

A New Photoacoustic Soot Spectrophone for Filter-Free Measurements of Black Carbon at 880 nm

 ,
, 
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Materials and Methods
2.1.1. Measurement of Particle Absorption by Photoacoustic Spectroscopy
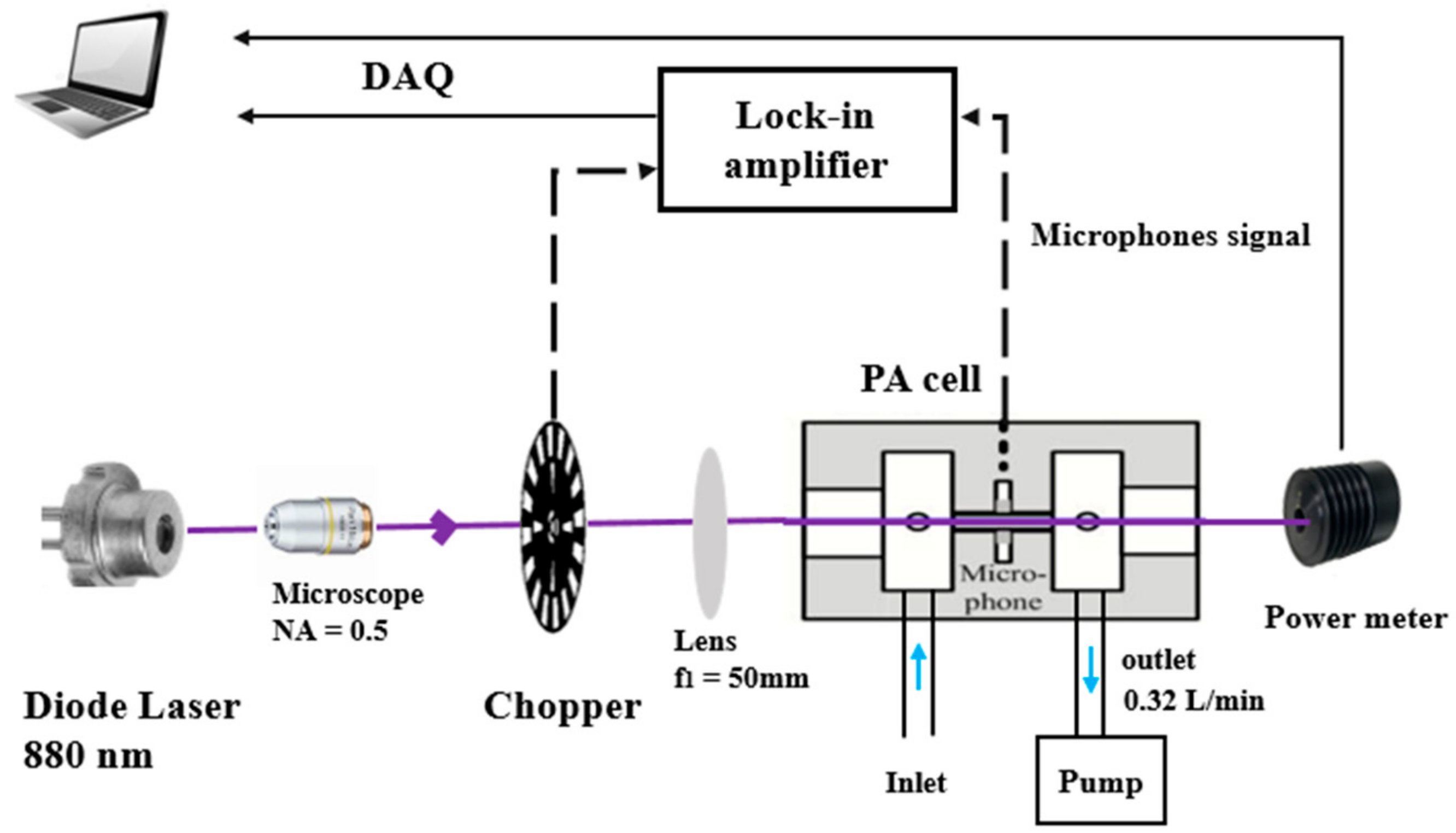
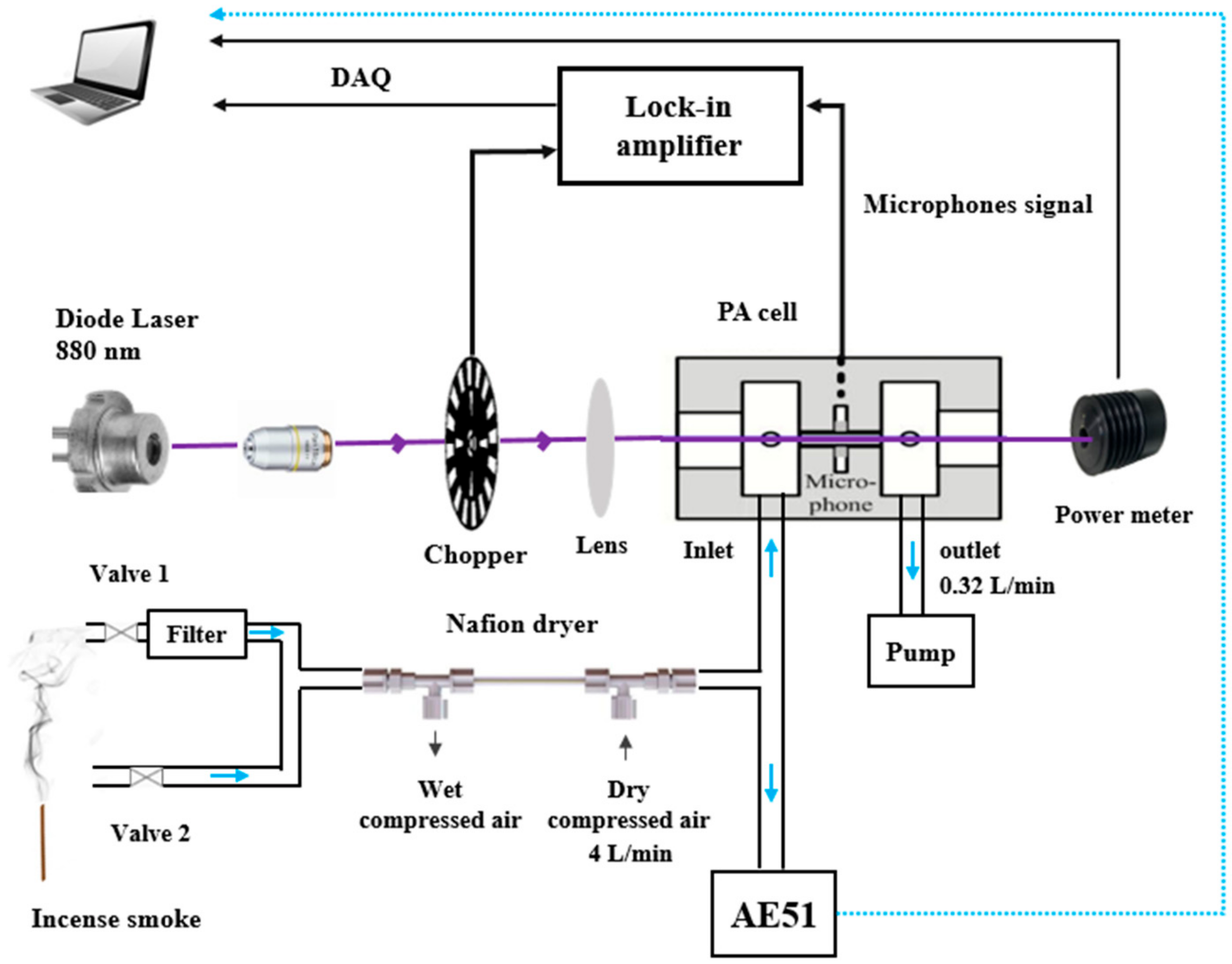
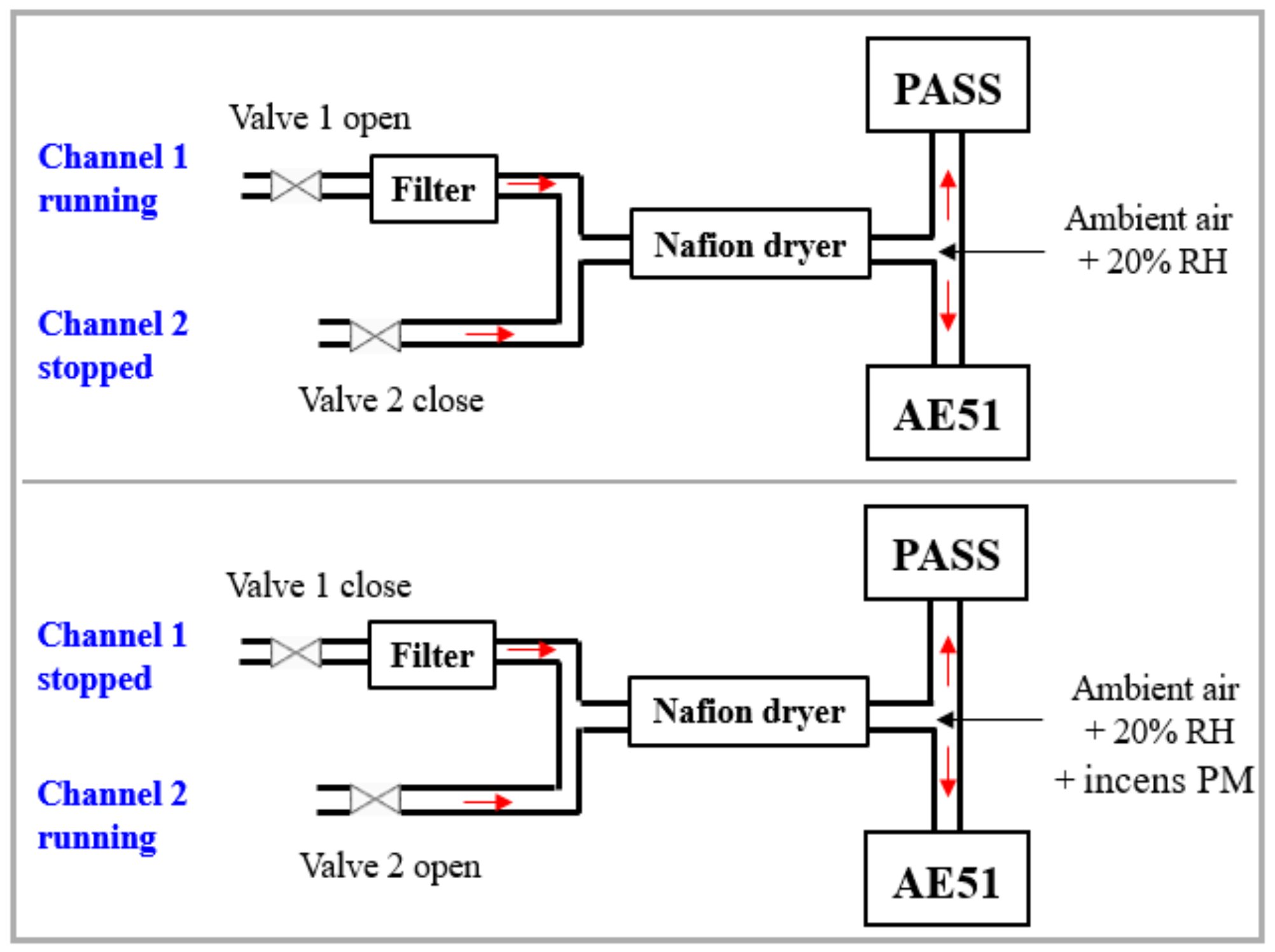
2.1.2. Experimental Setup
2.1.3. Calibration of the PA Cell
2.1.4. Modulation Frequency Optimization
- Nitrogen (N2) with low water content (RH = 9.5%, T = 27 °C)—Referred to as dry N2 in the following sections;
- Compressed air with low water content (RH = 12%, T = 27 °C)—Referred to as dry compressed air in the following sections;
- Humid filtered indoor ambient air (RH = 34%, T = 27 °C), filtered by a PTFE membrane filter (0.2 μm) to remove particles, assuming that the mass concentration of particles with a size less than 0.2 μm is negligible (<1 μg.m−3) given their small size; hence, their effects on the PA signal are also negligible.
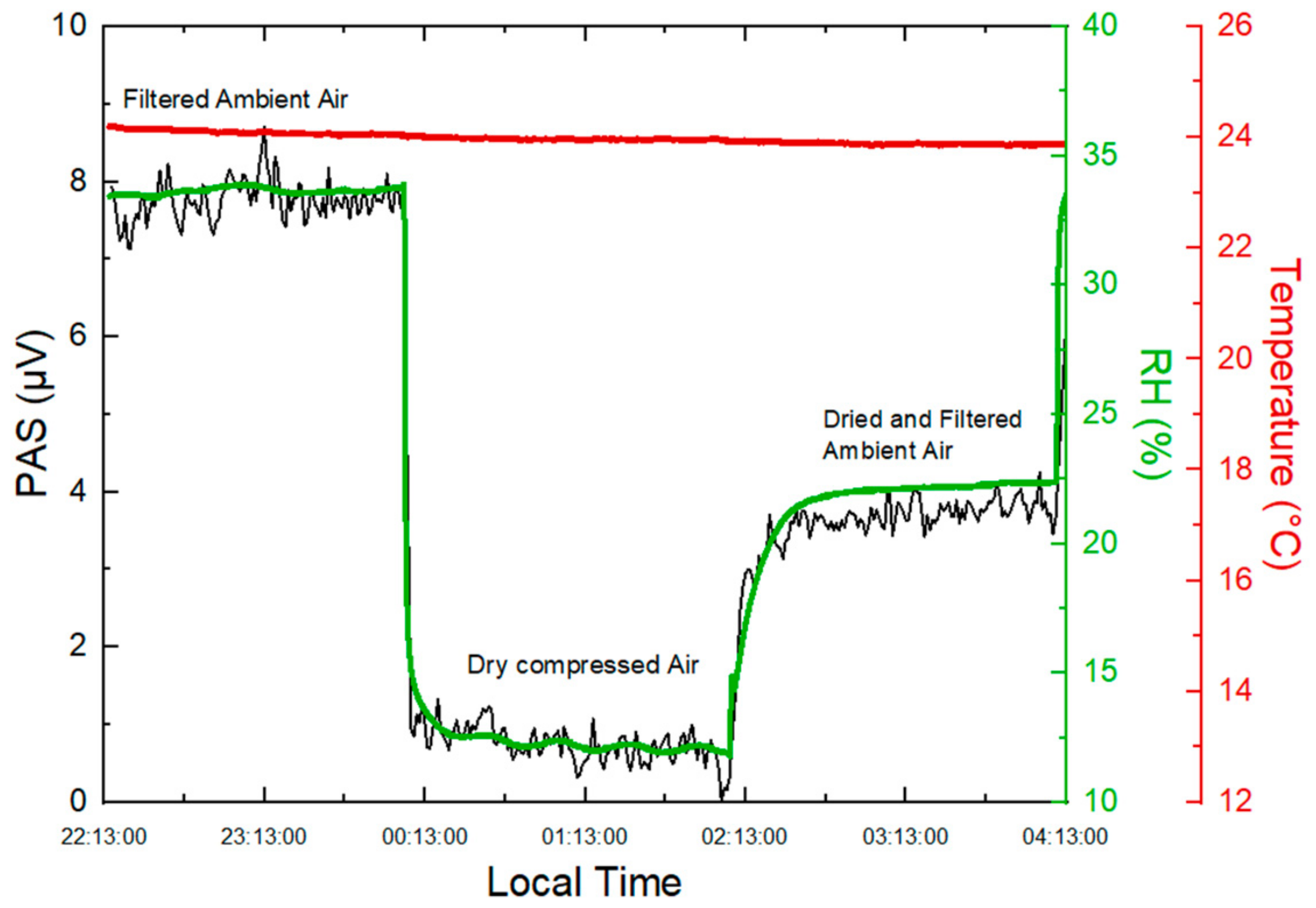
2.1.5. Impact of Relative Humidity on PA Signal
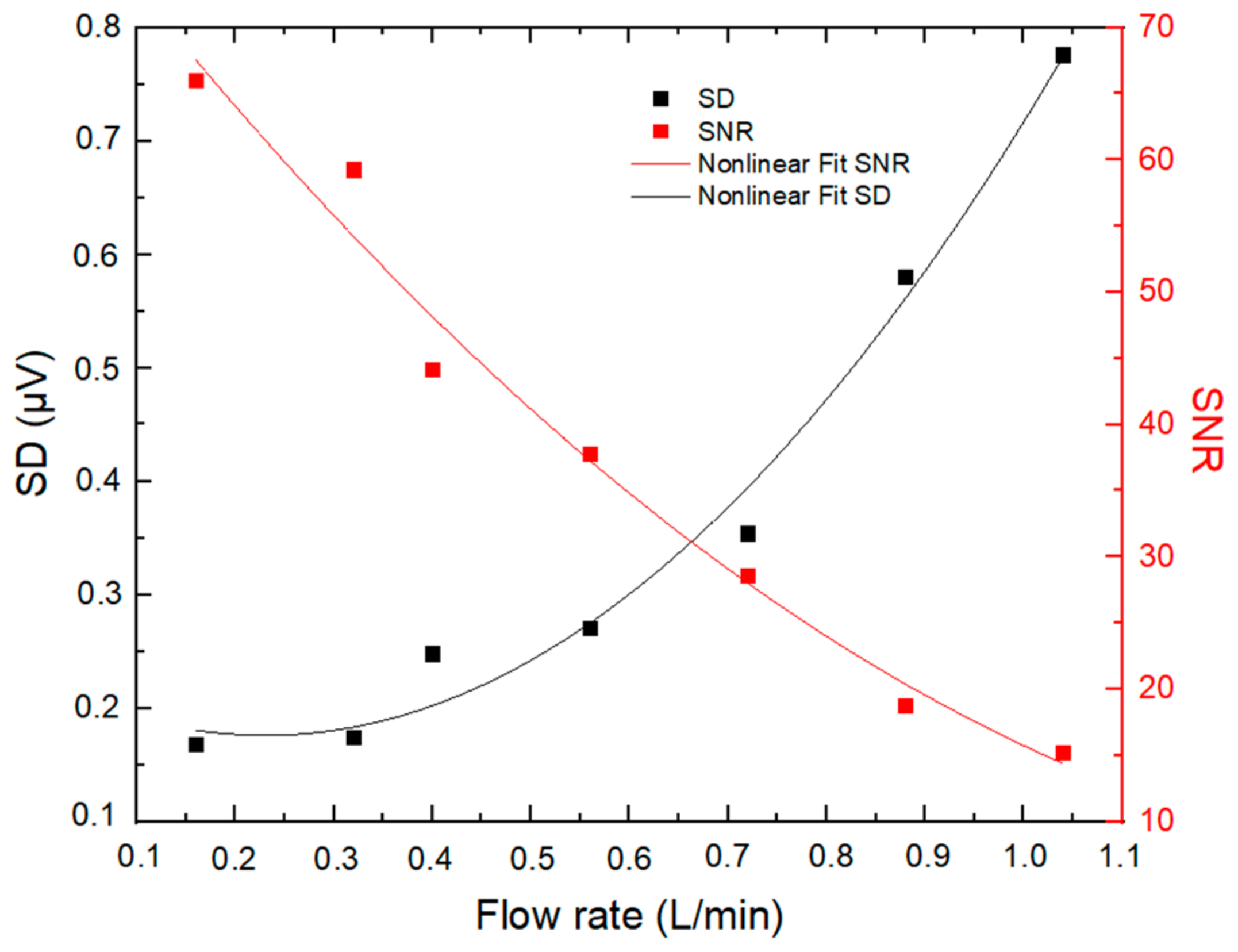
2.1.6. Sampling Flow Rate
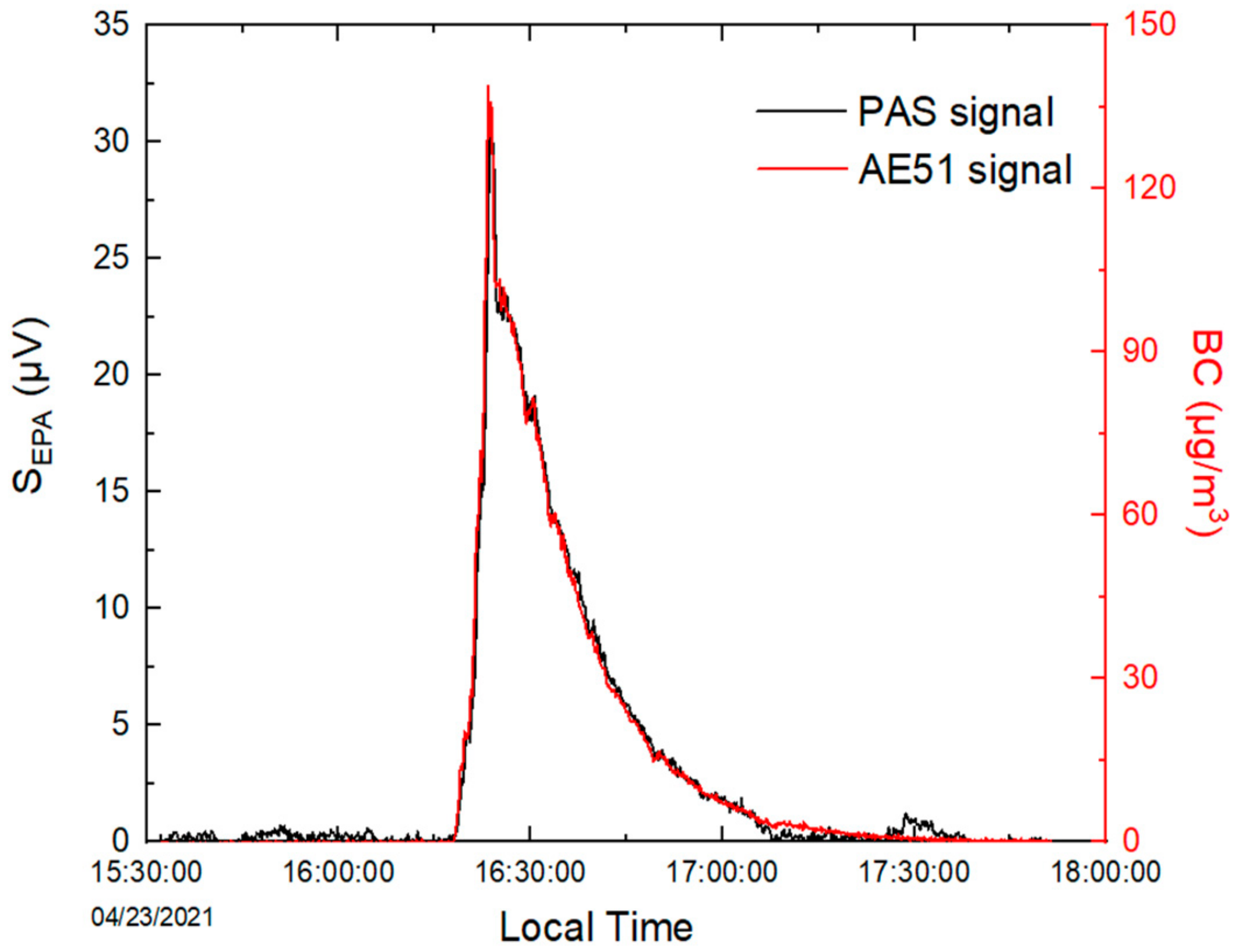
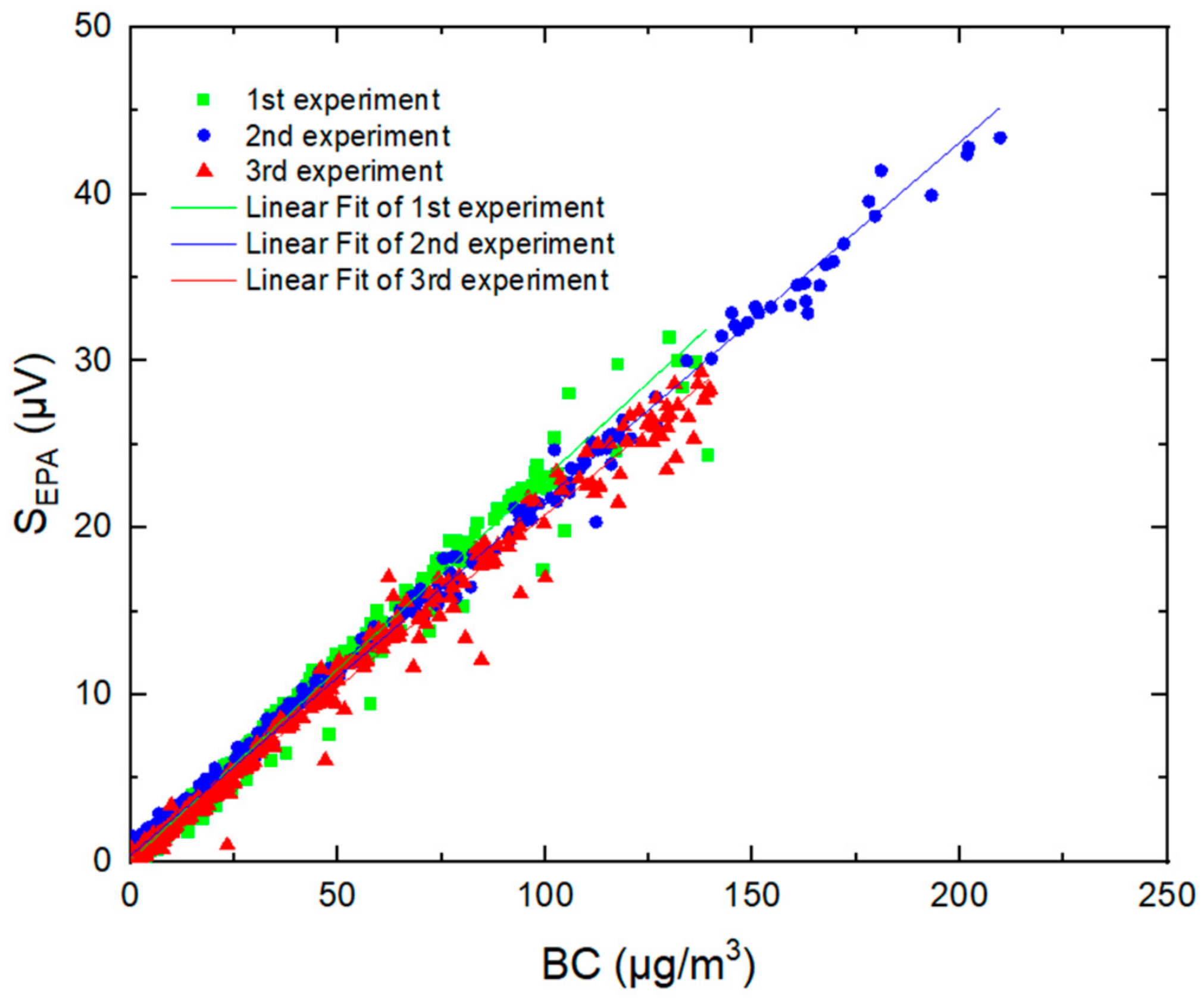
2.2. Evaluation of the PASS Performances
3. Conclusions and Outlooks
Author Contributions
Funding
Conflicts of Interest
References
- Zaelke, D. Primer on Short-Lived Climate Pollutants; IGSD: Washington, DC, USA, 2013. [Google Scholar]
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.; Kerminen, V.M.; Kondo, Y.; Liao, H.; Lohmann, U.; et al. Clouds and aerosols. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2013; pp. 572–657. [Google Scholar]
- Bond, T.C.; Doherty, S.J.; Fahey, D.W.; Foster, P.M.; Berntsen, T.; DeAngelo, B.J.; Flanner, M.G.; Ghan, S.; Kärcher, B.; Koch, D.; et al. Bounding the role of black carbon in the climate system: A scientific assessment: Black carbon in the climate system. J. Geophys. Res. Atmos. 2013, 118, 5380–5552. [Google Scholar] [CrossRef]
- Black Carbon, Climate & Clean Air Coalition. Available online: https://www.ccacoalition.org (accessed on 15 September 2022).
- Yamaguchi, M.; Izuta, T. Effects of Black Carbon and Ammonium Sulfate Particles on Plants. In Air Pollution Impacts on Plants in East Asia; Izuta, T., Ed.; Springer: Tokyo, Japan, 2017; pp. 295–308. [Google Scholar] [CrossRef]
- Wang, G.; Shen, F.; Yi, H.; Hubert, P.; Deguine, A.; Petitprez, D.; Maamary, R.; Augustin, P.; Fourmentin, M.; Fertein, E.; et al. Laser absorption spectroscopy applied to monitoring of hort-lived climate pollutants (SLCPs). J. Mol. Spectrosc. 2018, 348, 142–151. [Google Scholar] [CrossRef]
- Boparai, P.; Lee, J.; Bond, T.C. Revisiting Thermal-Optical Analyses of Carbonaceous Aerosol Using a Physical Model. Aerosol Sci. Technol. 2008, 42, 930–948. [Google Scholar] [CrossRef]
- Schulz, C.; Kock, B.F.; Hofmann, M.; Michelsen, H.; Will, S.; Bougie, B.; Suntz, R.; Smallwood, G. Laser-induced incandescence: Recent trends and current questions. Appl. Phys. B 2006, 83, 333–354. [Google Scholar] [CrossRef]
- Müller, T.; Henzing, J.S.; de Leeuw, G.; Wiedensohler, A.; Alastuey, A.; Angelov, H.; Bizjak, M.; Coen, M.C.; Engström, J.E.; Gruening, C.; et al. Characterization and intercomparison of aerosol absorption photometers: Result of two intercomparison workshops. Atmos. Meas. Tech. 2011, 4, 245–268. [Google Scholar] [CrossRef]
- Schmid, O.; Artaxo, P.; Arnott, W.P.; Chand, D.; Gatti, L.V.; Frank, G.P.; Hoffer, A.; Schnaiter, M.; Andreae, M.O. Spectral light absorption by ambient aerosols influenced by biomass burning in the Amazon Basin. I: Comparison and field calibration of absorption measurement techniques. Atmos. Chem. Phys. 2006, 20, 3443–3462. [Google Scholar] [CrossRef]
- Wang, G.; Kulinski, P.; Hubert, P.; Deguine, A.; Petitprez, D.; Crumeyrolle, S.; Fertein, E.; Deboudt, K.; Flament, P.; Sigrist, M.W.; et al. Filter-free light absorption measurement of volcanic ashes and ambient particulate matter using multi-wavelength photoacoustic spectroscopy. PIER 2019, 166, 59–74. [Google Scholar] [CrossRef]
- Petzold, A.; Niessner, R. Photoacoustic soot sensor for in-situ black carbon monitoring. Appl. Phys. B 1996, 63, 191–197. [Google Scholar] [CrossRef]
- Beck, H.A.; Niessner, R.; Haisch, C. Development and characterization of a mobile photoacoustic sensor for on-line soot emission monitoring in diesel exhaust gas. Anal. Bioanal. Chem. 2003, 375, 1136–1143. [Google Scholar] [CrossRef]
- Arnott, W.P.; Walker, J.W.; Moosmüller, H.; Elleman, R.A.; Jonsson, H.H.; Buzorius, G.; Conant, W.C.; Flagan, R.C.; Seinfeld, J.H. Photoacoustic insight for aerosol light absorption aloft from meteorological aircraft and comparison with particle soot absorption photometer measurements: DOE Southern Great Plains climate research facility and the coastal stratocumulus imposed perturbation experiments. J. Geophys. Res. 2006, 111, D05S02. [Google Scholar] [CrossRef]
- Lack, D.A.; Richardson, M.S.; Law, D.; Langridge, J.M.; Cappa, C.D.; McLanghlin, R.J.; Murphy, D.M. Aircraft Instrument for Comprehensive Characterization of Aerosol Optical Properties, Part 2: Black and Brown Carbon Absorption and Absorption Enhancement Measured with Photo Acoustic Spectroscopy. Aerosol Sci. Technol. 2012, 46, 555–568. [Google Scholar] [CrossRef]
- Lewis, K.; Arnott, W.P.; Moosmüller, H.; Wold, C.E. Strong spectral variation of biomass smoke light absorption and single scattering albedo observed with a novel dual-wavelength photoacoustic instrument. J. Geophys. Res. 2008, 113, D16203. [Google Scholar] [CrossRef]
- Hamasha, K.M.; Arnott, W.P. Photoacoustic measurements of black carbon light absorption coefficients in Irbid city, Jordan. Environ. Monit. Assess. 2010, 166, 485–494. [Google Scholar] [CrossRef]
- Kirchstetter, T.W.; Novakov, T.; Hobbs, P.V. Evidence that the spectral dependence of light absorption by aerosols is affected by organic carbon: Spectral light absorption by aerosols. J. Geophys. Res. 2004, 109, D21208. [Google Scholar] [CrossRef]
- Miklós, A.; Hess, P.; Bozóki, Z. Application of acoustic resonators in photoacoustic trace gas analysis and metrology. Rev. Sci. Instrum. 2001, 72, 1937–1955. [Google Scholar] [CrossRef]
- Ajtai, T.; Filep, Á.; Schnaiter, M.; Linke, C.; Vragel, M.; Bozóki, Z.; Szabó, G.; Leisner, T. A novel multi−wavelength photoacoustic spectrometer for the measurement of the UV–vis-NIR spectral absorption coefficient of atmospheric aerosols. J. Aerosol Sci. 2010, 41, 1020–1029. [Google Scholar] [CrossRef]
- Bozóki, Z.; Pogány, A.; Szabó, G. Photoacoustic Instruments for Practical Applications: Present, Potentials, and Future Challenges. Appl. Spectrosc. Rev. 2011, 46, 1–37. [Google Scholar] [CrossRef]
- Arnott, W.P.; Moosmüller, H.; Walker, J.W. Nitrogen dioxide and kerosene-flame soot calibration of photoacoustic instruments for measurement of light absorption by aerosols. Rev. Sci. Instrum. 2000, 71, 4545. [Google Scholar] [CrossRef]
- Nakayama, T.; Suzuki, H.; Kagamitani, S.; Ikeda, Y.; Uchiyama, A.; Matsumi, Y. Characterization of a Three Wavelength Photoacoustic Soot Spectrometer (PASS-3) and a Photoacoustic Extinctiometer (PAX). J. Meteorol. Soc. Jpn. 2015, 93, 285–308. [Google Scholar] [CrossRef]
- Linke, C.; Ibrahim, I.; Schleicher, N.; Hitzenberger, R.; Andreae, M.O.; Leisner, T.; Schnaiter, M. A novel single-cavity three-wavelength photoacoustic spectrometer for atmospheric aerosol research. Atmos. Meas. Tech. 2016, 9, 5331–5346. [Google Scholar] [CrossRef]
- Fischer, D.A.; Smith, G.D. A portable, four-wavelength, single-cell photoacoustic spectrometer for ambient aerosol absorption. Aerosol Sci. Technol. 2018, 52, 393–406. [Google Scholar] [CrossRef]
- Vandaele, A.C.; Hermans, C.; Simon, P.C.; Carleer, M.; Colin, R.; Fally, S.; Mérienne, M.F.; Jenouvrier, A.; Coquart, B. Measurements of the NO2 absorption cross-section from 42,000 cm−1 to 10,000 cm−1 (238–1000 nm) at 220 K and 294 K. J. Quant. Spectrosc. Radiat. Transf. 1998, 59, 171–184. [Google Scholar] [CrossRef]
- Kosterev, A.A.; Mosely, T.S.; Tittel, F.K. Impact of humidity on quartz-enhanced photoacoustic spectroscopy based detection of HCN. Appl. Phys. B 2006, 85, 295–300. [Google Scholar] [CrossRef]
- Lang, B.; Breitegger, P.; Brunnhofer, G.; Valero, J.P.; Schweighart, S.; Klug, A.; Hassler, W.; Bergmann, A. Molecular relaxation effects on vibrational water vapor photoacoustic spectroscopy in air. Appl. Phys. B. 2020, 126, 64. [Google Scholar] [CrossRef]
- Babu, S.S.; Moorthy, K.K.; Manchanda, R.K.; Sinha, P.R.; Satheesh, S.K.; Vajja, D.P.; Srinivasan, S.; Kumar, V.H.A. Free tropospheric black carbon aerosol measurements using high altitude balloon: Do BC layers build “their own homes” up in the atmosphere? Geophys. Res. Lett. 2011, 38, L08803. [Google Scholar] [CrossRef]
- Chilinski, M.T.; Markowicz, K.M.; Markowicz, J. Observation of vertical variability of black carbon concentration in lower troposphere on campaigns in Poland. Atmos. Environ. 2016, 137, 155–170. [Google Scholar] [CrossRef]
- Lee, J. Performance Test of MicroAeth® AE51 at Concentrations Lower than 2 μg/m3 in Indoor Laboratory. Appl. Sci. 2019, 9, 2766. [Google Scholar] [CrossRef]
- Bond, T.C.; Bergstrom, R.W. Light Absorption by Carbonaceous Particles: An Investigative Review. Aerosol Sci. Technol. 2006, 40, 27–67. [Google Scholar] [CrossRef]
- Radney, J.G.; Zangmeister, C.D. Measurement of Gas and Aerosol Phase Absorption Spectra across the Visible and Near-IR Using Supercontinuum Photoacoustic Spectroscopy. Anal. Chem. 2015, 87, 7356–7363. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abichou, G.; Ngagine, S.H.; Ba, T.N.; Wang, G.; Flament, P.; Deboudt, K.; Dusanter, S.; Sigrist, M.W.; Tomas, A.; Chen, W. A New Photoacoustic Soot Spectrophone for Filter-Free Measurements of Black Carbon at 880 nm. Molecules 2022, 27, 6065. https://doi.org/10.3390/molecules27186065
Abichou G, Ngagine SH, Ba TN, Wang G, Flament P, Deboudt K, Dusanter S, Sigrist MW, Tomas A, Chen W. A New Photoacoustic Soot Spectrophone for Filter-Free Measurements of Black Carbon at 880 nm. Molecules. 2022; 27(18):6065. https://doi.org/10.3390/molecules27186065
Chicago/Turabian StyleAbichou, Goufrane, Soulemane H. Ngagine, Tong N. Ba, Gaoxuan Wang, Pascal Flament, Karine Deboudt, Sébastien Dusanter, Markus W. Sigrist, Alexandre Tomas, and Weidong Chen. 2022. "A New Photoacoustic Soot Spectrophone for Filter-Free Measurements of Black Carbon at 880 nm" Molecules 27, no. 18: 6065. https://doi.org/10.3390/molecules27186065
APA StyleAbichou, G., Ngagine, S. H., Ba, T. N., Wang, G., Flament, P., Deboudt, K., Dusanter, S., Sigrist, M. W., Tomas, A., & Chen, W. (2022). A New Photoacoustic Soot Spectrophone for Filter-Free Measurements of Black Carbon at 880 nm. Molecules, 27(18), 6065. https://doi.org/10.3390/molecules27186065