
Detailed Structural Characterization of Oxidized Sucrose and Its Application in the Fully Carbohydrate-Based Preparation of a Hydrogel from Carboxymethyl Chitosan


Abstract
:1. Introduction
2. Results and Discussion
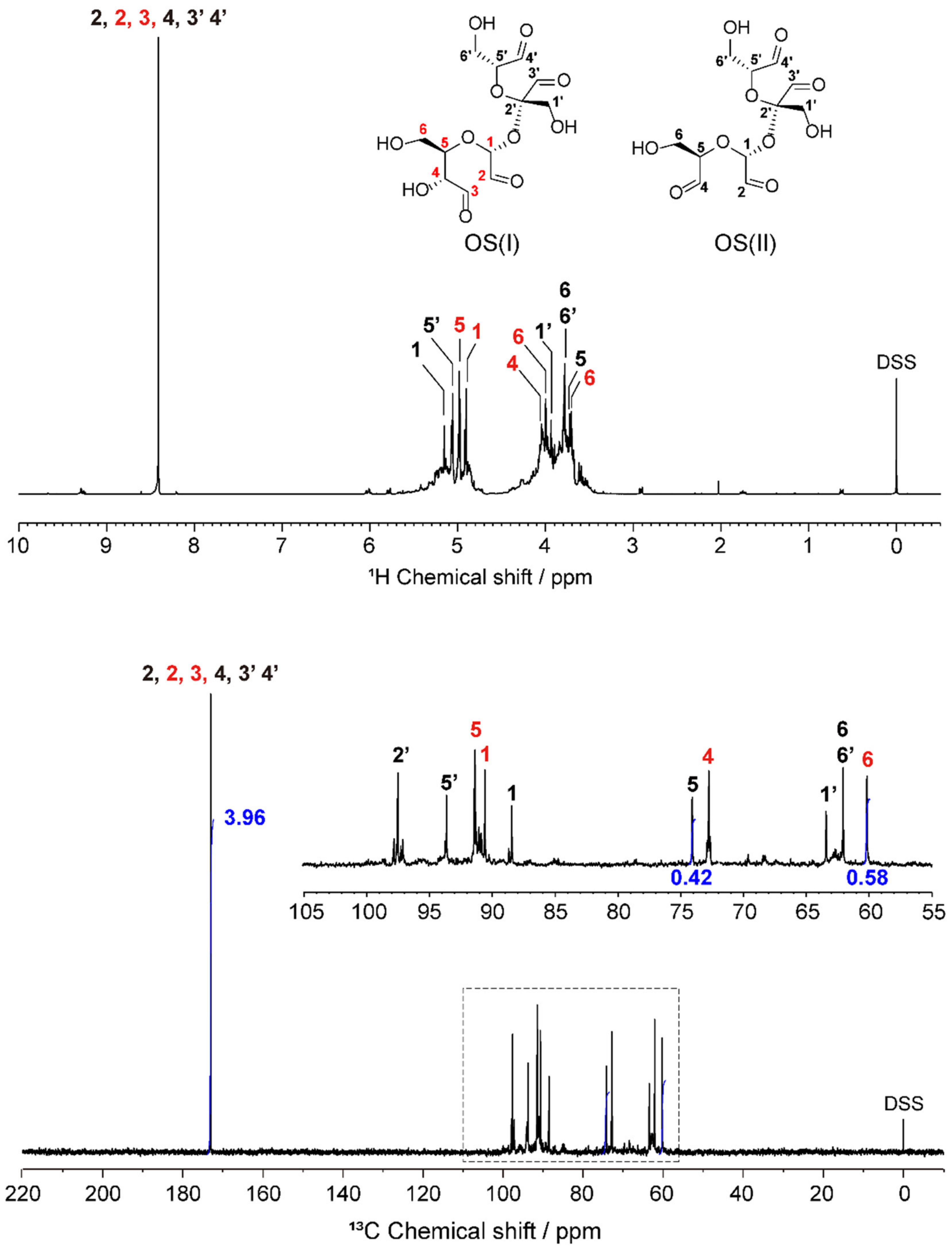
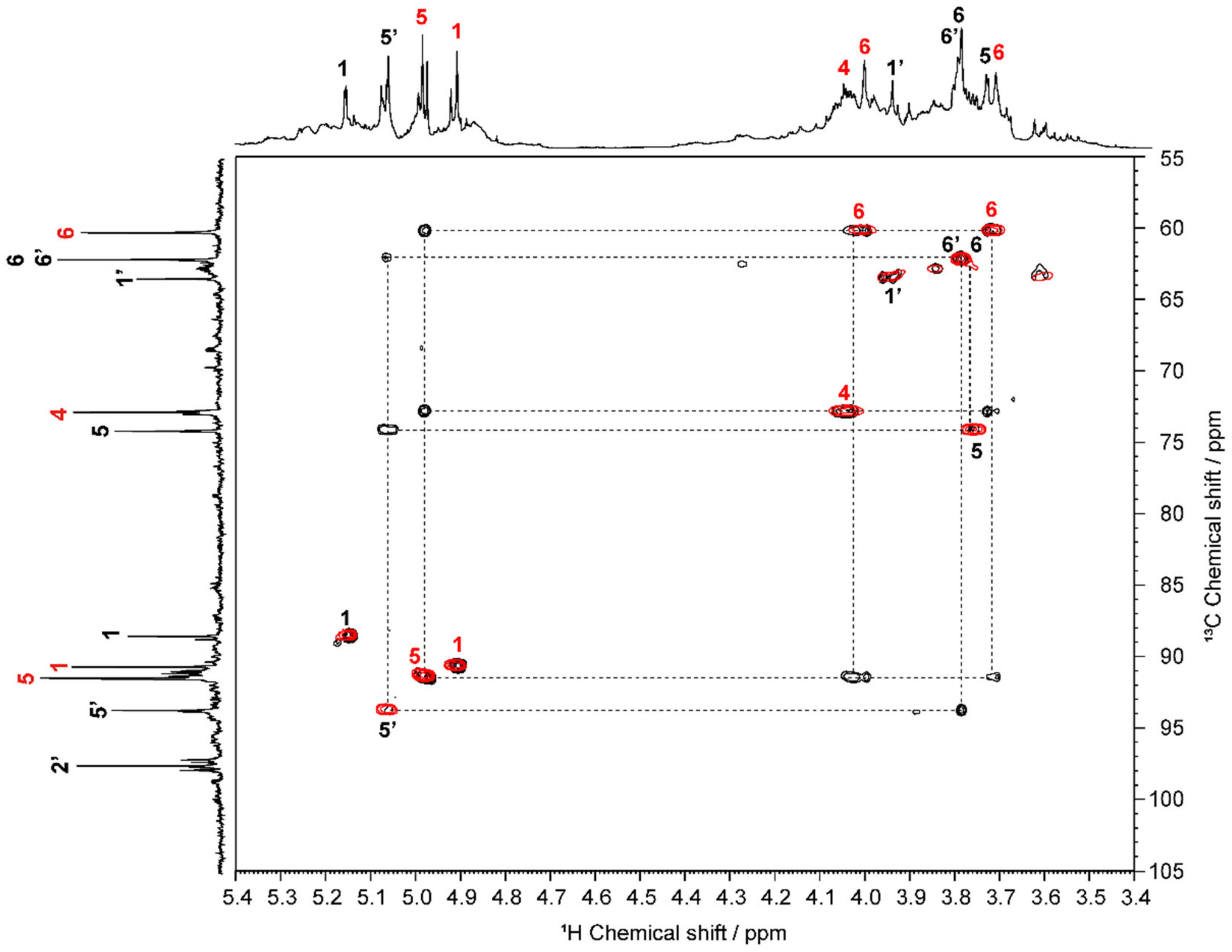
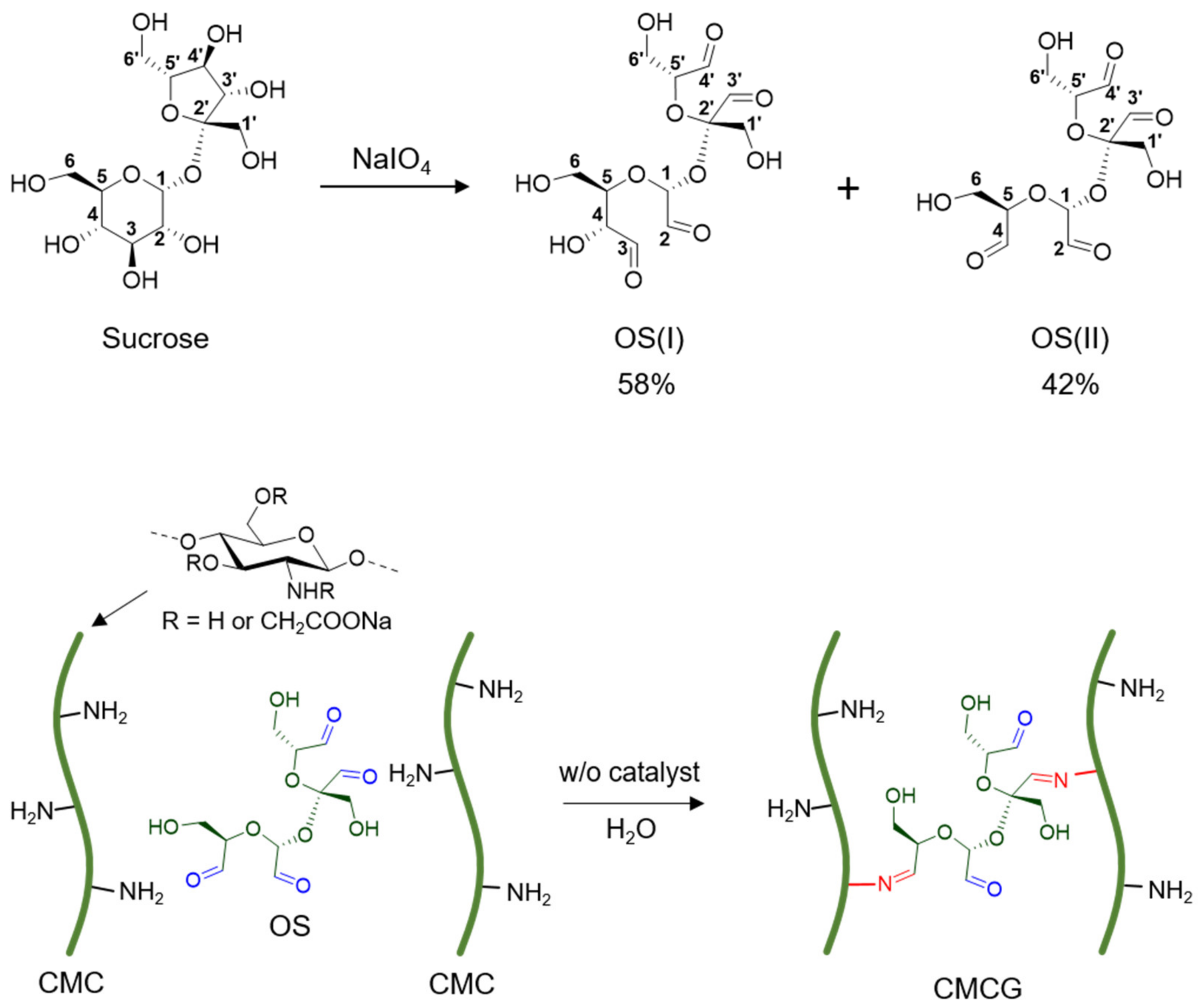
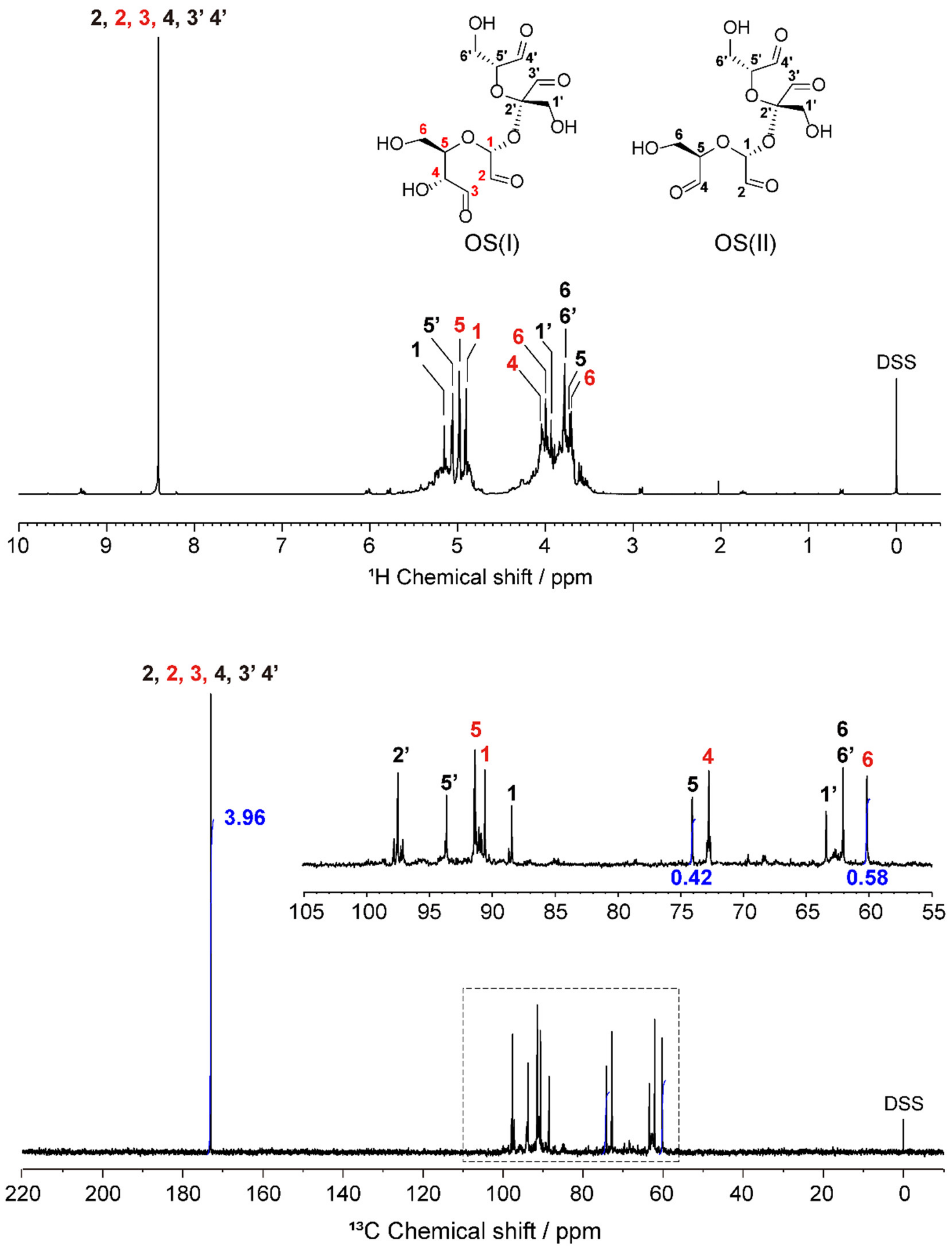
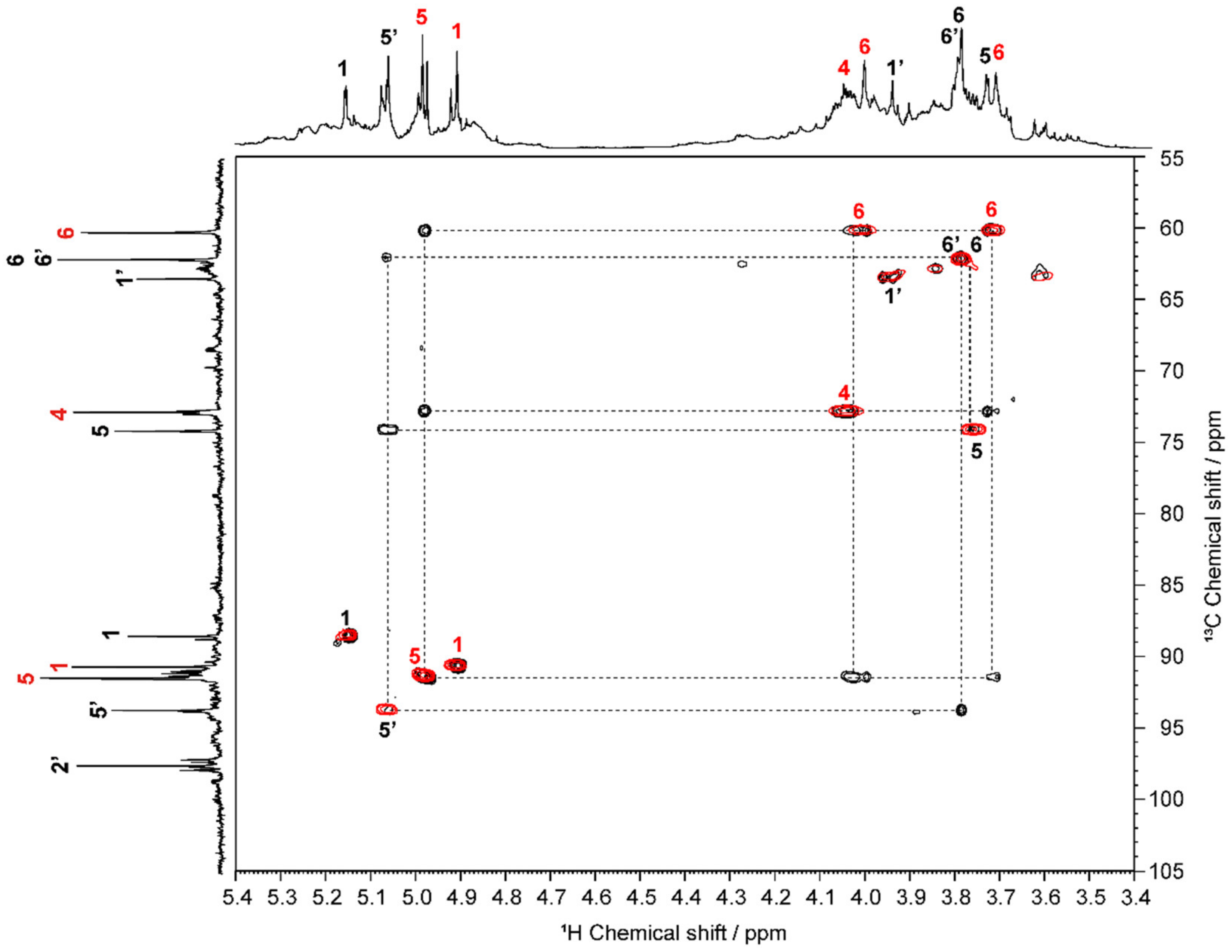
2.1. Structural Characterization of OS
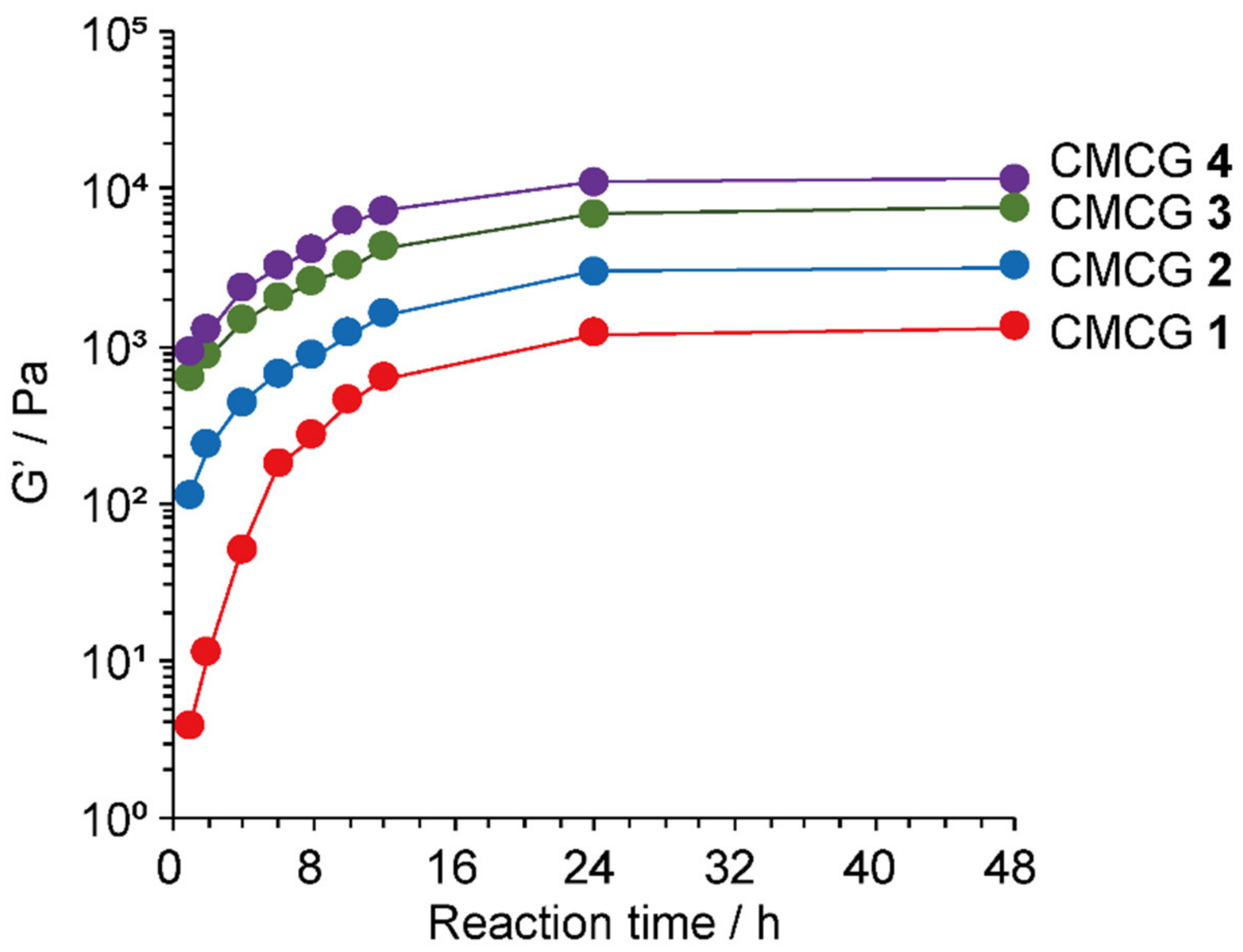
2.2. Preparation of CMC Hydrogels Using OS
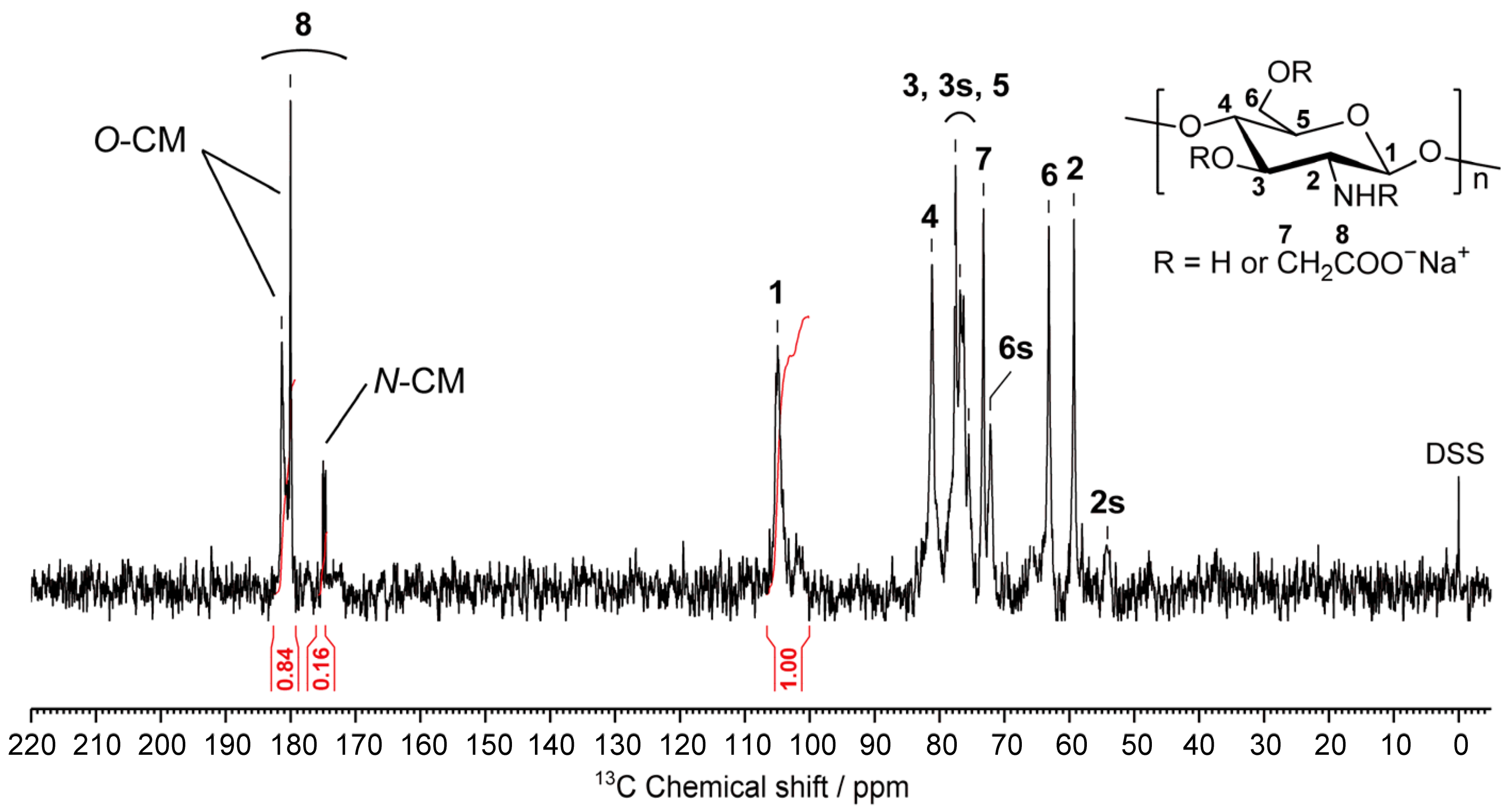
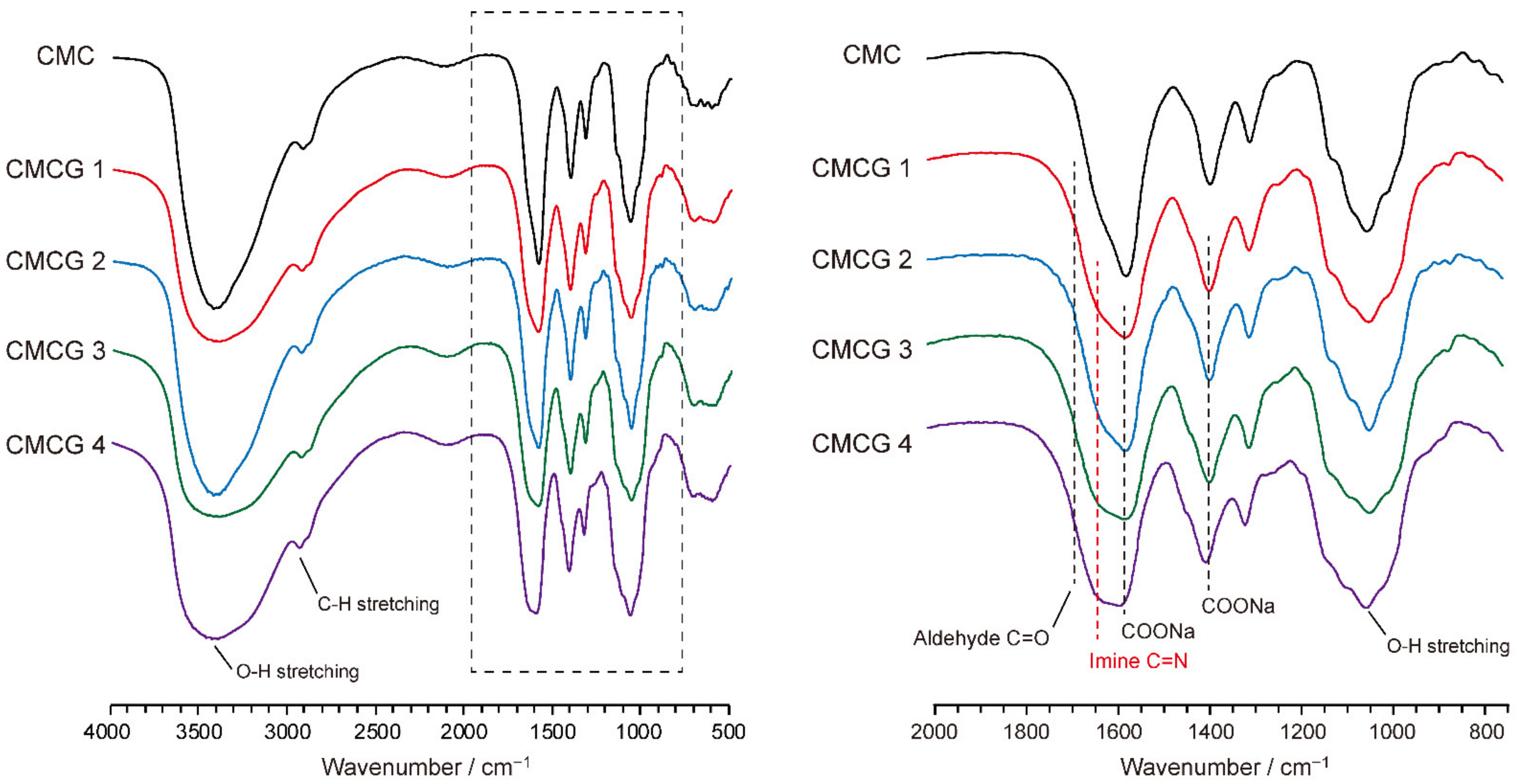
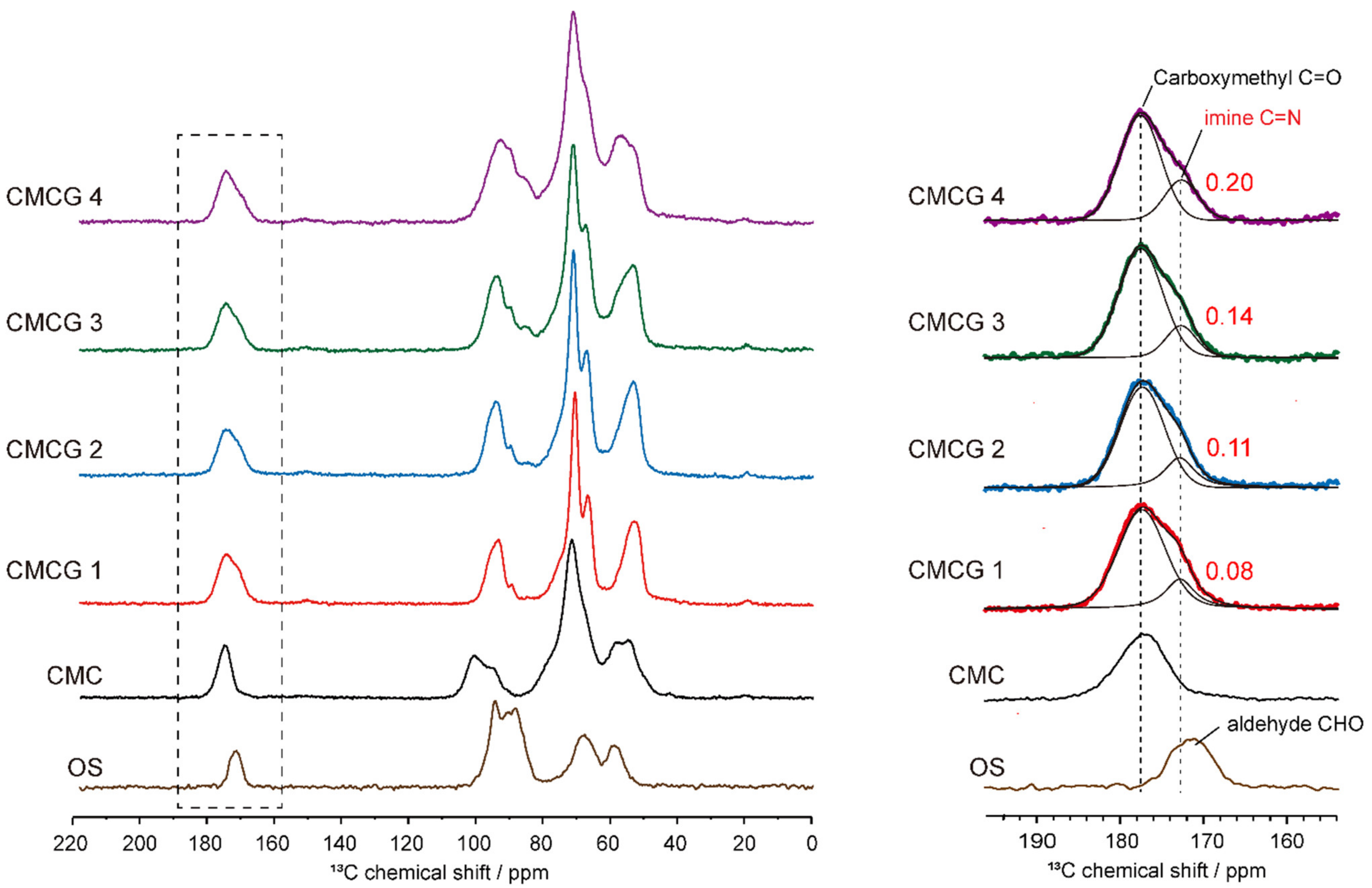
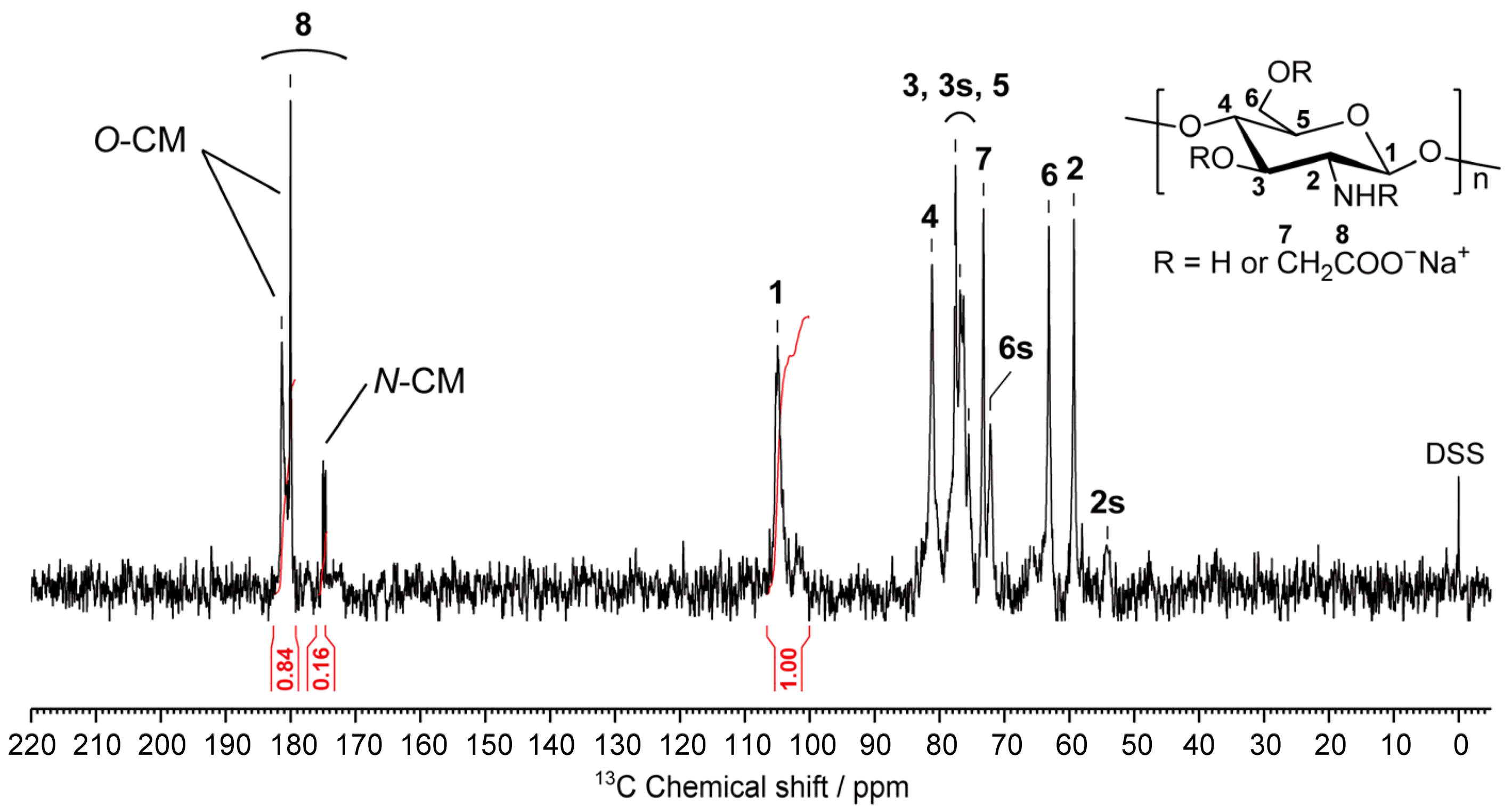
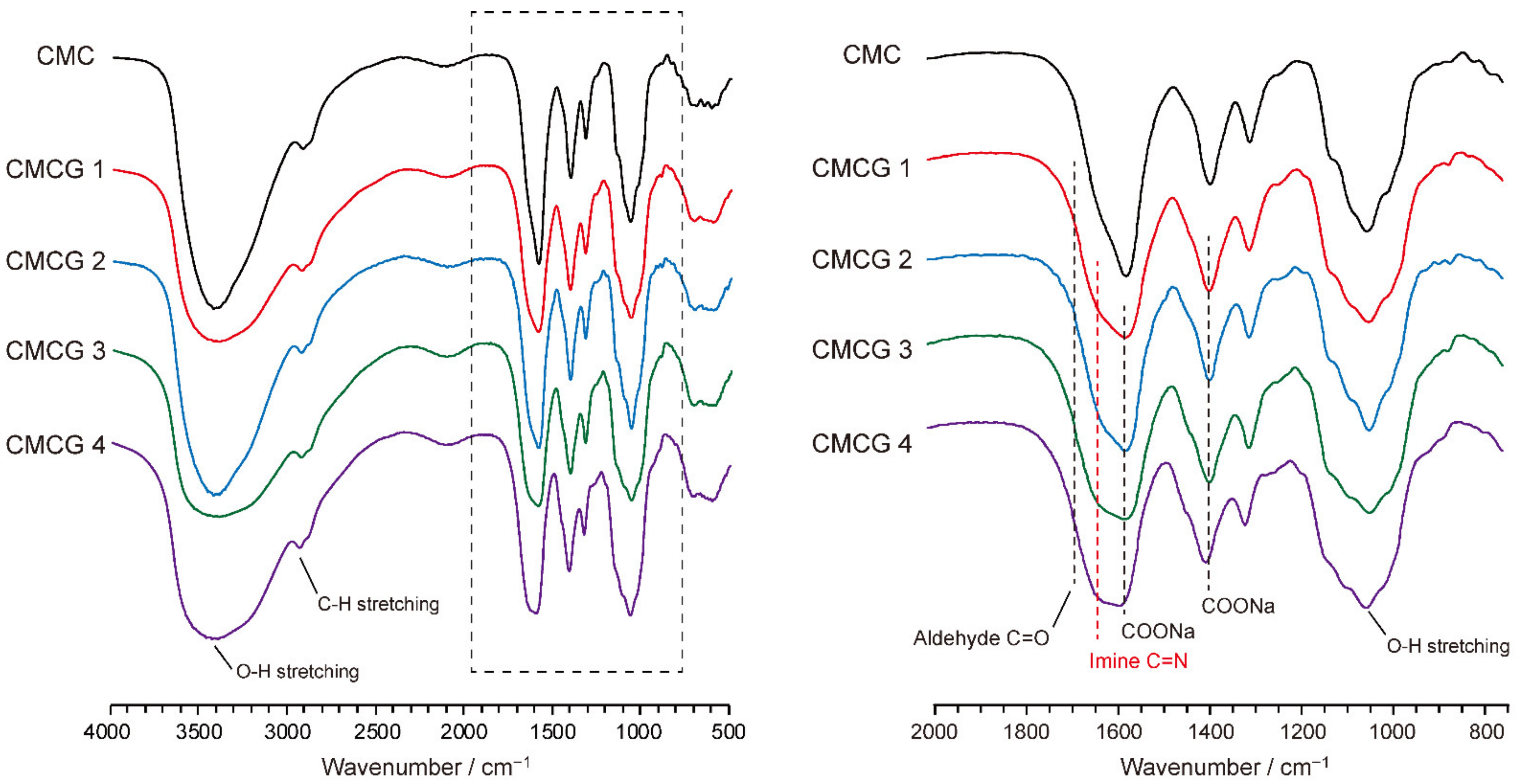
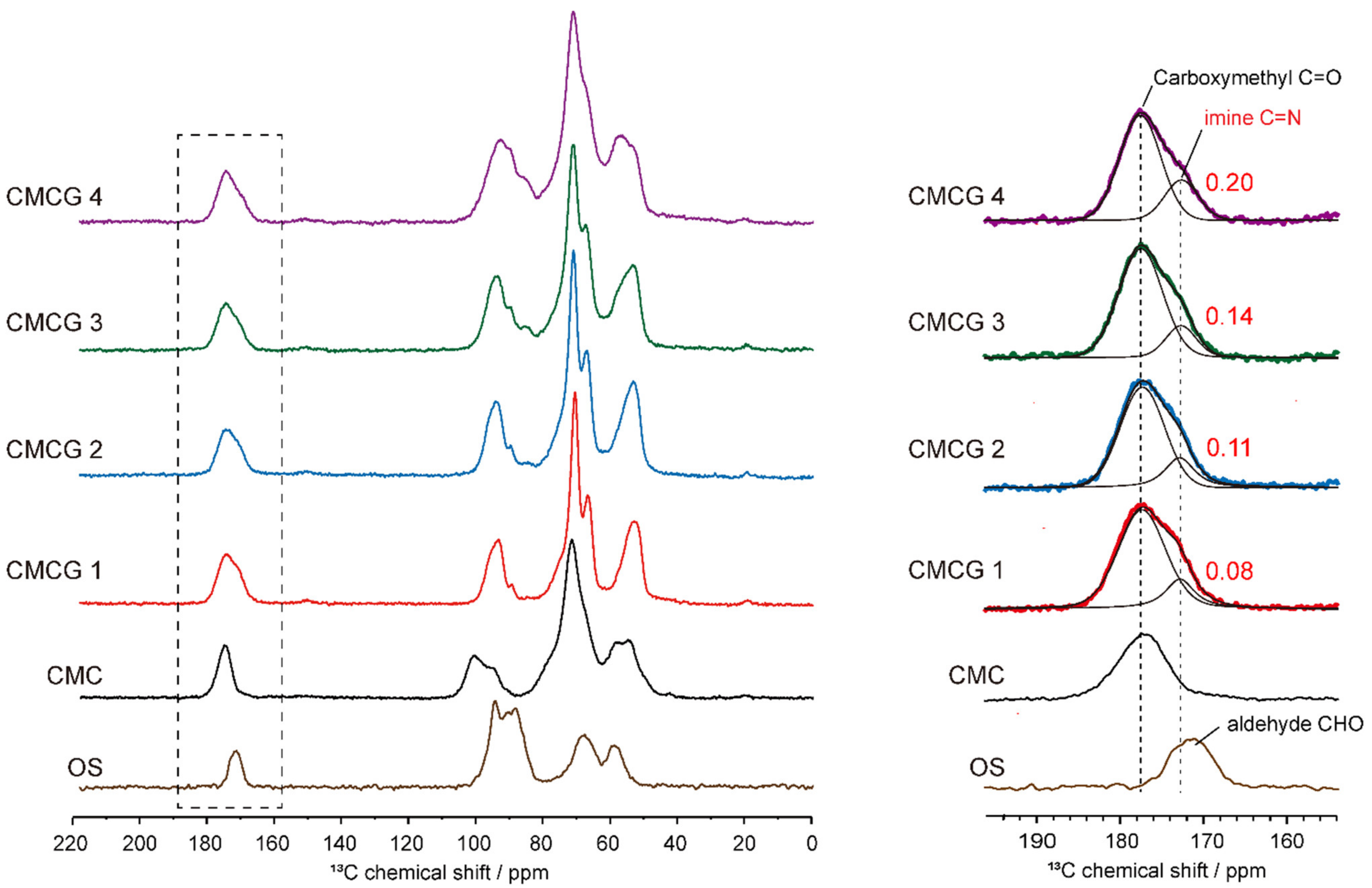
2.3. Structural Characterization of the Prepared CMCG Specimens
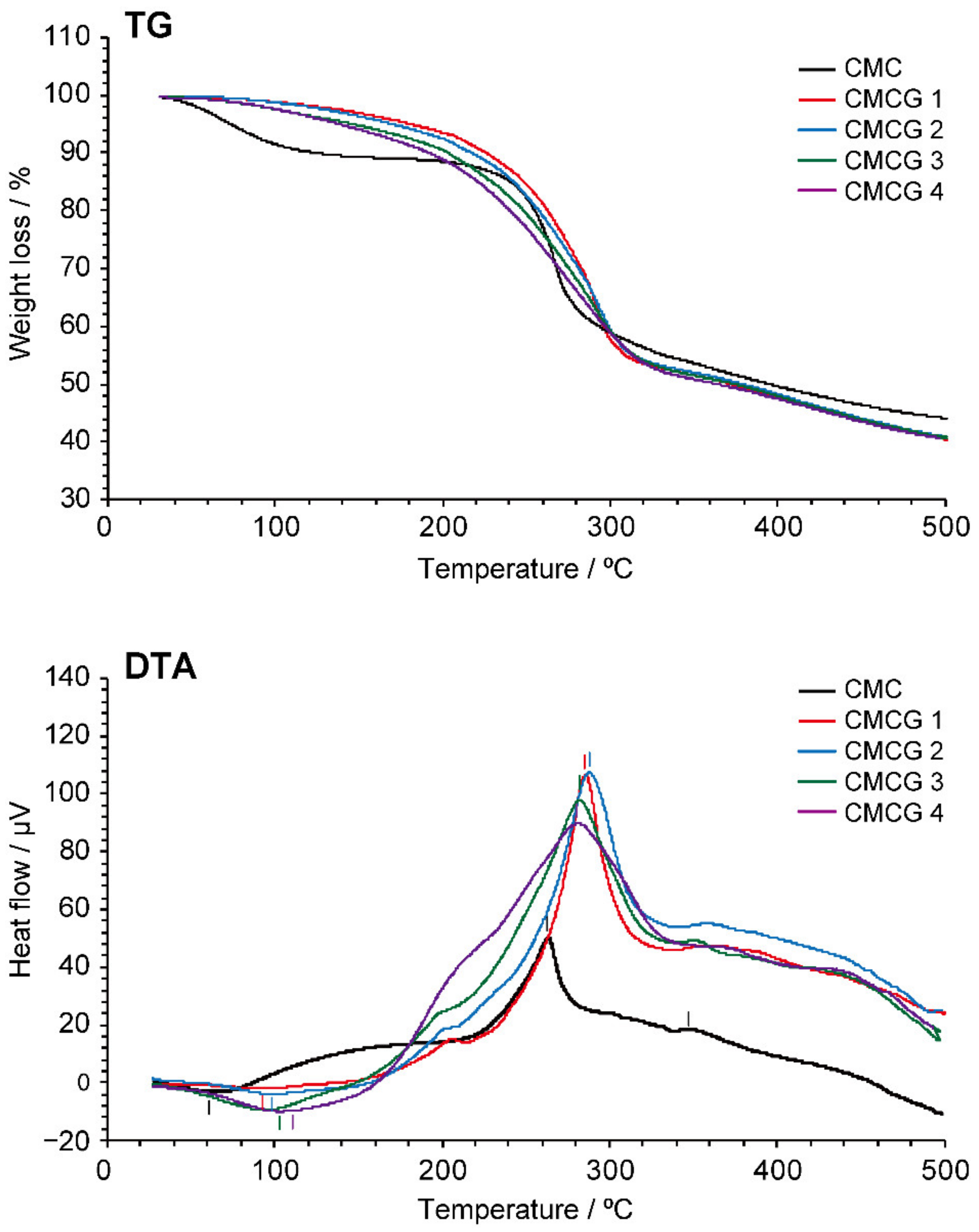
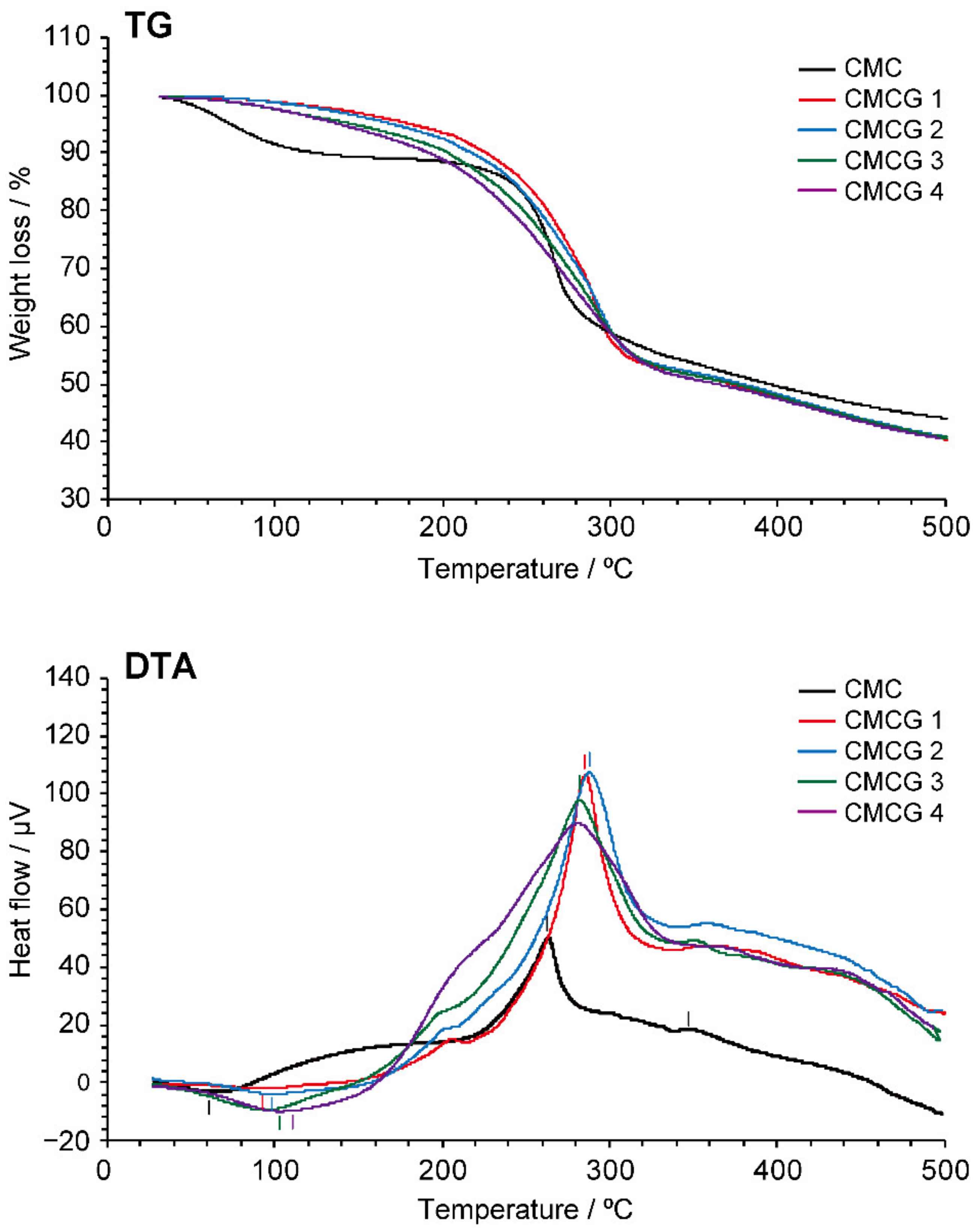
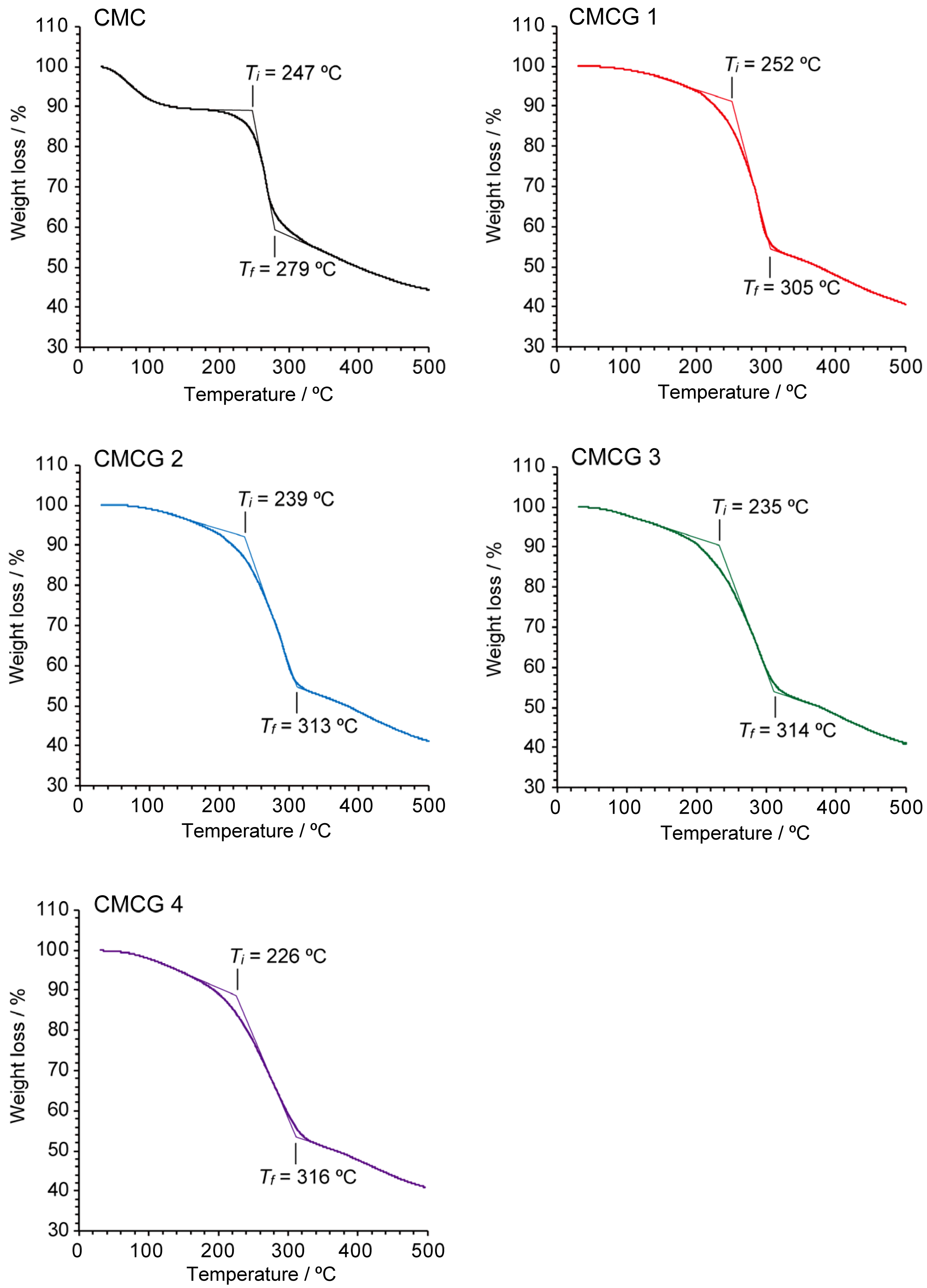
2.4. Thermal Properties of CMCG
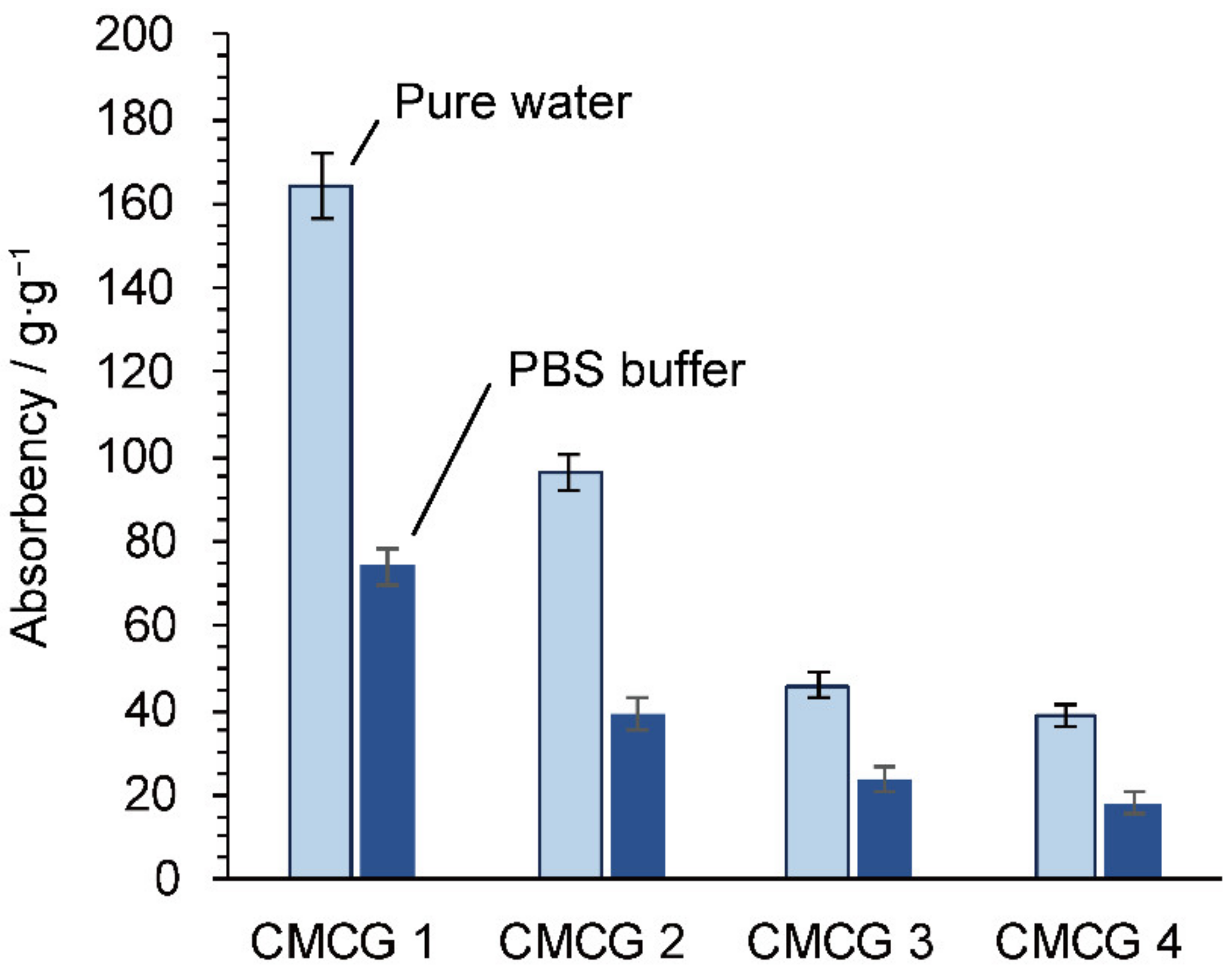
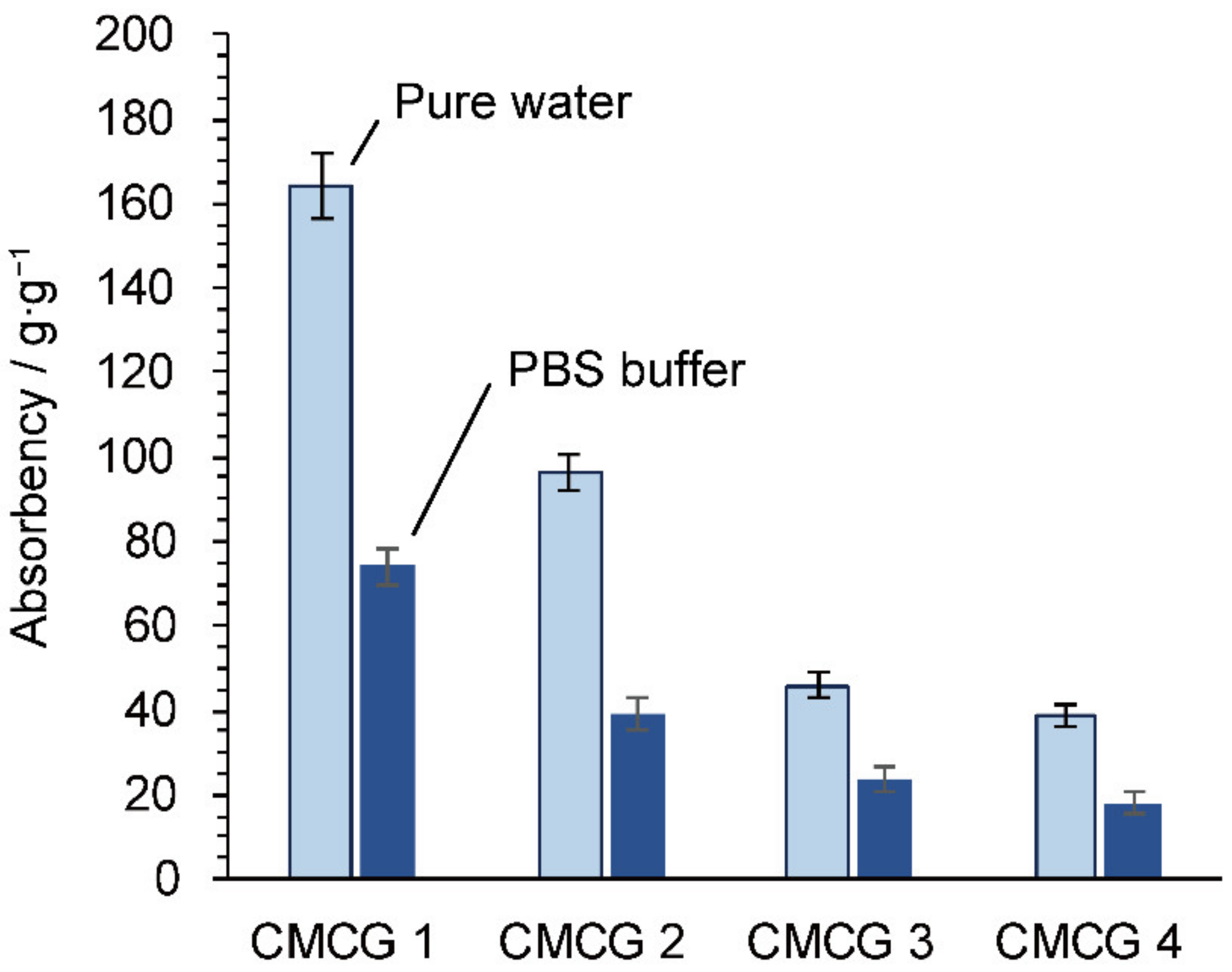
2.5. Water Absorbency of the CMCG Specimens
2.6. Scanning Electron Microscopy (SEM) Observations of CMCG
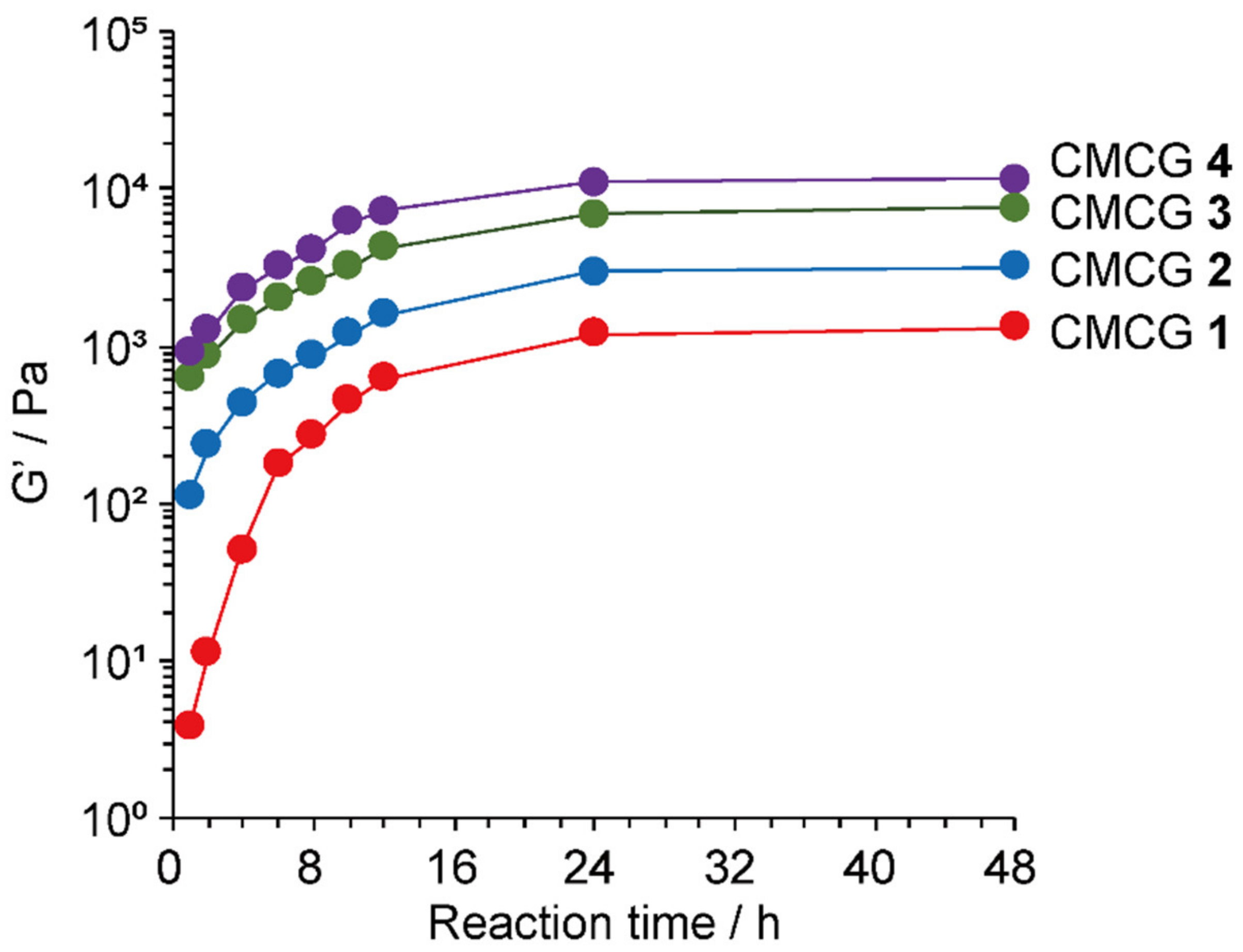
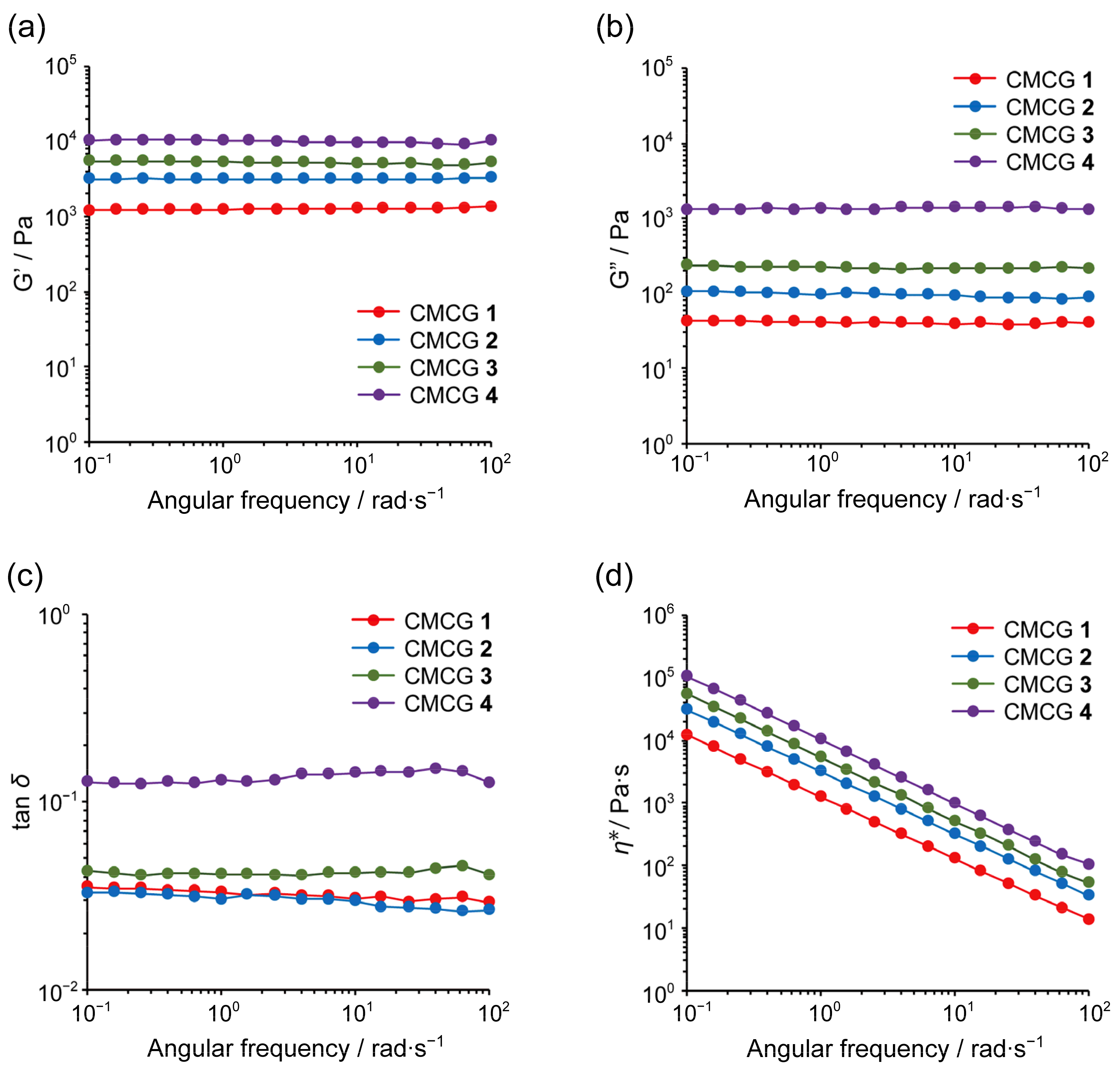
2.7. Rheological Properties of the CMCG Specimens
3. Materials and Methods
3.1. Materials
3.2. Preparation and Structural Characterization of OS
3.2.1. Preparation
3.2.2. Aldehyde Quantification Using the Titration Method
3.2.3. Structural Characterization
3.3. Preparation and Characterization of the CMCG Specimens
3.3.1. Preparation
3.3.2. Structural Characterization
3.3.3. TG/DTA
3.3.4. Water-Absorbency Tests
3.3.5. SEM Observations
3.3.6. Rheological Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kono, H. Characterization and properties of carboxymethyl cellulose hydrogels crosslinked by polyethylene glycol. Carbohydr. Polym. 2014, 106, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Kasai, R.D.; Radhika, D.; Archana, S.; Shanavaz, H.; Koutavarapu, R.; Lee, D.-Y.; Shim, J. A review on hydrogels classification and recent developments in biomedical applications. Int. J. Polym. Mater. Polym. Biomater. 2022; 1–11, in press. [Google Scholar] [CrossRef]
- Mitura, S.; Sionkowska, A.; Jaiswai, A. Biopolymers for hydrogels in cosmetics: A review. J. Mater. Sci. Mater. Med. 2020, 31, 50. [Google Scholar] [CrossRef] [PubMed]
- Pakdel, P.M.; Peighambardoust, S.J. Review on recent progress in chitosan-based hydrogels for wastewater treatment application. Carbohydr. Polym. 2018, 201, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Eivazzadeh-Keihan, R.; Noruzi, E.B.; Mehrban, S.F.; Aliabadi, H.A.M.; Karimi, M.; Mohammadi, A.; Maleki, A.; Mahdavi, M.; Larijani, B.; Shalan, A.E. Review: The latest advances in biomedical applications of chitosan hydrogel as a powerful natural structure with eye-catching biological properties. J. Mater. Sci. 2022, 57, 3855–3891. [Google Scholar] [CrossRef]
- Wang, W.; Meng, Q.; Li, Q.; Liu, J.; Zhou, M.; Jin, Z.; Zhao, K. Chitosan derivatives and their application in biomedicine. Int. J. Mol. Sci. 2020, 21, 487. [Google Scholar] [CrossRef]
- Zhao, D.; Yu, S.; Sun, B.; Gao, S.; Guo, S.; Zhao, K. Biomedical applications of chitosan and its derivative nanoparticles. Polymers 2018, 10, 462. [Google Scholar] [CrossRef]
- Hoell, I.A.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Structure and function of enzymes acting on chitin and chitosan. Biotechnol. Genet. Eng. Rev. 2010, 27, 331–366. [Google Scholar] [CrossRef]
- Wegrzynowska-Drzymalska, K.; Grebicka, P.; Mlynarczyk, D.T.; Chelminiak-Dudkiewicz, D.; Kaczmarek, H.; Goslinski, T.; Ziegler-Borowska, M. Crosslinking of chitosan with dialdehyde chitosan as a new approach for biomedical applications. Materials 2020, 13, 3413. [Google Scholar] [CrossRef]
- Uddin, M.S.; Ju, J. Effect of crosslinking agents on drug distribution in chitosan hydrogel for targeted drug delivery to treat cancer. J. Polym. Res. 2020, 27, 81. [Google Scholar] [CrossRef]
- Kono, H.; Teshirogi, T. Cyclodextrin-grafted chitosan hydrogels for controlled drug delivery. Int. J. Biol. Macromol. 2015, 72, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Song, M.; Hourston, D.J. Novel chitosan-based films cross-linked by genipin with improved physical properties. Biomacromolecules 2004, 5, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.T.N.; Huang, L.; Lemos, H.; Mellor, A.L.; Novakovic, K. Genipin-crosslinked chitosan hydrogels: Preliminary evaluation of the in vitro biocompatibility and biodegradation. J. Appl. Polym. Sci. 2021, 136, 50848. [Google Scholar] [CrossRef]
- Münster, L.; Fojtů, M.; Capáková, Z.; Muchová, M.; Musilová, L.; Vaculovič, T.; Balvan, J.; Kuřitka, I.; Masařík, M.; Vícha, J. Oxidized polysaccharides for anticancer-drug delivery: What is the role of structure? Carbohydr. Polym. 2021, 257, 117562. [Google Scholar] [CrossRef]
- Gomez, C.G.; Rinaudo, M.; Villar, M.A. Oxidation of sodium alginate and characterization of the oxidized derivatives. Carbohydr. Polym. 2007, 67, 296–304. [Google Scholar] [CrossRef]
- Cabral, J.D.; Roxburgh, M.; Shi, Z.; Liu, L.; McConnell, M.; Williams, G.; Evans, N.; Hanton, L.R.; Simpson, J.; Moratti, S.C.; et al. Synthesis, physiochemical characterization, and biocompatibility of a chitosan/dextran-based hydrogel for postsurgical adhesion prevention. J. Mater. Sci. Mater. Med. 2014, 25, 2743–2756. [Google Scholar] [CrossRef]
- Pandit, A.H.; Mazumdar, N.; Ahmad, S. Periodate oxidized hyaluronic acid-based hydrogel scaffolds for tissue engineering applications. Int. J. Biol. Macromol. 2019, 137, 853–869. [Google Scholar] [CrossRef]
- Takai, T.; Sato, M.; Iijima, H.; Kawakami, K. In situ gellable oxidized citrus pectin for localized delivery of anticancer drugs and prevention of homotypic cancer cell aggregation. Biomacromolecules 2010, 11, 3525–3530. [Google Scholar] [CrossRef]
- Liu, P.; Xu, H.; Mi, X.; Xu, L.; Yang, Y. Oxidized sucrose: A potent and biocompatible crosslinker for three-dimensional fibrous protein scaffolds. Macromol. Mater. Eng. 2015, 300, 414–422. [Google Scholar] [CrossRef]
- El-Feky, G.S.; Zayed, G.M.; Elshaier, Y.A.M.M.; Alsharif, F.M. Chitosan-gelatin hydrogel crosslinked with oxidized sucrose for the ocular delivery of timolol maleate. J. Pharm. Sci. 2018, 107, 3098–3104. [Google Scholar] [CrossRef]
- Wang, P.; Sheng, F.; Tang, W.S.; ud-Din, Z.; Chen, L.; Nawaz, A.; Hu, C.; Xiong, H. Synthesis and characterization of corn starch crosslinked with oxidized sucrose. Starch 2019, 71, 1800152. [Google Scholar] [CrossRef]
- Mousaviasl, S.; Saleh, T.; Shojaosadati, S.A.; Boddohi, S. Synthesis and characterization of schizophyllan nanogels via inverse emulsion using biobased materials. Int. J. Biol. Macromol. 2018, 120A, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Pourjavadi, A.; Aghajani, V.; Ghasemzadeh, H. Synthesis, characterization and swelling behavior of chitosan-sucrose as a novel full-polysaccharide superabsorbent hydrogel. J. Appl. Polym. Sci. 2008, 109, 2648–2655. [Google Scholar] [CrossRef]
- Liu, J.; Xu, Y.; Xia, T.; Xue, C.; Liu, L.; Chang, P.; Wang, D.; Sun, X. Oxidized oligosaccharides stabilize rehydrated sea cucumbers against high-temperature impact. Int. J. Mol. Sci. 2020, 21, 5204. [Google Scholar] [CrossRef] [PubMed]
- Jalaja, K.; James, N.R. Electrospun gelatin nanofibers: A facile cross-linking approach using oxidized sucrose. Int. J. Biol. Macromol. 2015, 73, 270–278. [Google Scholar] [CrossRef]
- Xu, H.; Liu, P.; Mi, X.; Xu, L.; Yang, Y. Potent and regularizable crosslinking of ultrafine fibrous protein scaffolds for tissue engineering using a cytocompatible disaccharide derivative. J. Mater. Chem. B 2015, 3, 3609–3616. [Google Scholar] [CrossRef]
- Xu, H.; Canisag, H.; Mu, B.; Yang, Y. Robust and flexible films from 100% starch cross-linked by biobased disaccharide derivative. ACS Sustain. Chem. Eng. 2015, 3, 2631–2639. [Google Scholar] [CrossRef]
- Feng, M.; Hu, X.; Yin, Y.; Liang, Y.; Niu, J.; Yao, J. Structural analysis of oxidized sucrose and its application as a crease-resistant crosslinking agent. Polymers 2022, 14, 2842. [Google Scholar] [CrossRef]
- Wit, D.D.; Berg, R.V.D.; Johanssen, L.A.M.; Rantwijk, F.V.; Maat, L.; Kieboom, A.P.G. The periodate oxidation of sucrose in aqueous N,N-dimethylformamide. Carbohyd. Res. 1992, 226, 253–260. [Google Scholar] [CrossRef]
- Lundborg, M.; Fontana, C.; Widmalm, G. Automatic structure determination of regular polysaccharides based solely on NMR spectroscopy. Biomacromolecules 2011, 12, 3851–3855. [Google Scholar] [CrossRef]
- Kono, H.; Kato, T. Elucidation of substituent distribution states for carboxymethyl chitosan by detailed NMR analysis. Carbohydr. Polym. Technol. Appl. 2021, 2, 100175. [Google Scholar] [CrossRef]
- Kono, H.; Kondo, N.; Hirabayashi, K.; Ogata, M.; Totani, K.; Ikematsu, S.; Osada, M. NMR spectroscopic structural characterization of a water-soluble β-(1→3, 1→6)-glucan from Aureobasidium pullulans. Carbohydr. Polym. 2017, 174, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Kono, H. Determination of mole fractions of ethyl-cellulose-containing monomers by NMR. Carbohydr. Res. 2017, 445, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Onishi, K.; Nakamura, T. Characterization and bisphenol A adsorption capacity of β-cyclodextrin-carboxymethylcellulose-based hydrogels. Carbohydr. Polym. 2013, 98, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Uno, T.; Tsujisaki, H.; Matsushima, T.; Tajima, K. Nanofibrillated bacterial cellulose modified with (3-aminopropyl)trimethoxysilane under aqueous conditions: Applications to poly(methyl methacrylate) fiber-reinforced nanocomposites. ACS Omega 2020, 5, 29561–29569. [Google Scholar] [CrossRef]
- Kono, H.; Otaka, F.; Ozaki, M. Preparation and characterization of guar gum hydrogels as carrier materials for controlled protein drug delivery. Carbohydr. Polym. 2014, 111, 830–840. [Google Scholar] [CrossRef]
- Katugampola, P.; Winstead, C.; Adeleke, A. Thermal stability of carboxymethyl chitosan varying the degree of substitution. Int. J. Pharm. Sci. Invent. 2014, 3, 42–48. [Google Scholar]
- Li, Z.; Lin, Z. Recent advances in polysaccharide-based hydrogels for synthesis and applications. Aggregate 2021, 2, e21. [Google Scholar] [CrossRef]
- Fujita, S.; Tazawa, T.; Kono, H. Preparation and enzyme degradability of spherical and water-absorbent gels from sodium carboxymethyl cellulose. Gels 2022, 8, 321. [Google Scholar] [CrossRef]
- Kono, H.; Ogasawara, K.; Kusumoto, R.; Oshima, K.; Hashimoto, H.; Shimizu, Y. Cationic cellulose hydrogels cross-linked by poly (ethylene glycol): Preparation, molecular dynamics, and adsorption of anionic dyes. Carbohydr. Polym. 2016, 152, 170–180. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OS | Residue 1 | H1/C1 | H2/C2 | H3/C3 | H4/C4 | H5/C5 | H6/C6 |
|---|---|---|---|---|---|---|---|
| OS(I) | Glc | 4.91/90.7 | 8.41/173.1 | 8.41/173.1 | 4.05/72.9 | 4.98/91.5 | 4.01, 3.71/60.3 |
| Fru | 3.94/63.5 | n.d./97.6 | 8.41/173.1 | 8.41/173.1 | 5.06/93.8 | 3.80/62.2 | |
| OS(II) | Glc | 5.16/88.5 | 8.41/173.1 | 8.41/173.1 | 3.73/74.2 | 3.79/62.2 | |
| Fru | 3.94/63.5 | n.d./97.6 | 8.41/173.1 | 8.41/173.1 | 5.06/93.8 | 3.80/62.2 |
| Sample | CMC/mg (Amine/mmol) | OS/mg (Aldehyde/mmol) | CR 1 |
|---|---|---|---|
| CMCG 1 | 400 (0.14) | 6.10 (0.07) | 0.08 |
| CMCG 2 | 400 (0.14) | 12.2 (0.14) | 0.11 |
| CMCG 3 | 400 (0.14) | 24.5 (0.28) | 0.14 |
| CMCG 4 | 400 (0.14) | 49.0 (0.56) | 0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kono, H.; Noda, J.; Wakamori, H. Detailed Structural Characterization of Oxidized Sucrose and Its Application in the Fully Carbohydrate-Based Preparation of a Hydrogel from Carboxymethyl Chitosan. Molecules 2022, 27, 6137. https://doi.org/10.3390/molecules27186137
Kono H, Noda J, Wakamori H. Detailed Structural Characterization of Oxidized Sucrose and Its Application in the Fully Carbohydrate-Based Preparation of a Hydrogel from Carboxymethyl Chitosan. Molecules. 2022; 27(18):6137. https://doi.org/10.3390/molecules27186137
Chicago/Turabian StyleKono, Hiroyuki, Junki Noda, and Haruki Wakamori. 2022. "Detailed Structural Characterization of Oxidized Sucrose and Its Application in the Fully Carbohydrate-Based Preparation of a Hydrogel from Carboxymethyl Chitosan" Molecules 27, no. 18: 6137. https://doi.org/10.3390/molecules27186137
APA StyleKono, H., Noda, J., & Wakamori, H. (2022). Detailed Structural Characterization of Oxidized Sucrose and Its Application in the Fully Carbohydrate-Based Preparation of a Hydrogel from Carboxymethyl Chitosan. Molecules, 27(18), 6137. https://doi.org/10.3390/molecules27186137