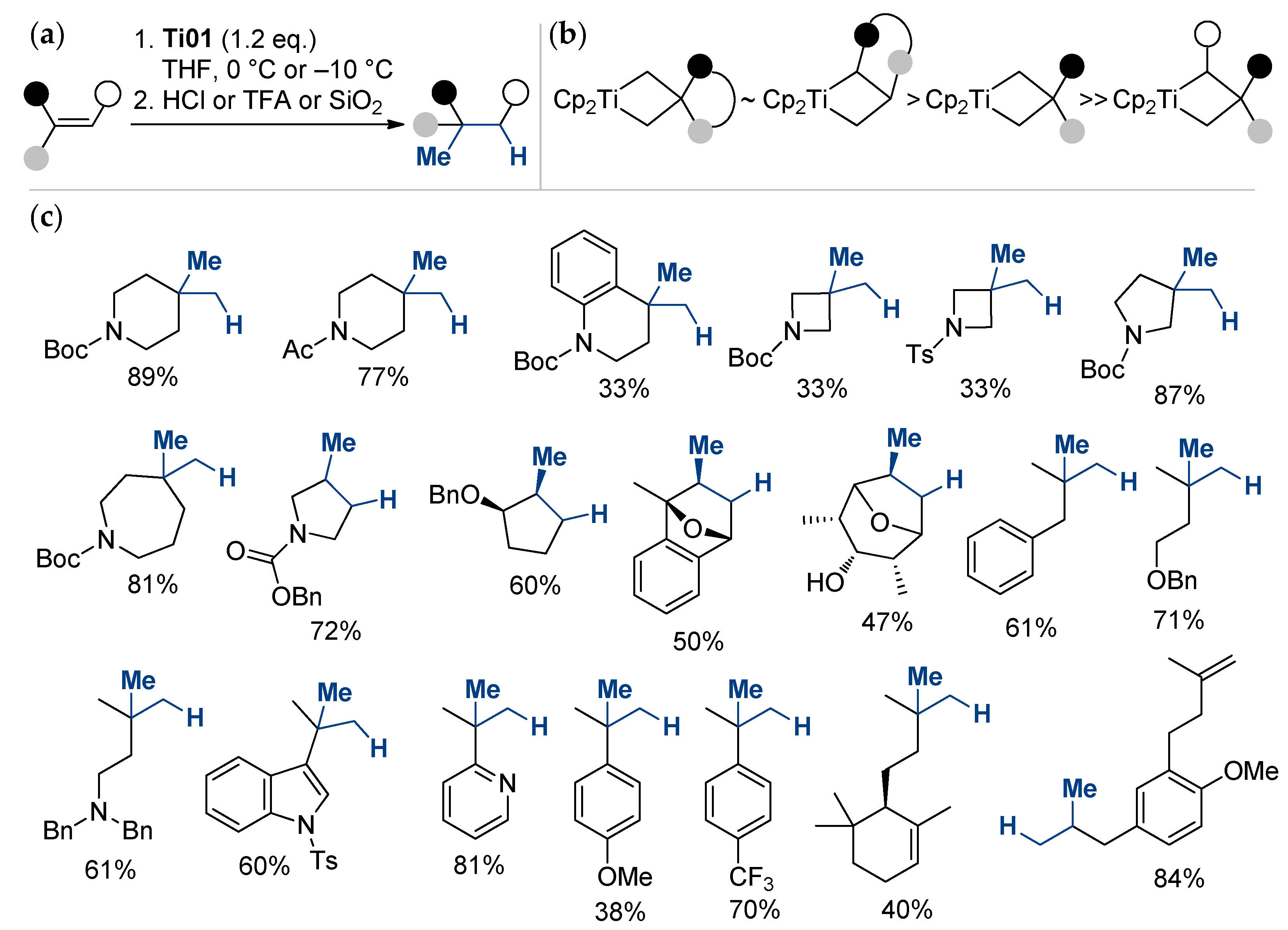
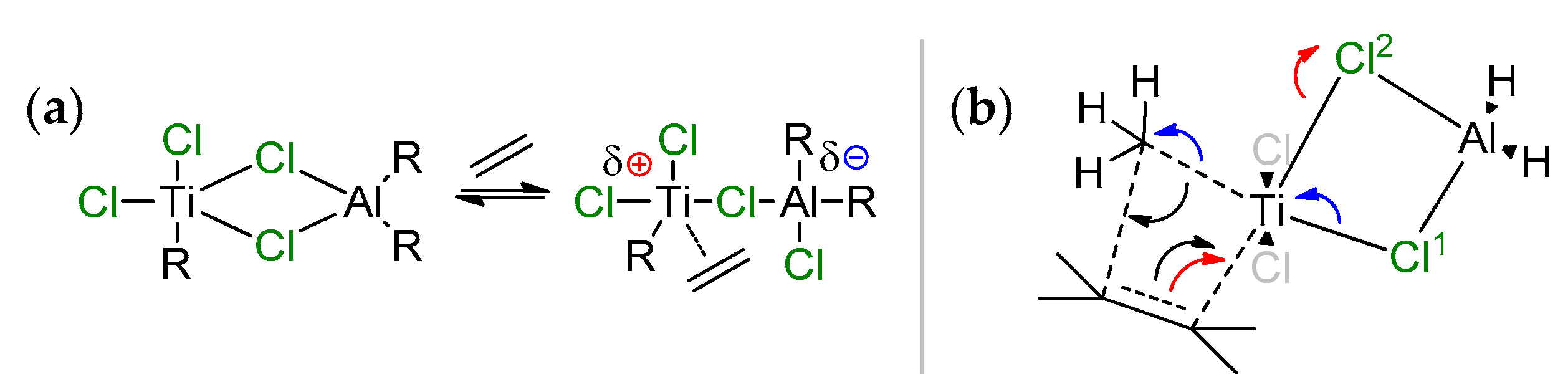
2.1. The Tebbe Reagent and Similar Ti Complexes
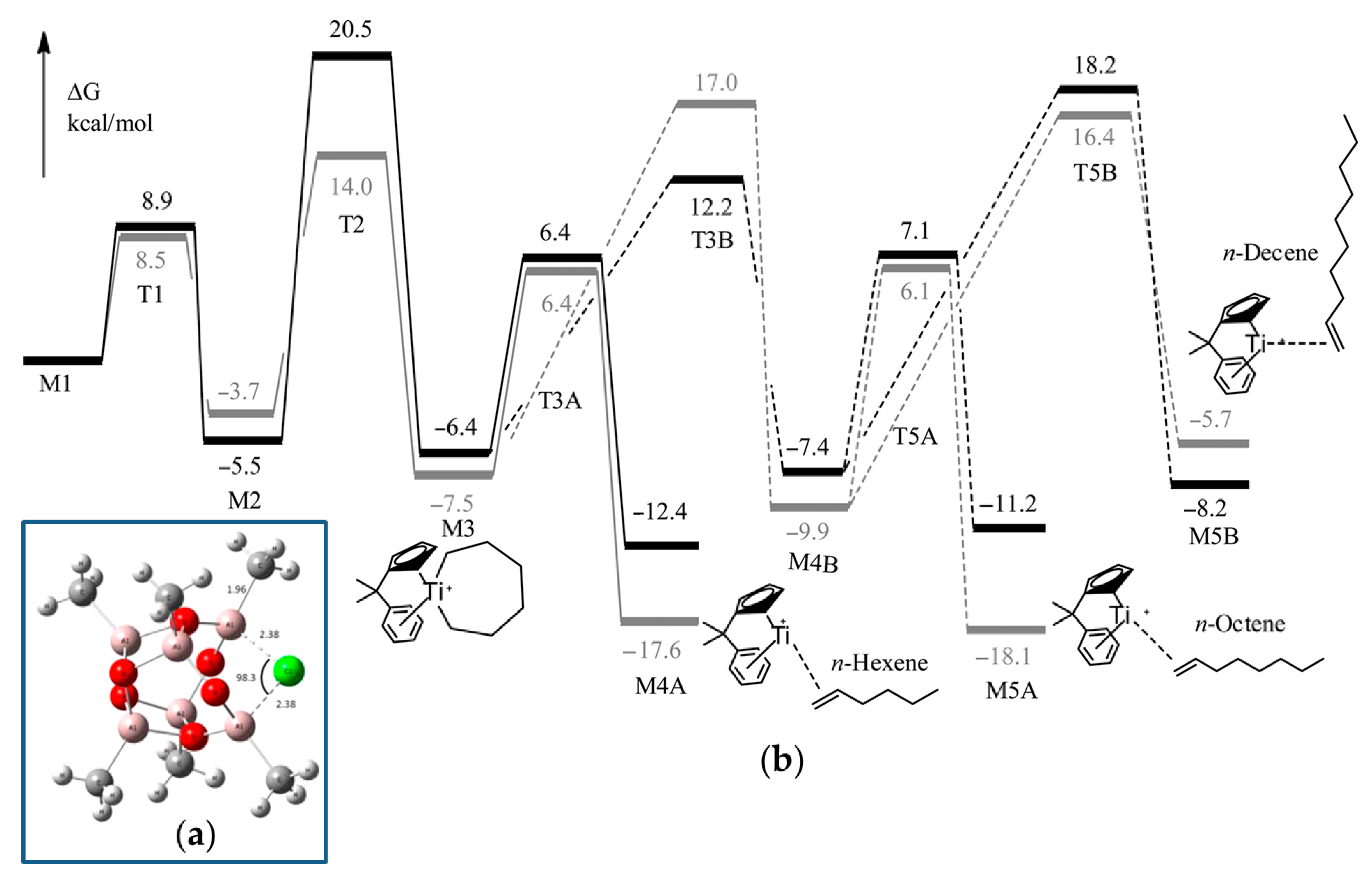
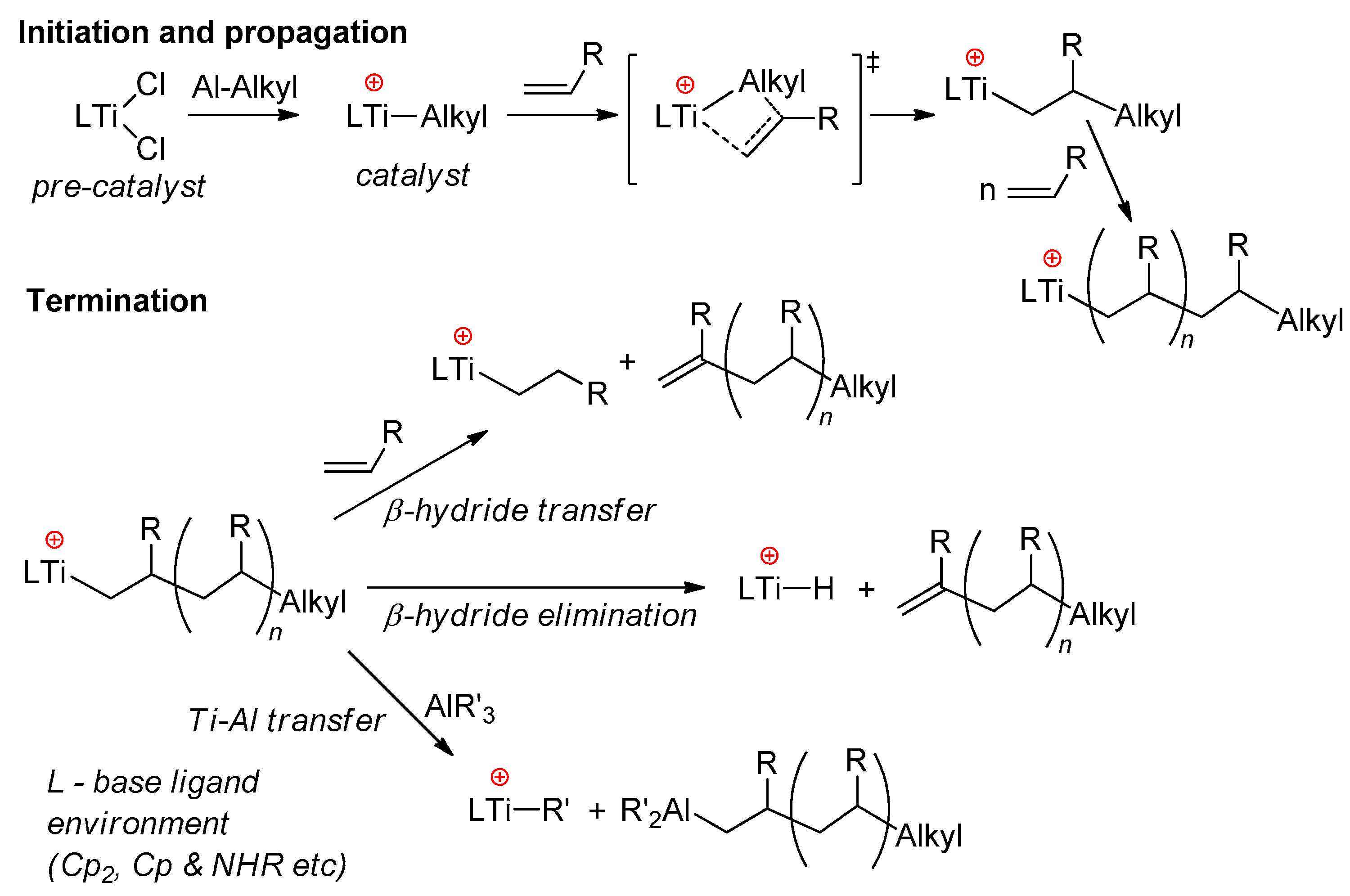
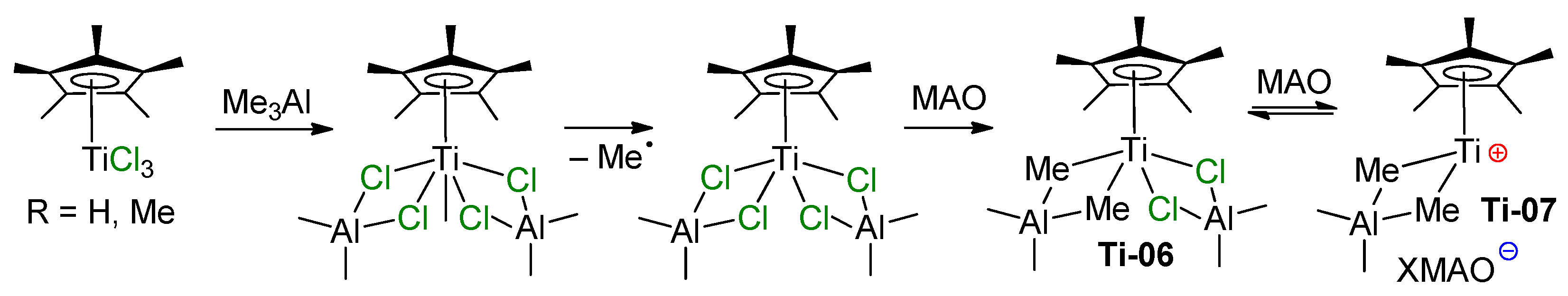
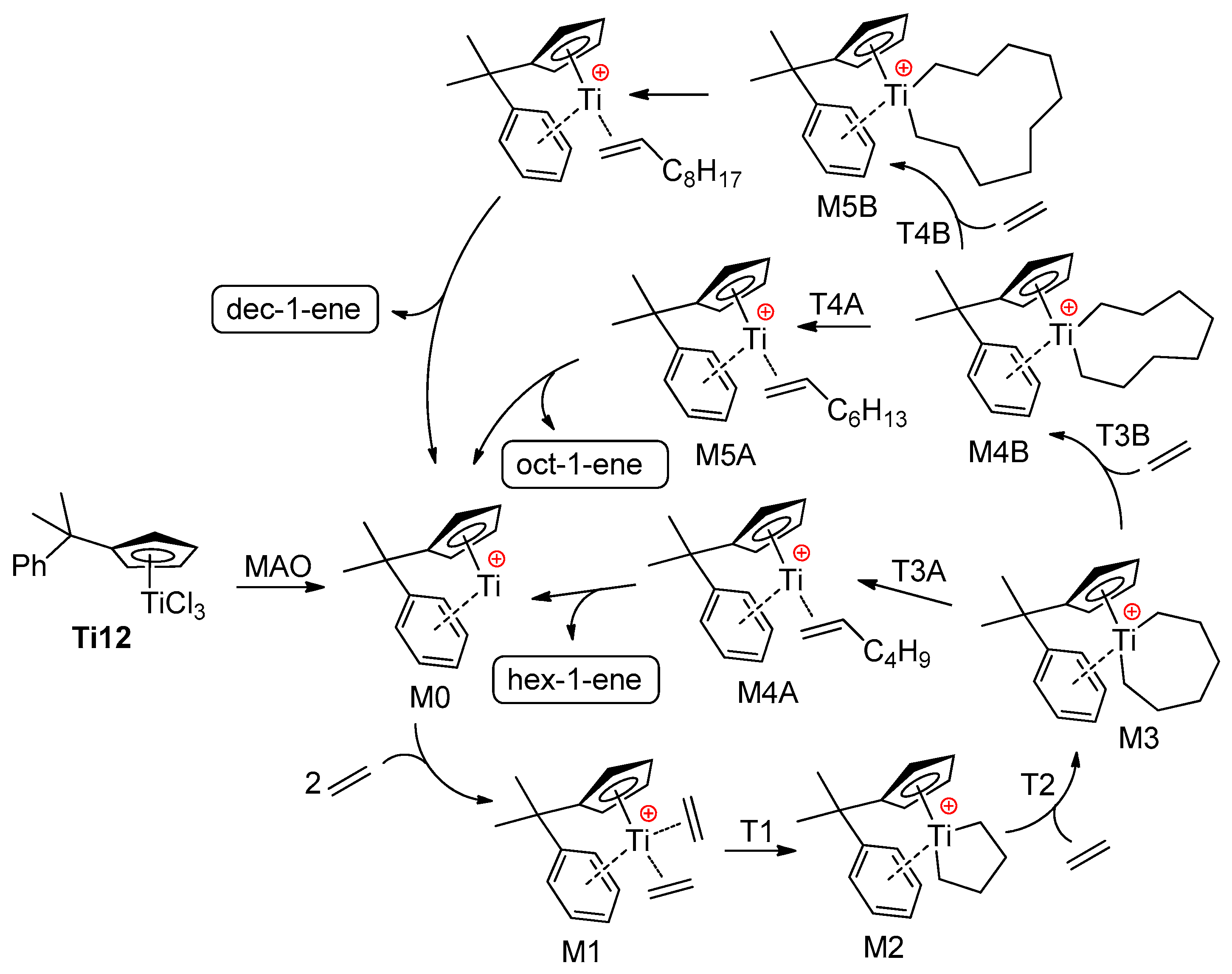
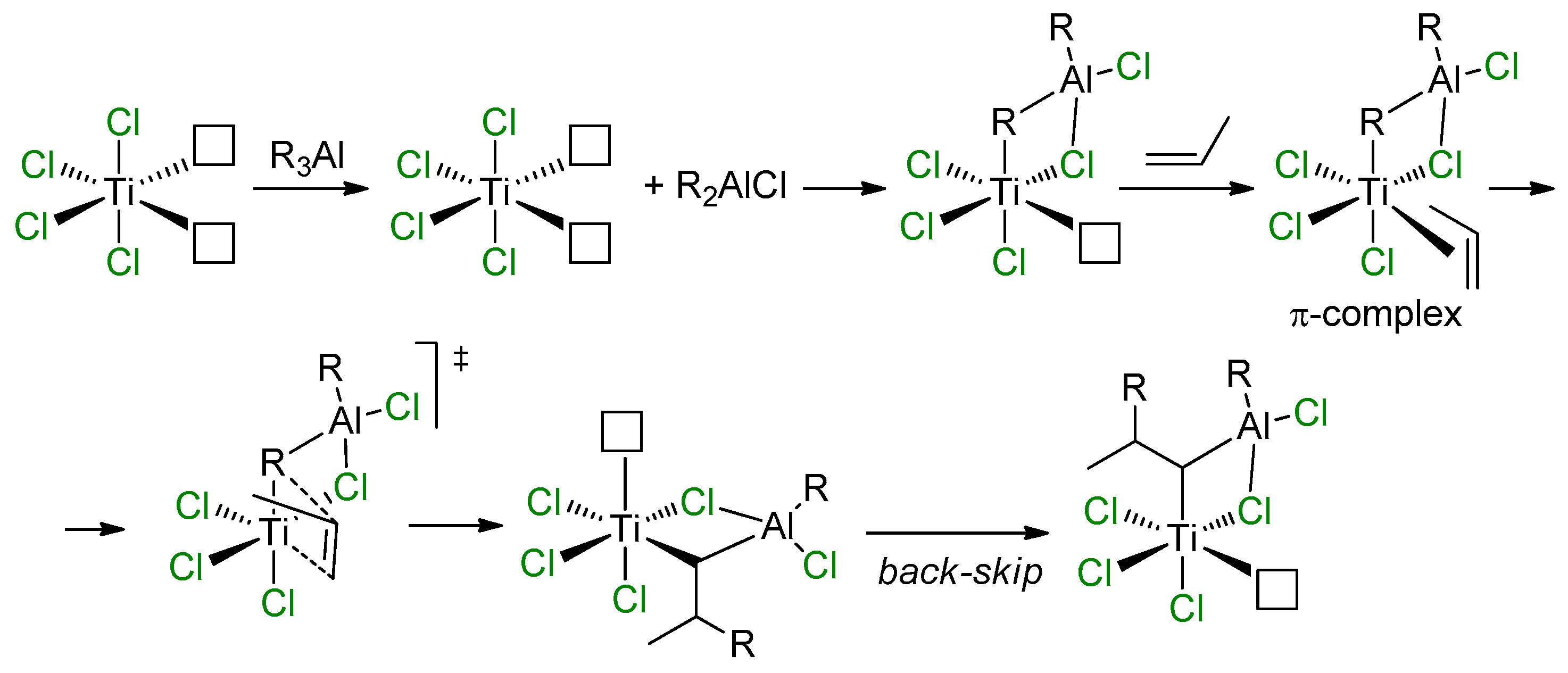
2.2. Single-Site Ti Catalysts of the Polymerization and Oligomerization of α-Olefins
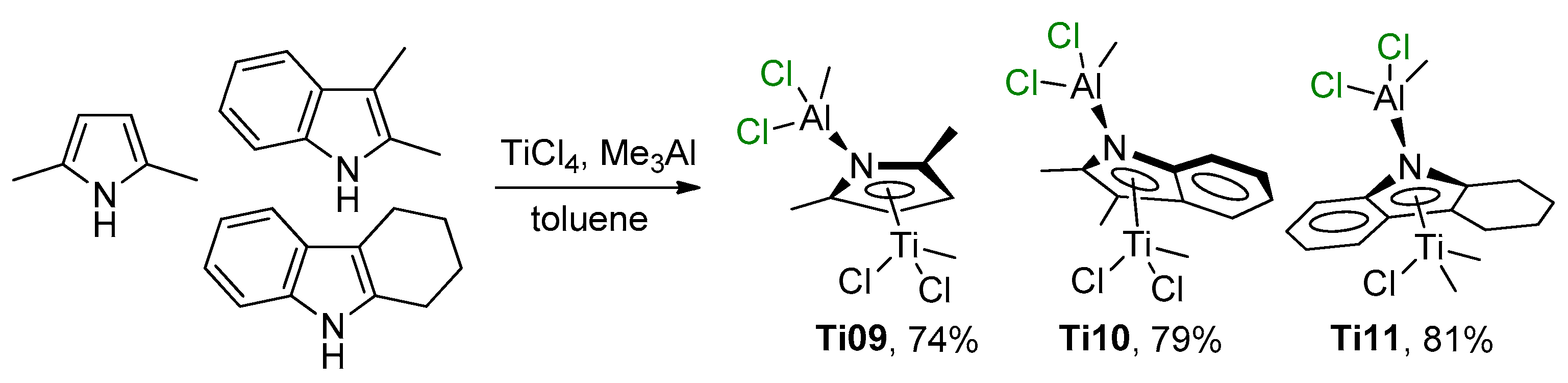
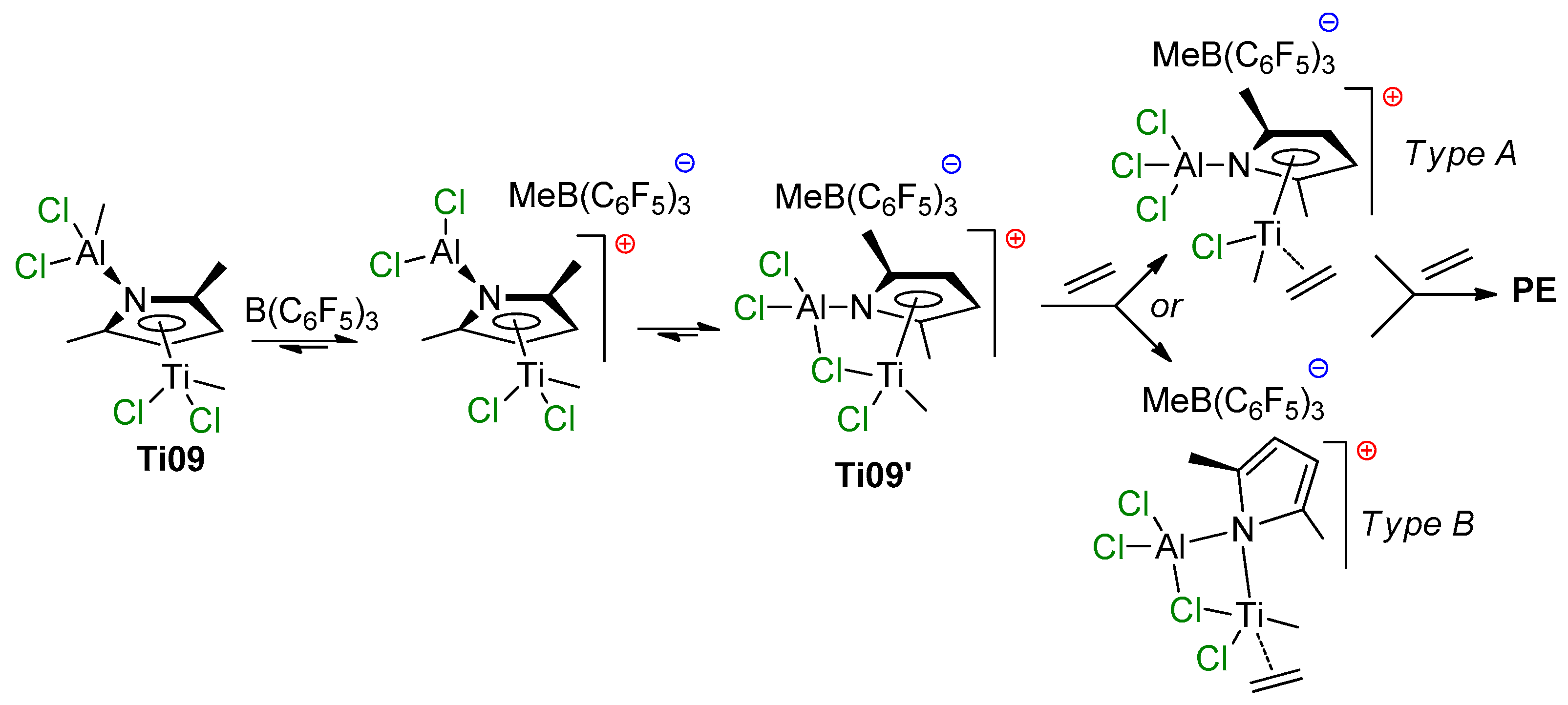
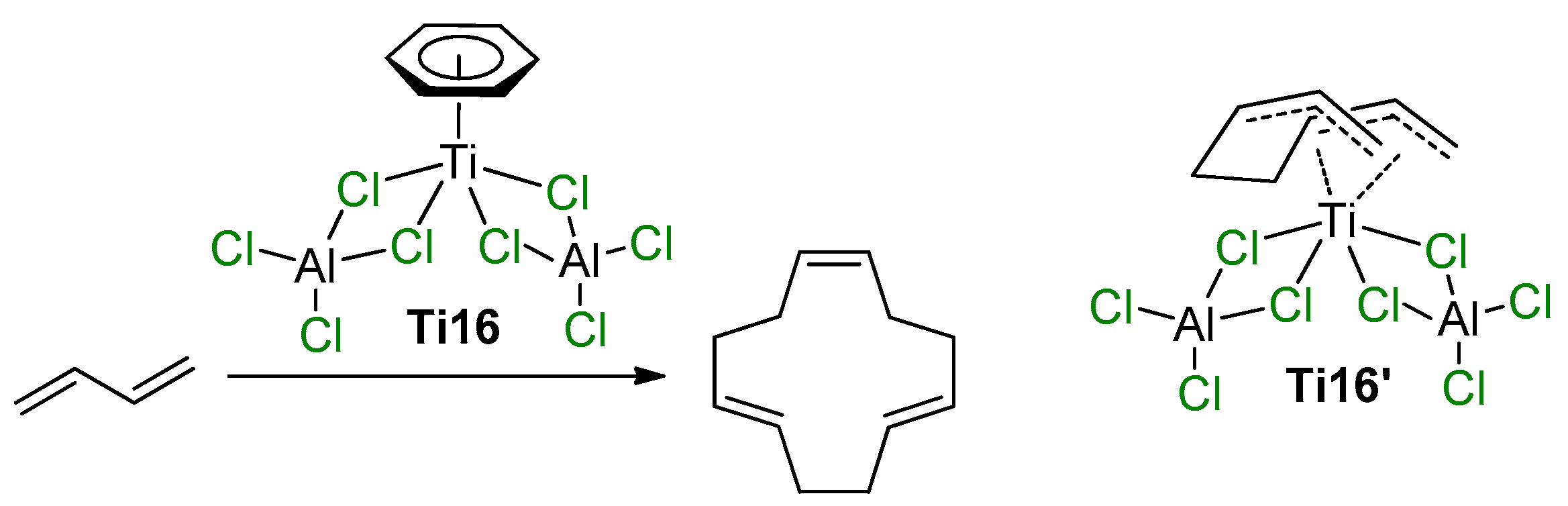
2.3. Single-Site Ti Catalysts of Polymerization of Dienes
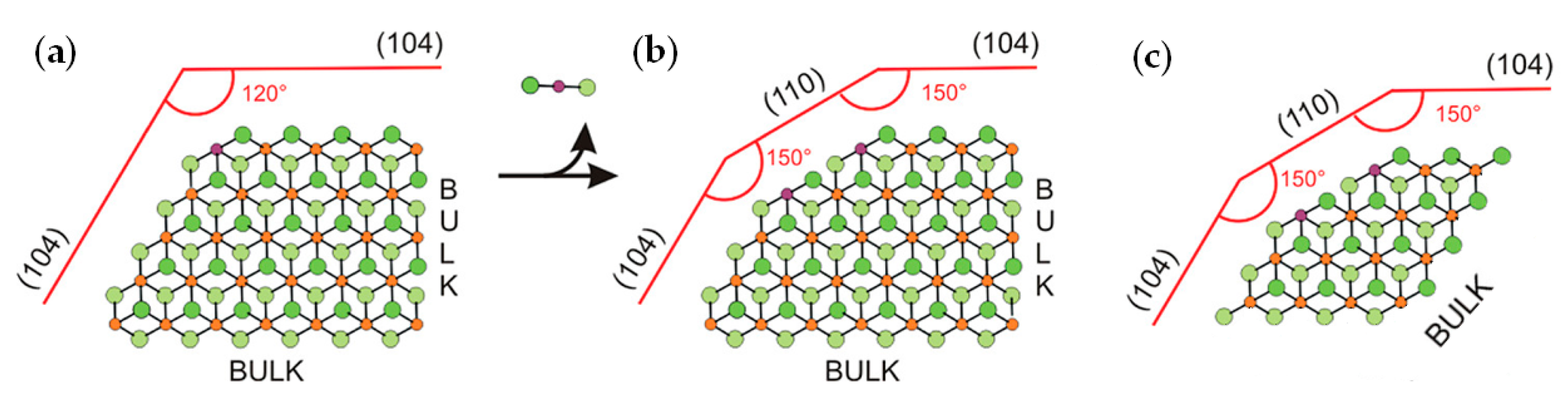
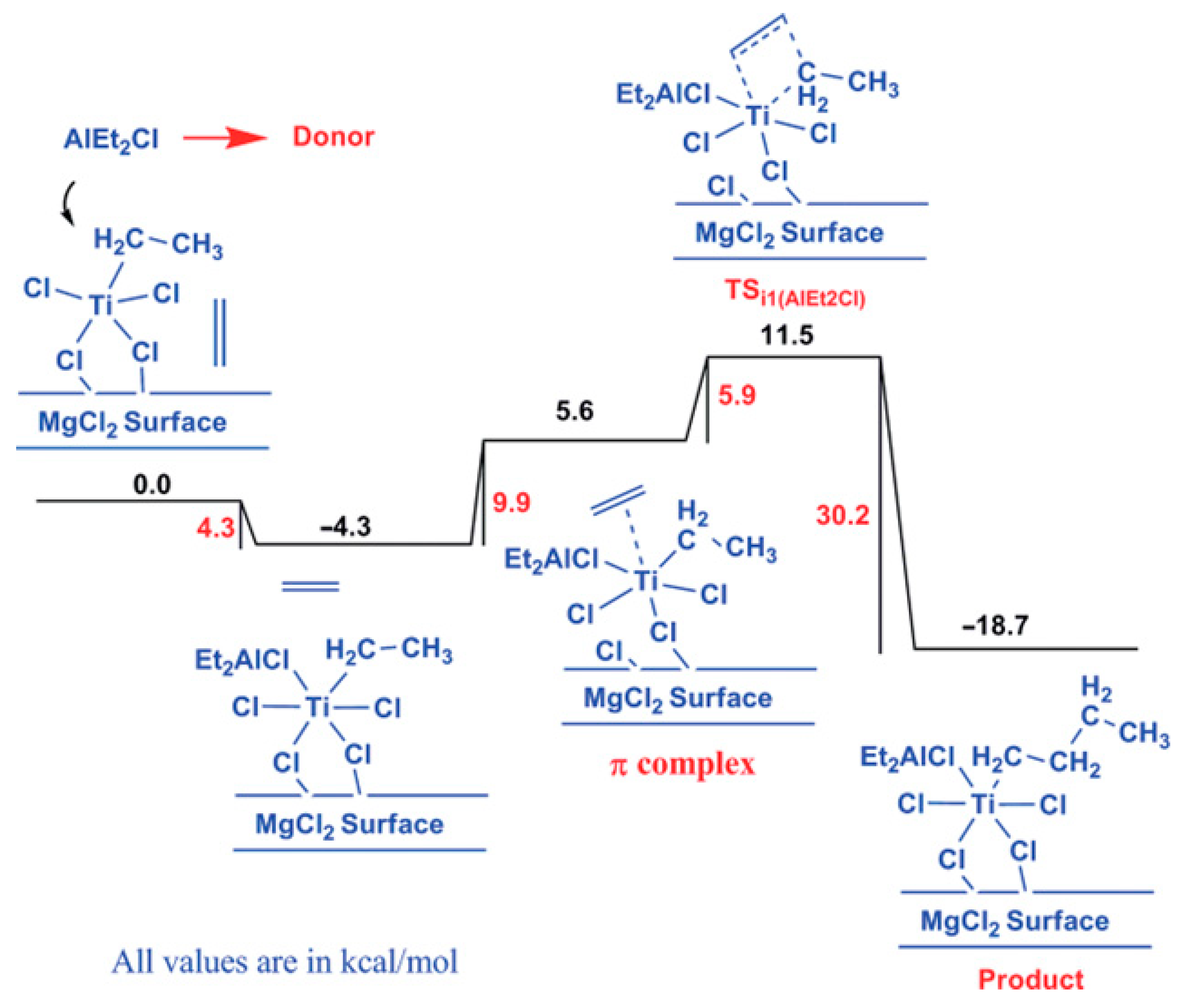
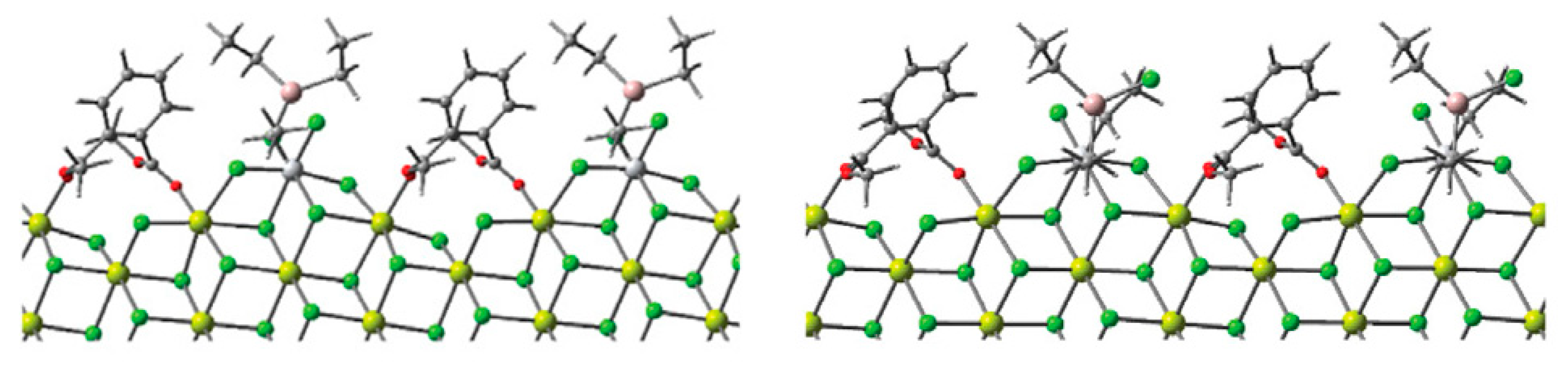
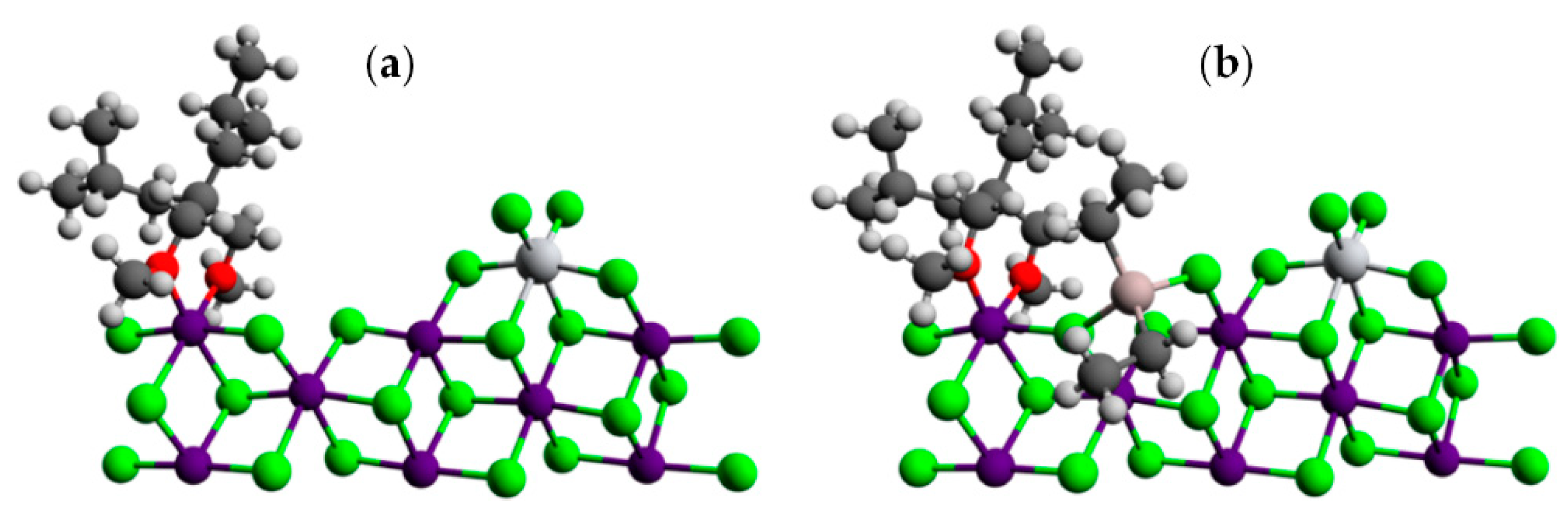
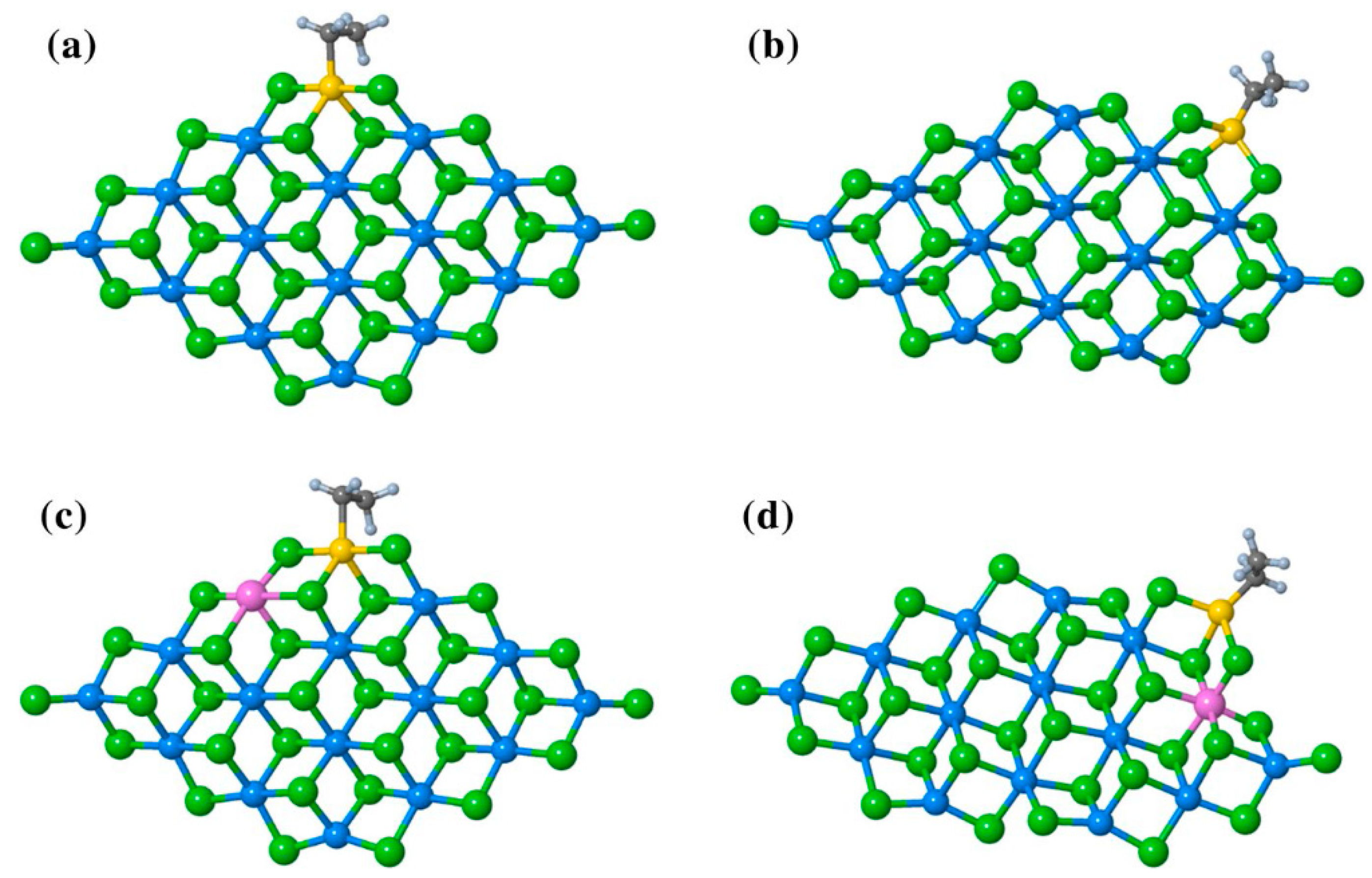
2.4. Heterogeneous Ti Ziegler–Natta Catalysts
3. Complexes of Zr
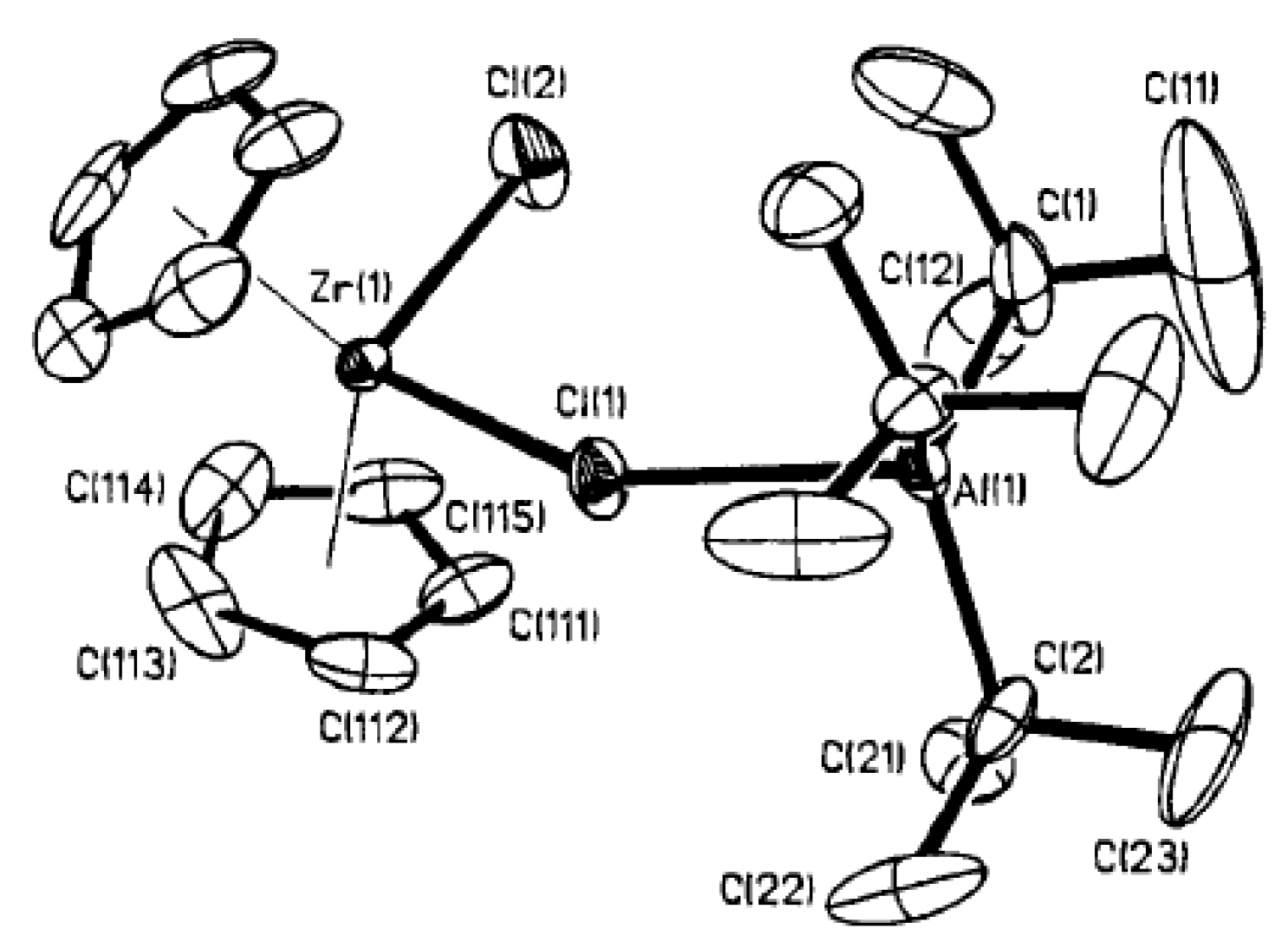
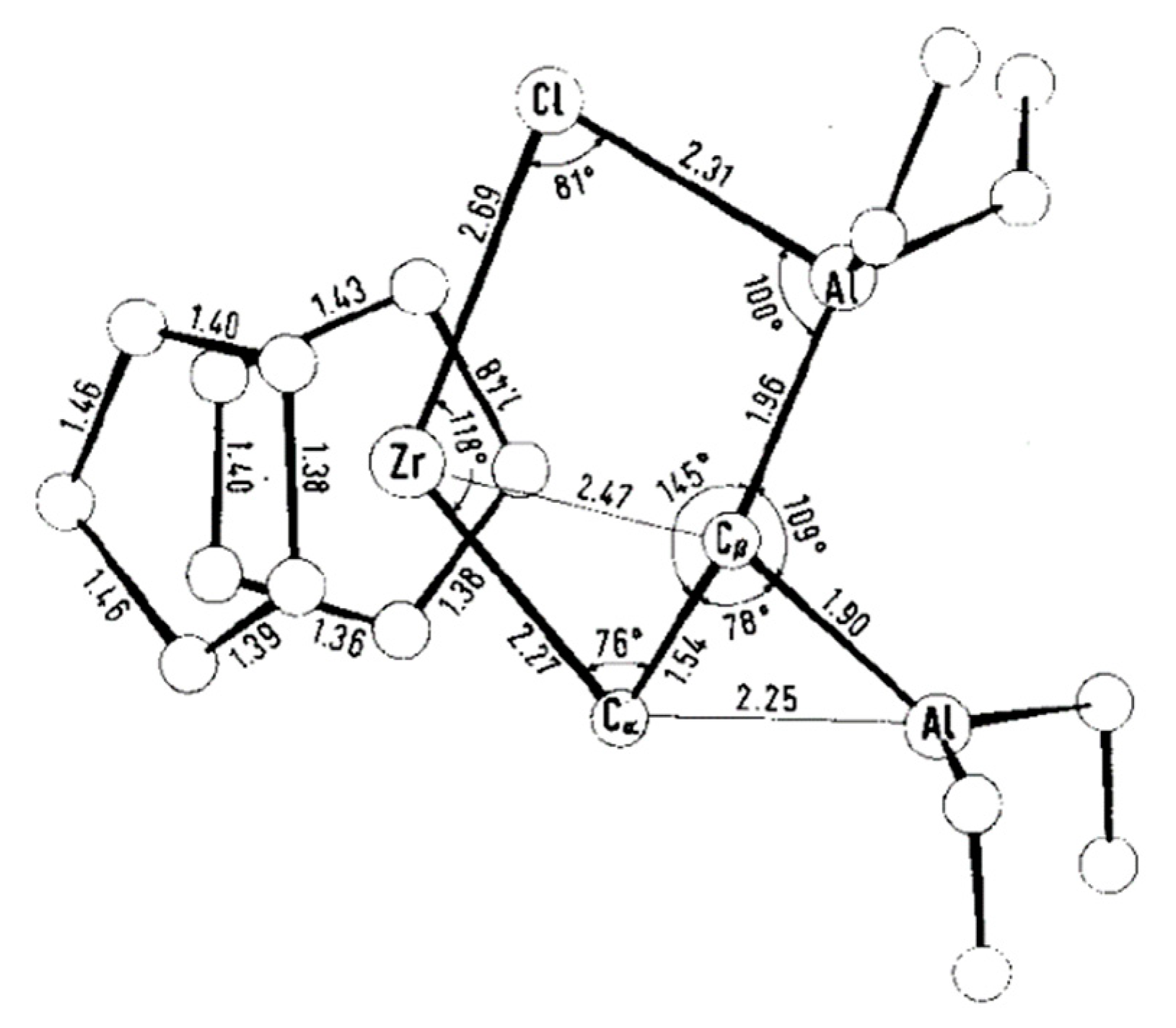
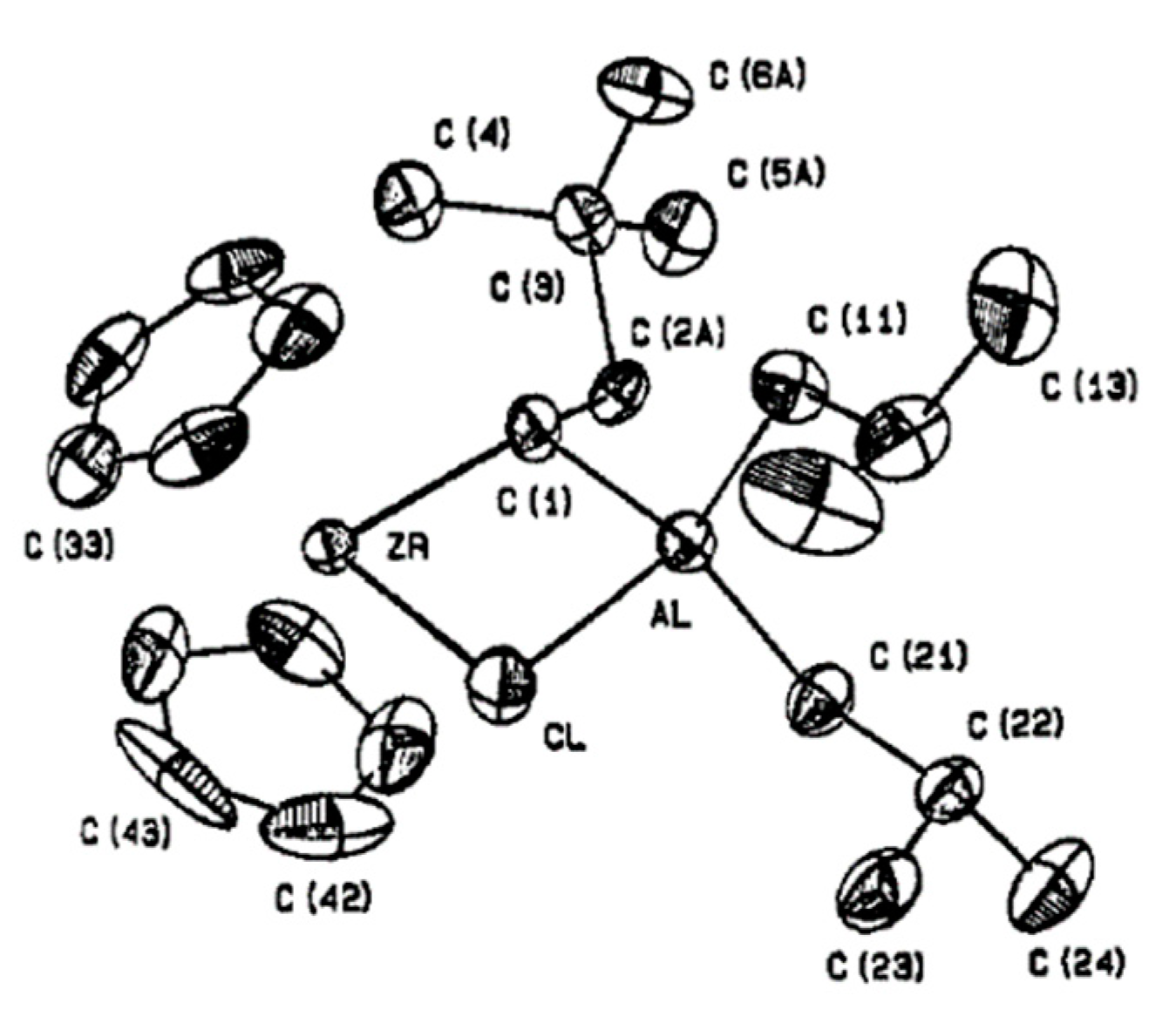
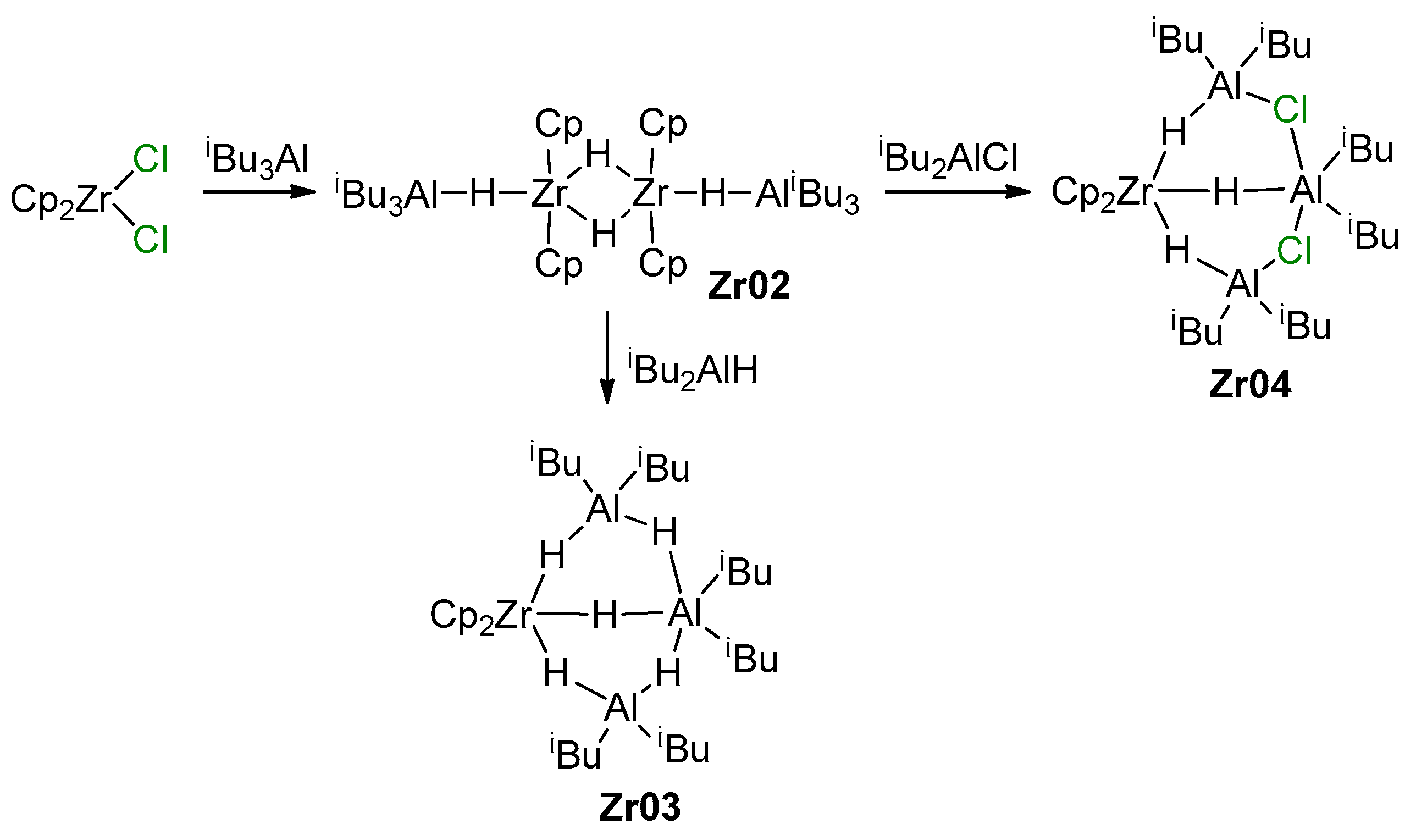
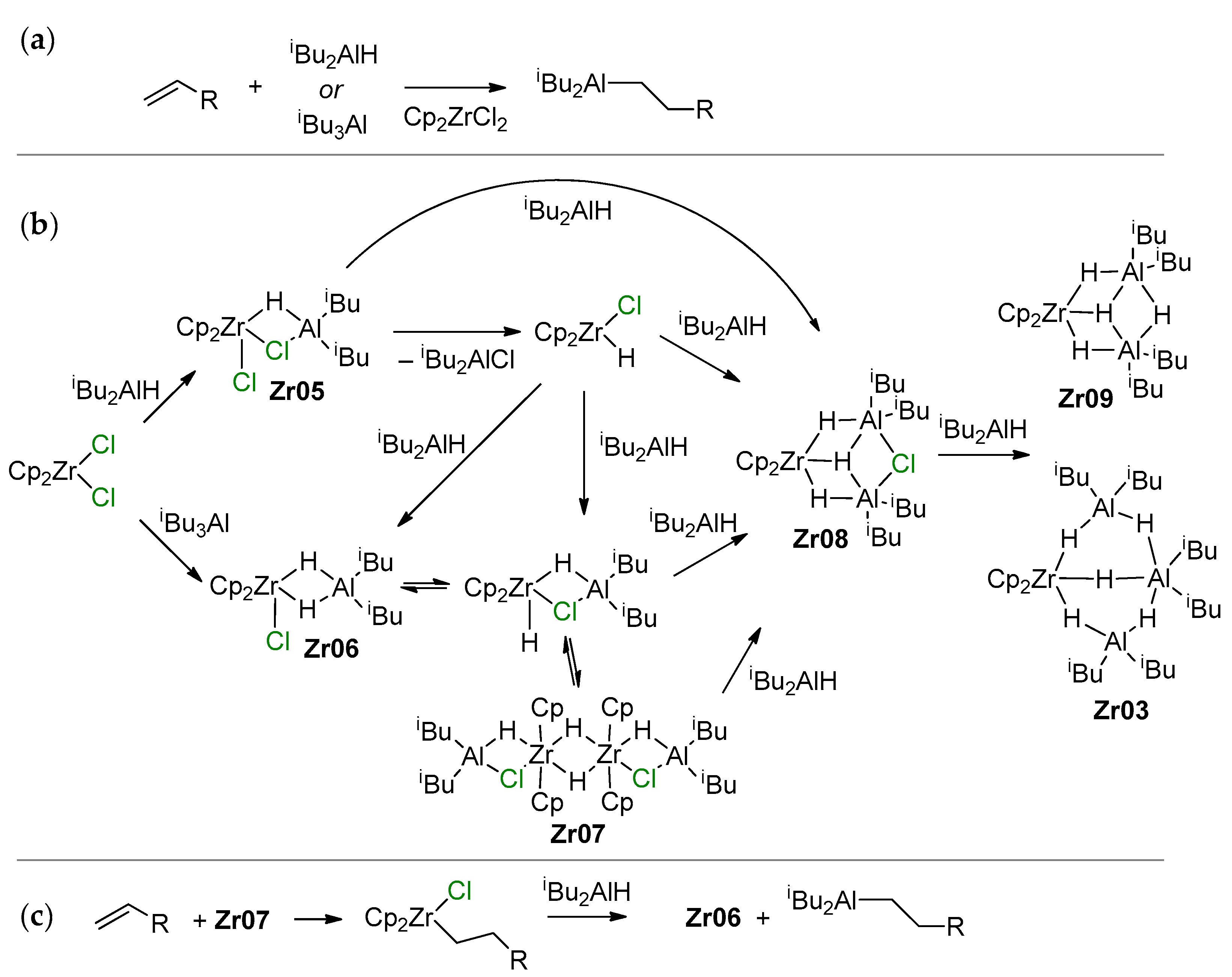
3.1. Zirconocene Chemistry: Non-Charged Complexes
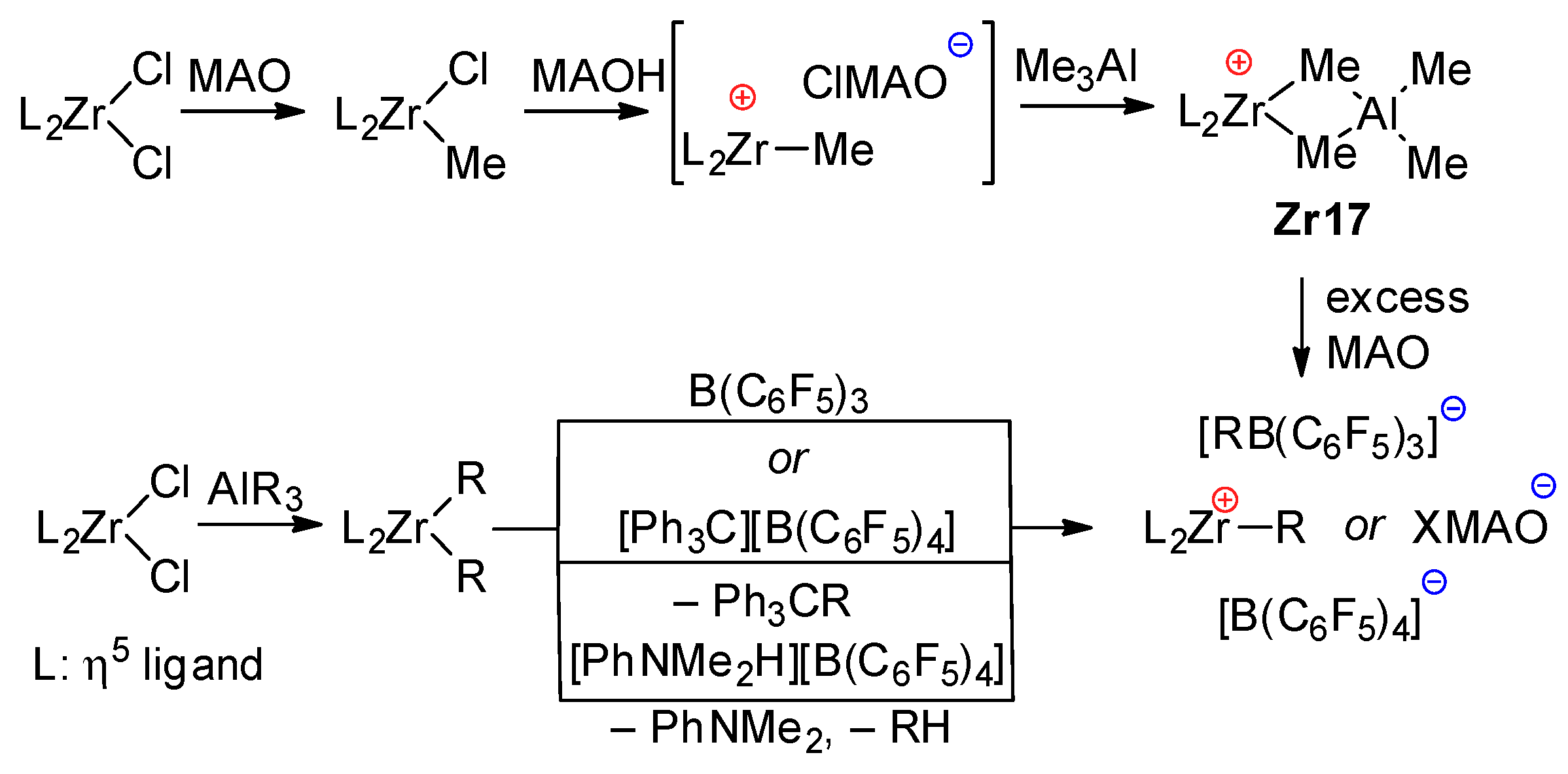
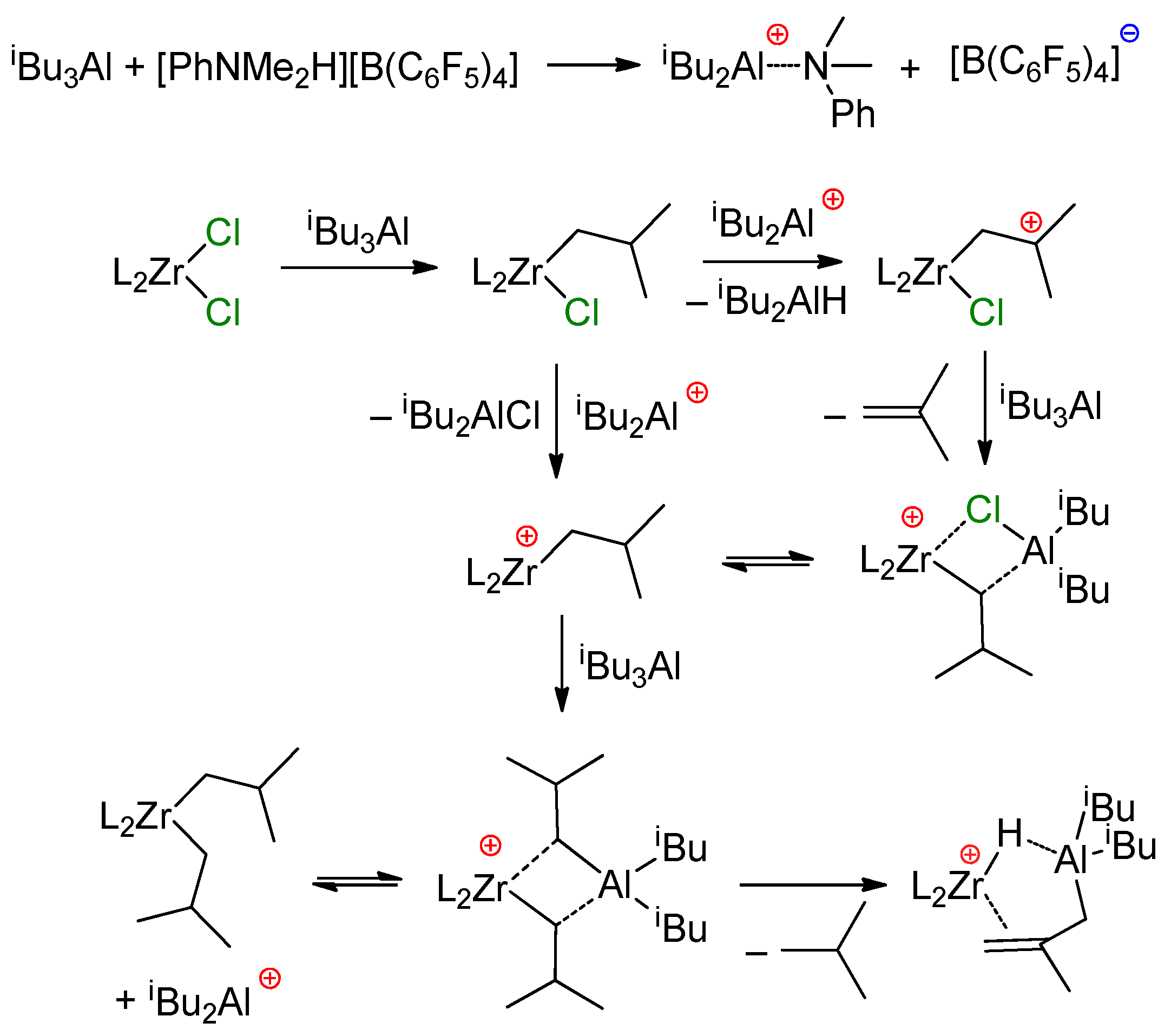
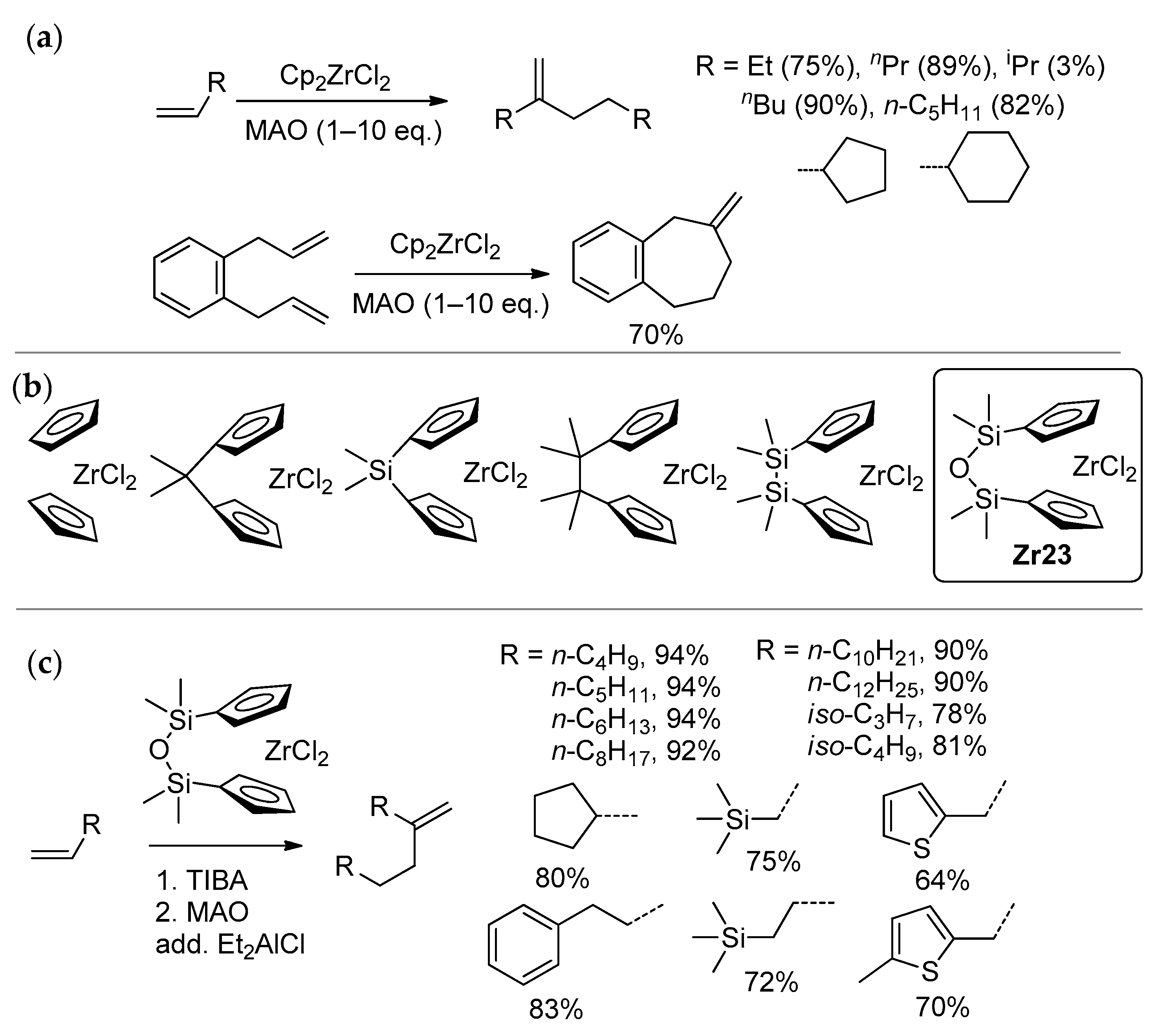
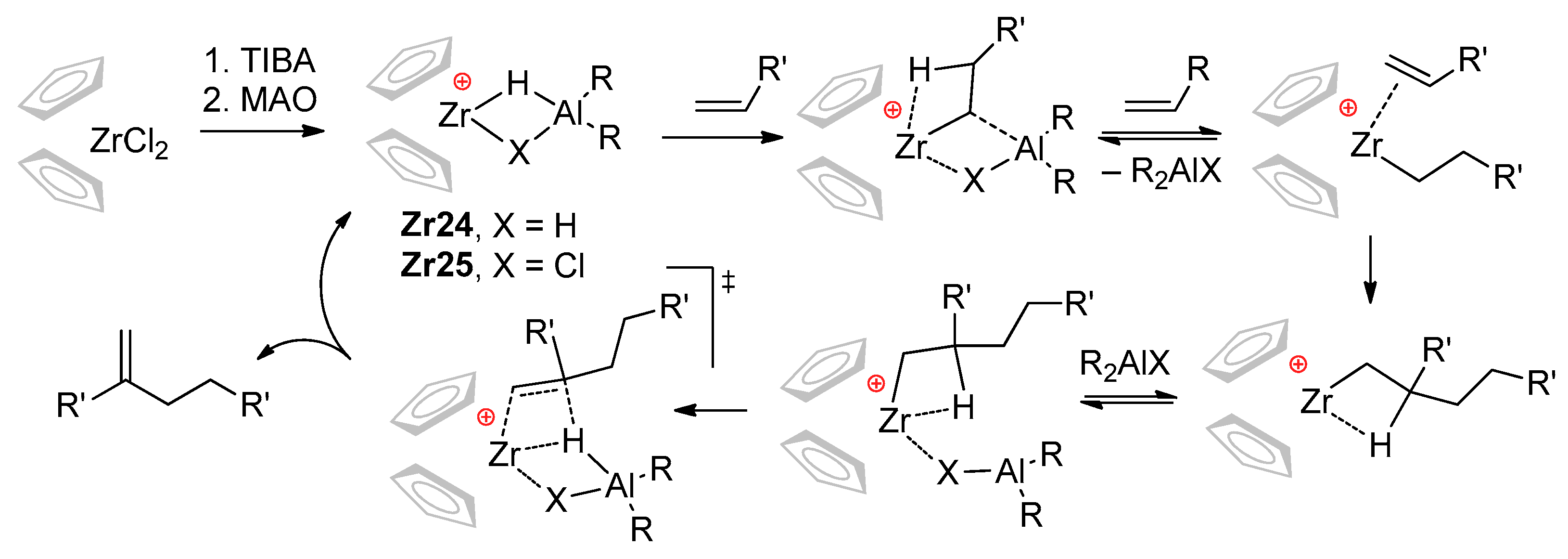
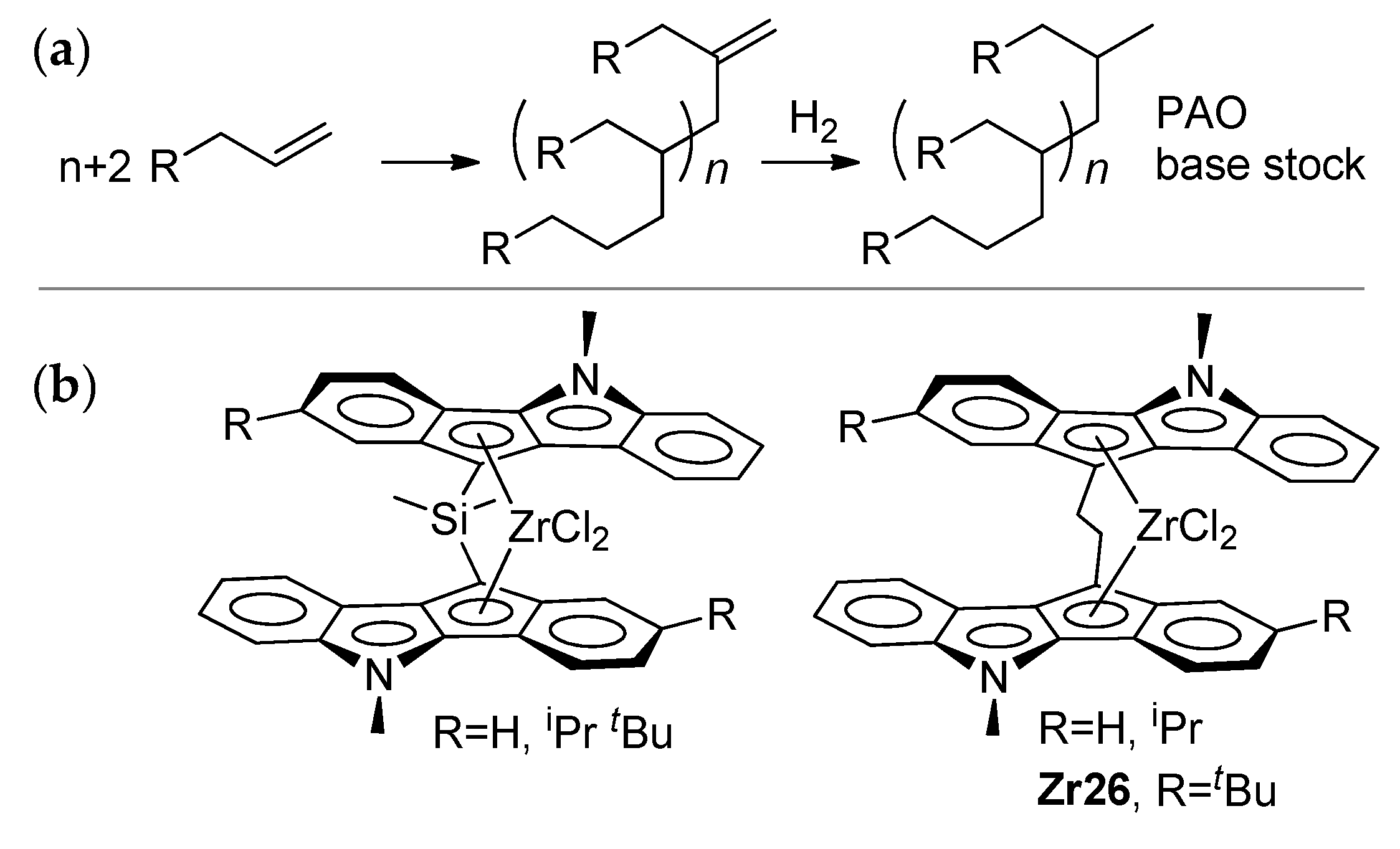
3.2. Zirconocene Cationic Complexes in Oligomerization and Polymerization of α-Olefins
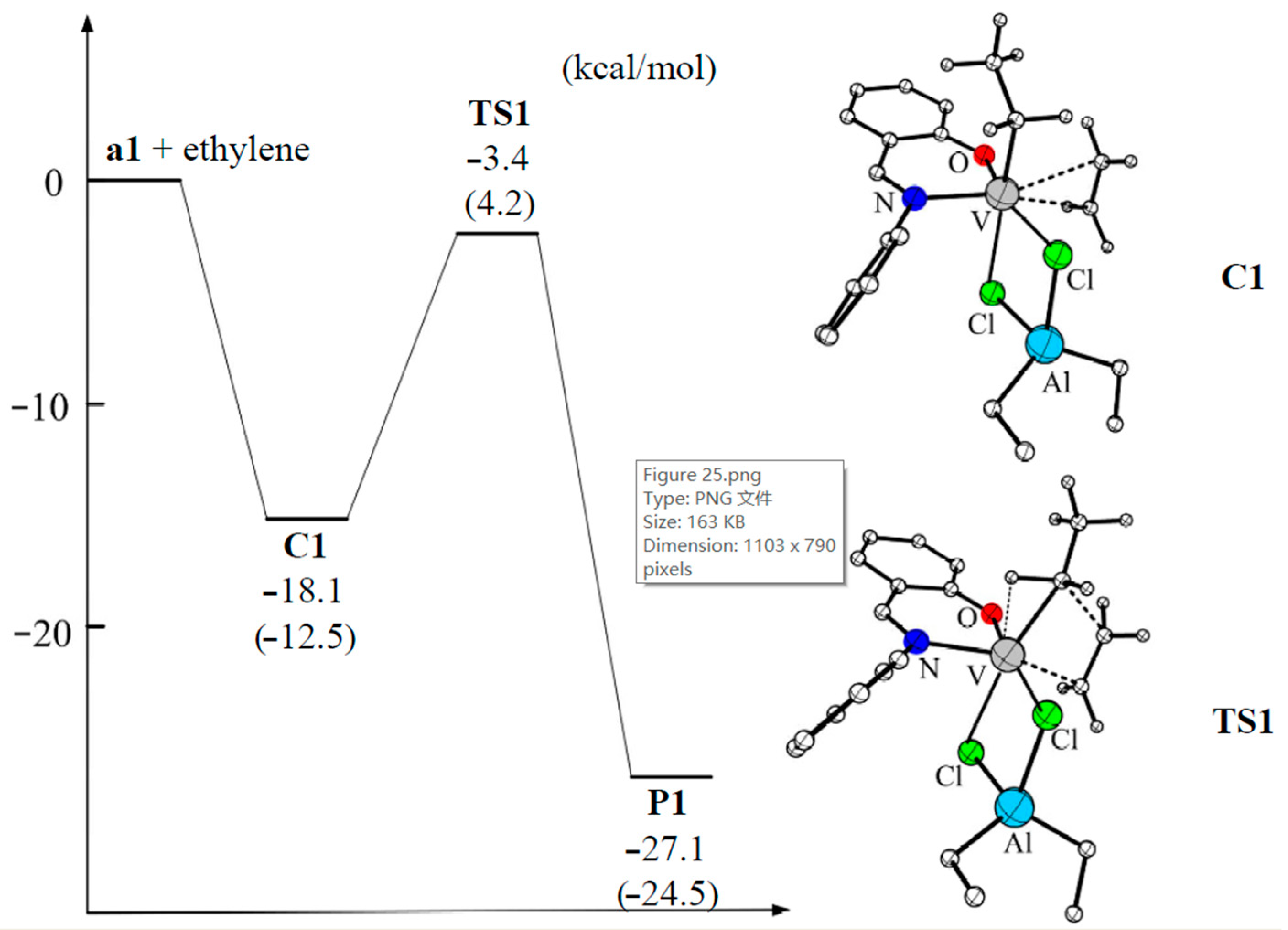
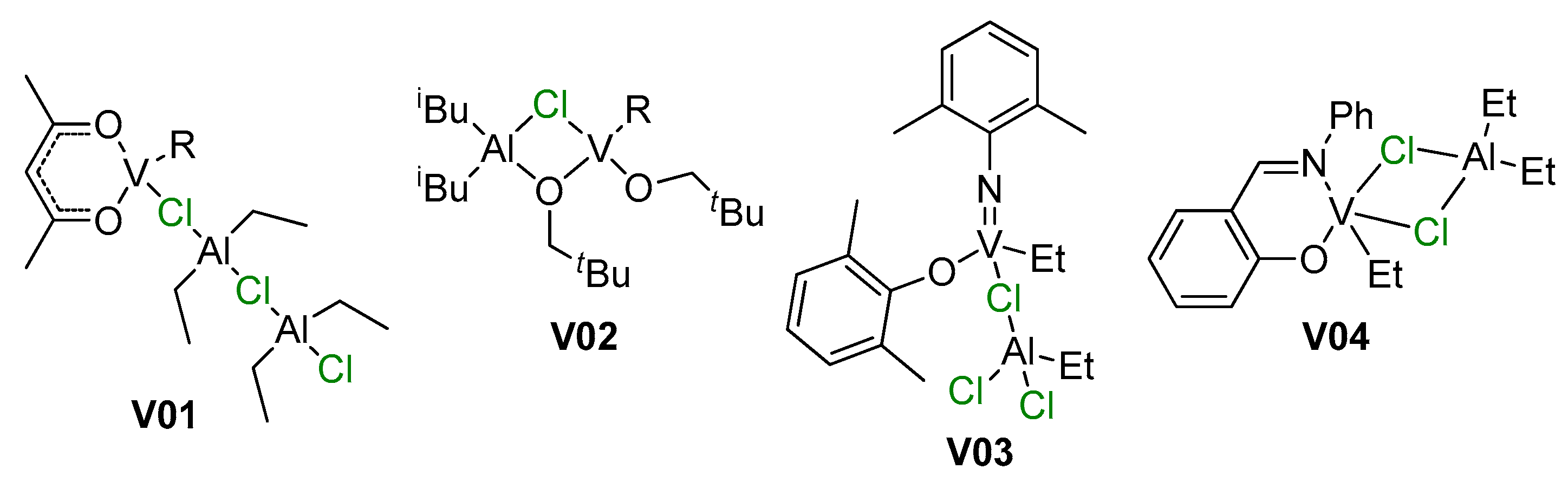
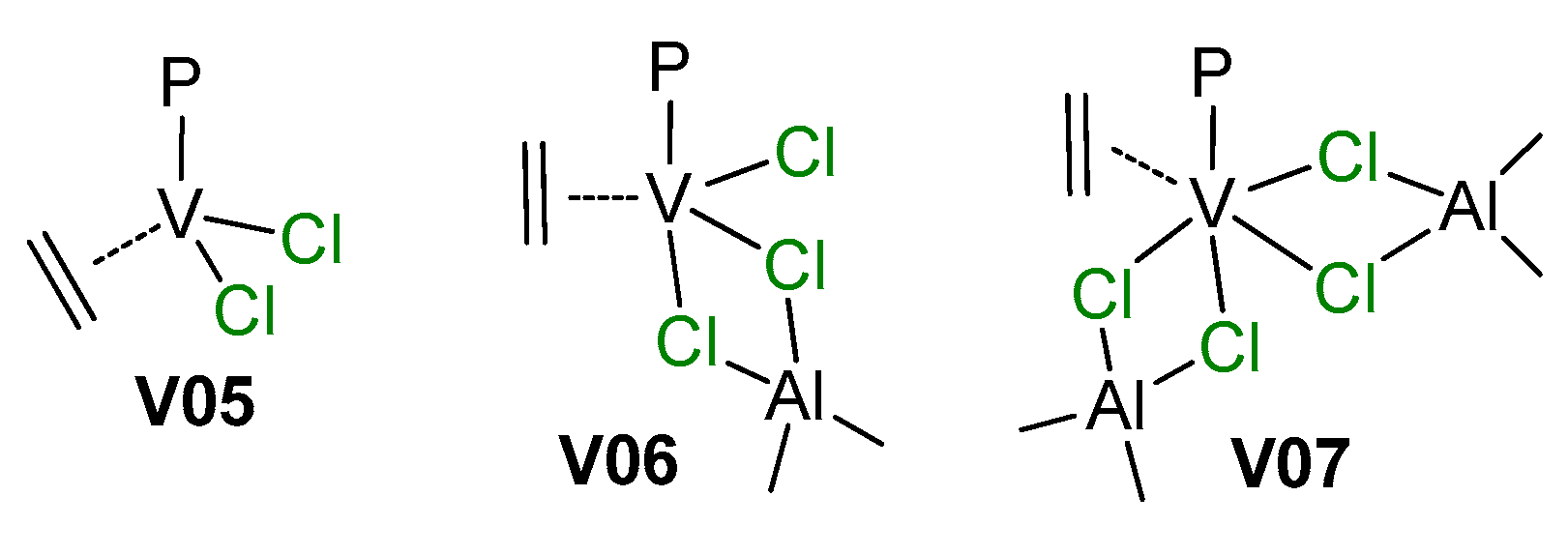
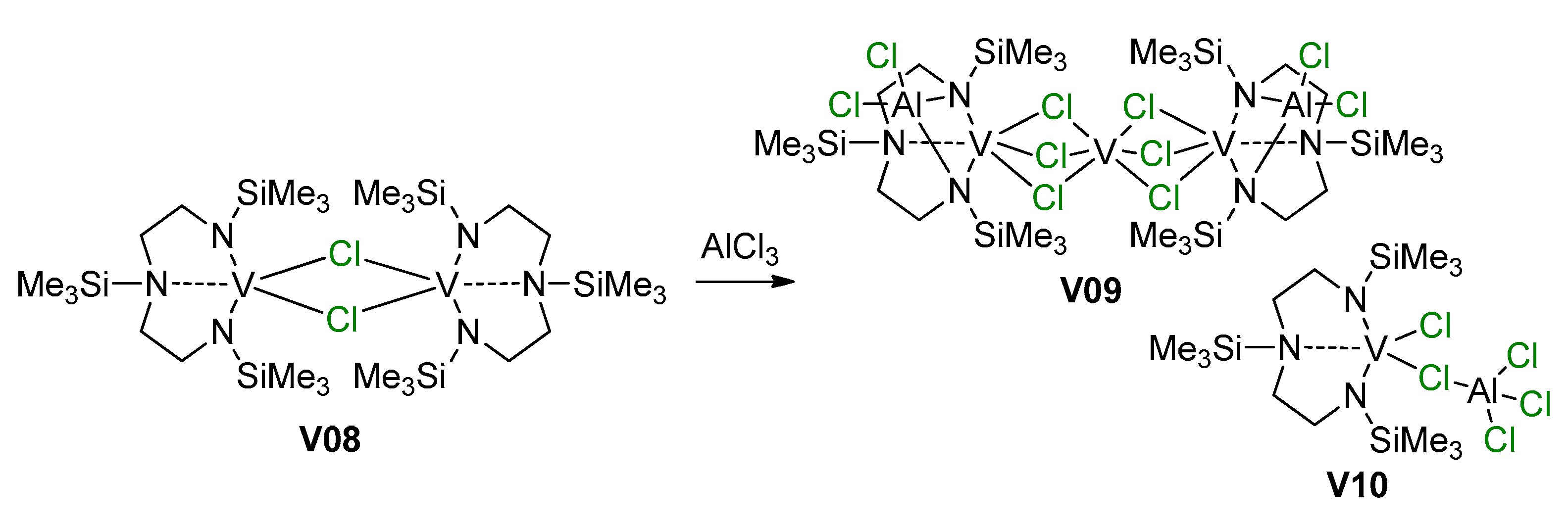
4. Complexes of V
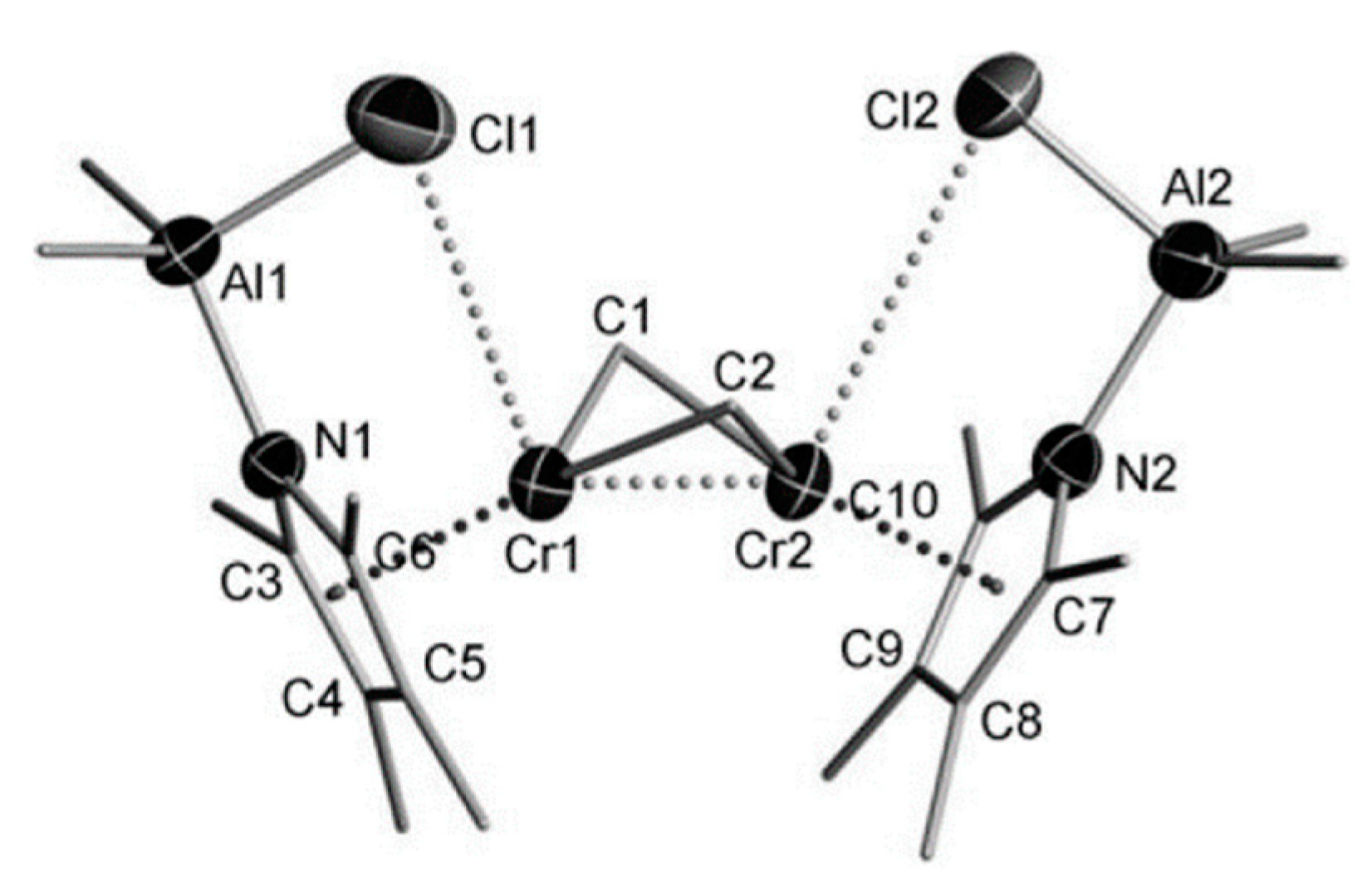
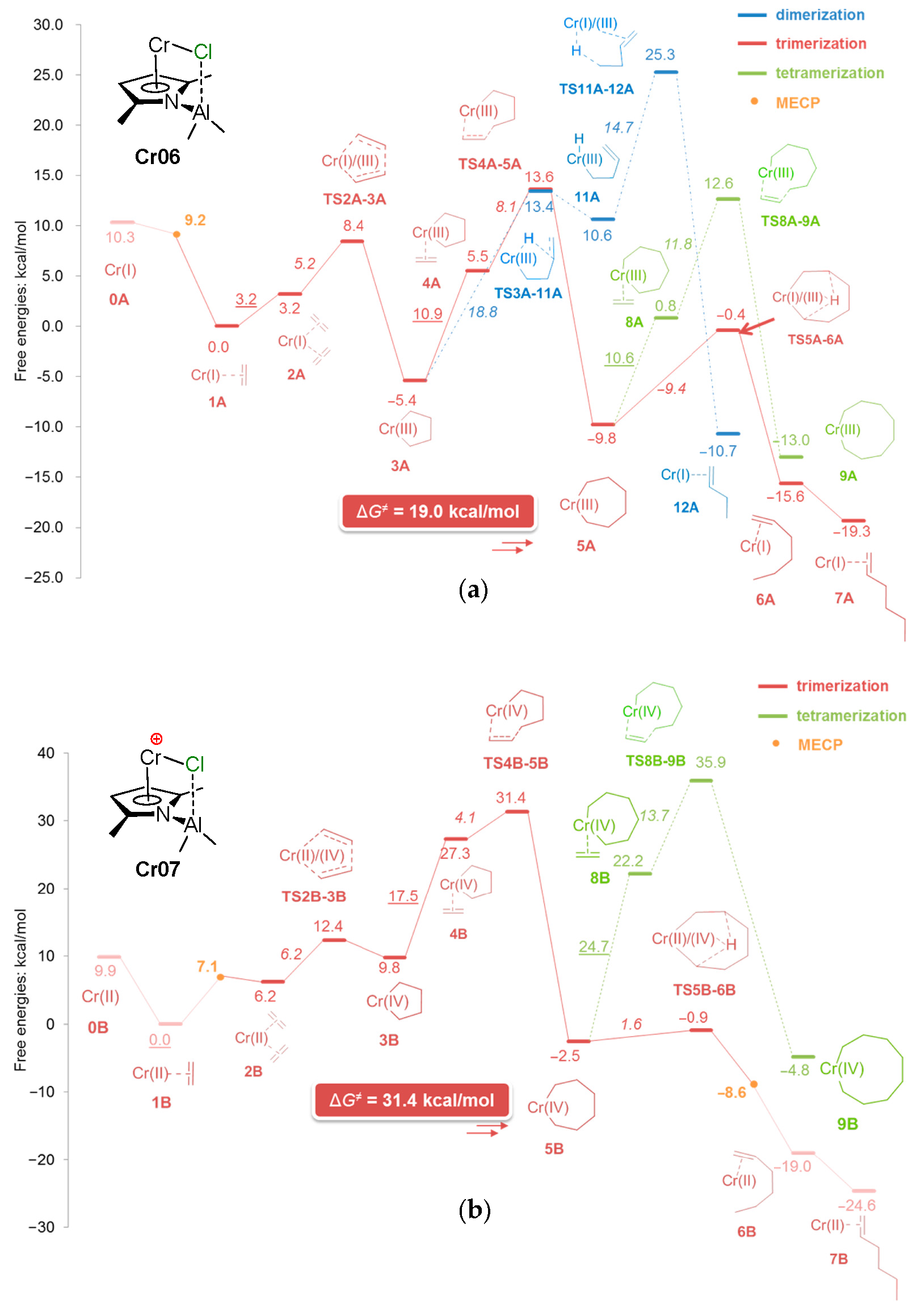
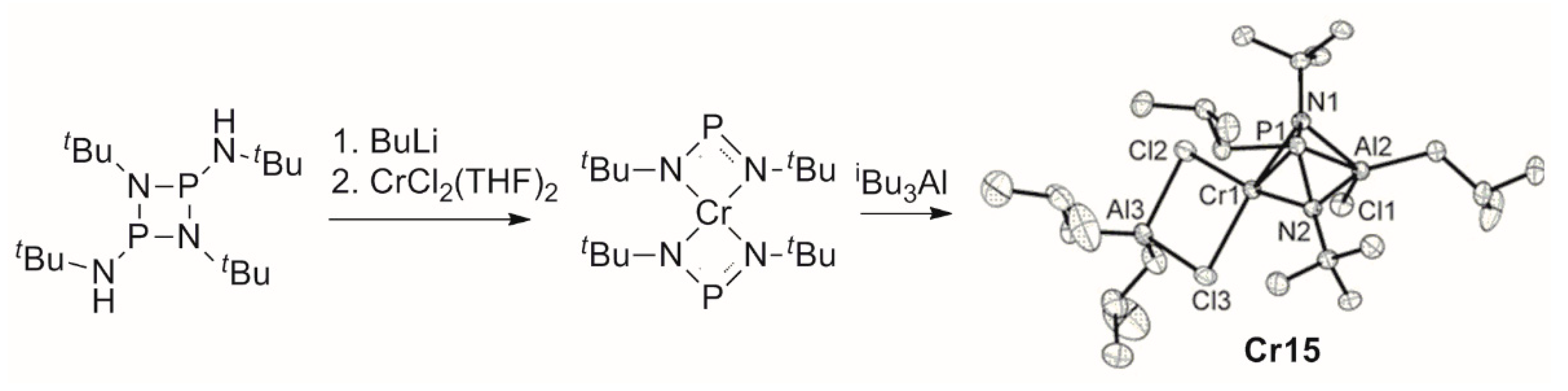
5. Complexes of Cr
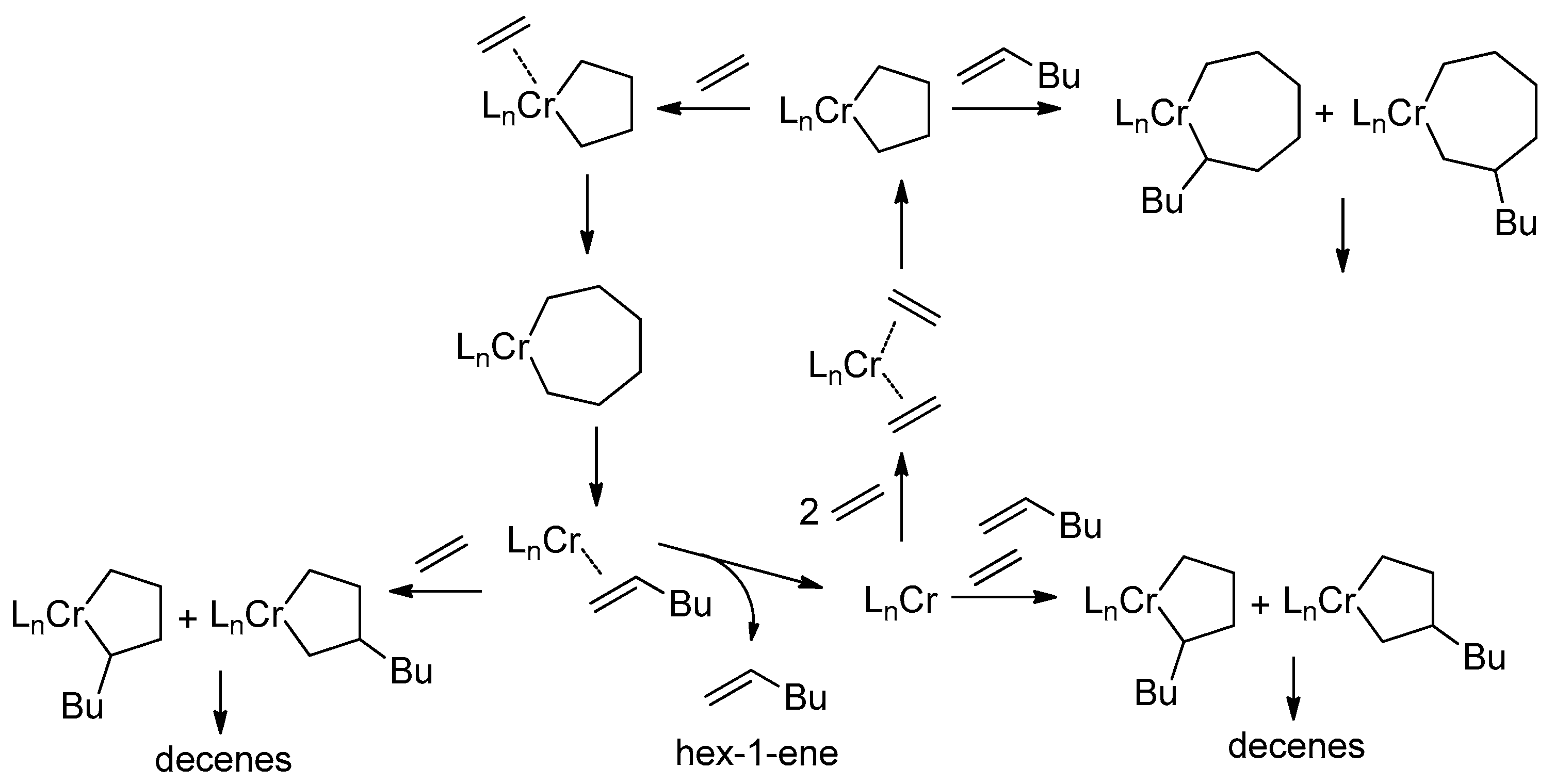
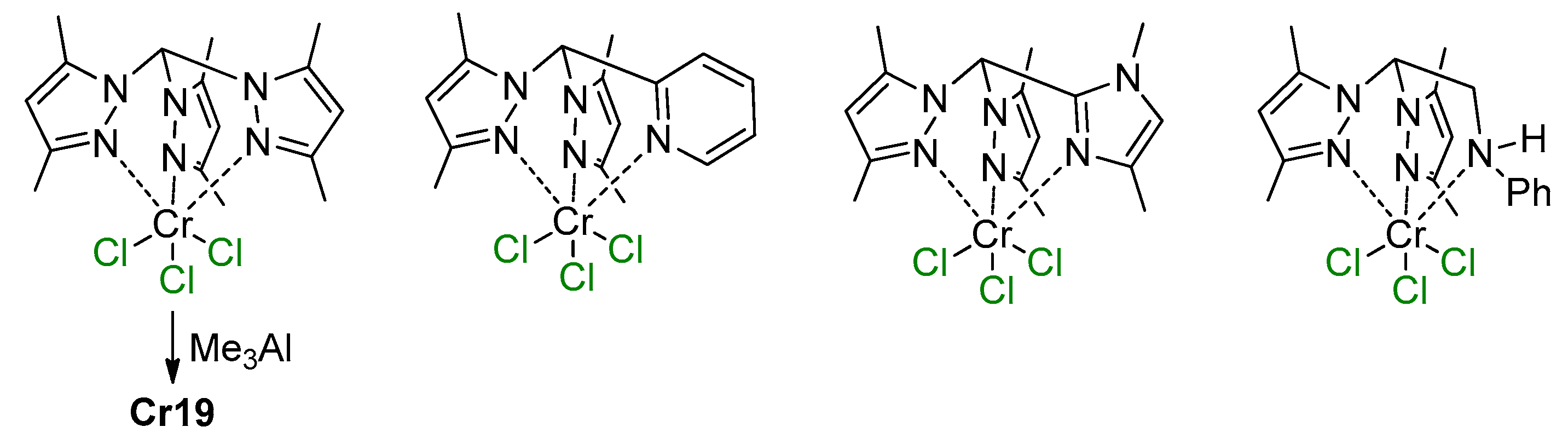
5.1. Selective Oligomerization of Ethylene Catalyzed by Cr Complexes
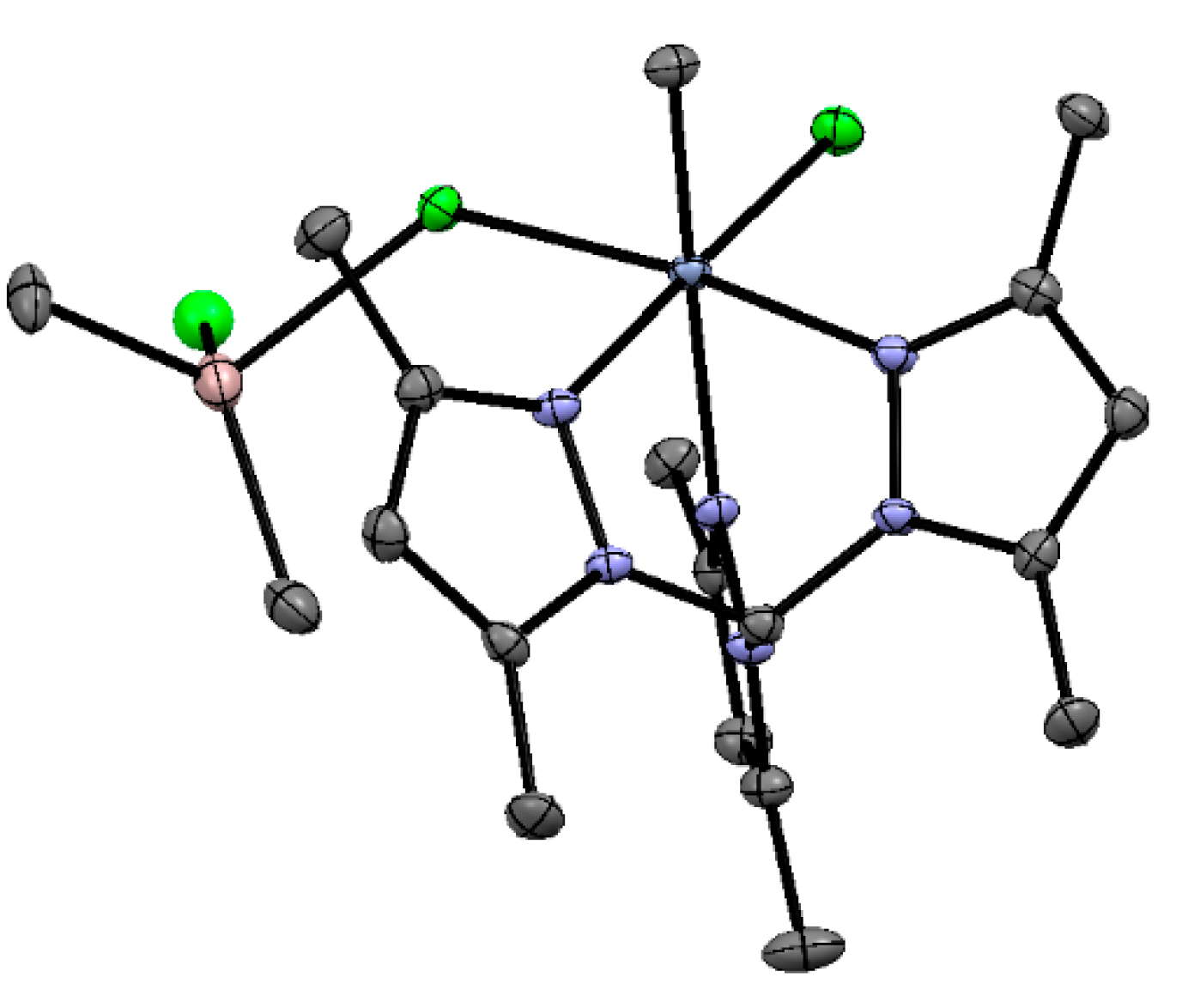
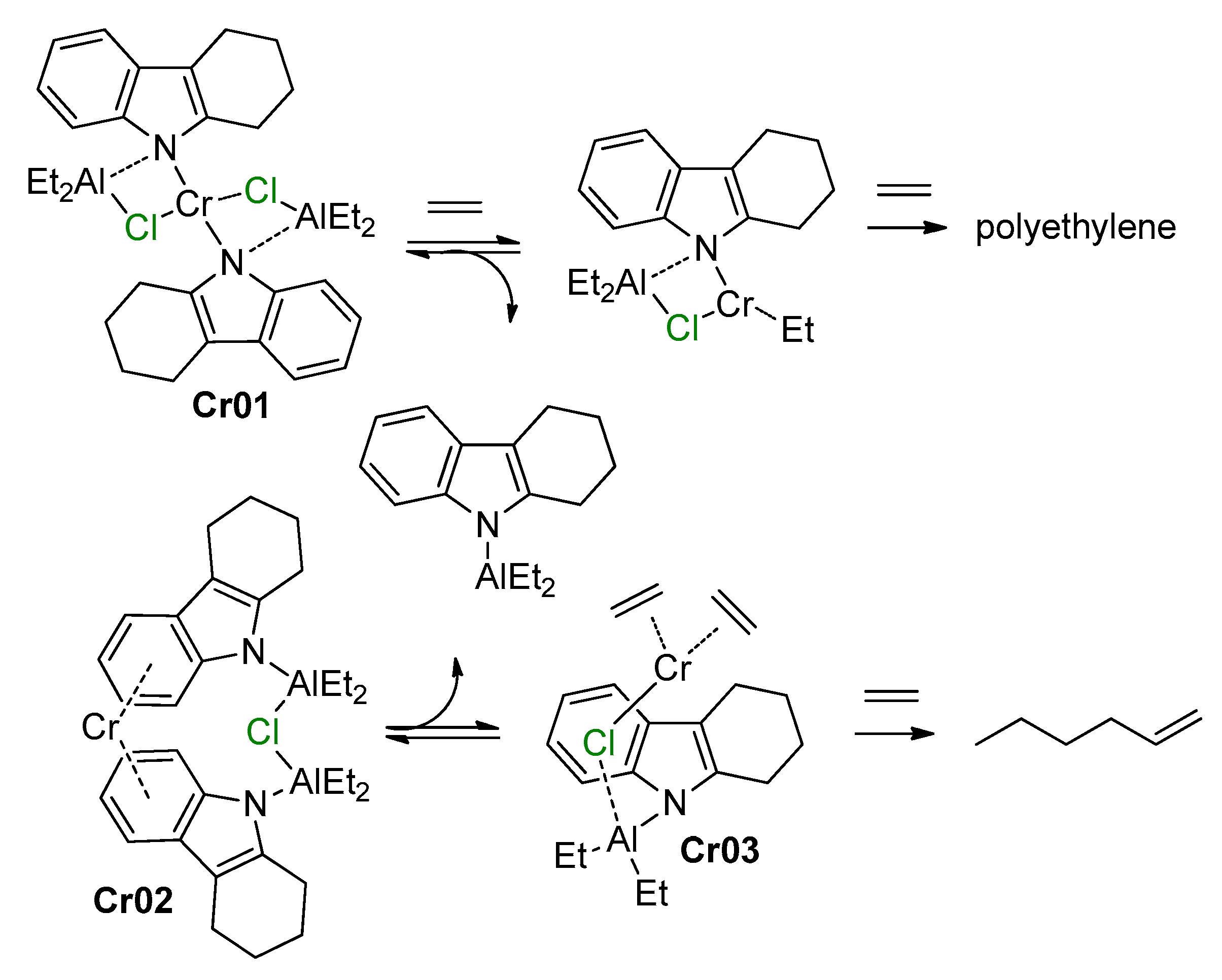
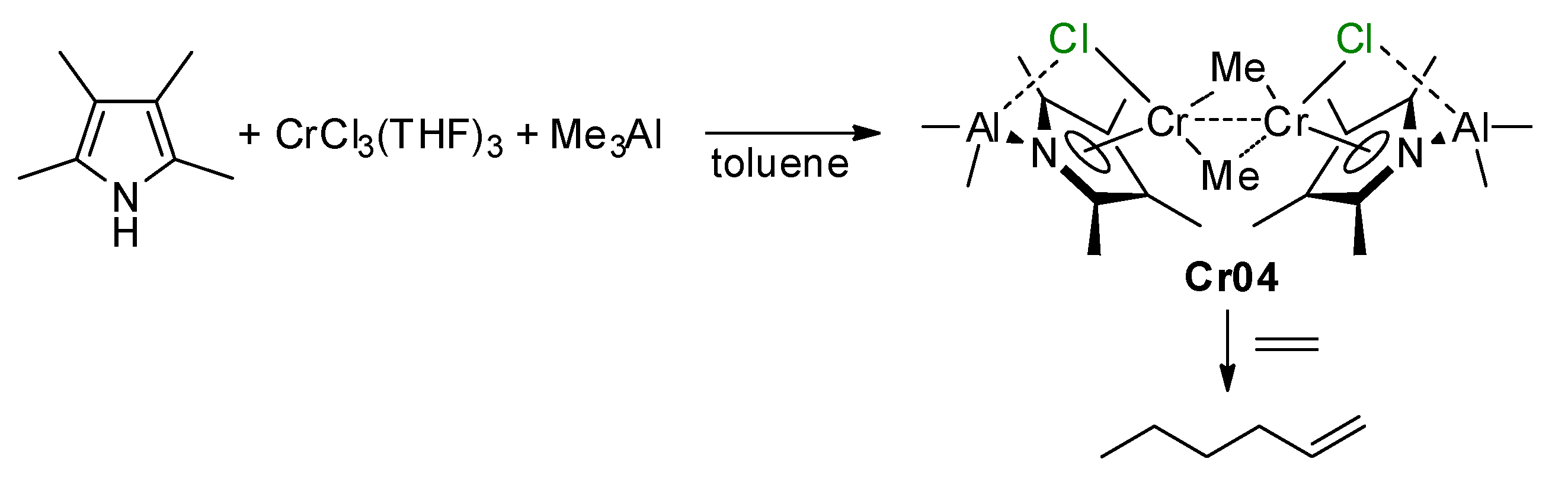
5.2. Cr Complexes with Pyrrole and Similar Ligands
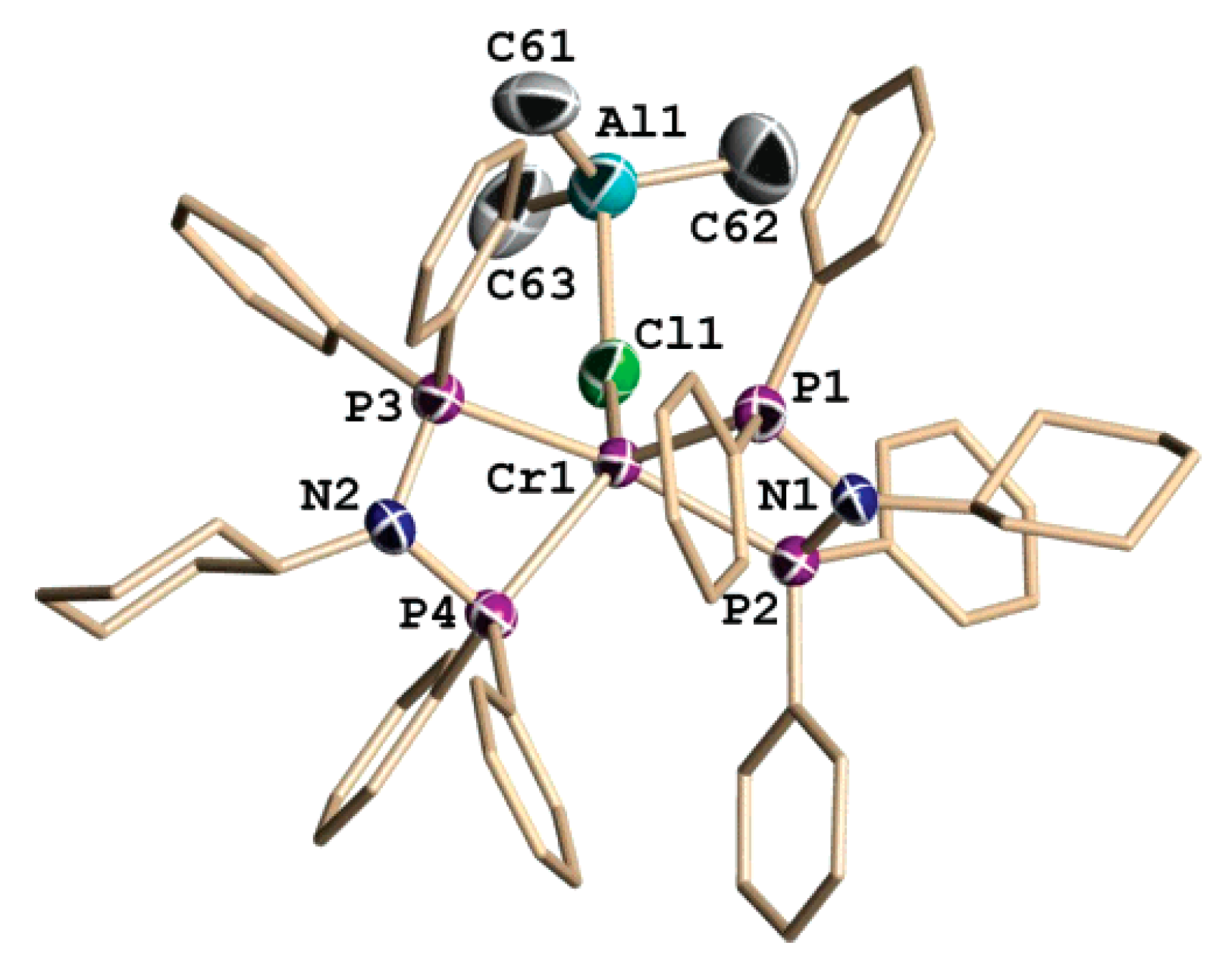
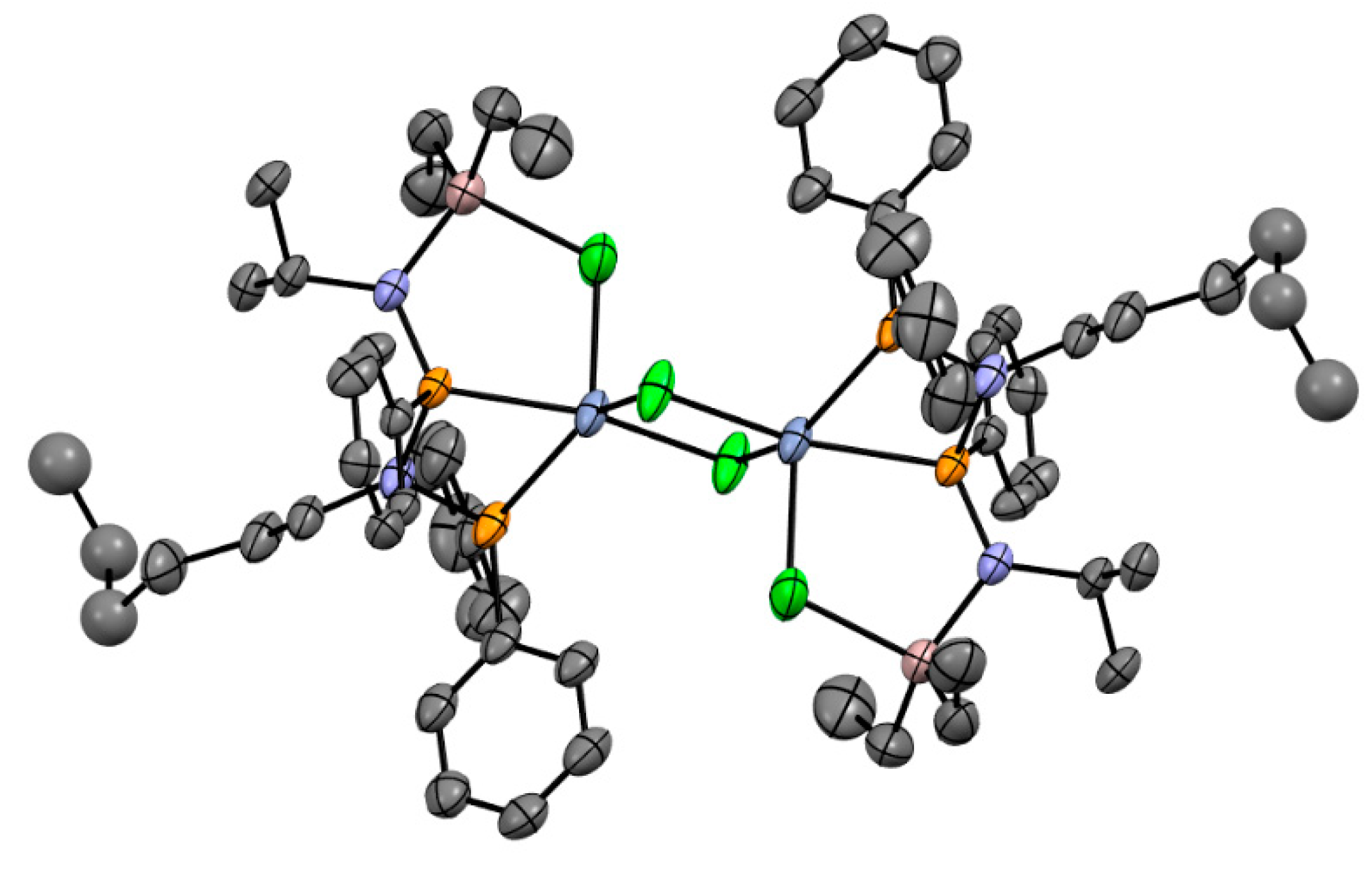
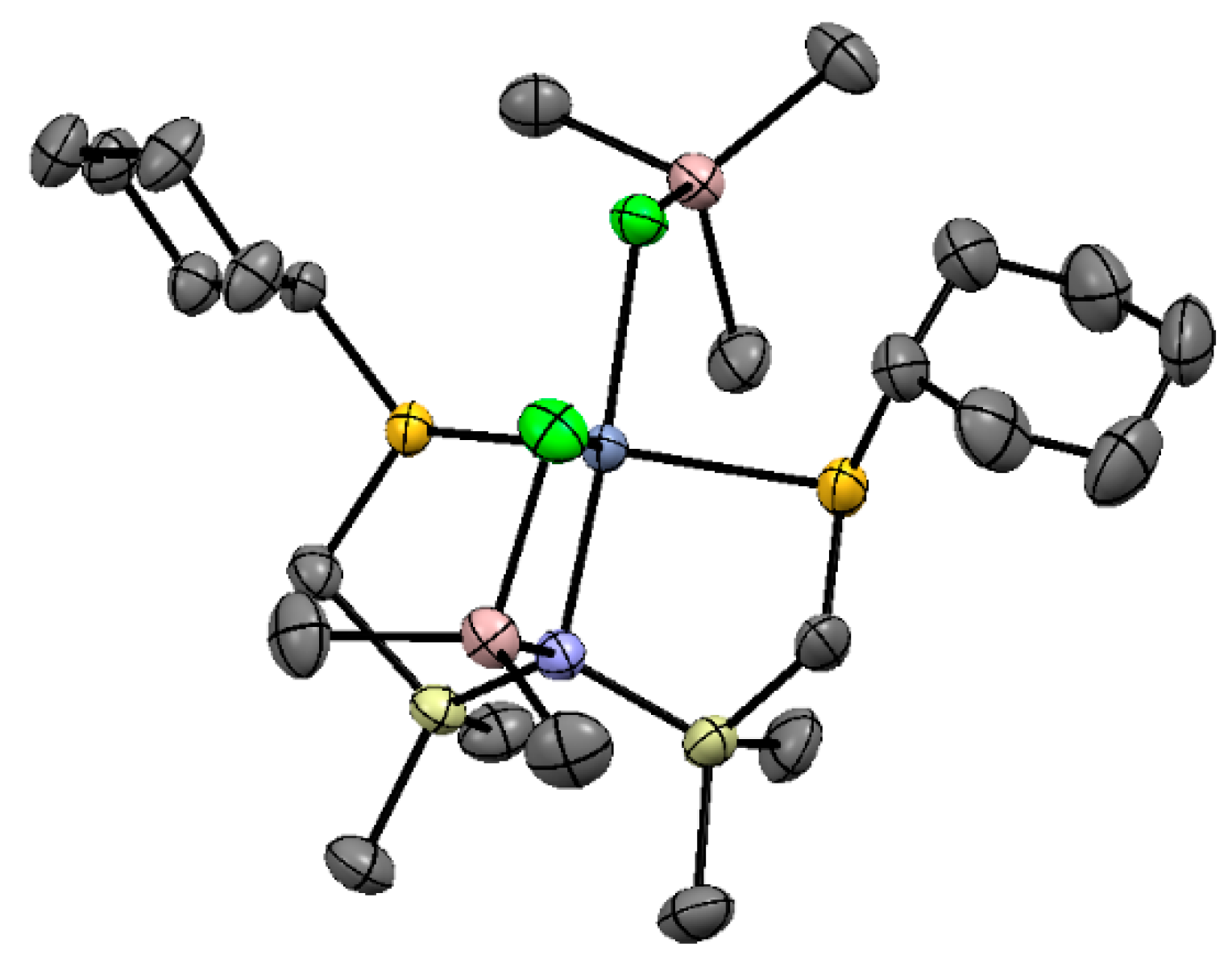
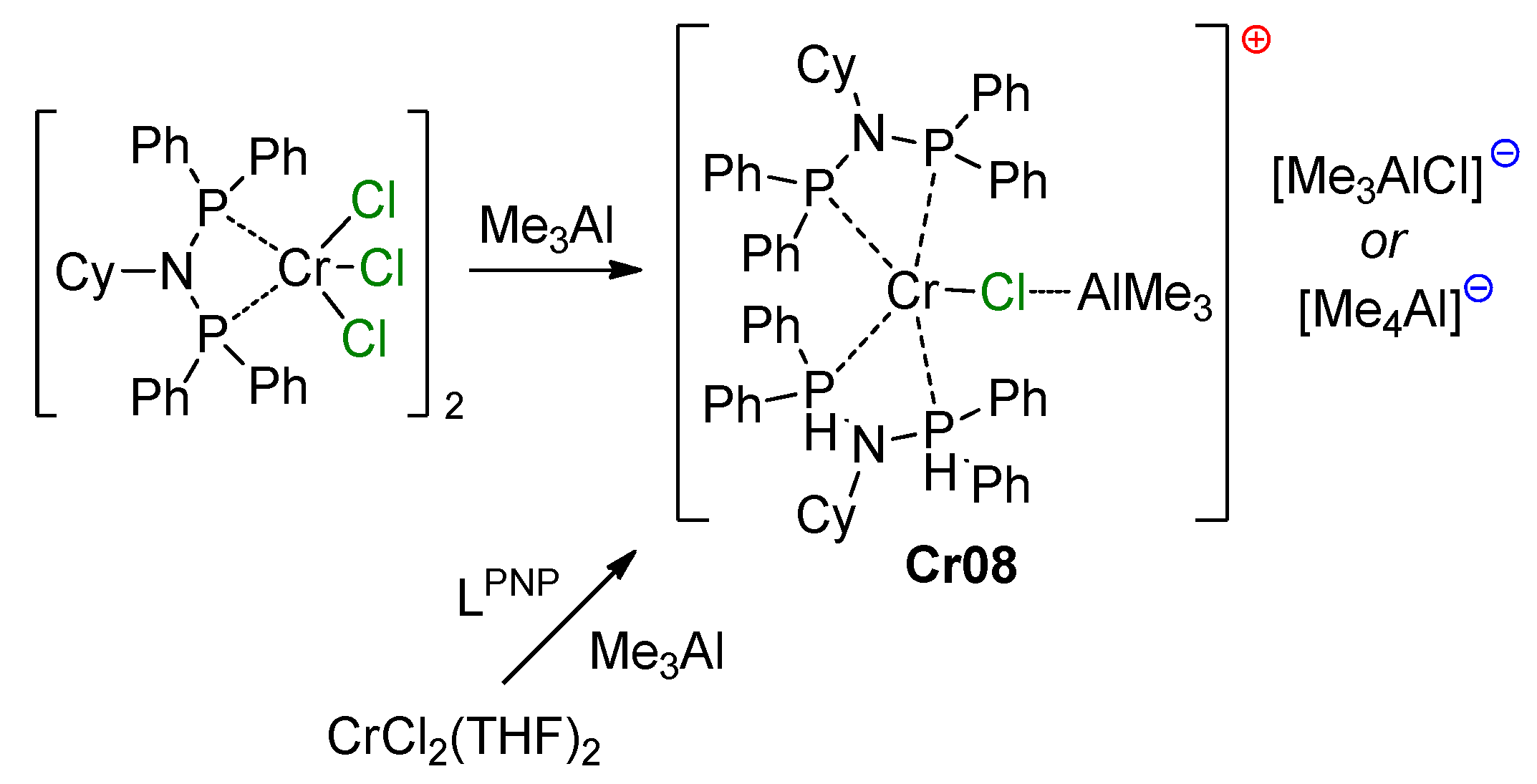
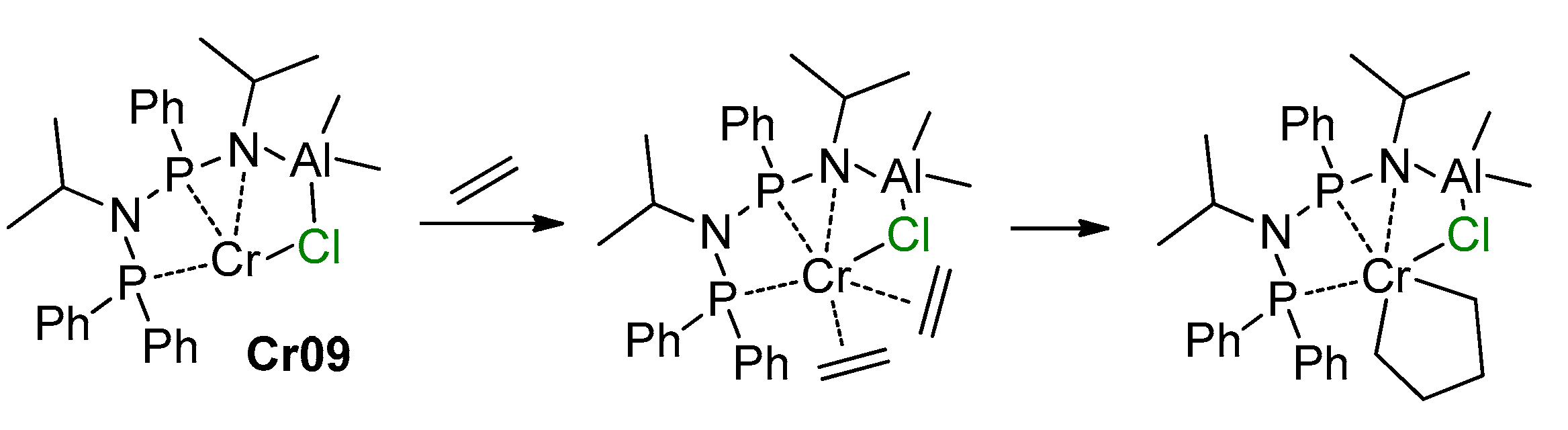
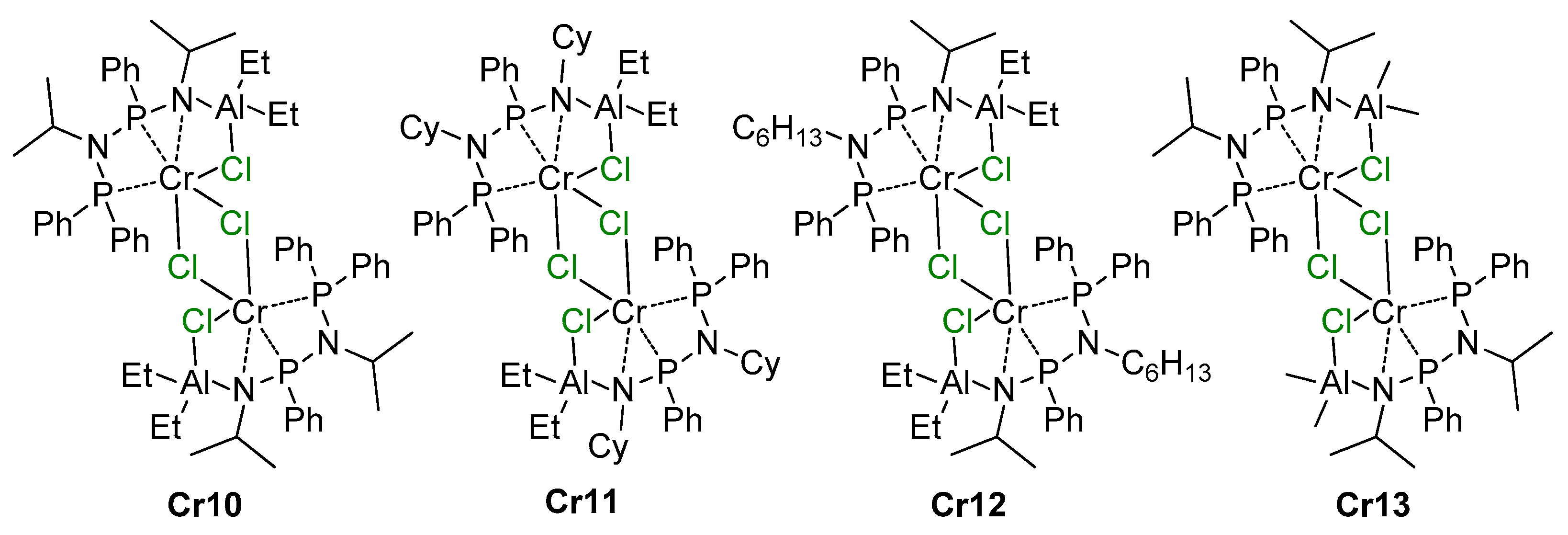
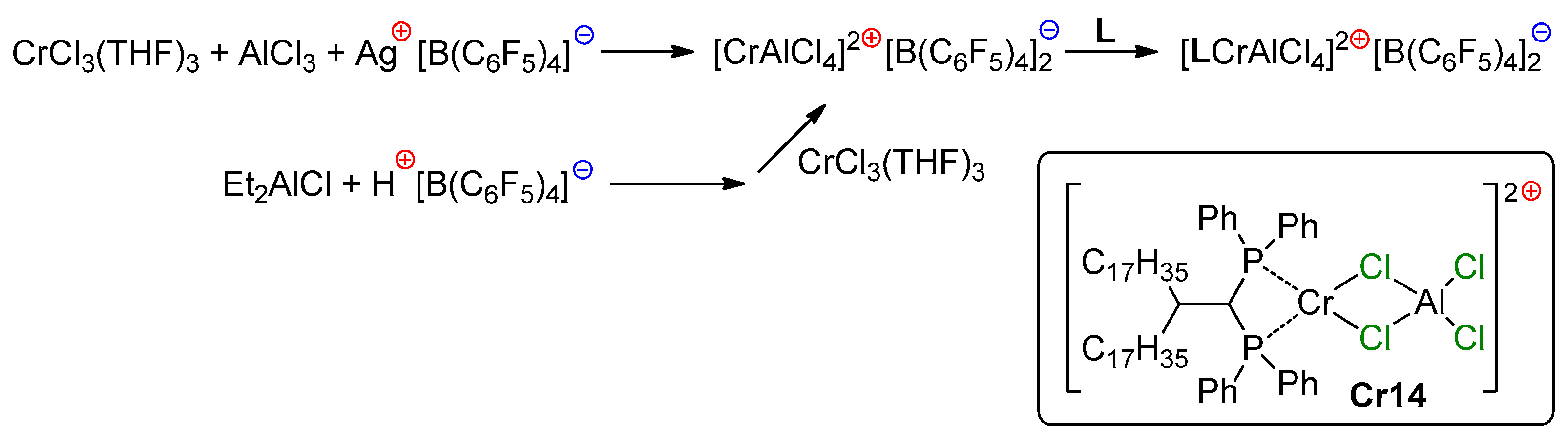
5.3. Cr Aminodiphosphine Complexes
5.4. Other Cr Complexes in the Single-Site Catalysis of Oligomerization and Polymerization
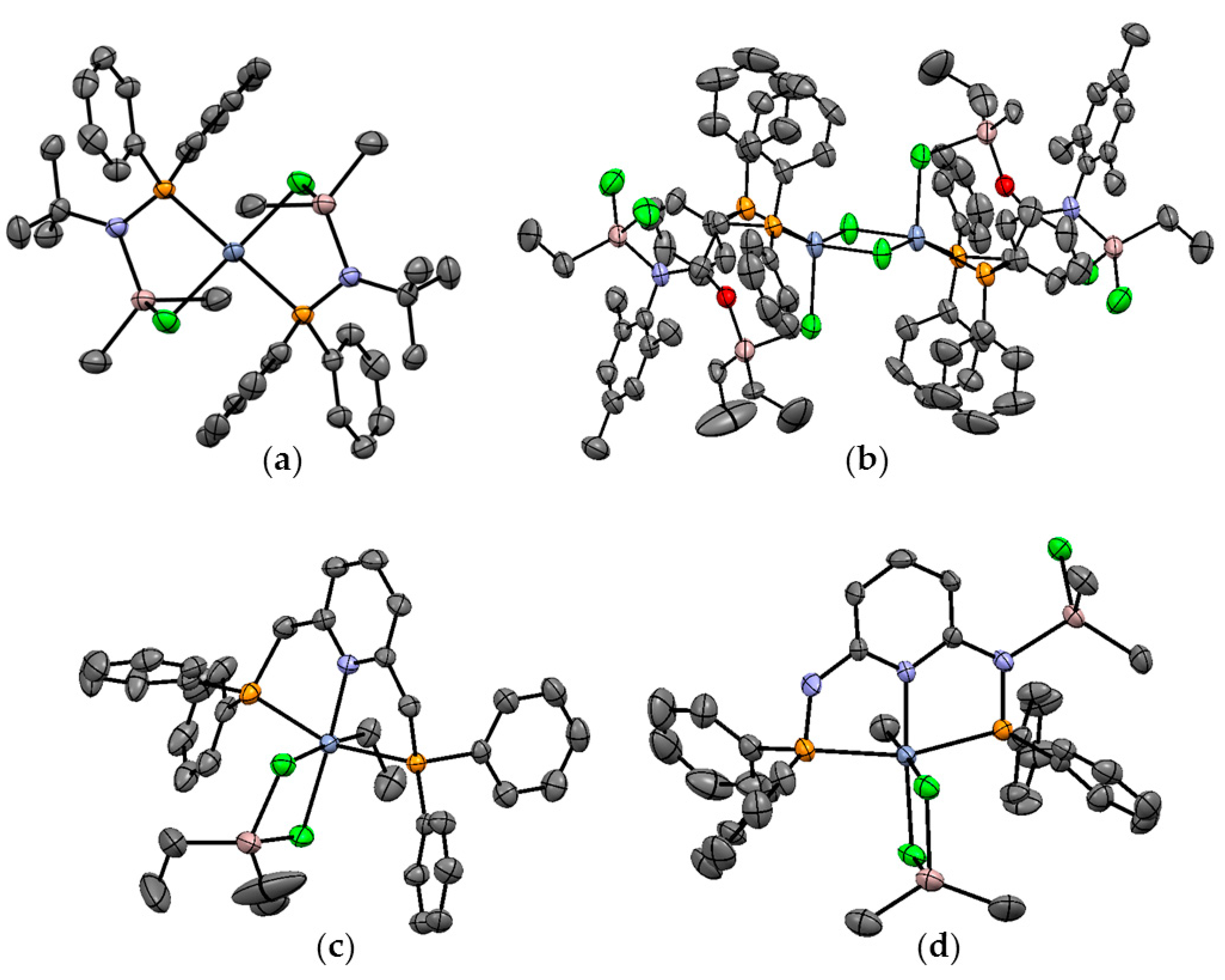
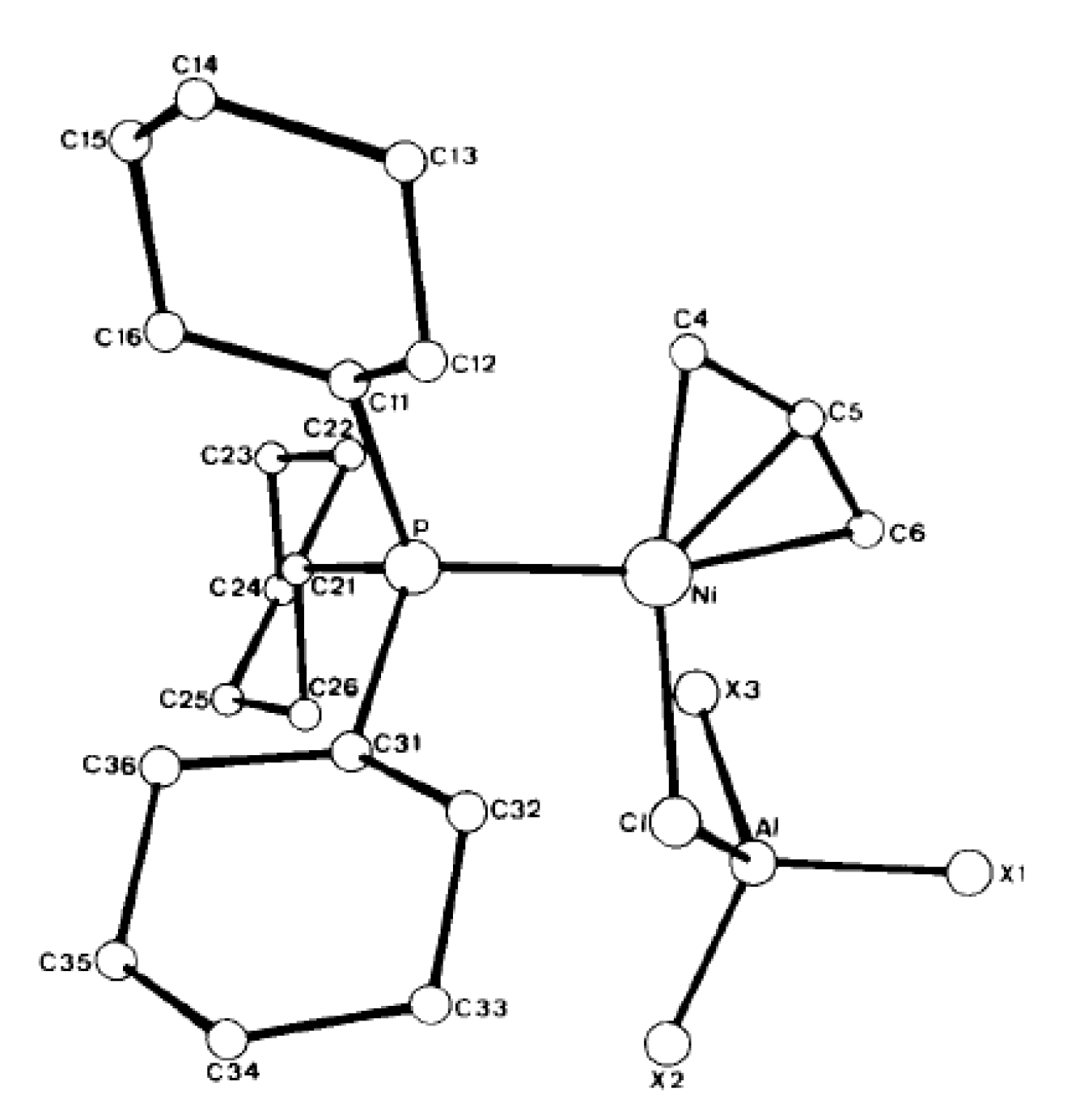
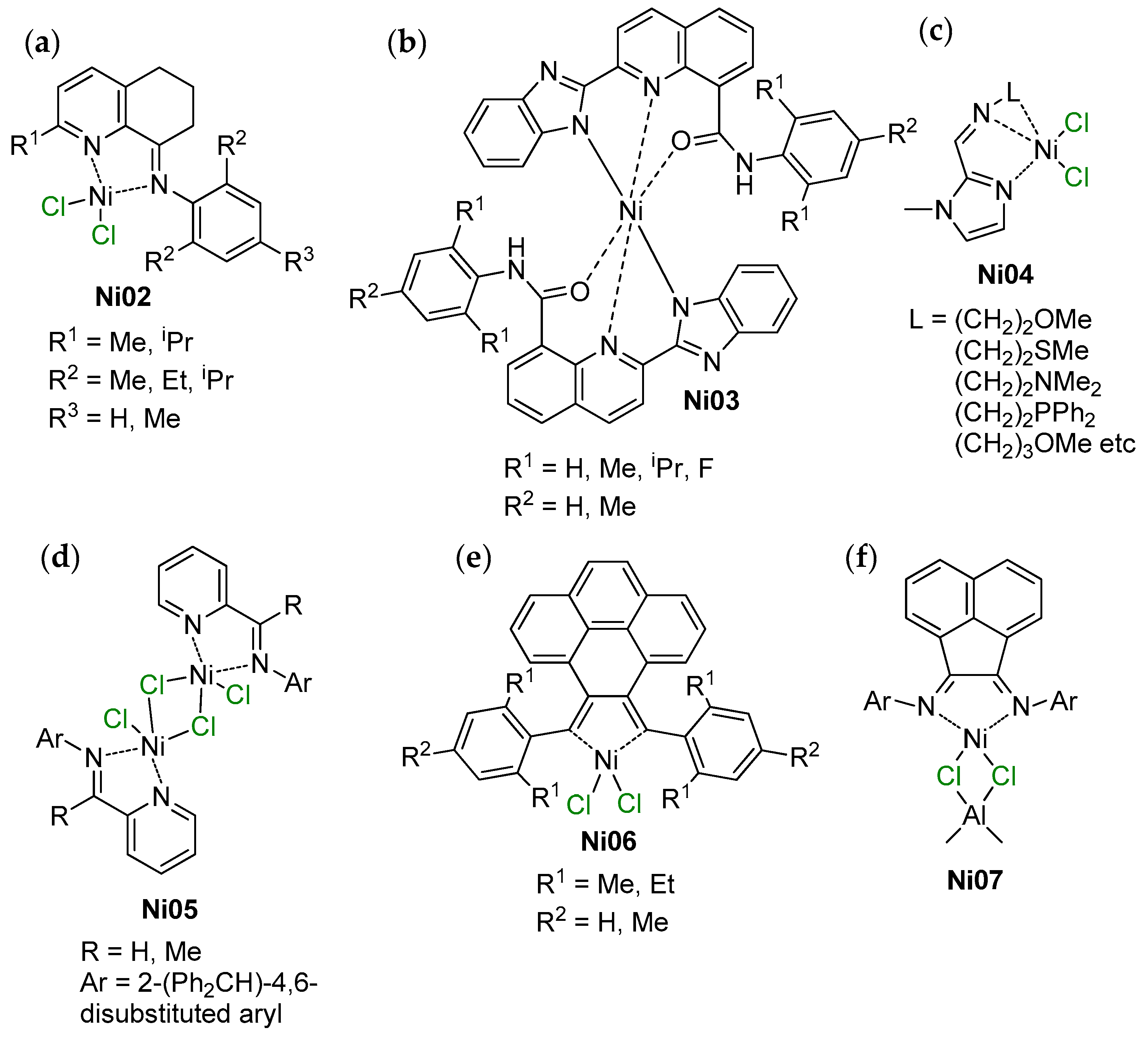
6. Complexes of Ni
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sauter, D.W.; Taoufik, M.; Boisson, C. Polyolefins, a Success Story. Polymers 2017, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Jubinville, D.; Esmizadeh, E.; Saikrishnan, S.; Tzoganakis, C.; Mekonnen, T. A comprehensive review of global production and recycling methods of polyolefin (PO) based products and their post-recycling applications. Sustain. Mater. Technol. 2020, 25, e00188. [Google Scholar] [CrossRef]
- Ricci, G.; Leone, G. Recent advances in the polymerization of butadiene over the last decade. Polyolefins J. 2014, 1, 43–60. [Google Scholar] [CrossRef]
- Ricci, G.; Pampaloni, G.; Sommazzi, A.; Masi, F. Dienes Polymerization: Where We Are and What Lies Ahead. Macromolecules 2021, 54, 5879–5914. [Google Scholar] [CrossRef]
- Baier, M.C.; Zuideveld, M.A.; Mecking, S. Post-Metallocenes in the Industrial Production of Polyolefins. Angew. Chem. Int. Ed. 2014, 53, 9722–9744. [Google Scholar] [CrossRef]
- Franssen, N.M.G.; Reek, J.N.H.; de Bruin, B. Synthesis of functional ‘polyolefins’: State of the art and remaining challenges. Chem. Soc. Rev. 2013, 42, 5809–5832. [Google Scholar] [CrossRef]
- Stürzel, M.; Mihan, S.; Mülhaupt, R. From Multisite Polymerization Catalysis to Sustainable Materials and All-Polyolefin Composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef]
- Stalzer, M.M.; Delferro, M.; Marks, T.J. Supported Single-Site Organometallic Catalysts for the Synthesis of High-Performance Polyolefins. Catal. Lett. 2015, 145, 3–14. [Google Scholar] [CrossRef]
- Mohite, A.S.; Rajpurkar, Y.D.; More, A.P. Bridging the gap between rubbers and plastics: A review on thermoplastic polyolefin elastomers. Polym. Bull. 2021, 79, 1309–1343. [Google Scholar] [CrossRef]
- Zanchin, G.; Leone, G. Polyolefin thermoplastic elastomers from polymerization catalysis: Advantages, pitfalls and future challenges. Prog. Polym. Sci. 2021, 113, 101342. [Google Scholar] [CrossRef]
- Kumawat, J.; Gupta, V.K. Fundamental aspects of heterogeneous Ziegler–Natta olefin polymerization catalysis: An experimental and computational overview. Polym. Chem. 2020, 11, 6107–6128. [Google Scholar] [CrossRef]
- Al-Jarallah, A.M.; Anabtawi, J.A.; Siddiqui, M.A.B.; Aitani, A.M.; Al-Sa’doun, A.W. Ethylene dimerization and oligomerization to butene-1 and linear α-olefins: A review of catalytic systems and processes. Catal. Today 1992, 14, 1–121. [Google Scholar] [CrossRef]
- Bariashir, C.; Huang, C.; Solan, G.A.; Sun, W.-H. Recent advances in homogeneous chromium catalyst design for ethylene tri-, tetra-, oligo- and polymerization. Coord. Chem. Rev. 2019, 385, 208–229. [Google Scholar] [CrossRef]
- Salian, S.M.; Bagui, M.; Jasra, R.V. Industrially relevant ethylene trimerization catalysts and processes. Appl. Petrochem. Res. 2021, 11, 267–279. [Google Scholar] [CrossRef]
- Tembe, G. Catalytic tri- and tetramerization of ethylene: A mechanistic overview. Catal. Rev. 2022, 1–56. [Google Scholar] [CrossRef]
- Breuil, P.-A.R.; Magna, L.; Olivier-Bourbigou, H. Role of Homogeneous Catalysis in Oligomerization of Olefins: Focus on Selected Examples Based on Group 4 to Group 10 Transition Metal Complexes. Catal. Lett. 2015, 145, 173–192. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Ivchenko, P. Fair Look at Coordination Oligomerization of Higher α-Olefins. Polymers 2020, 12, 1082. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Khalilov, L.M.; Dzhemilev, U.M. Mechanisms of reactions of organoaluminium compounds with alkenes and alkynes catalyzed by Zr complexes. Russ. Chem. Rev. 2012, 81, 524–548. [Google Scholar] [CrossRef]
- Lamberti, M.; Mazzeo, M.; Pappalardo, D.; Pellecchia, C. Mechanism of stereospecific polymerization of α-olefins by late-transition metal and octahedral group 4 metal catalysts. Coord. Chem. Rev. 2009, 253, 2082–2097. [Google Scholar] [CrossRef]
- Bryliakov, K.P.; Talsi, E.P. Frontiers of mechanistic studies of coordination polymerization and oligomerization of α-olefins. Coord. Chem. Rev. 2012, 256, 2994–3007. [Google Scholar] [CrossRef]
- Parveen, R.; Cundari, T.R.; Younker, J.M.; Rodriguez, G.; McCullough, L. DFT and QSAR Studies of Ethylene Polymerization by Zirconocene Catalysts. ACS Catal. 2019, 9, 9339–9349. [Google Scholar] [CrossRef]
- Chen, M.-C.; Roberts, J.A.S.; Marks, T.J. Marked Counteranion Effects on Single-Site Olefin Polymerization Processes. Correlations of Ion Pair Structure and Dynamics with Polymerization Activity, Chain Transfer, and Syndioselectivity. J. Am. Chem. Soc. 2004, 126, 4605–4625. [Google Scholar] [CrossRef] [PubMed]
- Kuklin, M.S.; Hirvi, J.T.; Bochmann, M.; Linnolahti, M. Toward Controlling the Metallocene/Methylaluminoxane-Catalyzed Olefin Polymerization Process by a Computational Approach. Organometallics 2015, 34, 3586–3597. [Google Scholar] [CrossRef]
- Tymińska, N.; Zurek, E. DFT-D Investigation of Active and Dormant Methylaluminoxane (MAO) Species Grafted onto a Magnesium Dichloride Cluster: A Model Study of Supported MAO. ACS Catal. 2015, 5, 6989–6998. [Google Scholar] [CrossRef]
- Laine, A.; Coussens, B.B.; Hirvi, J.T.; Berthoud, A.; Friederichs, N.; Severn, J.R.; Linnolahti, M. Effect of Ligand Structure on Olefin Polymerization by a Metallocene/Borate Catalyst: A Computational Study. Organometallics 2015, 34, 2415–2421. [Google Scholar] [CrossRef]
- Trefz, T.K.; Henderson, M.A.; Linnolahti, M.; Collins, S.; McIndoe, J.S. Mass Spectrometric Characterization of Methylaluminoxane-Activated Metallocene Complexes. Chem. Eur. J. 2015, 21, 2980–2991. [Google Scholar] [CrossRef]
- Theurkauff, G.; Bondon, A.; Dorcet, V.; Carpentier, J.-F.; Kirillov, E. Heterobi- and -trimetallic Ion Pairs of Zirconocene-Based Isoselective Olefin Polymerization Catalysts with AlMe3. Angew. Chem. Int. Ed. 2015, 54, 6343–6346. [Google Scholar] [CrossRef]
- Collins, S.; Linnolahti, M.; Garcia Zamora, M.; Zijlstra, H.S.; Rodríguez Hernández, M.T.; Perez-Camacho, O. Activation of Cp2ZrX2 (X = Me, Cl) by Methylaluminoxane As Studied by Electrospray Ionization Mass Spectrometry: Relationship to Polymerization Catalysis. Macromolecules 2017, 50, 8871–8884. [Google Scholar] [CrossRef]
- Oliva, L.; Oliva, P.; Galdi, N.; Pellecchia, C.; Sian, L.; Macchioni, A.; Zuccaccia, C. Solution Structure and Reactivity with Metallocenes of AlMe2F: Mimicking Cation–Anion Interactions in Metallocenium–Methylalumoxane Inner-Sphere Ion Pairs. Angew. Chem. Int. Ed. 2017, 56, 14227–14231. [Google Scholar] [CrossRef]
- Desert, X.; Proutiere, F.; Welle, A.; Den Dauw, K.; Vantomme, A.; Miserque, O.; Brusson, J.-M.; Carpentier, J.-F.; Kirillov, E. Zirconocene-Catalyzed Polymerization of α-Olefins: When Intrinsic Higher Activity Is Flawed by Rapid Deactivation. Organometallics 2019, 38, 2664–2673. [Google Scholar] [CrossRef]
- Sian, L.; Macchioni, A.; Zuccaccia, C. Understanding the Role of Metallocenium Ion-Pair Aggregates on the Rate of Olefin Insertion into the Metal–Carbon Bond. ACS Catal. 2020, 10, 1591–1606. [Google Scholar] [CrossRef]
- Parveen, R.; Cundari, T.R.; Younker, J.M.; Rodriguez, G. Computational Assessment of Counterion Effect of Borate Anions on Ethylene Polymerization by Zirconocene and Hafnocene Catalysts. Organometallics 2020, 39, 2068–2079. [Google Scholar] [CrossRef]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Budzelaar, P.H.M.; Vittoria, A.; Macchioni, A.; Busico, V.; Ehm, C. Methylaluminoxane’s Molecular Cousin: A Well-defined and “Complete” Al-Activator for Molecular Olefin Polymerization Catalysts. ACS Catal. 2021, 11, 4464–4475. [Google Scholar] [CrossRef]
- Collins, S.; Linnolahti, M. Activation of Substituted Metallocene Catalysts Using Methylaluminoxane. ChemCatChem 2022, 14, e202101918. [Google Scholar] [CrossRef]
- Culver, D.B.; Dorn, R.W.; Venkatesh, A.; Meeprasert, J.; Rossini, A.J.; Pidko, E.A.; Lipton, A.S.; Lief, G.R.; Conley, M.P. Active Sites in a Heterogeneous Organometallic Catalyst for the Polymerization of Ethylene. ACS Central Sci. 2021, 7, 1225–1231. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K.; Palatov, E.R. Catalytic Systems Based on Cp2ZrX2 (X = Cl, H), Organoaluminum Compounds and Perfluorophenylboranes: Role of Zr,Zr- and Zr,Al-Hydride Intermediates in Alkene Dimerization and Oligomerization. Catalysts 2021, 11, 39. [Google Scholar] [CrossRef]
- Christoffers, J.; Bergman, R.G. Catalytic Dimerization Reactions of α-Olefins and α,ω-Dienes with Cp2ZrCl2/Poly(methylalumoxane): Formation of Dimers, Carbocycles, and Oligomers. J. Am. Chem. Soc. 1996, 118, 4715–4716. [Google Scholar] [CrossRef]
- Christoffers, J.; Bergman, R.G. Zirconocene-alumoxane (1:1)—A catalyst for the selective dimerization of α-olefins. Inorg. Chim. Acta 1998, 270, 20–27. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Ivchenko, P.V. Zirconocene-catalyzed dimerization of 1-hexene: Two-stage activation and structure–catalytic performance relationship. Catal. Commun. 2016, 79, 6–10. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Ivchenko, P.; Tavtorkin, A.; Vinogradov, A.; Vinogradov, A. Non-traditional Ziegler-Natta catalysis in α-olefin transformations: Reaction mechanisms and product design. Pure Appl. Chem. 2017, 89, 1017–1032. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Vinogradov, A.; Vinogradov, A.; Karchevsky, S.; Ivchenko, P. Zirconocene-Catalyzed Dimerization of α-Olefins: DFT Modeling of the Zr-Al Binuclear Reaction Mechanism. Molecules 2019, 24, 3565. [Google Scholar] [CrossRef] [PubMed]
- Nifant’ev, I.; Vinogradov, A.; Vinogradov, A.; Karchevsky, S.; Ivchenko, P. Experimental and Theoretical Study of Zirconocene-Catalyzed Oligomerization of 1-Octene. Polymers 2020, 12, 1590. [Google Scholar] [CrossRef] [PubMed]
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Bagrov, V.V.; Churakov, A.V.; Minyaev, M.E.; Kiselev, A.V.; Salakhov, I.I.; Ivchenko, P.V. A competetive way to low-viscosity PAO base stocks via heterocene-catalyzed oligomerization of dec-1-ene. Mol. Catal. 2022, 529, 112542. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Suo, H.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L.; Sun, W.-H. Recent developments in vanadium-catalyzed olefin coordination polymerization. Coord. Chem. Rev. 2020, 416, 213332. [Google Scholar] [CrossRef]
- Olivier-Bourbigou, H.; Breuil, P.A.R.; Magna, L.; Michel, T.; Pastor, M.F.E.; Delcroix, D. Nickel Catalyzed Olefin Oligomerization and Dimerization. Chem. Rev. 2020, 120, 7919–7983. [Google Scholar] [CrossRef]
- Bekmukhamedov, G.E.; Sukhov, A.V.; Kuchkaev, A.M.; Yakhvarov, D.G. Ni-Based Complexes in Selective Ethylene Oligomerization Processes. Catalysts 2020, 10, 498. [Google Scholar] [CrossRef]
- Alferov, K.A.; Belov, G.P.; Meng, Y. Chromium catalysts for selective ethylene oligomerization to 1-hexene and 1-octene: Recent results. Appl. Catal. A Gen. 2017, 542, 71–124. [Google Scholar] [CrossRef]
- Petit, J.; Magna, L.; Mézailles, N. Alkene oligomerization via metallacycles: Recent advances and mechanistic insights. Coord. Chem. Rev. 2022, 450, 214227. [Google Scholar] [CrossRef]
- Tebbe, F.N.; Parshall, G.W.; Reddy, G.S. Olefin homologation with titanium methylene compounds. J. Am. Chem. Soc. 1978, 100, 3611–3613. [Google Scholar] [CrossRef]
- Thompson, R.; Nakamaru-Ogiso, E.; Chen, C.-H.; Pink, M.; Mindiola, D.J. Structural Elucidation of the Illustrious Tebbe Reagent. Organometallics 2014, 33, 429–432. [Google Scholar] [CrossRef]
- Kurogi, T.; Kuroki, K.; Moritani, S.; Takai, K. Structural elucidation of a methylenation reagent of esters: Synthesis and reactivity of a dinuclear titanium(III) methylene complex. Chem. Sci. 2021, 12, 3509–3515. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.; Mindiola, D.J. A tribute to Frederick Nye Tebbe. Lewis acid stabilized alkylidyne, alkylidene, and imides of 3D early transition metals. Dalton Trans. 2009, 40, 8463–8472. [Google Scholar] [CrossRef] [PubMed]
- Finch, W.C.; Anslyn, E.V.; Grubbs, R.H. Substituent effects on the cleavage rates of titanocene metallacyclobutanes. J. Am. Chem. Soc. 1988, 110, 2406–2413. [Google Scholar] [CrossRef]
- Hartner, F.W., Jr.; Schwartz, J. Synthesis and Characterization of “Long-Chain” Alkylidene-Bridged Hetero Bimetallic Complexes. J. Am. Chem. Soc. 1981, 103, 4979–4981. [Google Scholar] [CrossRef]
- Halterman, R.L.; Ramsey, T.M. Conversion of meso-alkenes to chiral alkenes via titanocene-catalyzed ring-opening/ring-closing olefin metathesis. J. Organomet. Chem. 1997, 547, 41–48. [Google Scholar] [CrossRef]
- Pine, S.H.; Pettit, R.J.; Geib, G.D.; Cruz, S.G.; Gallego, C.H.; Tijerina, T.; Pine, R.D. Carbonyl Methylenation Using a Titanium-Aluminum (Tebbe) Complex. J. Org. Chem. 1985, 50, 1212–1216. [Google Scholar] [CrossRef]
- Nasrallah, D.J.; Zehnder, T.E.; Ludwig, J.R.; Steigerwald, D.C.; Kiernicki, J.J.; Szymczak, N.K.; Schindler, C.S. Hydrazone and Oxime Olefination via Ruthenium Alkylidenes. Angew. Chem. Int. Ed. 2022, 61, e202112101. [Google Scholar] [CrossRef]
- Howard, T.R.; Lee, J.B.; Grubbs, R.H. Titanium metallacarbene-metallacyclobutane reactions: Stepwise metathesis. J. Am. Chem. Soc. 1980, 102, 6876–6878. [Google Scholar] [CrossRef]
- Brown-Wensley, K.A.; Buchwald, S.L.; Cannizzo, L.; Clawson, L.; Ho, S.; Meinhardt, D.; Stille, J.R.; Straus, D.; Grubbs, R.H. Cp2TiCH2 complexes in synthetic applications. Pure Appl. Chem. 1983, 55, 1733–1744. [Google Scholar] [CrossRef]
- Ikariya, T.; Ho, S.C.H.; Grubbs, R.H. Mechanism of rearrangement of titanacyclobutanes. Organometallics 1985, 4, 199–200. [Google Scholar] [CrossRef]
- Lee, J.B.; Ott, K.C.; Grubbs, R.H. Kinetics and stereochemistry of the titanacyclobutane-titanaethylene interconversion. Investigation of a degenerate olefin metathesis reaction. J. Am. Chem. Soc. 1982, 104, 7491–7496. [Google Scholar] [CrossRef]
- Law, J.A.; Bartfield, N.M.; Frederich, J.H. Site-Specific Alkene Hydromethylation via Protonolysis of Titanacyclobutanes. Angew. Chem. Int. Ed. 2021, 60, 14360–14364. [Google Scholar] [CrossRef] [PubMed]
- Zefirova, A.K.; Shilov, A.E. The kinetics and mechanism of the interaction of aluminium alkyls with titanium halides. Dokl. Akad. Nauk SSSR 1961, 136, 599–602. [Google Scholar]
- Dyachkovskii, F.S.; Shilova, A.K.; Shilov, A.E. The role of free ions in reactions of olefins with soluble complex catalysts. J. Polym. Sci. Part C Polym. Symp. 1967, 16, 2333–2339. [Google Scholar] [CrossRef]
- Bryliakov, K.P.; Babushkin, D.E.; Talsi, E.P.; Voskoboynikov, A.Z.; Gritzo, H.; Schroder, L.; Damrau, H.-R.H.; Wieser, U.; Schaper, F.; Brintzinger, H.H. ansa-Titanocene Catalysts for α-Olefin Polymerization. Syntheses, Structures, and Reactions with Methylaluminoxane and Boron-Based Activators. Organometallics 2005, 24, 894–904. [Google Scholar] [CrossRef]
- Machat, M.R.; Jandl, C.; Rieger, B. Titanocenes in Olefin Polymerization: Sustainable Catalyst System or an Extinct Species? Organometallics 2017, 36, 1408–1418. [Google Scholar] [CrossRef]
- Williams, T.J.; Smith, A.D.H.; Buffet, J.-C.; Turner, Z.R.; O’Hare, D. Group 4 constrained geometry complexes for olefin (co)polymerisation. Mol. Catal. 2020, 486, 110872. [Google Scholar] [CrossRef]
- Klosin, J.; Fontaine, P.P.; Figueroa, R. Development of Group IV Molecular Catalysts for High Temperature Ethylene-α-Olefin Copolymerization Reactions. Acc. Chem. Res. 2015, 48, 2004–2016. [Google Scholar] [CrossRef]
- Yuan, S.-F.; Yan, Y.; Solan, G.A.; Ma, Y.; Sun, W.-H. Recent advancements in N-ligated group 4 molecular catalysts for the (co)polymerization of ethylene. Coord. Chem. Rev. 2020, 411, 213254. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Ivchenko, P.V.; Vinogradov, A.A. Heterocycle-fused cyclopentadienyl metal complexes: Heterocene synthesis, structure and catalytic applications. Coord. Chem. Rev. 2021, 426, 213515. [Google Scholar] [CrossRef]
- Walsh, D.J.; Hyatt, M.G.; Miller, S.A.; Guironnet, D. Recent Trends in Catalytic Polymerizations. ACS Catal. 2019, 9, 11153–11188. [Google Scholar] [CrossRef]
- Mach, K.; Antropiusová, H.; Poláček, J. Ethyl-substituted (π5-cyclopentadienyl)-bis(dihaloalanedi-μ-halo)titanium(III) and (η6-benzene)bis(dihaloalanedi-μ-halo)titanium(II) chloro and bromo complexes. J. Organomet. Chem. 1980, 194, 285–295. [Google Scholar] [CrossRef]
- Mach, K.; Varga, V.; Antropiusová, H.; Poláček, J. Effects of methyl substituents at the cyclopentadienyl ligand on the properties of C5H5TiCl3 and C5H5TiAl2Cl8−x(C2H5)x (x = 0–4) complexes. J. Organomet. Chem. 1987, 333, 205–215. [Google Scholar] [CrossRef]
- Bonoldi, L.; Abis, L.; Fiocca, L.; Fusco, R.; Longo, L.; Simone, F.; Spera, S. Monotitanocene catalysts: An ESR study of Ti(III) derivatives formed in presence of MAO and other organoaluminium compounds. J. Mol. Catal. A Chem. 2004, 219, 47–56. [Google Scholar] [CrossRef]
- Jensen, V.R.; Ystenes, M.; Waernmark, K.; Aakermark, B.; Svennson, M.; Siegbahn, P.E.M.; Blomberg, M.R.A. Strength of the metal-olefin bond in titanium complexes related to Ziegler-Natta catalysis. A theoretical model study of a square-pyramidal active center postulated to be found in titanium halide-based catalysts. Organometallics 1994, 13, 282–288. [Google Scholar] [CrossRef]
- Jensen, V.R.; Borve, K.J.; Westberg, N.; Ystenes, M. Titanium-Ethylene Complexes Proposed To Be Intermediates in Ziegler-Natta Catalysis. Can They Be Detected through Vibrational Spectroscopy? Organometallics 1995, 14, 4349–4358. [Google Scholar] [CrossRef]
- Jensen, V.R.; Borve, K.J.; Ystenes, M. Ziegler-Natta Ethylene Insertion Reaction for a Five-Coordinate Titanium Chloride Complex Bridged to an Aluminum Hydride Cocatalyst. J. Am. Chem. Soc. 1995, 117, 4109–4117. [Google Scholar] [CrossRef]
- Sakai, S. Ab Initio Studies on the Ziegler-Natta Polymerization Reaction Mechanisms. The Role of Cocatalysis. J. Phys. Chem. 1994, 98, 12053–12058. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, L.; Ma, L.; Fu, Z.; Yang, W. Synthesis and characterization of oligomer from 1-decene catalyzed by supported Ziegler–Natta catalysts. Eur. Polym. J. 2005, 41, 2909–2915. [Google Scholar] [CrossRef]
- Huang, Q.; Chen, L.; Sheng, Y.; Ma, L.; Fu, Z.; Yang, W. Synthesis and characterization of oligomer from 1-decene catalyzed by AlCl3/TiCl4/SiO2/Et2AlCl. J. Appl. Polym. Sci. 2006, 101, 584–590. [Google Scholar] [CrossRef]
- Gagieva, S.C.; Kurmaev, D.A.; Tuskaev, V.A.; Khrustalev, V.N.; Churakov, A.V.; Golubev, E.K.; Sizov, A.I.; Zvukova, T.M.; Buzin, M.I.; Nikiforova, G.G.; et al. First example of cationic titanium (III) complexes with crown ether as catalysts for ethylene polymerization. Eur. Polym. J. 2022, 170, 111166. [Google Scholar] [CrossRef]
- Kulangara, S.V.; Jabri, A.; Yang, Y.; Korobkov, I.; Gambarotta, S.; Duchateau, R. Synthesis, X-ray Structural Analysis, and Ethylene Polymerization Studies of Group IV Metal Heterobimetallic Aluminum-Pyrrolyl Complexes. Organometallics 2012, 31, 6085–6094. [Google Scholar] [CrossRef]
- Pasha, F.A.; Basset, J.-M.; Toulhoat, H.; de Bruin, T. DFT Study on the Impact of the Methylaluminoxane Cocatalyst in Ethylene Oligomerization Using a Titanium-Based Catalyst. Organometallics 2015, 34, 426–431. [Google Scholar] [CrossRef]
- McGuinness, D.S. Olefin Oligomerization via Metallacycles: Dimerization, Trimerization, Tetramerization, and Beyond. Chem. Rev. 2011, 111, 2321–2341. [Google Scholar] [CrossRef]
- Linnolahti, M.; Collins, S. Formation, Structure, and Composition of Methylaluminoxane. ChemPhysChem 2017, 18, 3369–3374. [Google Scholar] [CrossRef]
- Tyumkina, T.V.; Islamov, D.N.; Kovyazin, P.V.; Parfenova, L.V. Chain and cluster models of methylaluminoxane as activators of zirconocene hydride, alkyl and metallacyclopropane intermediates in alkene transformations. Mol. Catal. 2021, 512, 111768. [Google Scholar] [CrossRef]
- Barr, J.L.; Kumar, A.; Lionetti, D.; Cruz, C.A.; Blakemore, J.D. Understanding the Roles of Triethylaluminum in Phosphinimide-Supported Titanium Catalyst Systems for Ethylene Polymerization. Organometallics 2019, 38, 2150–2155. [Google Scholar] [CrossRef]
- Kumar, A.; Barr, J.L.; Cruz, C.A.; Blakemore, J.D. Heterobimetallic [Ti, Al] Complexes: Divergent Synthesis, Redox Properties, and Ethylene Polymerization Catalysis. Organometallics 2021, 40, 2139–2148. [Google Scholar] [CrossRef]
- Porri, L.; Giarrusso, A. 5—Conjugated Diene Polymerization. In Comprehensive Polymer Science and Supplements; Allen, G., Bevington, J.C., Eds.; Pergamon Press: Oxford, UK, 1989; pp. 53–108. [Google Scholar] [CrossRef]
- Pragliola, S.; Botta, A.; Longo, P. Solvent effect in 1,3-butadiene polymerization by cyclopentadienyl titanium trichloride (CpTiCl3)/methylaluminoxane (MAO) and pentamethylcyclopentadienyl titanium trichloride (Cp*TiCl3)/MAO catalysts. Eur. Polym. J. 2019, 111, 20–27. [Google Scholar] [CrossRef]
- Poláček, J.; Antropiusová, H.; Hanuš, V.; Petrusová, L.; Mach, K. Titanium-catalyzed cyclotrimerization of butadiene: I. Arenetitanium(II) chloro- and bromo-alane complexes. J. Mol. Catal. 1985, 29, 165–180. [Google Scholar] [CrossRef]
- Poláček, J.; Antropiusová, H.; Petrusová, L.; Mach, K. Titanium-catalyzed cyclotrimerization of butadiene: Part II. The (C6H6)TiII(AlCl4)2-EtxAlCl3−x(x = 1–3) systems. J. Mol. Catal. 1990, 58, 53–73. [Google Scholar] [CrossRef]
- Troyanov, S.I.; Poláček, J.; Antropiusová, H.; Mach, K. The crystal structure of (η6-C6Me6)Ti[(μ-Cl)2(AlClEt)]2 and the catalytic activity of the (C6Me6)TiAl2Cl8−xEtx (x = 0–4) complexes towards butadiene. J. Organomet. Chem. 1992, 430, 317–325. [Google Scholar] [CrossRef]
- Thewalt, U.; Österle, F. Strukturchemie titanorganischer Verbindungen: Die Struktur von π-(CH3)6C6Ti(Cl2AlCl2)2 · C6H6. J. Organomet. Chem. 1979, 172, 317–324. [Google Scholar] [CrossRef]
- Thewalt, U.; Stollmaier, F. Structurchemie titanorganischer verbindungen: Die structur von η6-C6H6Ti(Cl2AlCl2)2. J. Organomet. Chem. 1982, 228, 149–152. [Google Scholar] [CrossRef]
- Wilke, G. Fifty Years of Ziegler Catalysts: Consequences and Development of an Invention. Angew. Chem. Int. Ed. 2003, 42, 5000–5008. [Google Scholar] [CrossRef]
- Cossee, P. Ziegler-Natta catalysis I. Mechanism of polymerization of α-olefins with Ziegler-Natta catalysts. J. Catal. 1964, 3, 80–88. [Google Scholar] [CrossRef]
- Arlman, E.J.; Cossee, P. Ziegler-Natta catalysis III. Stereospecific polymerization of propene with the catalyst system TiCl3-AlEt3. J. Catal. 1964, 3, 99–104. [Google Scholar] [CrossRef]
- Rodriguez, L.A.M.; van Looy, H.M. Studies on Ziegler-Natta catalysts. Part V. Stereospecificity of the active center. J. Polym. Sci. Part A-1 Polym. Chem. 1966, 4, 1971–1992. [Google Scholar] [CrossRef]
- Ziegler, K.; Holzkamp, E.; Breil, H.; Martin, H. Polymerisation von Äthylen und anderen Olefinen. Angew. Chem. 1955, 67, 426. [Google Scholar] [CrossRef]
- Natta, G. Stereospezifische Katalysen und isotaktische Polymere. Angew. Chem. 1956, 68, 393–403. [Google Scholar] [CrossRef]
- Kumawat, J.; Gupta, V.K.; Vanka, K. The Nature of the Active Site in Ziegler–Natta Olefin Polymerization Catalysis Systems—A Computational Investigation. Eur. J. Inorg. Chem. 2014, 2014, 5063–5076. [Google Scholar] [CrossRef]
- Credendino, R.; Liguori, D.; Fan, Z.; Morini, G.; Cavallo, L. Toward a Unified Model Explaining Heterogeneous Ziegler–Natta Catalysis. ACS Catal. 2015, 5, 5431–5435. [Google Scholar] [CrossRef]
- Bahri-Laleh, N. Interaction of different poisons with MgCl2/TiCl4 based Ziegler-Natta catalysts. Appl. Surf. Sci. 2016, 379, 395–401. [Google Scholar] [CrossRef]
- Potapov, A.G. Titanium-magnesium Ziegler-Natta catalysts: New insight on the active sites precursor. Mol. Catal. 2017, 432, 155–159. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Smetannikov, O.V.; Tavtorkin, A.N.; Chinova, M.S.; Ivchenko, P.V. Titanium–Magnesium Nanocatalysts of Polymerization (Review). Pet. Chem. 2016, 56, 480–490. [Google Scholar] [CrossRef]
- Klaue, A.; Kruck, M.; Friederichs, N.; Bertola, F.; Wu, H.; Morbidelli, M. Insight into the Synthesis Process of an Industrial Ziegler–Natta Catalyst. Ind. Eng. Chem. Res. 2019, 58, 886–896. [Google Scholar] [CrossRef]
- Zorve, P.; Linnolahti, M. Adsorption of Titanium Tetrachloride on Magnesium Dichloride Clusters. ACS Omega 2018, 3, 9921–9928. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Piemontesi, F.; Morini, G.; Cavallo, L. Key Elements in the Structure and Function Relationship of the MgCl2/TiCl4/Lewis Base Ziegler–Natta Catalytic System. Macromolecules 2007, 40, 9181–9189. [Google Scholar] [CrossRef]
- Correa, A.; Credendino, R.; Pater, J.T.M.; Morini, G.; Cavallo, L. Theoretical Investigation of Active Sites at the Corners of MgCl2 Crystallites in Supported Ziegler–Natta Catalysts. Macromolecules 2012, 45, 3695–3701. [Google Scholar] [CrossRef]
- Kashiwa, N.; Yoshitake, J. The influence of the valence state of titanium in MgCl2-supported titanium catalysts on olefin polymerization. Makrokol. Chem. 1984, 185, 1133–1138. [Google Scholar] [CrossRef]
- Piovano, A.; Signorile, M.; Braglia, L.; Torelli, P.; Martini, A.; Wada, T.; Takasao, G.; Taniike, T.; Groppo, E. Electronic Properties of Ti Sites in Ziegler–Natta Catalysts. ACS Catal. 2021, 11, 9949–9961. [Google Scholar] [CrossRef]
- Potapov, A.G.; Terskikh, V.V.; Zakharov, V.A.; Bukatov, G.D. 27Al NMR MAS study of the surface Al complexes formed in reaction of organoaluminium compounds with supported TiCl4/MgCl2 catalyst. J. Mol. Catal. A Chem. 1999, 145, 147–152. [Google Scholar] [CrossRef]
- Linnolahti, M.; Pakkanen, T.A.; Bazhenova, A.S.; Denifl, P.; Leinonen, T.; Pakkanen, A. Alkylation of titanium tetrachloride on magnesium dichloride in the presence of Lewis bases. J. Catal. 2017, 353, 89–98. [Google Scholar] [CrossRef]
- Yu, Y.; McKenna, T.F.L.; Boisson, C.; Lacerda Miranda, M.S.; Martins Jr., O. Engineering Poly(ethylene-co-1-butene) through Modulating the Active Species by Alkylaluminum. ACS Catal. 2020, 10, 7216–7229. [Google Scholar] [CrossRef]
- Vittoria, A.; Meppelder, A.; Friederichs, N.; Busico, V.; Cipullo, R. Demystifying Ziegler–Natta Catalysts: The Origin of Stereoselectivity. ACS Catal. 2017, 7, 4509–4518. [Google Scholar] [CrossRef]
- Abazari, M.; Jamjah, R.; Bahri-Laleh, N.; Hanifpour, A. Synthesis and evaluation of a new three-metallic high-performance Ziegler–Natta catalyst for ethylene polymerization: Experimental and computational studies. Polym. Bull. 2022, 79, 7265–7280. [Google Scholar] [CrossRef]
- Masoori, M.; Nekoomanesh, M.; Posada-Pérez, S.; Rashedi, R.; Bahri-Laleh, N. Exploring cocatalyst type effect on the Ziegler–Natta catalyzed ethylene polymerizations: Experimental and DFT studies. J. Polym. Res. 2022, 29, 1–11. [Google Scholar] [CrossRef]
- Kaminsky, W.; Funck, A.; Hähnsen, H. New application for metallocene catalysts in olefin polymerization. Dalton Trans. 2009, 41, 8803–8810. [Google Scholar] [CrossRef]
- Kaminsky, W. Production of Polyolefins by Metallocene Catalysts and Their Recycling by Pyrolysis. Macromol. Symp. 2016, 360, 10–22. [Google Scholar] [CrossRef]
- Busico, V. Metal-catalysed olefin polymerisation into the new millennium: A perspective outlook. Dalton Trans. 2009, 41, 8794–8802. [Google Scholar] [CrossRef]
- Beck, S.; Brintzinger, H.H. Alkyl exchange between aluminium trialkyls and zirconocene dichloride complexes—A measure of electron densities at the Zr center. Inorg. Chim. Acta 1998, 270, 376–381. [Google Scholar] [CrossRef]
- Laine, A.; Linnolahti, M.; Pakkanen, T.A. Alkylation and activation of metallocene polymerization catalysts by reactions with trimethylaluminum: A computational study. J. Organomet. Chem. 2012, 716, 79–85. [Google Scholar] [CrossRef]
- Kumawat, J.; Gupta, V.K. Catalyst Activation and Chain Transfer Agents in Metallocene Catalysts for Ethylene Polymerization: A Computational Investigation. Eur. J. Inorg. Chem. 2021, 2021, 3332–3343. [Google Scholar] [CrossRef]
- Eilertsen, J.L.; Støvneng, J.A.; Ystenes, M.; Rytter, E. Activation of Metallocenes for Olefin Polymerization As Monitored by IR Spectroscopy. Inorg. Chem. 2005, 44, 4843–4851. [Google Scholar] [CrossRef] [PubMed]
- Parfenova, L.V.; Kovyazin, P.V.; Gabdrakhmanov, V.Z.; Istomina, G.P.; Ivchenko, P.V.; Nifant’ev, I.E.; Khalilov, L.M.; Dzhemilev, U.M. Ligand exchange processes in zirconocene dichloride–trimethylaluminum bimetallic systems and their catalytic properties in reaction with alkenes. Dalton Trans. 2018, 47, 16918–16937. [Google Scholar] [CrossRef]
- Harlan, C.J.; Bott, S.G.; Barron, A.R. Three-Coordinate Aluminum Is Not a Prerequisite for Catalytic Activity in the Zirconocene-Alumoxane Polymerization of Ethylene. J. Am. Chem. Soc. 1995, 117, 6465–6474. [Google Scholar] [CrossRef]
- Götz, C.; Rau, A.; Luft, G. Ternary metallocene catalyst systems based on metallocene dichlorides and AlBu3i/[PhNMe2H][B(C6F5)4]: NMR investigations of the influence of Al/Zr ratios on alkylation and on formation of the precursor of the active metallocene species. J. Mol. Catal. A Chem. 2002, 184, 95–110. [Google Scholar] [CrossRef]
- Baldwin, S.M.; Bercaw, J.E.; Brintzinger, H.H. Alkylaluminum-Complexed Zirconocene Hydrides: Identification of Hydride-Bridged Species by NMR Spectroscopy. J. Am. Chem. Soc. 2008, 130, 17423–17433. [Google Scholar] [CrossRef]
- Negishi, E.; Yoshida, T. A novel zirconium-catalyzed hydroalumination of olefins. Tetrahedron Lett. 1980, 21, 1501–1504. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Pechatkina, S.V.; Khalilov, L.M.; Dzhemilev, U.M. Mechanism of Cp2ZrCl2-catalyzed olefin hydroalumination by alkylalanes. Russ. Chem. Bull. 2005, 54, 316–327. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Balaev, A.V.; Gubaidullin, I.M.; Abzalilova, L.R.; Pechatkina, S.V.; Khalilov, L.M.; Spivak, S.I.; Dzhemilev, U.M. Kinetic model of olefin hydroalumination by HAlBui2 and AlBui3 in the presence of Cp2ZrCl2 catalyst. Int. J. Chem. Kinet. 2007, 39, 333–339. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Vil’danova, R.F.; Pechatkina, S.V.; Khalilov, L.M.; Dzhemilev, U.M. New effective reagent [Cp2ZrH2 · ClAlEt2]2 for alkene hydrometallation. J. Organomet. Chem. 2007, 692, 3424–3429. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Kovyazin, P.V.; Nifant’ev, I.E.; Khalilov, L.M.; Dzhemilev, U.M. Role of Zr,Al Hydride Intermediate Structure and Dynamics in Alkene Hydroalumination with XAlBui2 (X = H, Cl, Bui), Catalyzed by Zr η5 Complexes. Organometallics 2015, 34, 3559–3570. [Google Scholar] [CrossRef]
- Pankratyev, E.Y.; Tyumkina, T.V.; Parfenova, L.V.; Khalilov, L.M.; Khursan, S.L.; Dzhemilev, U.M. DFT Study on Mechanism of Olefin Hydroalumination by XAlBui2 in the Presence of Cp2ZrCl2 Catalyst. I. Simulation of Intermediate Formation in Reaction of HAlBui2 with Cp2ZrCl2. Organometallics 2009, 28, 968–977. [Google Scholar] [CrossRef]
- Nurislamova, L.F.; Gubaydullin, I.M.; Koledina, K.F. Kinetic model of isolated reactions of the catalytic hydroalumination of olefins. Reac. Kinet. Mech. Catal. 2015, 116, 79–93. [Google Scholar] [CrossRef]
- Nurislamova, L.F.; Gubaydullin, I.M.; Koledina, K.F.; Safin, R.R. Kinetic model of the catalytic hydroalumination of olefins with organoaluminum compounds. Reac. Kinet. Mech. Catal. 2015, 117, 1–14. [Google Scholar] [CrossRef]
- Pankratyev, E.Y.; Tyumkina, T.V.; Parfenova, L.V.; Khalilov, L.M.; Khursan, S.L.; Dzhemilev, U.M. DFT and Ab Initio Study on Mechanism of Olefin Hydroalumination by XAlBui2 in the Presence of Cp2ZrCl2 Catalyst. II. Olefin Interaction with Catalytically Active Centers. Organometallics 2011, 30, 6078–6089. [Google Scholar] [CrossRef]
- Pankratyev, E.Y.; Khursan, S.L.; Dzhemilev, U.M. DFT and ab initio study on mechanism of olefin hydroalumination by XAlBui2 in the presence of Cp2ZrCl2 catalyst. III. Efficiency of transmetallation in Cp2ZrRCl–XAlBui2 system. J. Organomet. Chem. 2012, 718, 117–123. [Google Scholar] [CrossRef]
- Culver, D.B.; Corieri, J.; Lief, G.; Conley, M.P. Reactions of Triisobutylaluminum with Unbridged or Bridged Group IV Metallocene Dichlorides. Organometallics 2022, 41, 892–899. [Google Scholar] [CrossRef]
- Van Horn, D.E.; Negishi, E. Selective carbon-carbon bond formation via transition metal catalysts. 8. Controlled carbometalation. Reaction of acetylenes with organoalane-zirconocene dichloride complexes as a route to stereo- and regio-defined trisubstituted olefins. J. Am. Chem. Soc. 1978, 100, 2252–2254. [Google Scholar] [CrossRef]
- Xu, S.; Negishi, E. Zirconium-Catalyzed Asymmetric Carboalumination of Unactivated Terminal Alkenes. Acc. Chem. Res. 2016, 49, 2158–2168. [Google Scholar] [CrossRef]
- Kondakov, D.Y.; Negishi, E. Zirconium-catalyzed enantioselective methylalumination of monosubstituted alkenes. J. Am. Chem. Soc. 1995, 117, 10771–10772. [Google Scholar] [CrossRef]
- Kondakov, D.Y.; Negishi, E. Zirconium-Catalyzed Enantioselective Alkylalumination of Monosubstituted Alkenes Proceeding via Noncyclic Mechanism. J. Am. Chem. Soc. 1996, 118, 1577–1578. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Zakirova, I.V.; Kovyazin, P.V.; Karchevsky, S.G.; Istomina, G.P.; Khalilov, L.M.; Dzhemilev, U.M. Intramolecular mobility of η5-ligands in chiral zirconocene complexes and the enantioselectivity of alkene functionalization by organoaluminum compounds. Dalton Trans. 2016, 45, 12814–12826. [Google Scholar] [CrossRef] [PubMed]
- Negishi, E.; Kondakov, D.Y.; Choueiry, D.; Kasai, K.; Takahashi, T. Multiple Mechanistic Pathways for Zirconium-Catalyzed Carboalumination of Alkynes. Requirements for Cyclic Carbometalation Processes Involving C-H Activation. J. Am. Chem. Soc. 1996, 118, 9577–9588. [Google Scholar] [CrossRef]
- Kaminsky, W.; Kopf, J.; Thirase, G. Notiz über die Röntgenstrukturanalyse von Al,Zr-μ-Chloro-1-[bis(cyclopentadienyl)zirkonio(IV)]-2,2-bis(diäthylaluminio)äthan. Eur. J. Org. Chem. 1974, 1531–1533. [Google Scholar] [CrossRef]
- Khalilov, L.M.; Parfenova, L.V.; Rusakov, S.V.; Ibragimov, A.G.; Dzhemilev, U.M. Synthesis and conversions of metallocycles. 22. NMR studies of the mechanism of Cp2ZrCl2-catalyzed cycloalumination of olefins with triethylaluminium to form aluminacyclopentanes. Bull. Acad. Sci. USSR Div. Chem. Sci. 2000, 49, 2051–2058. [Google Scholar] [CrossRef]
- Rusakov, S.V.; Khalilov, L.M.; Parfenova, L.V.; Ibragimov, A.G.; Ponomarev, O.A.; Dzhemilev, U.M. Synthesis and transformations of metallacycles. 27. Quantum-chemical study of the mechanism of styrene catalytic cyclometallation with triethylaluminum in the presence of Cp2ZrCl2. Bull. Acad. Sci. USSR Div. Chem. Sci. 2001, 50, 2336–2345. [Google Scholar] [CrossRef]
- Tyumkina, T.V.; Islamov, D.N.; Parfenova, L.V.; Whitby, R.J.; Khalilov, L.M.; Dzhemilev, U.M. Mechanistic aspects of chemo- and regioselectivity in Cp2ZrCl2-catalyzed alkene cycloalumination by AlEt3. J. Organomet. Chem. 2016, 822, 135–143. [Google Scholar] [CrossRef]
- Tyumkina, T.V.; Islamov, D.N.; Parfenova, L.V.; Karchevsky, S.G.; Khalilov, L.M.; Dzhemilev, U.M. Mechanism of Cp2ZrCl2-Catalyzed Olefin Cycloalumination with AlEt3: Quantum Chemical Approach. Organometallics 2018, 37, 2406–2418. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Kovyazin, P.V.; Mukhamadeeva, O.V.; Ivchenko, P.V.; Nifant’ev, I.E.; Khalilov, L.M.; Dzhemilev, U.M. Zirconocene dichlorides as catalysts in alkene carbo- and cyclometalation by AlEt3: Intermediate structures and dynamics. Dalton Trans. 2021, 50, 15802–15820. [Google Scholar] [CrossRef] [PubMed]
- Erker, G.; Noe, R.; Wingbermühle, D. Seven-Membered Heterodimetallic Ring Systems from (Conjugated Diene) Group 4 Metallocene Complexes and Organoaluminium Reagents. Eur. J. Inorg. Chem. 1994, 127, 805–811. [Google Scholar] [CrossRef]
- Hartner Jr., F. W.; Schwartz, J.; Clift, S.M. Synthesis of carbene complexes of Group IV metals from alkylidene-bridged heterobimetallic precursors. J. Am. Chem. Soc. 1983, 105, 640–641. [Google Scholar] [CrossRef]
- Hartner, F.M.; Clift, S.M.; Schwartz, J.; Tulip, T.H. Synthesis and characterization of alkylidene-bridged zirconium-aluminum complexes. Organometallics 1987, 6, 1346–1350. [Google Scholar] [CrossRef]
- Pédeutour, J.-N.; Cramail, H.; Deffieux, A. Influence of X ligand nature in the activation process of racEt(Ind)2ZrX2 by methylaluminoxane. J. Mol. Catal. A Chem. 2001, 176, 87–94. [Google Scholar] [CrossRef]
- Pédeutour, J.-N.; Cramail, H.; Deffieux, A. The negative role of chloride counter-anion in the activation process of zirconocene dichloride by methylaluminoxane. J. Mol. Catal. A Chem. 2001, 174, 81–87. [Google Scholar] [CrossRef]
- Franceschini, F.C.; Tavares, T.T.R.; dos Santos, J.H.Z.; Ferreira, M.L.; Soares, J.B.P. Ethylene and Propylene Polymerization Using In Situ Supported Me2Si(Ind)2ZrCl2 Catalyst: Experimental and Theoretical Study. Macromol. Mater. Eng. 2006, 291, 279–287. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Kovyazin, P.V.; Tyumkina, T.V.; Islamov, D.N.; Lyapina, A.R.; Karchevsky, S.G.; Ivchenko, P.V. Reactions of bimetallic Zr,Al-hydride complexes with methylaluminoxane: NMR and DFT study. J. Organomet. Chem. 2017, 851, 30–39. [Google Scholar] [CrossRef]
- Stahl, N.G.; Salata, M.R.; Marks, T.J. B(C6F5)3- vs Al(C6F5)3-Derived Metallocenium Ion Pairs. Structural, Thermochemical, and Structural Dynamic Divergences. J. Am. Chem. Soc. 2005, 127, 10898–10909. [Google Scholar] [CrossRef]
- Velthoen, M.E.Z.; Munoz-Murillo, A.; Bouhmadi, A.; Cecius, M.; Diefenbach, S.; Weckhuysen, B.M. The multifaceted role of methylaluminoxane in metallocene-based olefin polymerization catalysis. Macromolecules 2018, 51, 343–355. [Google Scholar] [CrossRef]
- Velthoen, M.E.Z.; Boereboom, J.M.; Bulo, R.E.; Weckhuysen, B.M. Insights into the activation of silica-supported metallocene olefin polymerization catalysts by methylaluminoxane. Catal. Today 2019, 334, 223–230. [Google Scholar] [CrossRef]
- Baldwin, S.M.; Bercaw, J.E.; Henling, L.M.; Day, M.W.; Brintzinger, H.H. Cationic Alkylaluminum-Complexed Zirconocene Hydrides: NMR-Spectroscopic Identification, Crystallographic Structure Determination, and Interconversion with Other Zirconocene Cations. J. Am. Chem. Soc. 2011, 133, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Lenton, T.N.; Bercaw, J.E.; Panchenko, V.N.; Zakharov, V.A.; Babushkin, D.E.; Soshnikov, I.E.; Talsi, E.P.; Brintzinger, H.H. Formation of Trivalent Zirconocene Complexes from ansa-Zirconocene-Based Olefin-Polymerization Precatalysts: An EPR- and NMR-Spectroscopic Study. J. Am. Chem. Soc. 2013, 135, 10710–10719. [Google Scholar] [CrossRef]
- Thorn, M.G.; Diefenbach, S.P. Book of Abstracts; Advances in Polyolefins X: Santa Rosa, CA, USA; American Chemical Society, Division of Polymer Chemistry, Inc.: Blacksburg, VA, USA, 2015. [Google Scholar]
- Thorn, M.G.; Marcel, L. SPE International Polyolefins Conference 2016; Global Interdependence: Houston, TX, USA; SPE South Texas Section, Society of Plastics Engineers: Bethel, CT, USA, 2016. [Google Scholar]
- Slaugh, L.H.; Schoenthal, G.W. Vinylidene Olefin Process. U.S. Patent 4658078, 14 April 1987. [Google Scholar]
- Thiele, S.; Erker, G. Adjusting the Features of Active Metallocene Ziegler Systems for Their Potential Use as Carbon–Carbon Coupling Catalysts in Organic Synthesis. Eur. J. Inorg. Chem. 1997, 130, 201–207. [Google Scholar] [CrossRef]
- Janiak, C.; Blank, F. Metallocene Catalysts for Olefin Oligomerization. Macromol. Symp. 2006, 236, 14–22. [Google Scholar] [CrossRef]
- Janiak, C.; Lange, K.C.H.; Marquardt, P. α-Olefin oligomers with narrow molar mass distributions from zirconocene/methylaluminoxane catalysts—An examination of the structure-reactivity relationship. Macromol. Rapid Commun. 1995, 16, 643–650. [Google Scholar] [CrossRef]
- Kissin, Y.V.; Schwab, F.C. Post-Oligomerization of alpha-Olefin Oligomers: A Route to Single-Component and Multicomponent Synthetic Lubricating Oils. J. Appl. Polym. Sci. 2009, 111, 273–280. [Google Scholar] [CrossRef]
- Kissin, Y.V. Detailed Kinetics of 1-Hexene Oligomerization Reaction with (n-Bu-Cp)2ZrCl2–MAO Catalyst. Macromol. Chem. Phys. 2009, 210, 1241–1246. [Google Scholar] [CrossRef]
- Boccia, A.C.; Costabile, C.; Pragliola, S.; Longo, P. Selective Dimerization of γ-Branched α-Olefins in the Presence of C2v Group-4 Metallocene-Based Catalysts. Macromol. Chem. Phys. 2004, 205, 1320–1326. [Google Scholar] [CrossRef]
- Salmela, M.; Lehtinen, T.; Efimova, E.; Santala, S.; Santala, V. Towards bioproduction of poly-α-olefins from lignocellulose. Green Chem. 2020, 22, 5067–5076. [Google Scholar] [CrossRef]
- Bagheri, V.; Moore, L.D.; Di Giacinto, P.M.; Sanchezrivas, M. Low Viscosity Oligomer Oil Product, Process and Composition. U.S. Patent 20130225459, 29 August 2013. [Google Scholar]
- Sato, H.; Kamimura, H. Process for Producing Saturated Aliphatic Hydrocarbon Compound, and Lubricant Composition. U.S. Patent 8373011, 12 February 2013. [Google Scholar]
- Patil, A.; Lewis, K.G.; Bodige, S.; Zushma, S. Ester Compounds, Lubricating Oil Composition Containing Same and Process for Making Same. U.S. Patent 2019062663, 28 February 2019. [Google Scholar]
- Sato, H.; Kashiwamura, T.; Okamoto, T.; Yokota, K. Carbonyl Compound Containing Long-Chain Branched Alkyl Group. U.S. Patent 7402610, 22 July 2008. [Google Scholar]
- Harvey, B.G.; Quintana, R.L. Synthesis of renewable jet and diesel fuels from 2-ethyl-1-hexene. Energy Environ. Sci. 2010, 3, 352–357. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Minyaev, M.E.; Tavtorkin, A.N.; Vinogradov, A.A.; Ivchenko, P.V. Branched alkylphosphinic and disubstituted phosphinic and phosphonic acids: Effective synthesis based on α-olefin dimers and applications in lanthanide extraction and separation. RSC Adv. 2017, 7, 24122–24128. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Ivchenko, P.V. Production Method of Vinylidene Olefins. RU Patent RU2652118, 25 April 2018. [Google Scholar]
- Takeuchi, K.; Fujikawa, S. Base Oil for Oil Drilling Fluid and Oil Drilling Fluid Composition. U.S. Patent 2011251445, 13 October 2011. [Google Scholar]
- Fujikawa, S.; Okamoto, T.; Yokota, K. Process for Producing Unsaturated Hydrocarbon Compound. U.S. Patent 8119850, 21 February 2012. [Google Scholar]
- Kovyazin, P.V.; Bikmeeva, A.K.; Islamov, D.N.; Yanybin, V.M.; Tyumkina, T.V.; Parfenova, L.V. Ti Group Metallocene-Catalyzed Synthesis of 1-Hexene Dimers and Tetramers. Molecules 2021, 26, 2775. [Google Scholar] [CrossRef] [PubMed]
- Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K. Bimetallic Zr,Zr-Hydride Complexes in Zirconocene Catalyzed Alkene Dimerization. Molecules 2020, 25, 2216. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jang, Y.E.; Jeon, J.Y.; Go, M.J.; Lee, J.; Kim, S.K.; Lee, S.-I.; Lee, B.Y. Preparation of ansa-metallocenes for production of poly(α-olefin) lubricants. Dalton Trans. 2014, 43, 10132–10138. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Bezzubov, S.I.; Ivchenko, P.V. Catalytic oligomerization of α-olefins in the presence of two-stage activated zirconocene catalyst based on 6,6-dimethylfulvene ‘dimer’. Mendeleev Commun. 2017, 27, 35–37. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, K. Catalytic polymerization of 1-decene using a silicon-bridged metallocene system. Petrol. Sci. Technol. 2017, 35, 1451–1456. [Google Scholar] [CrossRef]
- Dong, S.Q.; Mi, P.K.; Xu, S.; Zhang, J.; Zhao, R.D. Preparation and Characterization of Single-Component Poly-α-olefin Oil Base Stocks. Energy Fuels 2019, 33, 9796–9804. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Sedov, I.V.; Dorokhov, V.G.; Lyadov, A.S.; Ivchenko, P.V. Structurally uniform 1-hexene, 1-octene, and 1-decene oligomers: Zirconocene/MAO-catalyzed preparation, characterization, and prospects of their use as low-viscosity low-temperature oil base stocks. Appl. Catal. A Gen. 2018, 549, 40–50. [Google Scholar] [CrossRef]
- Shao, H.; Wang, R.; Li, H.; Guo, X.; Jiang, T. Synthesis of low-molecular-weight poly-α-olefins using silicon-bridged zirconocene catalyst for lubricant basestock. Arab. J. Chem. 2020, 13, 2715–2721. [Google Scholar] [CrossRef]
- Jalali, A.; Nekoomanehs-Haghighi, M.; Dehghani, S.; Bahri-Laleh, N. Effect of metal type on the metallocene-catalyzed oligomerization of 1-hexene and 1-octene to produce polyα-olefin-based synthetic lubricants. Appl. Organomet. Chem. 2020, 34, e5338. [Google Scholar] [CrossRef]
- Lamb, J.V.; Buffet, J.-C.; Turner, Z.R.; Khamnaen, T.; O’Hare, D. Metallocene Polyethylene Wax Synthesis. Macromolecules 2020, 53, 5847–5856. [Google Scholar] [CrossRef]
- Tritto, I.; Boggioni, L.; Sacchi, M.C.; Dall’Occo, T. Novel aluminum based cocatalysts for metallocene catalyzed olefin polymerization. J. Mol. Catal. A Chem. 2003, 204, 305–314. [Google Scholar] [CrossRef]
- Hagen, H.; Boersma, J.; van Koten, G. Homogeneous vanadium-based catalysts for the Ziegler–Natta polymerization of α-olefins. Chem. Soc. Rev. 2002, 31, 357–364. [Google Scholar] [CrossRef]
- Gambarotta, S. Vanadium-based Ziegler–Natta: Challenges, promises, problems. Coord. Chem. Rev. 2003, 237, 229–243. [Google Scholar] [CrossRef]
- Redshaw, C. Vanadium procatalysts bearing chelating aryloxides: Structure–activity trends in ethylenepolymerisation. Dalton Trans. 2010, 39, 5595–5604. [Google Scholar] [CrossRef]
- Wu, J.-Q.; Li, Y.-S. Well-defined vanadium complexes as the catalysts for olefin polymerization. Coord. Chem. Rev. 2011, 255, 2303–2314. [Google Scholar] [CrossRef]
- Nomura, K.; Zhang, S. Design of Vanadium Complex Catalysts for Precise Olefin Polymerization. Chem. Rev. 2011, 111, 2342–2362. [Google Scholar] [CrossRef]
- Zambelli, A.; Sessa, I.; Grisi, F.; Fusco, R.; Accomazzi, P. Syndiotactic Polymerization of Propylene: Single-Site Vanadium Catalysts in Comparison with Zirconium and Nickel. Macromol. Rapid Commun. 2001, 22, 297–310. [Google Scholar] [CrossRef]
- Feghali, K.; Harding, D.J.; Reardon, D.; Gambarotta, S.; Yap, G.; Wang, Q. Stability of Metal-Carbon Bond versus Metal Reduction during Ethylene Polymerization Promoted by a Vanadium Complex: The Role of the Aluminum Cocatalyst. Organometallics 2002, 21, 968–976. [Google Scholar] [CrossRef]
- Liguori, D.; Centore, R.; Csok, Z.; Tuzi, A. Polymerization of Propene and 1,3-Butadiene with Vanadyl(V) Monoamidinate Precatalysts and MAO or Dialkylaluminum Chloride Cocatalysts. Macromol. Chem. Phys. 2004, 205, 1058–1063. [Google Scholar] [CrossRef]
- Wang, W.; Nomura, K. Notable Effects of Aluminum Alkyls and Solvents for Highly Efficient Ethylene (Co)polymerizations Catalyzed by (Arylimido)- (aryloxo)vanadium Complexes. Adv. Synth. Catal. 2006, 348, 743–750. [Google Scholar] [CrossRef]
- Soshnikov, I.E.; Semikolenova, N.V.; Shubin, A.A.; Bryliakov, K.P.; Zakharov, V.A.; Redshaw, C.; Talsi, E.P. EPR Monitoring of Vanadium(IV) Species Formed upon Activation of Vanadium(V) Polyphenolate Precatalysts with AlR2Cl and AlR2Cl/Ethyltrichloroacetate (R = Me, Et). Organometallics 2009, 28, 6714–6720. [Google Scholar] [CrossRef]
- Xu, B.-C.; Hu, T.; Wu, J.-Q.; Hu, N.-H.; Li, Y.-S. Novel vanadium(III) complexes with bidentate N,N-chelating iminopyrrolide ligands: Synthesis, characterization and catalytic behaviour of ethylene polymerization and copolymerization with 10-undecen-1-ol. Dalton Trans. 2009, 41, 8854–8863. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.-S.; Shi, X.-C.; Li, Y.-S. Ethylene homopolymerizaton and copolymerizaton by vanadium(III) complexes containing tridentate or tetradentate iminopyrrolyl ligands. J. Polym. Sci. A Polym. Chem. 2011, 49, 2700–2708. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Lu, L.-P.; Li, B.-X.; Li, Y.-S. Synthesis, structural characterization, and olefin polymerization behavior of vanadium(III) complexes bearing bidentate phenoxy-phosphine ligands. J. Polym. Sci. A Polym. Chem. 2012, 50, 4721–4731. [Google Scholar] [CrossRef]
- Kurmaev, D.A.; Kolosov, N.A.; Gagieva, S.C.; Borissova, A.O.; Tuskaev, V.A.; Bravaya, N.M.; Bulychev, B.M. Coordination compounds of chromium (+3) and vanadium (+3) and (+5) with 2,6-bis(diphenylhydroxymethyl)pyridyl ligand: Synthesis and study of catalytic activity in the polymerization of ethylene. Inorg. Chim. Acta 2013, 396, 136–143. [Google Scholar] [CrossRef]
- Wu, J.-Q.; Pan, L.; Hu, N.-H.; Li, Y.-S. Synthesis, Structural Characterization, and Ethylene Polymerization Behavior of the Vanadium(III) Complexes Bearing Salicylaldiminato Ligands. Organometallics 2008, 27, 3840–3848. [Google Scholar] [CrossRef]
- Wu, J.-Q.; Pan, L.; Li, Y.-G.; Liu, S.-R.; Li, Y.-S. Synthesis, Structural Characterization, and Olefin Polymerization Behavior of Vanadium(III) Complexes Bearing Tridentate Schiff Base Ligands. Organometallics 2009, 28, 1817–1825. [Google Scholar] [CrossRef]
- Wang, Y.; Zuo, M.; Li, Y. Theoretical investigation of the mechanism of ethylene polymerization with salicylaldiminato vanadium(III) complexes. Chin. J. Catal. 2015, 36, 657–666. [Google Scholar] [CrossRef]
- Białek, M.; Bisz, E. Dichlorovanadium(IV) diamine-bis(phenolate) complexes for ethylene (co)polymerization and 1-olefin isospecific polymerization. J. Catal. 2018, 362, 65–73. [Google Scholar] [CrossRef]
- Soshnikov, I.E.; Semikolenova, N.V.; Bryliakov, K.P.; Zakharov, V.A.; Talsi, E.P. Vanadium olefin polymerization catalysts: NMR spectroscopic characterization of V(III) intermediates. J. Organomet. Chem. 2018, 867, 4–13. [Google Scholar] [CrossRef]
- Yi, J.; Nakatani, N.; Nomura, K.; Hada, M. Time-dependent DFT study of the K-edge spectra of vanadium and titanium complexes: Effects of chloride ligands on pre-edge features. Phys. Chem. Chem. Phys. 2020, 22, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Nakatani, N.; Nomura, K. Solution XANES and EXAFS analysis of active species of titanium, vanadium complex catalysts in ethylene polymerisation/dimerisation and syndiospecific styrene polymerisation. Dalton Trans. 2020, 49, 8008–8028. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Izawa, I.; Kuboki, M.; Inoue, K.; Aoki, H.; Tsutsumi, K. Solution XAS Analysis for Reactions of Phenoxide-Modified (Arylimido)vanadium(V) Dichloride and (Oxo)vanadium(V) Complexes with Al Alkyls: Effect of Al Cocatalyst in Ethylene (Co)polymerization. Catalysts 2022, 12, 198. [Google Scholar] [CrossRef]
- Liu, K.; Liu, Z.; Cheng, R.; He, X.; Liu, B. Mechanistic study on the effects of co-catalyst on ethylene polymerization over supported vanadocene catalyst. Mol. Catal. 2020, 486, 110852. [Google Scholar] [CrossRef]
- Do, L.H.; Labinger, J.A.; Bercaw, J.E. Mechanistic Studies of Ethylene and α-Olefin Co-Oligomerization Catalyzed by Chromium–PNP Complexes. Organometallics 2012, 31, 5143–5149. [Google Scholar] [CrossRef][Green Version]
- Zilbershtein, T.M.; Kardash, V.A.; Suvorova, V.V.; Golovko, A.K. Decene formation in ethylene trimerization reaction catalyzed by Cr-pyrrole system. Appl. Catal. A Gen. 2014, 475, 371–378. [Google Scholar] [CrossRef]
- Gong, M.; Liu, Z.; Li, Y.; Ma, Y.; Sun, Q.; Zhang, J.; Liu, B. Selective Co-Oligomerization of Ethylene and 1-Hexene by Chromium-PNP Catalysts: A DFT Study. Organometallics 2016, 35, 972–981. [Google Scholar] [CrossRef]
- Dixon, J.T.; Green, M.J.; Hess, F.M.; Morgan, D.H. Advances in selective ethylene trimerization—A critical overview. J. Organomet. Chem. 2004, 689, 3641–3668. [Google Scholar] [CrossRef]
- Wass, D.F. Chromium-catalysed ethene trimerisation and tetramerization—Breaking the rules in olefin oligomerisation. Dalton. Trans. 2007, 8, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, P.W.; Clément, N.D.; Tschan, M.J.-L. New processes for the selective production of 1-octene. Coord. Chem. Rev. 2011, 255, 1499–1517. [Google Scholar] [CrossRef]
- Belov, G.P. Tetramerization of Ethylene to Octene-1 (A Review). Pet. Chem. 2012, 52, 139–154. [Google Scholar] [CrossRef]
- Agapie, T. Selective ethylene oligomerization: Recent advances in chromium catalysis and mechanistic investigations. Coord. Chem. Rev. 2011, 255, 861–880. [Google Scholar] [CrossRef]
- Forestière, A.; Olivier-Bourbigou, H.; Saussine, L. Oligomerization of Monoolefins by Homogeneous Catalysts. Oil. Gas Sci. Technol. 2009, 64, 649–667. [Google Scholar] [CrossRef]
- Belov, G.P.; Matkovsky, P.E. Processes for the Production of Higher Linear α-Olefins. Pet. Chem. 2010, 50, 296–302. [Google Scholar] [CrossRef]
- Reagen, W.K. Process for Olefin Polymerization. European Patent 0417477, 9 August 1990. [Google Scholar]
- Knudsen, R.D.; Freeman, J.W.; Lashier, M.E. Olefin Production. U.S. Patent 5563312, 8 October 1996. [Google Scholar]
- Lashier, M.E. Process for the Oligomerization of Olefins. European Patent 0780353, 25 June 1997. [Google Scholar]
- Freeman, J.W.; Buster, J.L.; Knudsen, R.D. Olefin Production. U.S. Patent 5856257, 5 January 1999. [Google Scholar]
- Tanaka, E.; Urata, H.; Oshiki, T.; Aoshima, T.; Kawashima, R.; Iwade, S.; Nakamura, H.; Katsuki, S.; Okano, T. Process for Producing Alpha-Olefin Oligomer Compositions. European Patent 0611743, 24 August 1994. [Google Scholar]
- Yoshitaka, A.; Hirofumi, N.; Yoshaki, N.; Takeshi, O. Process for Producing Alpha-Olefin Oligomer. U.S. Patent 5856612, 5 January 1999. [Google Scholar]
- Van Rensburg, W.J.; Grové, C.; Steynberg, J.P.; Stark, K.B.; Huyser, J.J.; Steynberg, P.J. A DFT Study toward the Mechanism of Chromium-Catalyzed Ethylene Trimerization. Organometallics 2004, 23, 1207–1222. [Google Scholar] [CrossRef]
- Jabri, A.; Mason, C.B.; Sim, Y.; Gambarotta, S.; Burchell, T.J.; Duchateau, R. Isolation of Single-Component Trimerization and Polymerization Chromium Catalysts: The Role of the Metal Oxidation State. Angew. Chem. Int. Ed. 2008, 47, 9717–9721. [Google Scholar] [CrossRef]
- Vidyaratne, I.; Nikiforov, G.B.; Gorelsky, S.I.; Gambarotta, S.; Duchateau, R.; Korobkov, I. Isolation of a self-activating ethylene trimerization catalyst. Angew. Chem. Int. Ed. 2009, 48, 6552–6566. [Google Scholar] [CrossRef]
- Budzelaar, P.H.M. Ethene trimerization at CrI/CrIII – A density functional theory (DFT) study. Can. J. Chem. 2009, 87, 832–837. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Z.; Cheng, R.; He, X.; Liu, B. Mechanistic DFT Study on Ethylene Trimerization of Chromium Catalysts Supported by a Versatile Pyrrole Ligand System. Organometallics 2014, 33, 2599–2607. [Google Scholar] [CrossRef]
- Venderbosch, B.; Oudsen, J.-P.H.; Wolzak, L.A.; Martin, D.J.; Korstanje, T.J.; Tromp, M. Spectroscopic Investigation of the Activation of a Chromium-Pyrrolyl Ethene Trimerization Catalyst. ACS Catal. 2019, 9, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.; Cohen, S.A.; Cooley, N.A.; Murphy, A.; Scutt, J.; Wass, D.F. High activity ethylene trimerisation catalysts based on diphosphine ligands. Chem. Commun. 2002, 8, 858–859. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, H.M.; Park, H.S.; Kim, S.D.; Kwon, S.J.; Tahara, A.; Nagashima, H.; Lee, B.Y. MAO-free and extremely active catalytic system for ethylene tetramerization. Appl. Organomet. Chem. 2019, 33, e4829. [Google Scholar] [CrossRef]
- Jabri, A.; Crewdson, P.; Gambarotta, S.; Korobkov, I.; Duchateau, R. Isolation of a Cationic Chromium(II) Species in a Catalytic System for Ethylene Tri- and Tetramerization. Organometallics 2006, 25, 715–718. [Google Scholar] [CrossRef]
- Müller, B.H.; Fritz, P.M.; Bölt, H.; Wöhl, A.; Müller, W.; Winkler, F.; Wellenhoffer, A.; Rosenthal, U.; Hapke, M.; Peulecke, N.; et al. Catalyst Composition and Process for Di-, Tri- and/or Tetramerization of Ethylene. WO Patent 2009006979, 15 January 2009. [Google Scholar]
- Peitz, S.; Peulecke, N.; Aluri, B.R.; Hansen, S.; Müller, B.H.; Spannenberg, A.; Rosenthal, U.; Al-Hazmi, M.H.; Mosa, F.M.; Wöhl, A.; et al. A Selective Chromium Catalyst System for the Trimerization of Ethene and Its Coordination Chemistry. Eur. J. Inorg. Chem. 2010, 2010, 1167–1171. [Google Scholar] [CrossRef]
- Müller, B.H.; Peulecke, N.; Peitz, S.; Aluri, B.R.; Rosenthal, U.; Al-Hazmi, M.H.; Mosa, F.M.; Wöhl, A.; Müller, W. Activity Enhancement of a Catalyst System for the Selective Trimerization of Ethene to 1-Hexene by Modification of the Chromium to Chloride to Aluminium Ratio. Chem. Eur. J. 2011, 17, 6935–6938. [Google Scholar] [CrossRef]
- Peitz, S.; Peulecke, N.; Müller, B.H.; Spannenberg, A.; Drexler, H.-J.; Rosenthal, U.; Al-Hazmi, M.H.; Al-Eidan, K.E.; Wöhl, A.; Müller, W. Heterobimetallic Al-Cl-Cr Intermediates with Relevance to the Selective Catalytic Ethene Trimerization Systems Consisting of CrCl3(THF)3, the Aminophosphorus Ligands Ph2PNP(Ph)NH, and Triethylaluminum. Organometallics 2011, 30, 2364–2370. [Google Scholar] [CrossRef]
- Bartlett, S.A.; Moulin, J.; Tromp, M.; Reid, G.; Dent, A.J.; Cibin, G.; McGuinness, D.S.; Evans, J. Activation of [CrCl3{PPh2N(iPr)PPh2}] for the selective oligomerisation of ethene: A Cr K-edge XAFS study. Cat. Sci. Technol. 2016, 6, 6237–6246. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Roznyatovsky, V.A.; Grishin, Y.K.; Ivanyukl, A.V.; Sedov, I.V.; Churakov, A.V.; Ivchenko, P.V. 5,6-Dihydrodibenzo[c,e][1,2]azaphosphinine-based PNP ligands, Cr coordination and Cr(III) precatalysts for ethylene oligomerization. Organometallics 2018, 37, 2660–2664. [Google Scholar] [CrossRef]
- Lee, D.G.; Baek, J.W.; Lee, J.H.; Lee, H.J.; Seo, Y.H.; Lee, J.; Lee, C.G.; Lee, B.Y. Replacement of the Common Chromium Source CrCl3(thf)3 with Well-Defined [CrCl2(μ-Cl)(thf)2]2. Molecules 2021, 26, 1167. [Google Scholar] [CrossRef]
- Albahily, K.; Koç, E.; Al-Baldawi, D.; Savard, D.; Gambarotta, S.; Burchell, T.J.; Duchateau, R. Chromium Catalysts Supported by a Nonspectator NPN Ligand: Isolation of Single-Component Chromium Polymerization Catalysts. Angew. Chem. Int. Ed. 2008, 47, 5816–5819. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, D.S.; Brown, D.B.; Tooze, R.P.; Hess, F.M.; Dixon, J.T.; Slawin, A.M.Z. Ethylene Trimerization with Cr-PNP and Cr-SNS Complexes: Effect of Ligand Structure, Metal Oxidation State, and Role of Activator on Catalysis. Organometallics 2006, 25, 3605–3610. [Google Scholar] [CrossRef]
- Temple, C.; Jabri, A.; Crewdson, P.; Gambarotta, S.; Korobkov, I.; Duchateau, R. The Question of the Cr Oxidation State in the {Cr(SNS)} Catalyst for Selective Ethylene Trimerization: An Unanticipated Re-Oxidation Pathway. Angew. Chem. Int. Ed. 2006, 45, 7050–7053. [Google Scholar] [CrossRef] [PubMed]
- Albahily, K.; Gambarotta, S.; Duchateau, R. Ethylene Oligomerization Promoted by a Silylated-SNS Chromium System. Organometallics 2011, 30, 4655–4664. [Google Scholar] [CrossRef]
- Zhang, J.; Li, A.; Andy Hor, T.S. Crystallographic Revelation of the Role of AlMe3 (in MAO) in Cr [NNN] Pyrazolyl Catalyzed Ethylene Trimerization. Organometallics 2009, 28, 2935–2937. [Google Scholar] [CrossRef]
- Thapa, I.; Gambarotta, S.; Korobkov, I.; Duchateau, R.; Kulangara, S.V.; Chevalier, R. Switchable Chromium(II) Complexes of a Chelating Amidophosphine (N-P) for Selective and Nonselective Ethylene Oligomerization. Organometallics 2010, 29, 4080–4089. [Google Scholar] [CrossRef]
- Kulangara, S.V.; Mason, C.; Juba, M.; Yang, Y.; Thapa, I.; Gambarotta, S.; Korobkov, I.; Duchateau, R. Synthesis and Catalytic Oligomerization Activity of Chromium Catalysts of Ligand Systems with Switchable Connectivity. Organometallics 2012, 31, 6438–6449. [Google Scholar] [CrossRef]
- Alzamly, A.; Gambarotta, S.; Korobkov, I. Polymer-Free Ethylene Oligomerization Using a Pyridine-Based Pincer PNP-Type of Ligand. Organometallics 2013, 32, 7204–7212. [Google Scholar] [CrossRef]
- Alzamly, A.; Gambarotta, S.; Korobkov, I. Synthesis, Structures, and Ethylene Oligomerization Activity of Bis(phosphanylamine)pyridine Chromium/Aluminate Complexes. Organometallics 2013, 32, 7107–7115. [Google Scholar] [CrossRef]
- Alzamly, A.; Gambarotta, S.; Korobkov, I.; Murugesu, M.; Le Roy, J.J.H.; Budzelaar, P.H.M. Isolation of a Hexanuclear Chromium Cluster with a Tetrahedral Hydridic Core and Its Catalytic Behavior for Ethylene Oligomerization. Inorg. Chem. 2014, 53, 6073–6081. [Google Scholar] [CrossRef] [PubMed]
- Kulangara, S.V.; Haveman, D.; Vidjayacoumar, B.; Korobkov, I.; Gambarotta, S.; Duchateau, R. Effect of Cocatalysts and Solvent on Selective Ethylene Oligomerization. Organometallics 2015, 34, 1203–1210. [Google Scholar] [CrossRef]
- Keim, W. Oligomerization of Ethylene to α-Olefins: Discovery and Development of the Shell Higher Olefin Process (SHOP). Angew. Chem. Int. Ed. 2013, 52, 12492–12496. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Sémeril, D.; Matt, D.; Chetcuti, M.J.; Lutz, P. Structure–reactivity relationships in SHOP-type complexes: Tunable catalysts for the oligomerisation and polymerisation of ethylene. Dalton Trans. 2007, 5, 515–528. [Google Scholar] [CrossRef]
- Ittel, S.D.; Johnson, L.K.; Brookhart, M. Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chem. Rev. 2000, 100, 1169–1203. [Google Scholar] [CrossRef]
- Qasim, M.; Bashir, M.S.; Iqbal, S.; Mahmood, Q. Recent advancements in α-diimine-nickel and -palladium catalysts for ethylene polymerization. Eur. Polym. J. 2021, 160, 110783. [Google Scholar] [CrossRef]
- Wu, R.; Wu, W.K.; Stieglitz, L.; Gaan, S.; Rieger, B.; Heuberger, M. Recent advances on α-diimine Ni and Pd complexes for catalyzed ethylene (Co)polymerization: A comprehensive review. Coord. Chem. Rev. 2023, 474, 214844. [Google Scholar] [CrossRef]
- Mu, H.; Pan, L.; Song, D.; Li, Y. Neutral Nickel Catalysts for Olefin Homo- and Copolymerization: Relationships between Catalyst Structures and Catalytic Properties. Chem. Rev. 2015, 115, 12091–12137. [Google Scholar] [CrossRef]
- Mu, H.; Zhou, G.; Hu, X.; Jian, Z. Recent advances in nickel mediated copolymerization of olefin with polar monomers. Coord. Chem. Rev. 2021, 435, 213802. [Google Scholar] [CrossRef]
- Wang, F.; Chen, C. A continuing legend: The Brookhart-type α-diimine nickel and palladium catalysts. Polym. Chem. 2019, 10, 2354–2369. [Google Scholar] [CrossRef]
- Bogdanovlć, B. Catalysis and Organic Syntheses. Selectivity Control in Nickel-Catalyzed Olefin Oligomerization. In Advances in Organometallic Chemistry; Stone, F.G.A., West, R., Eds.; Elsevier: Amsterdam, The Netherlands, 1979; Volume 17, pp. 105–140. [Google Scholar] [CrossRef]
- Corker, J.M.; Evans, J. EXAFS studies of the activation of homogeneous nickel catalysts for propene dimerisation by aluminium reagents. J. Chem. Soc. Chem. Commun. 1991, 16, 1104–1106. [Google Scholar] [CrossRef]
- De Souza, R.F.; Simon, L.C.; do Carmo, M.; Alves, M. XAS study of the nickel(α-diimine) catalyst for olefin polymerization. J. Catal. 2003, 214, 165–168. [Google Scholar] [CrossRef]
- Chai, W.; Yu, J.; Wang, L.; Hu, X.; Redshaw, C.; Sun, W.-H. Synthesis, characterization and ethylene oligomerization behavior of N-(2-alkyl-5,6,7-trihydroquinolin-8-ylidene)arylaminonickel(II) dichlorides. Inorg. Chim. Acta 2012, 385, 21–26. [Google Scholar] [CrossRef]
- He, F.; Hao, X.; Cao, X.; Redshaw, C.; Sun, W.-H. Nickel halide complexes bearing 2-benzimidazolyl-N-arylquinoline-8-carboxamide derived ligands: Synthesis, characterization and catalytic behavior towards ethylene oligomerization and the vinyl polymerization of norbornene. J. Organomet. Chem. 2012, 712, 46–51. [Google Scholar] [CrossRef]
- Boudier, A.; Breuil, P.-A.R.; Magna, L.; Olivier-Bourbigou, H.; Braunstein, P. Nickel(II) complexes with imino-imidazole chelating ligands bearing pendant donor groups (SR, OR, NR2, PR2) as precatalysts in ethylene oligomerization. J. Organomet. Chem. 2012, 718, 31–37. [Google Scholar] [CrossRef]
- Sun, W.-H.; Song, S.; Li, B.; Redshaw, C.; Hao, X.; Li, Y.-S.; Wang, F. Ethylene polymerization by 2-iminopyridylnickel halide complexes: Synthesis, characterization and catalytic influence of the benzhydryl group. Dalton Trans. 2012, 41, 11999–12010. [Google Scholar] [CrossRef]
- Song, K.; Yang, W.; Li, B.; Liu, Q.; Redshaw, C.; Li, Y.; Sun, W.-H. Nickel(II) complexes bearing 4,5-bis(arylimino)-pyrenylidenes: Synthesis, characterization, and ethylene polymerization behaviour. Dalton Trans. 2013, 42, 9166–9175. [Google Scholar] [CrossRef]
- Soshnikov, I.E.; Semikolenova, N.V.; Bryliakov, K.P.; Antonov, A.A.; Talsi, E.P. Ni(I) Intermediates Formed upon Activation of a Ni(II) α-Diimine Ethylene Polymerization Precatalyst with AlR3 (R = Me, Et, and iBu), AlR2Cl (R = Me, Et), and MMAO: A Comparative Study. Organometallics 2022, 41, 1015–1024. [Google Scholar] [CrossRef]
- Obuah, C.; Omondi, B.; Nozaki, K.; Darkwa, J. Solvent and co-catalyst dependent pyrazolylpyridinamine and pyrazolylpyrroleamine nickel(II) catalyzed oligomerization and polymerization of ethylene. J. Mol. Catal. A Chem. 2014, 382, 31–40. [Google Scholar] [CrossRef]
- Singh, A.; Maji, A.; Mohanty, A.; Ghosh, K. Design and synthesis of base-metal nickel(II) based catalysts: Studies on nearly selective formation of 1-butene from ethylene. J. Organomet. Chem. 2021, 934, 121631. [Google Scholar] [CrossRef]
- Leone, G.; Losio, S.; Piovani, D.; Sommazzi, A.; Ricci, G. Living copolymerization of ethylene with 4-methyl-1-pentene by an α-diimine Ni(II)/Et2AlCl catalyst: Synthesis of diblock copolymers via sequential monomer addition. Polym. Chem. 2012, 3, 1987–1990. [Google Scholar] [CrossRef]















































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
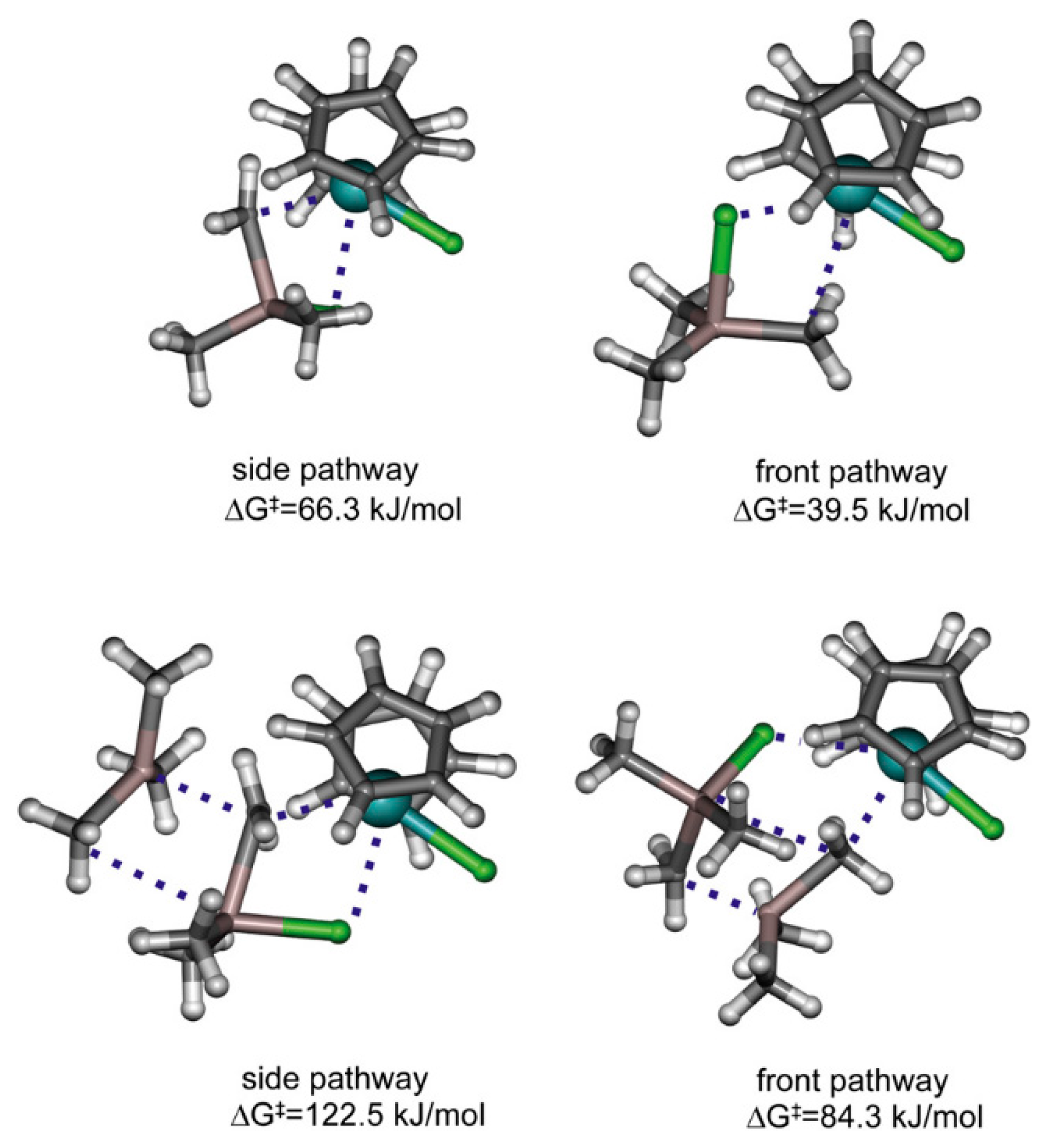{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
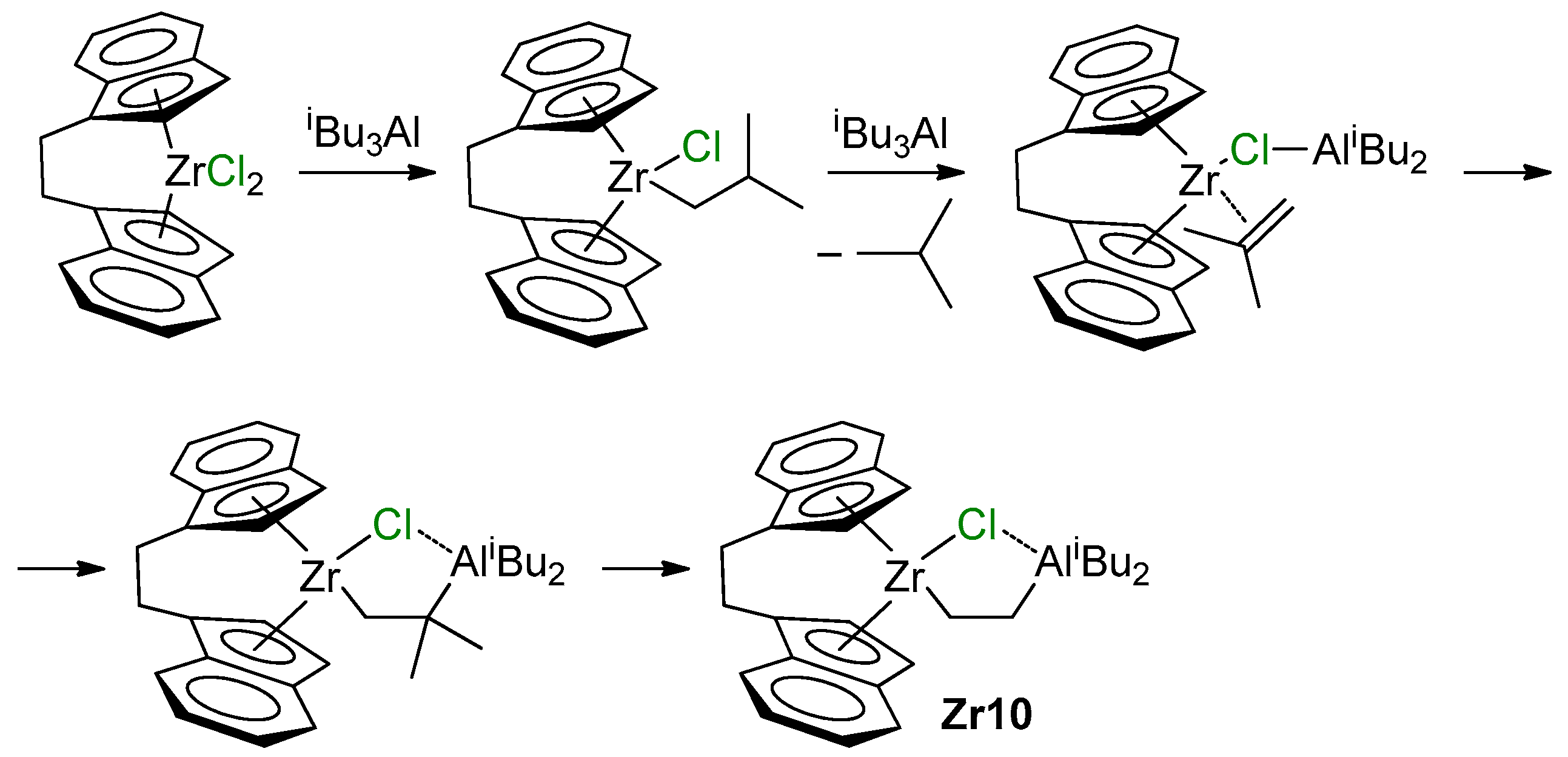{kind=link}
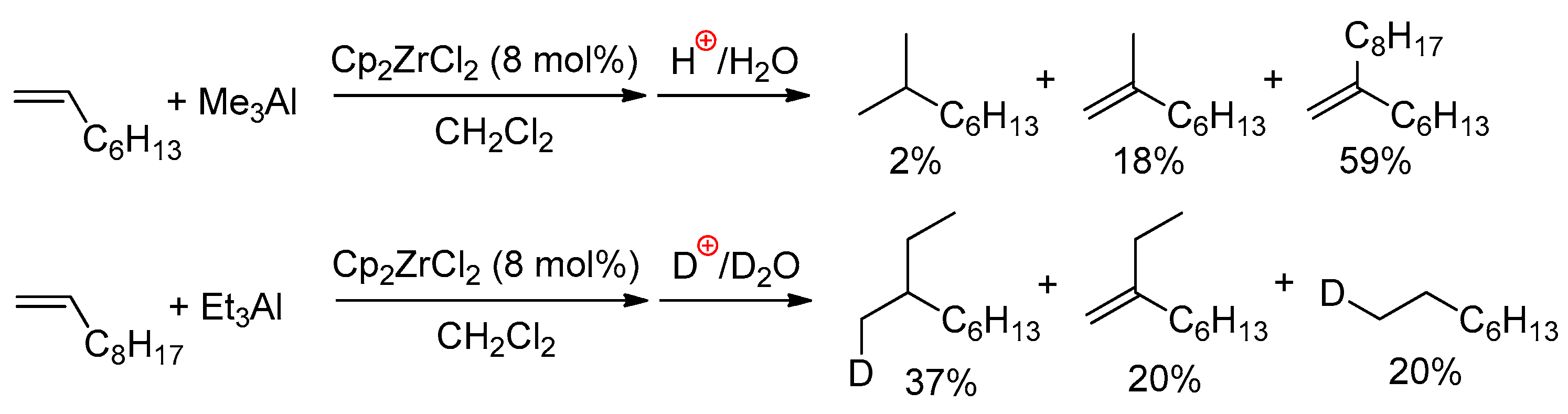{kind=link}
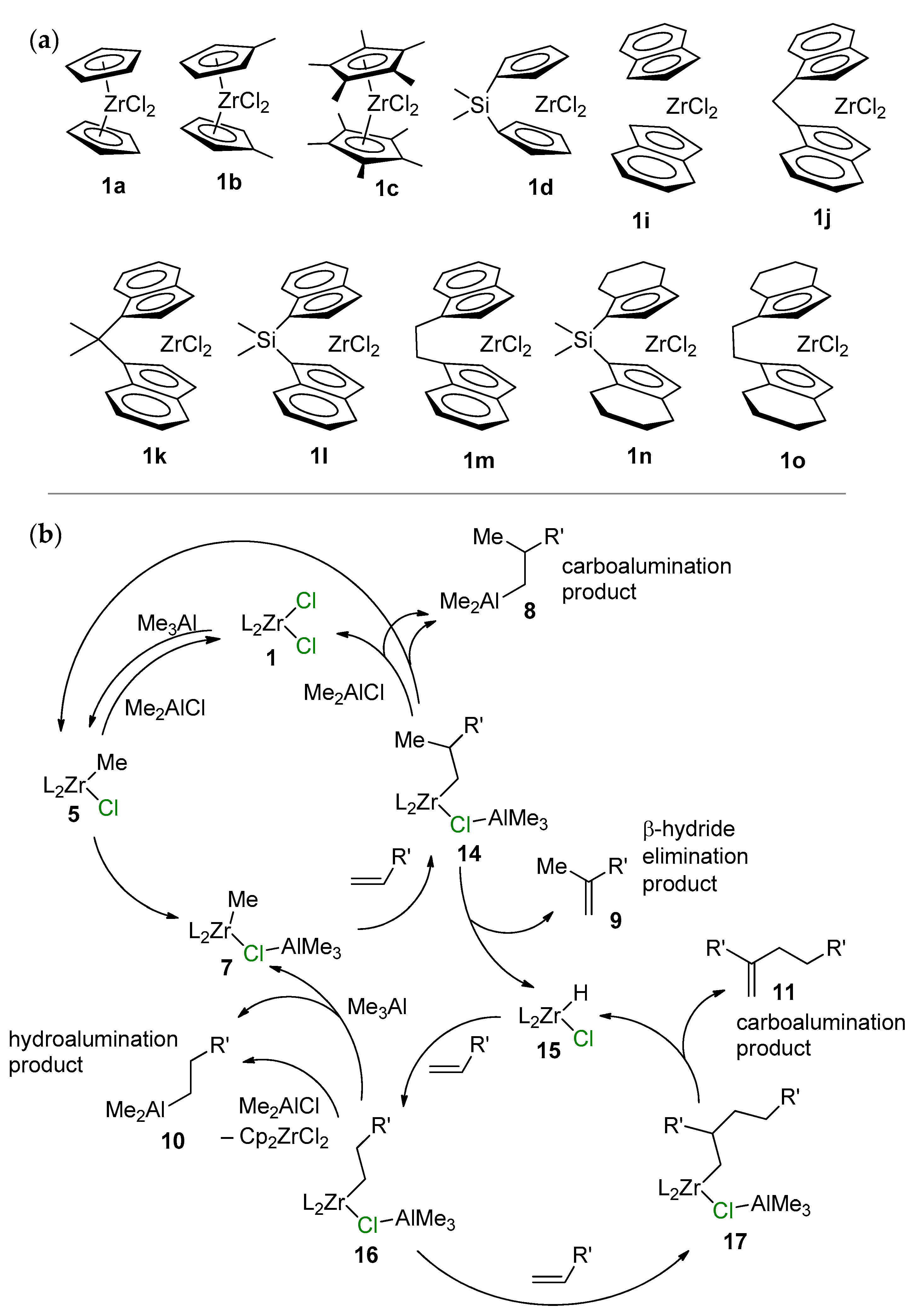{kind=link}
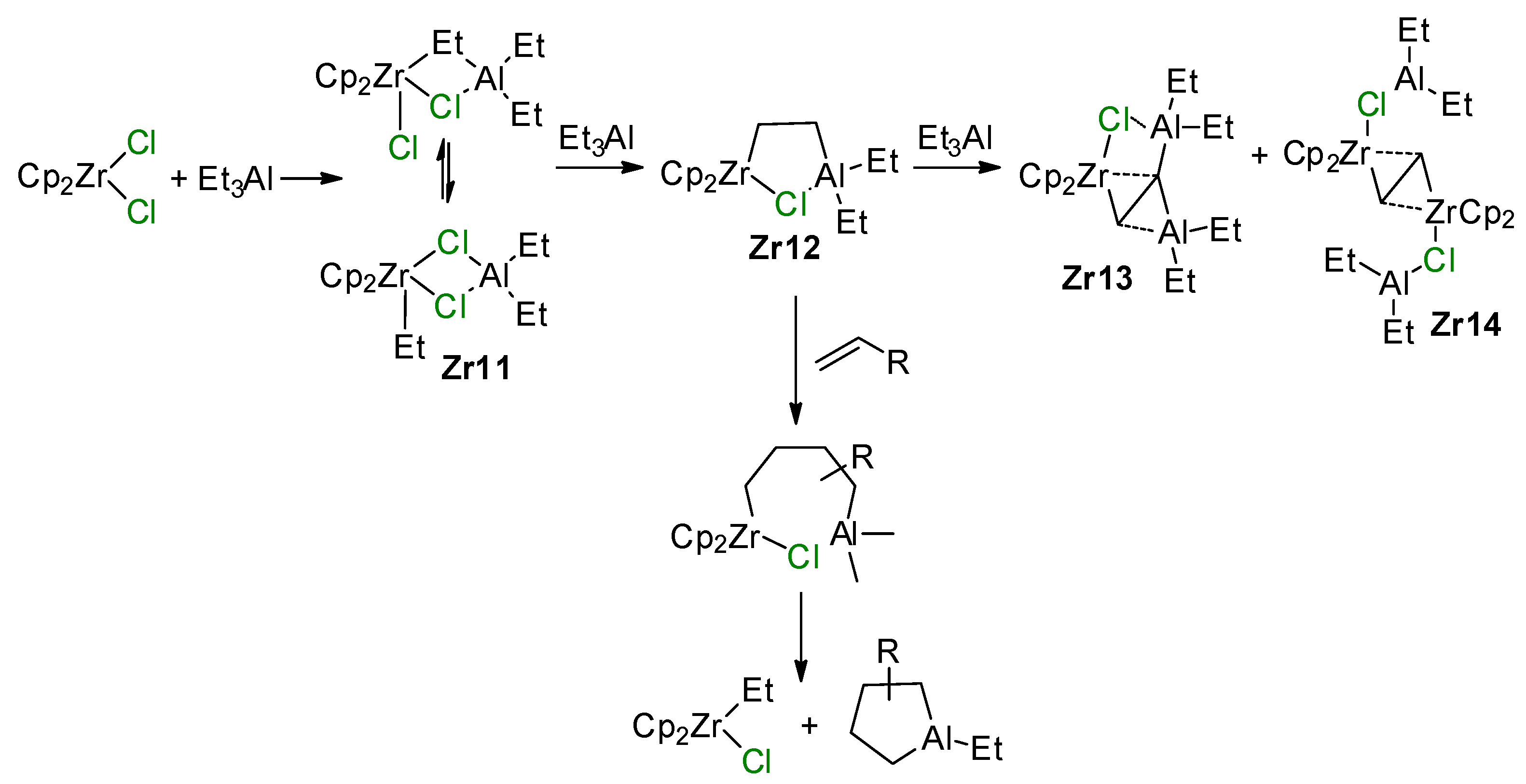{kind=link}
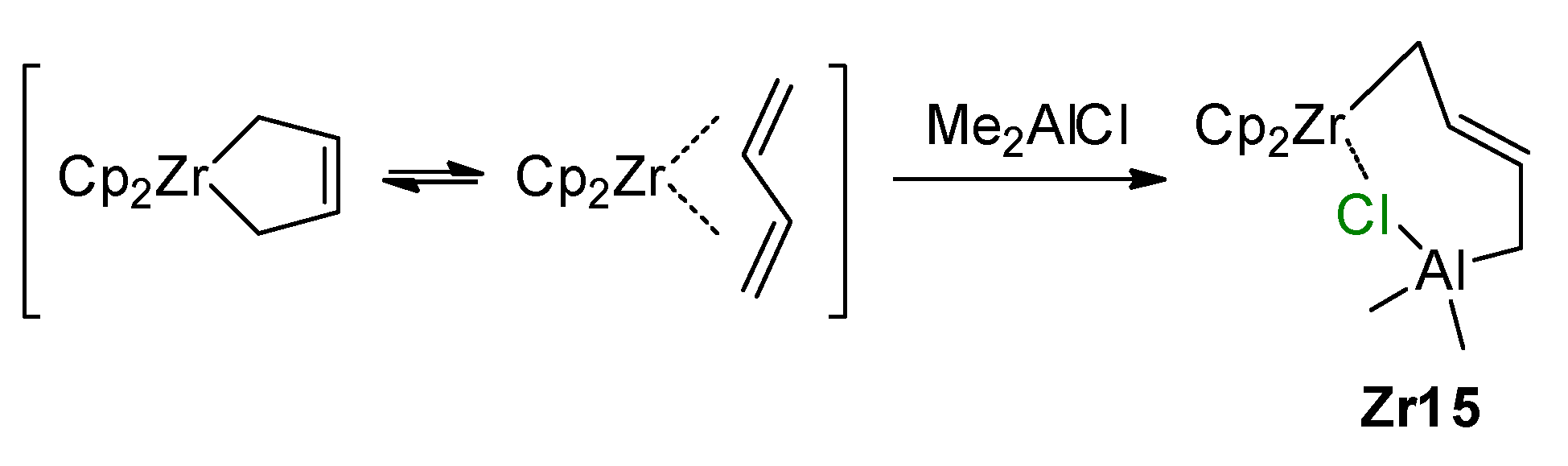{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L2ZrCl2 | Solvent | Conversion, % | Product Yield, % | |||
|---|---|---|---|---|---|---|
| 8 | 9 | 10 | 11 | |||
| 1a | CH2Cl2 | 92 | 3 | 14 | 7 | 68 |
| toluene | 69 | 3 | 21 | 7 | 38 | |
| 1b | CH2Cl2 | 84 | 11 | 14 | 7 | 52 |
| toluene | 39 | 9 | 9 | 9 | 12 | |
| 1c | CH2Cl2 | 68 | 53 | 8 | 7 | - |
| toluene | 44 | 15 | 14 | 14 | 1 | |
| 1d | CH2Cl2 | 24 | <1 | 5 | 5 | 13 |
| toluene | 13 | <1 | 4 | 6 | 2 | |
| 1i | CH2Cl2 | 89 | 31 | 19 | - | 39 |
| toluene | 70 | 38 | 14 | 10 | 8 | |
| 1j | CH2Cl2 | 0 | - | - | - | - |
| toluene | 11 | 9 | - | - | 2 | |
| 1k | CH2Cl2 | 2 | 0.6 | 0.3 | 0.7 | |
| toluene | 4 | <1 | 1.6 | 1 | 1 | |
| 1l | CH2Cl2 | 30 | 20 | 1 | <1 | 9 |
| toluene | 99 | 63 | 2 | - | 34 | |
| 1m | CH2Cl2 | 16 | 15 | - | <1 | <1 |
| toluene | 26 | 22 | - | 1 | 3 | |
| 1n | CH2Cl2 | 3 | 1 | - | - | - |
| toluene | 35 1 | 13 | - | - | - | |
| 1o | CH2Cl2 | 17 | 16 | <1 | <1 | - |
| toluene | 2 | 46 | 2 | <1 | 4 | |
| Cp2ZrMe2 | CH2Cl2 | 16 | 6 | 4 | 5 | 1 |
| toluene | 9 | 3 | 3 | <1 | <1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nifant’ev, I.E.; Salakhov, I.I.; Ivchenko, P.V. Transition Metal–(μ-Cl)–Aluminum Bonding in α-Olefin and Diene Chemistry. Molecules 2022, 27, 7164. https://doi.org/10.3390/molecules27217164
Nifant’ev IE, Salakhov II, Ivchenko PV. Transition Metal–(μ-Cl)–Aluminum Bonding in α-Olefin and Diene Chemistry. Molecules. 2022; 27(21):7164. https://doi.org/10.3390/molecules27217164
Chicago/Turabian StyleNifant’ev, Ilya E., Ildar I. Salakhov, and Pavel V. Ivchenko. 2022. "Transition Metal–(μ-Cl)–Aluminum Bonding in α-Olefin and Diene Chemistry" Molecules 27, no. 21: 7164. https://doi.org/10.3390/molecules27217164
APA StyleNifant’ev, I. E., Salakhov, I. I., & Ivchenko, P. V. (2022). Transition Metal–(μ-Cl)–Aluminum Bonding in α-Olefin and Diene Chemistry. Molecules, 27(21), 7164. https://doi.org/10.3390/molecules27217164