
Insights on Aggregation of Hen Egg-White Lysozyme from Raman Spectroscopy and MD Simulations

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
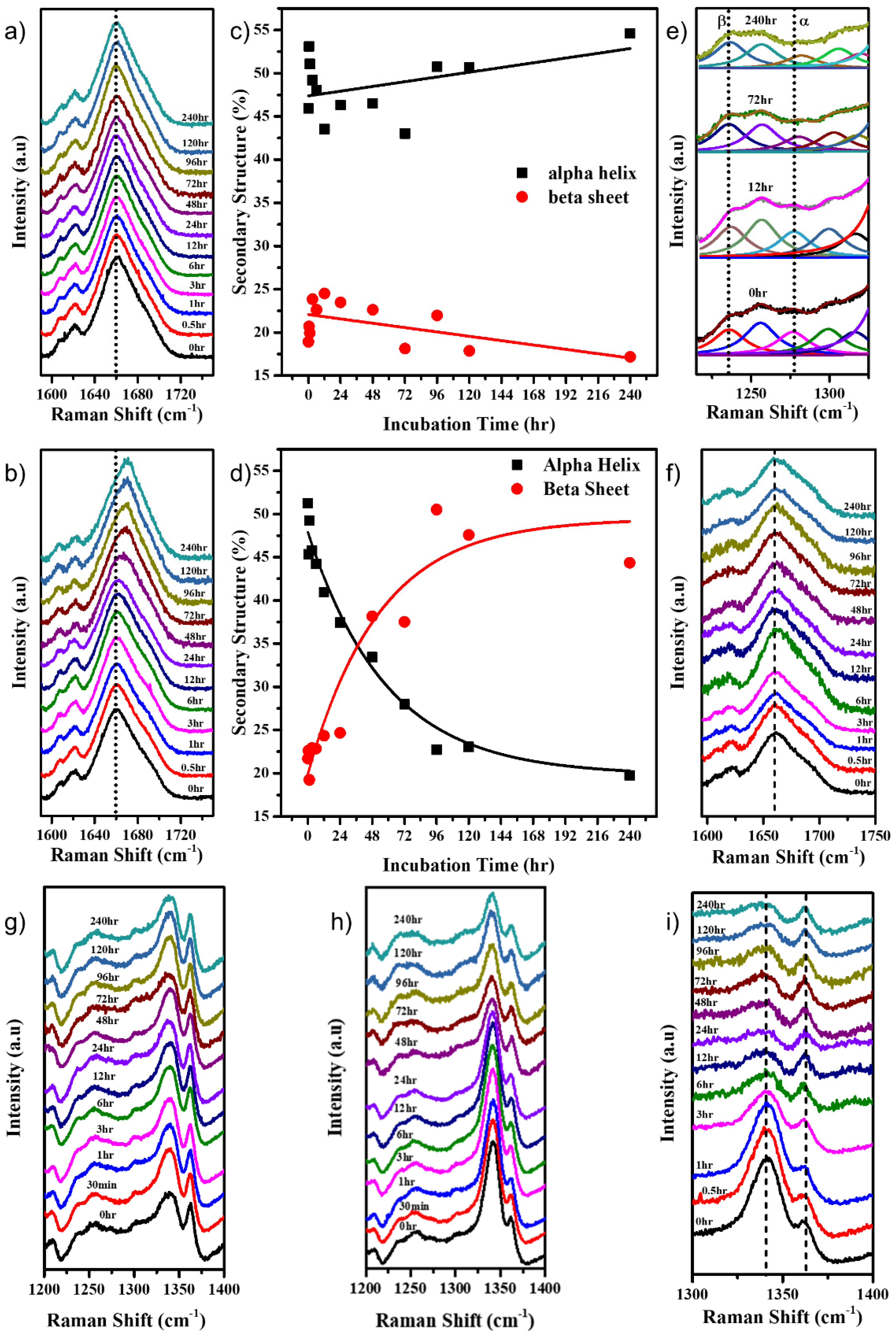
2. Results
2.1. Protein Unfolding
2.2. Protein Interactions and Aggregation
3. Discussion
3.1. Protein Unfolding
3.2. Protein Interactions and Aggregation
4. Materials and Methods
4.1. Materials
4.2. Lysozyme Sample Preparation and Incubation
4.3. Lysozyme Sample Preparation for Aggregation Inhibition Studies
4.4. Raman Spectroscopy
4.5. Raman Data Analysis: (Baseline Correction, Normalization and Curve Fitting)
4.6. UV-Vis Absorption Spectroscopy
4.7. DLS
4.8. Molecular Dynamics Simulations
4.8.1. Structure of the Hen Egg-White Lysozyme
4.8.2. Molecular Dynamic Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.M.; Yamamoto, A. Chapter 7—Macroautophagy of Aggregation-Prone Proteins in Neurodegenerative Disease. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2015; pp. 117–137. [Google Scholar]
- Cohen, F.E.; Prusiner, S.B. Pathologic conformations of prion proteins. Annu. Rev. Biochem. 1998, 67, 793–819. [Google Scholar] [CrossRef] [PubMed]
- Philo, J.; Arakawa, T. Mechanisms of Protein Aggregation. Curr. Pharm. Biotechnol. 2009, 10, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Bellotti, V.; Andreola, A.; Palladini, G.; Obici, L.; Casarini, S.; Perfetti, V. Protein Aggregation. Clin. Chem. Lab. Med. 2001, 39, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Housmans, J.A.J.; Wu, G.; Schymkowitz, J.; Rousseau, F. A guide to studying protein aggregation. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Arnaudov, L.N.; de Vries, R. Thermally Induced Fibrillar Aggregation of Hen Egg White Lysozyme. Biophys. J. 2005, 88, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Frieden, C. Protein aggregation processes: In search of the mechanism. Protein Sci. 2007, 16, 2334–2344. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hawkins, P.N.; Booth, D.R.; Vigushin, D.M.; Tennent, G.A.; Soutar, A.K.; Totty, N.; Nguyen, O.; Blake, C.C.F.; Terry, C.J.; et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 1993, 362, 553–557. [Google Scholar] [CrossRef]
- Valleix, S.; Drunat, S.; Philit, J.-B.; Adoue, D.; Piette, J.-C.; Droz, D.; MacGregor, B.; Canet, D.; Delpech, M.; Grateau, G. Hereditary renal amyloidosis caused by a new variant lysozyme W64R in a French family. Kidney Int. 2002, 61, 907–912. [Google Scholar] [CrossRef]
- Yazaki, M.; Farrell, S.A.; Benson, M.D. A novel lysozyme mutation Phe57Ile associated with hereditary renal amyloidosis. Kidney Int. 2003, 63, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, R.; Ravi, V.K.; Kumar, S.; Kumar, M.V.S.; Chandra, N. Lysozyme: A model protein for amyloid research. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: St. Salt Lake City, UT, USA, 2011; Volume 84, pp. 63–111. [Google Scholar]
- Dumetz, A.C.; Chockla, A.M.; Kaler, E.W.; Lenhoff, A.M. Effects of pH on protein–protein interactions and implications for protein phase behavior. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2008, 1784, 600–610. [Google Scholar] [CrossRef]
- Dolui, S.; Mondal, A.; Roy, A.; Pal, U.; Das, S.; Saha, A.; Maiti, N.C. Order, Disorder, and Reorder State of Lysozyme: Aggregation Mechanism by Raman Spectroscopy. J. Phys. Chem. B 2020, 124, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Ettah, I.; Ashton, L. Engaging with Raman Spectroscopy to Investigate Antibody Aggregation. Antibodies 2018, 7, 24. [Google Scholar] [CrossRef]
- Kurouski, D.; Van Duyne, R.P.; Lednev, I.K. Exploring the structure and formation mechanism of amyloid fibrils by Raman spectroscopy: A review. Analyst 2015, 140, 4967–4980. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.R.H.; Wilkins, D.K.; Chung, E.W.; Pitkeathly, M.C.; Chamberlain, A.K.; Zurdo, J.; Robinson, C.V.; Dobson, C.M. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain 11Edited by P. E. Wright. J. Mol. Biol. 2000, 300, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Sophianopoulos, A.J.; Van Holde, K.E. Evidence for Dimerization of Lysozyme in Alkaline Solution. J. Biol. Chem. 1961, 236, PC82–PC83. [Google Scholar] [CrossRef]
- Homchaudhuri, L.; Kumar, S.; Swaminathan, R. Slow aggregation of lysozyme in alkaline pH monitored in real time employing the fluorescence anisotropy of covalently labelled dansyl probe. FEBS Lett. 2006, 580, 2097–2101. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.K.; Swain, T.; Chandra, N.; Swaminathan, R. On the Characterization of Intermediates in the Isodesmic Aggregation Pathway of Hen Lysozyme at Alkaline pH. PLoS ONE 2014, 9, e87256. [Google Scholar] [CrossRef]
- Lewis, E.N.; Qi, W.; Kidder, L.H.; Amin, S.; Kenyon, S.M.; Blake, S. Combined Dynamic Light Scattering and Raman Spectroscopy Approach for Characterizing the Aggregation of Therapeutic Proteins. Molecules 2014, 19, 20888. [Google Scholar] [CrossRef] [PubMed]
- Bagriantsev, S.; Kushnirov, V.; Liebman, S. Analysis of Amyloid Aggregates Using Agarose Gel Electrophoresis. Methods Enzymol. 2006, 412, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Adawy, A.; Groves, M.R. The Use of Size Exclusion Chromatography to Monitor Protein Self-Assembly. Crystals 2017, 7, 331. [Google Scholar] [CrossRef]
- Fekete, S.; Beck, A.; Veuthey, J.-L.; Guillarme, D. Theory and practice of size exclusion chromatography for the analysis of protein aggregates. J. Pharm. Biomed. Anal. 2014, 101, 161–173. [Google Scholar] [CrossRef]
- Hong, P.; Koza, S.; Bouvier, E.S.P. Size-Exclusion Chromatography for the Analysis of Protein Biotherapeutics and their Aggregates. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2923–2950. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Chen, N.; Fan, W.; Li, M.; Zhou, X.; Liu, S. Double-edged effects of aluminium ions on amyloid fibrillation of hen egg-white lysozyme. Int. J. Biol. Macromol. 2019, 132, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim-Habibi, A.; Morshedi, D.; Rezaei-Ghaleh, N.; Sabbaghian, M.; Nemat-Gorgani, M. Protein-Protein interactions leading to aggregation: Perspectives on mechanism, significance and control. J. Iran. Chem. Soc. 2010, 7, 521–544. [Google Scholar] [CrossRef]
- Karamanos, T.K.; Kalverda, A.P.; Thompson, G.S.; Radford, S.E. Mechanisms of amyloid formation revealed by solution NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2015, 88–89, 86–104. [Google Scholar] [CrossRef] [PubMed]
- Bronsoms, S.; Trejo, S.A. Applications of Mass Spectrometry to the Study of Protein Aggregation. In Insoluble Proteins. Methods in Molecular Biology (Methods and Protocols); García-Fruitós, E., Ed.; Humana Press: New York, NY, USA, 2015; Volume 1258, pp. 331–345. [Google Scholar]
- Chaari, A.; Fahy, C.; Chevillot-Biraud, A.; Rholam, M. Insights into Kinetics of Agitation-Induced Aggregation of Hen Lysozyme under Heat and Acidic Conditions from Various Spectroscopic Methods. PLoS ONE 2015, 10, e0142095. [Google Scholar] [CrossRef] [PubMed]
- Eugenia, P.; Evgenia, P.; Evgenii, M.; Nikolai, K.; Brazhe, N.; Irina, T.; Fiordoro, S.; Claudio, N. Raman Spectroscopy of Protein Crystal Nucleation and Growth. Am. J. Biochem. Biotechnol. 2014, 10, 202–207. [Google Scholar] [CrossRef][Green Version]
- Niaura, G. Raman Spectroscopy in Analysis of Biomolecules. Encycl. Anal. Chem. 2014, 1–34. [Google Scholar] [CrossRef]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. Atomic-level description of ubiquitin folding. Proc. Natl. Acad. Sci. USA 2013, 110, 5915. [Google Scholar] [CrossRef] [PubMed]
- Prigozhin, M.B.; Gruebele, M. Microsecond folding experiments and simulations: A match is made. Phys. Chem. Chem. Phys. 2013, 15, 3372–3388. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Kukol, A. Molecular Dynamics Simulations. In Molecular Modeling of Proteins. Methods in molecular biology (Methods and Protocols); Humana Press/Springer: Berlin, Germany, 2015; Volume 1215, pp. 3–26. [Google Scholar]
- Sultan, M.M.; Denny, R.A.; Unwalla, R.; Lovering, F.; Pande, V.S. Millisecond dynamics of BTK reveal kinome-wide conformational plasticity within the apo kinase domain. Sci. Rep. 2017, 7, 15604. [Google Scholar] [CrossRef]
- Shaw, D.E.; Dror, R.O.; Salmon, J.K.; Grossman, J.P.; Mackenzie, K.M.; Bank, J.A.; Young, C.; Deneroff, M.M.; Batson, B.; Bowers, K.J.; et al. Millisecond-scale molecular dynamics simulations on Anton. In Proceedings of the Conference on High Performance Computing Networking, Oregon, Poland, 14–20 November 2009; pp. 1–11. [Google Scholar]
- Redler, R.L.; Shirvanyants, D.; Dagliyan, O.; Ding, F.; Kim, D.N.; Kota, P.; Proctor, E.A.; Ramachandran, S.; Tandon, A.; Dokholyan, N.V. Computational approaches to understanding protein aggregation in neurodegeneration. J. Mol. Cell Biol. 2014, 6, 104–115. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Mach, H.; Middaugh, C.R.; Denslow, N. Determining the identity and purity of recombinant proteins by UV absorption spectroscopy. Curr. Protoc. Protein Sci. 1995, 1, 7.2.1–7.2.21. [Google Scholar] [CrossRef]
- Filik, J.; Stone, N. Drop coating deposition Raman spectroscopy of protein mixtures. Analyst 2007, 132, 544–550. [Google Scholar] [CrossRef]
- Zhang, D.; Xie, Y.; Mrozek, M.F.; Ortiz, C.; Davisson, V.J.; Ben-Amotz, D. Raman Detection of Proteomic Analytes. Anal. Chem. 2003, 75, 5703–5709. [Google Scholar] [CrossRef]
- Day, R.; Bennion, B.J.; Ham, S.; Daggett, V. Increasing Temperature Accelerates Protein Unfolding Without Changing the Pathway of Unfolding. J. Mol. Biol. 2002, 322, 189–203. [Google Scholar] [CrossRef]
- Florence, T.M. Degradation of protein disulphide bonds in dilute alkali. Biochem. J. 1980, 189, 507–520. [Google Scholar] [CrossRef]
- Eleftheriou, M.; Germain, R.S.; Royyuru, A.K.; Zhou, R. Thermal Denaturing of Mutant Lysozyme with Both the OPLSAA and the CHARMM Force Fields. J. Am. Chem. Soc. 2006, 128, 13388–13395. [Google Scholar] [CrossRef]
- Kazmirski, S.L.; Daggett, V. Non-native interactions in protein folding intermediates: Molecular dynamics simulations of hen lysozyme1 1Edited by B. Honig. J. Mol. Biol. 1998, 284, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Meersman, F.; Atilgan, C.; Miles, A.J.; Bader, R.; Shang, W.; Matagne, A.; Wallace, B.A.; Koch, M.H.J. Consistent Picture of the Reversible Thermal Unfolding of Hen Egg-White Lysozyme from Experiment and Molecular Dynamics. Biophys. J. 2010, 99, 2255–2263. [Google Scholar] [CrossRef]
- Uversky, N.V. Amyloidogenesis of Natively Unfolded Proteins. Curr. Alzheimer Res. 2008, 5, 260–287. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Fan, W.; Chen, N.; Li, M.; Zhou, X.; Liu, S. Amyloid formation kinetics of hen egg white lysozyme under heat and acidic conditions revealed by Raman spectroscopy. J. Raman Spectrosc. 2019, 50, 629–640. [Google Scholar] [CrossRef]
- Smythe, C.V. The reaction of iodoacetate and of iodoacetamide with various sulfhydryl groups, with urease, and with yeast preparations. J. Biol. Chem. 1936, 114, 601–612. [Google Scholar] [CrossRef]
- Ravi, V.K.; Goel, M.; Kotamarthi, H.C.; Ainavarapu, S.R.K.; Swaminathan, R. Preventing Disulfide Bond Formation Weakens Non-Covalent Forces among Lysozyme Aggregates. PLoS ONE 2014, 9, e87012. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Lin, Y.; Yagi, H.; Lee, Y.-H.; Kitayama, H.; Sakurai, K.; So, M.; Ogi, H.; Naiki, H.; Goto, Y. Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc. Natl. Acad. Sci. USA 2012, 109, 14446–14451. [Google Scholar] [CrossRef]
- Halling, P.J. Proteins: Structures and molecular properties (2nd edition). J. Chem. Technol. Biotechnol. 1995, 62, 105. [Google Scholar] [CrossRef]
- Velev, O.D.; Kaler, E.W.; Lenhoff, A.M. Protein Interactions in Solution Characterized by Light and Neutron Scattering: Comparison of Lysozyme and Chymotrypsinogen. Biophys. J. 1998, 75, 2682–2697. [Google Scholar] [CrossRef]
- Verheul, M.; Roefs, S.P.F.M.; de Kruif, K.G. Kinetics of Heat-Induced Aggregation of β-Lactoglobulin. J. Agric. Food Chem. 1998, 46, 896–903. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef]
- Kumar, S.; Ravi Vijay, K.; Swaminathan, R. How do surfactants and DTT affect the size, dynamics, activity and growth of soluble lysozyme aggregates? Biochem. J. 2008, 415, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ravi, V.K.; Swaminathan, R. Suppression of lysozyme aggregation at alkaline pH by tri-N-acetylchitotriose. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2009, 1794, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Janek, T.; Rodrigues, L.R.; Gudiña, E.J.; Burger, J. Synergistic effect of hen egg white lysozyme and lysosomotropic surfactants on cell viability and membrane permeability. Colloids Surf. B Biointerfaces 2020, 185, 110598. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, J.C.; Artymiuk, P.J.; Phillips, D.C. Refinement of an enzyme complex with inhibitor bound at partial occupancy: Hen egg-white lysozyme and tri-N-acetylchitotriose at 1.75 Å resolution. J. Mol. Biol. 1992, 224, 613–628. [Google Scholar] [CrossRef]
- Schrödinger Release; Version 2022-2; Core Hopping: New York, NY, USA, 2021.
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Schrödinger Release; Version 2021; Desmond: New York, NY, USA, 2021.
- Chuang, G.-Y.; Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. DARS (Decoys As the Reference State) Potentials for Protein-Protein Docking. Biophys. J. 2008, 95, 4217–4227. [Google Scholar] [CrossRef]
- Fernandez-Recio, J.; Sternberg, M.J.E. The 4th meeting on the Critical Assessment of PRedicted Interaction (CAPRI) held at the Mare Nostrum, Barcelona. Proteins-Struct. Funct. Bioinform. 2010, 78, 3065–3066. [Google Scholar] [CrossRef]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef]
- Wang, J.; Hou, T.; Xu, X. Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr. Comput.-Aided Drug Des. 2006, 2, 287–306. [Google Scholar] [CrossRef]
- Teizo Kitagawa, S.H. Raman Spectroscopy of Proteins. In Handbook of Vibrational Spectroscopy; Peter Griffiths, J.M.C., Ed.; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Talari, A.C.S.; Movasaghi, Z.; Rehman, S.; Rehman, I.u. Raman Spectroscopy of Biological Tissues. Appl. Spectrosc. Rev. 2015, 50, 46–111. [Google Scholar] [CrossRef]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar] [CrossRef]
- Wang, C.-H.; Huang, C.-C.; Lin, L.-L.; Chen, W. The effect of disulfide bonds on protein folding, unfolding, and misfolding investigated by FT–Raman spectroscopy. J. Raman Spectrosc. 2016, 47, 940–947. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chalapathi, D.; Kumar, A.; Behera, P.; Sathi, S.N.; Swaminathan, R.; Narayana, C. Insights on Aggregation of Hen Egg-White Lysozyme from Raman Spectroscopy and MD Simulations. Molecules 2022, 27, 7122. https://doi.org/10.3390/molecules27207122
Chalapathi D, Kumar A, Behera P, Sathi SN, Swaminathan R, Narayana C. Insights on Aggregation of Hen Egg-White Lysozyme from Raman Spectroscopy and MD Simulations. Molecules. 2022; 27(20):7122. https://doi.org/10.3390/molecules27207122
Chicago/Turabian StyleChalapathi, Divya, Amrendra Kumar, Pratik Behera, Shijulal Nelson Sathi, Rajaram Swaminathan, and Chandrabhas Narayana. 2022. "Insights on Aggregation of Hen Egg-White Lysozyme from Raman Spectroscopy and MD Simulations" Molecules 27, no. 20: 7122. https://doi.org/10.3390/molecules27207122
APA StyleChalapathi, D., Kumar, A., Behera, P., Sathi, S. N., Swaminathan, R., & Narayana, C. (2022). Insights on Aggregation of Hen Egg-White Lysozyme from Raman Spectroscopy and MD Simulations. Molecules, 27(20), 7122. https://doi.org/10.3390/molecules27207122