
Improving Catalytic Activity of “Janus” MoSSe Based on Surface Interface Regulation


Abstract
:
1. Introduction
2. Computational Methods
3. Results and Discussion
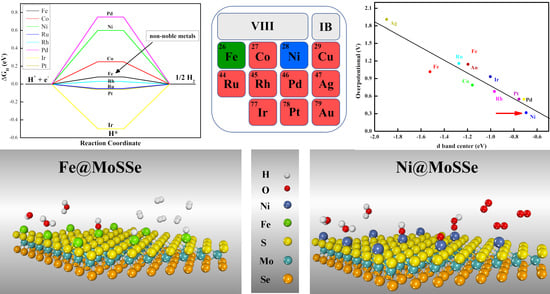
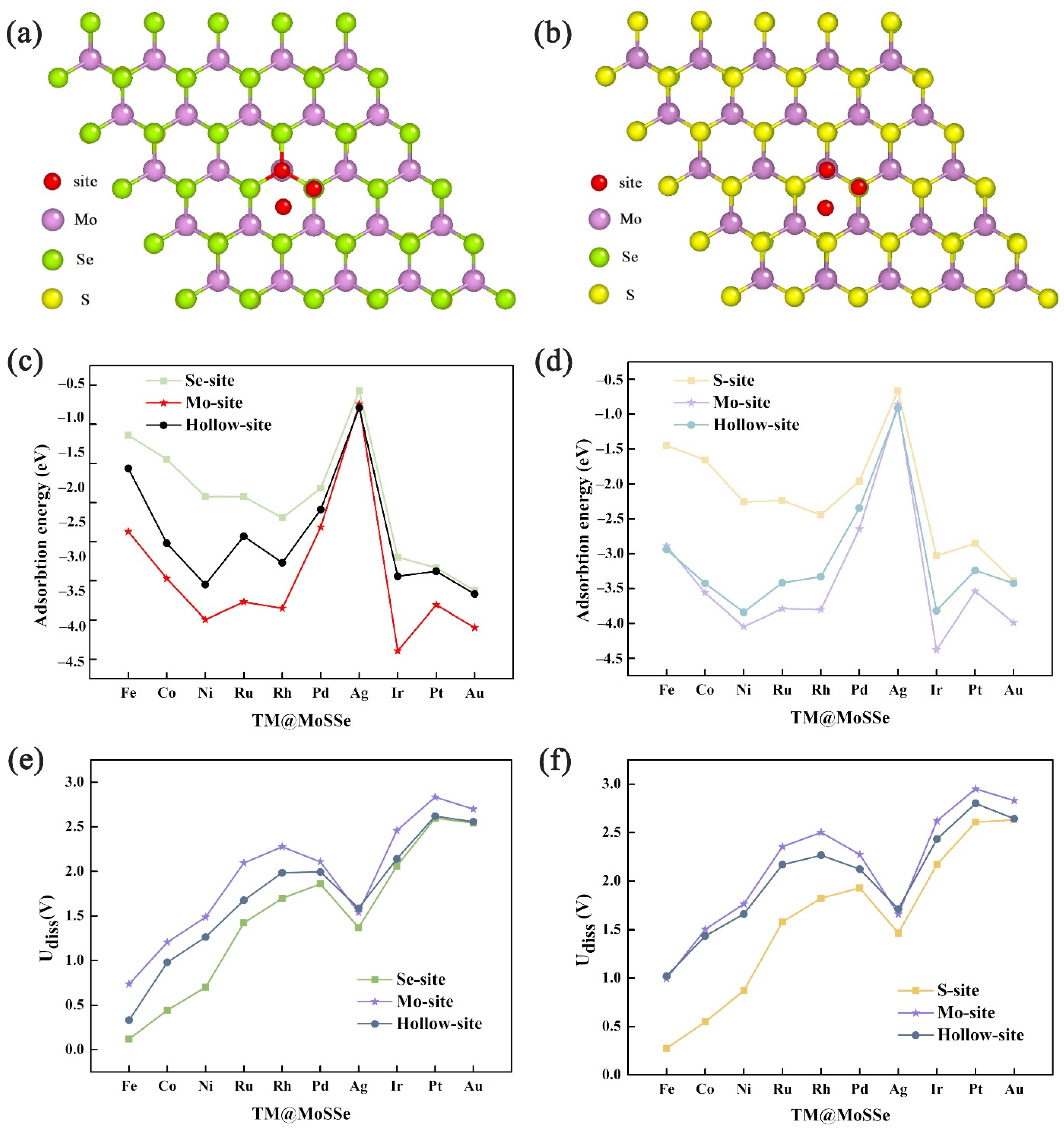
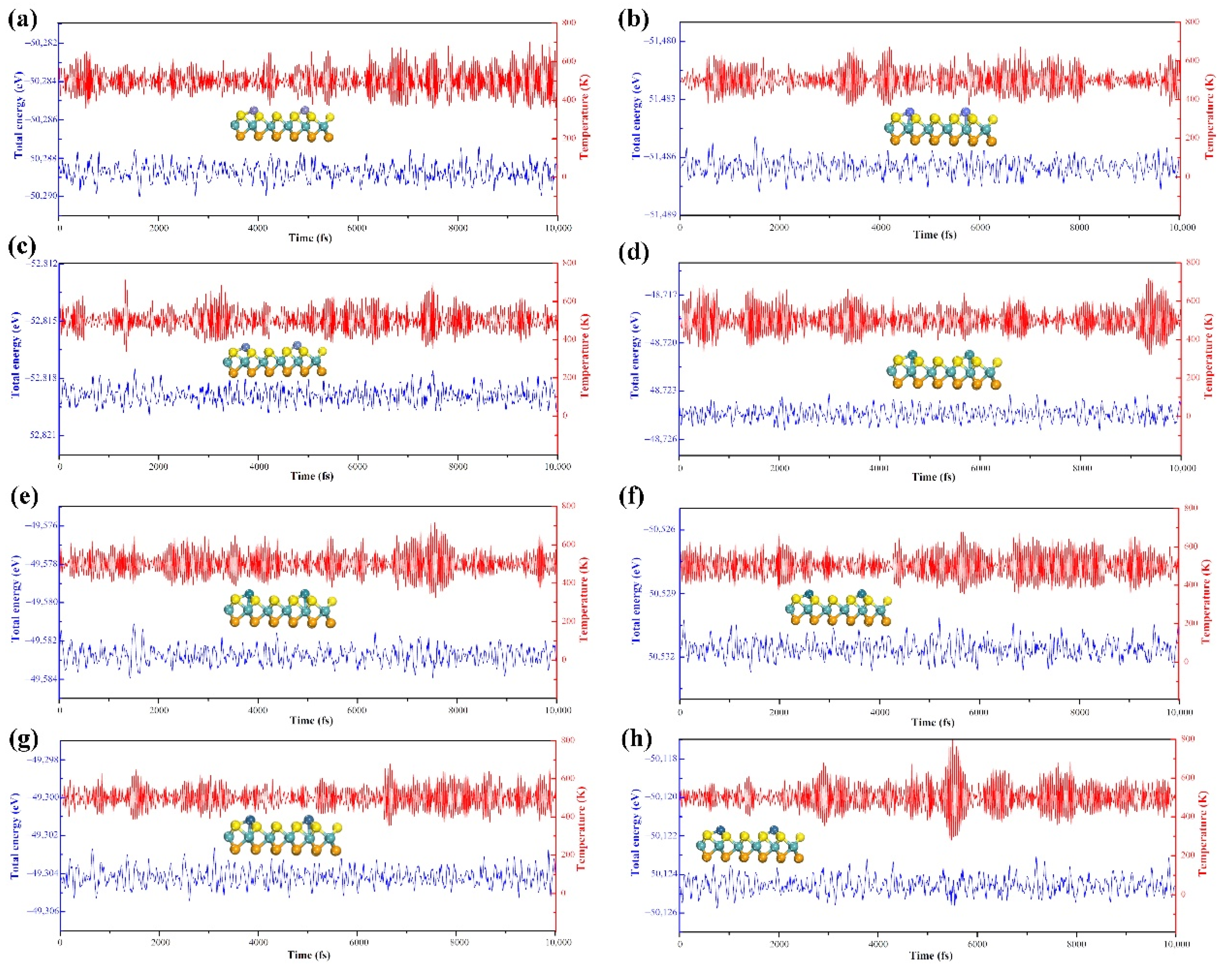
3.1. Geometric Structure and Stability of TM@MoSSe
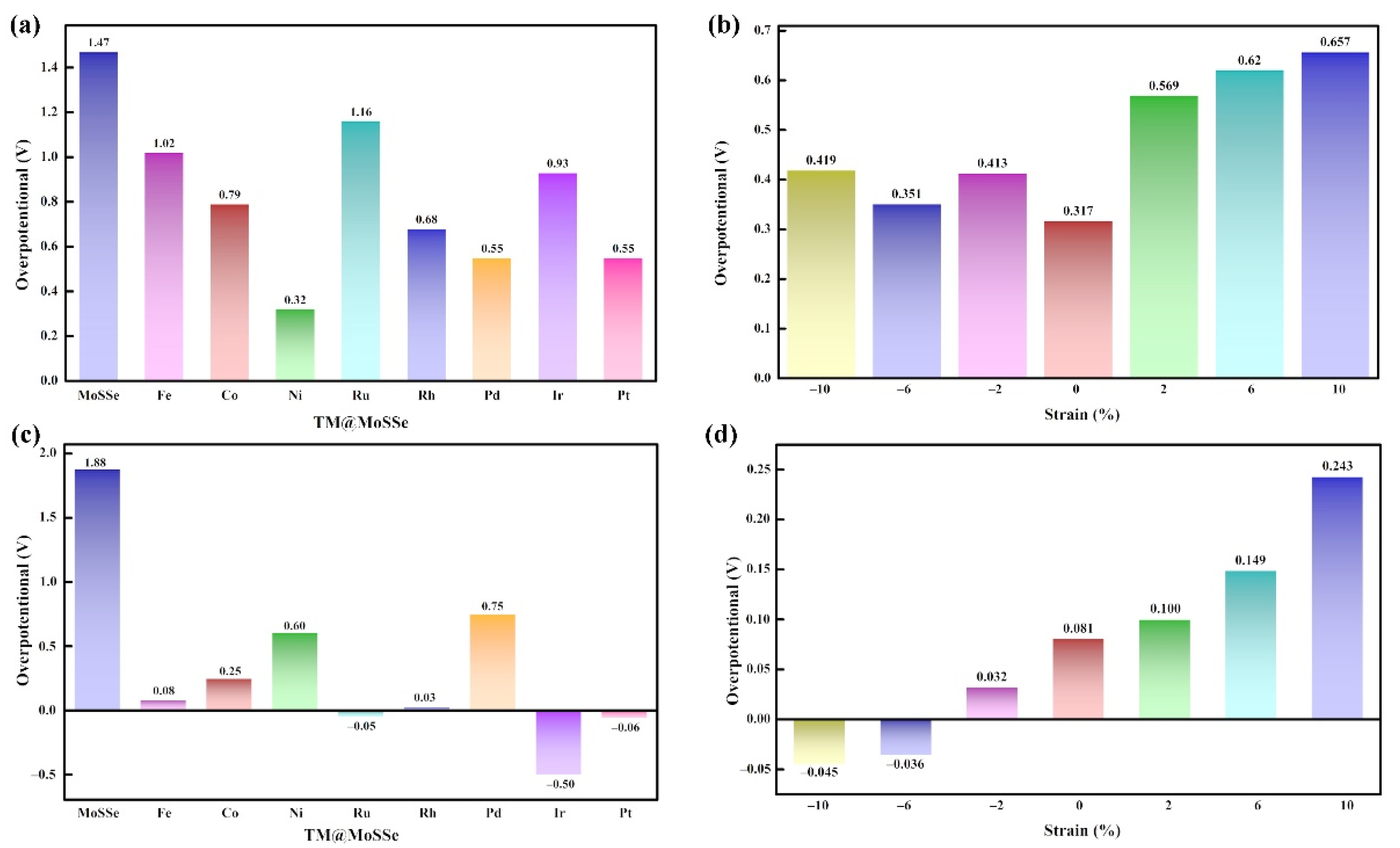
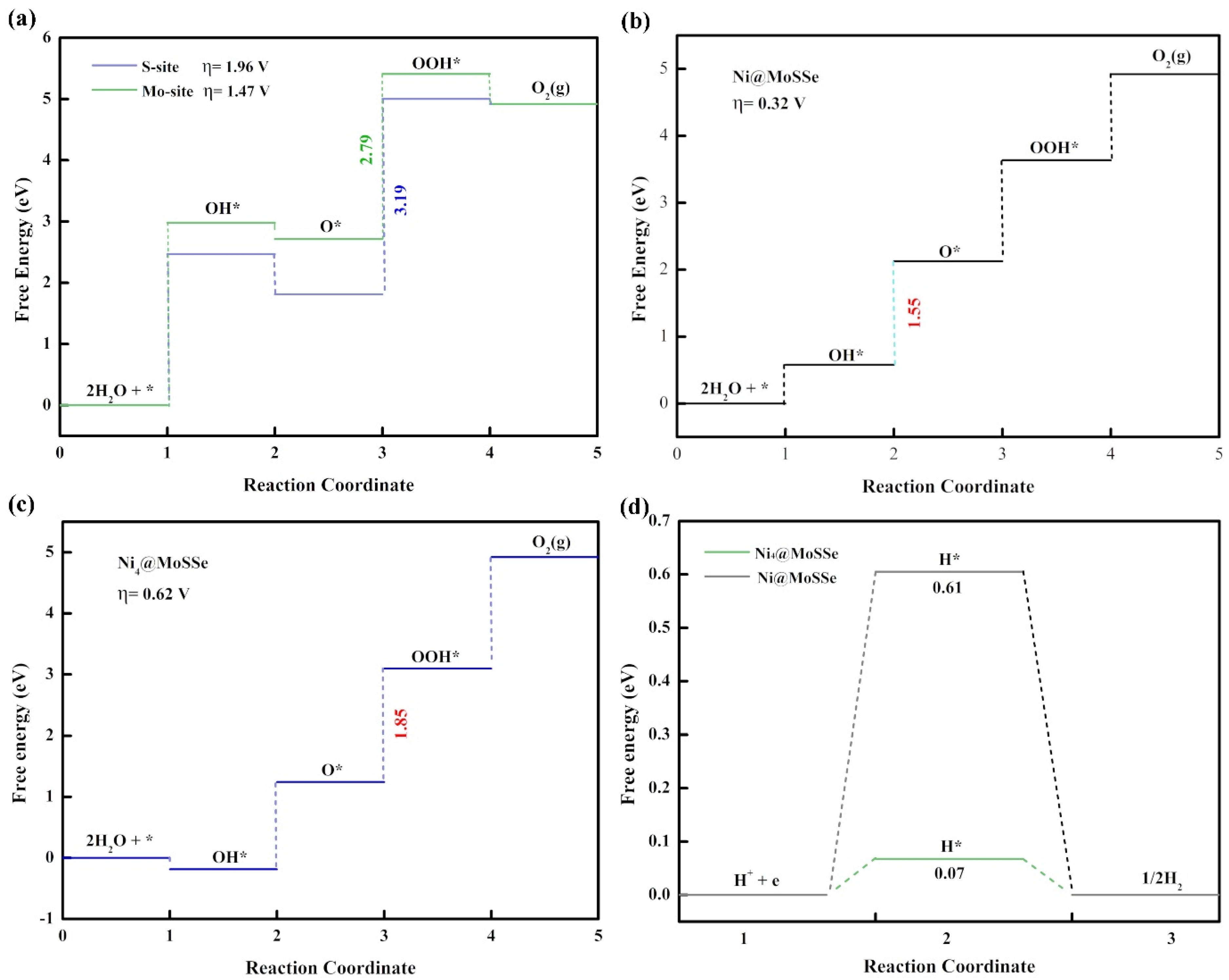
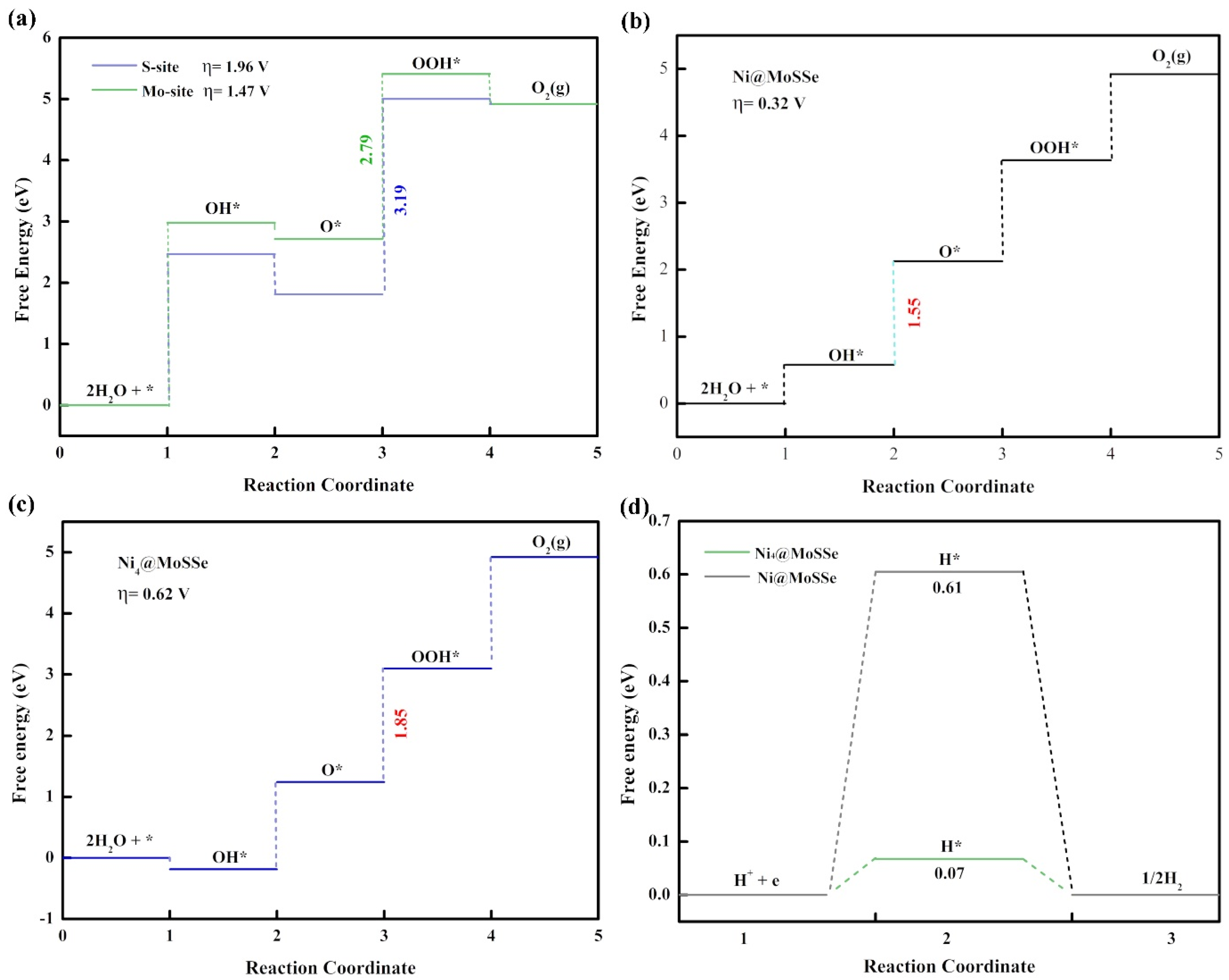
3.2. HER and OER Catalytic Activity
3.3. The Origin of Catalytic Activity of TM@MoSSe
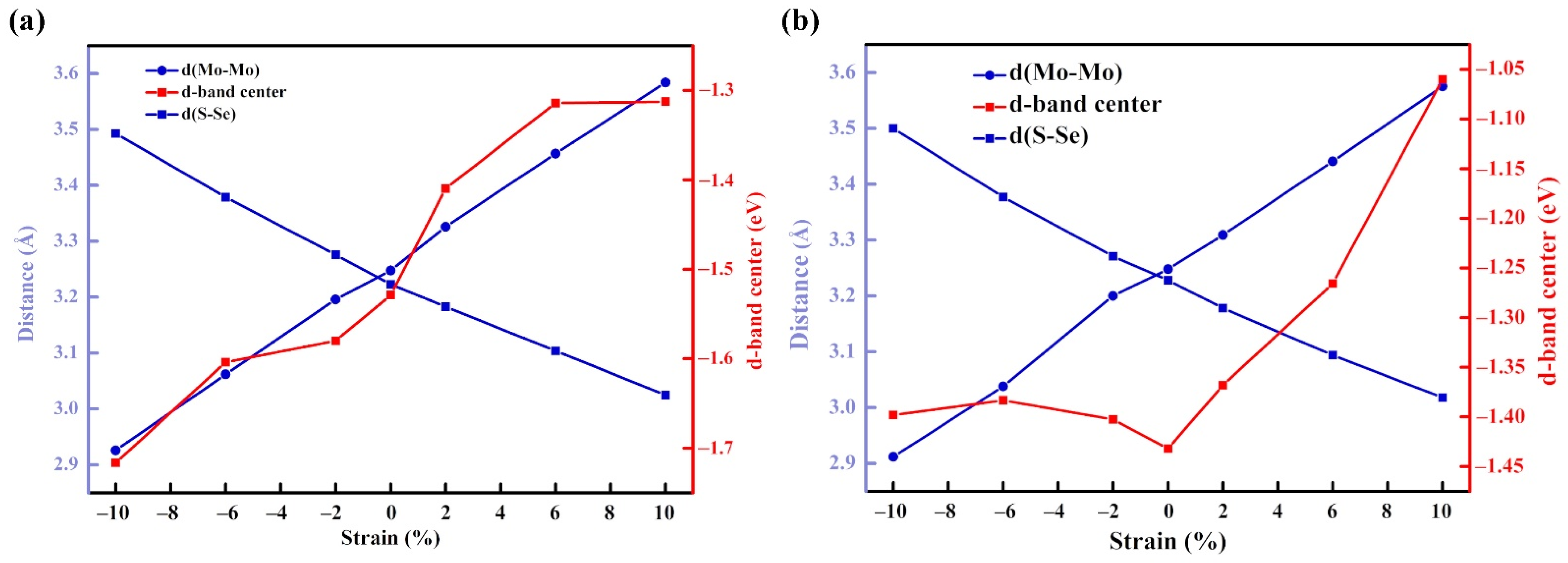
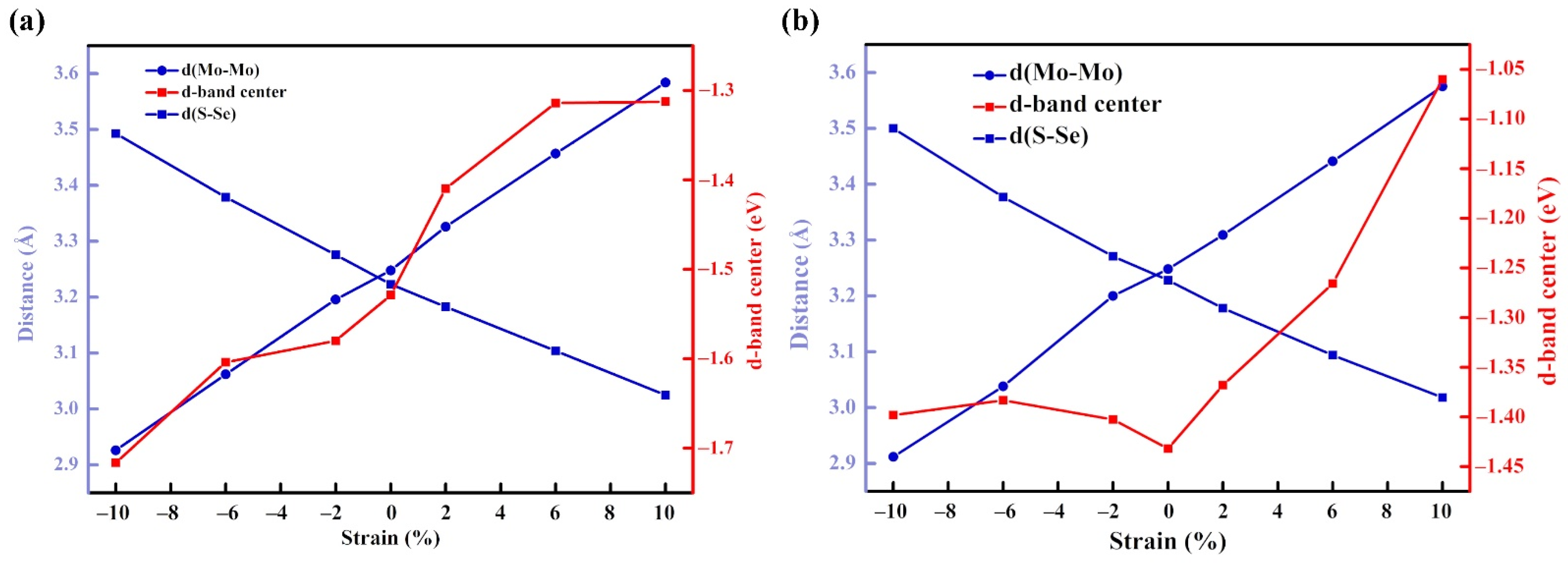
3.4. The Influence of Strain Effect
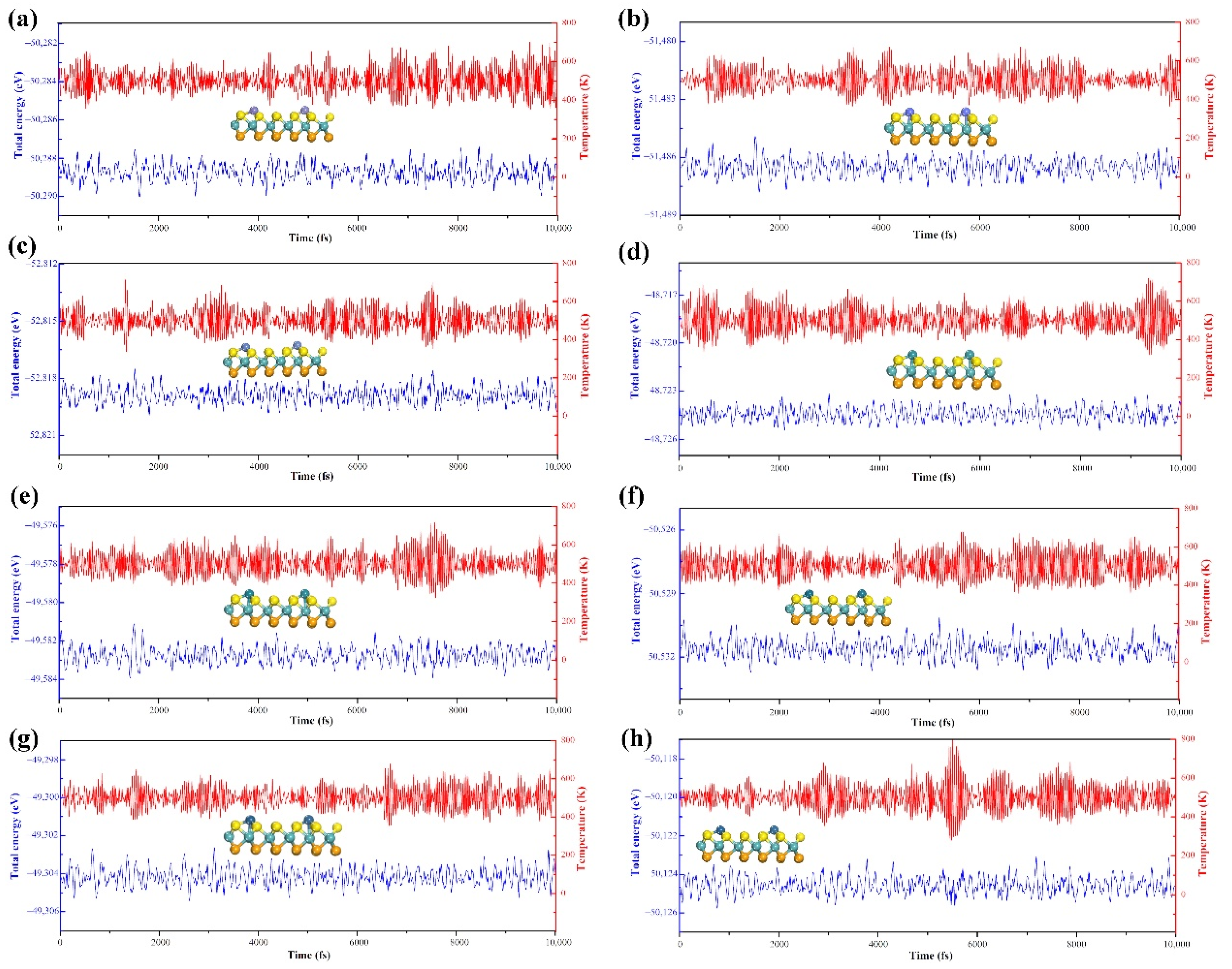
3.4.1. Structure and Stability
3.4.2. OER and HER Performance of Materials under Strain Effect
3.4.3. Mechanism Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledge
Conflicts of Interest
References
- Ball, M.; Weeda, M. The hydrogen economy—Vision or reality? Int. J. Hydrog. Energy 2015, 40, 7903–7919. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Thomas, I.L. Alternative energy technologies. Nature 2001, 414, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Najjar, Y.S.H. Hydrogen safety: The road toward green technology. Int. J. Hydrog. Energy 2013, 38, 10716–10728. [Google Scholar] [CrossRef]
- Turner, J.A. Sustainable hydrogen production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Wang, Y.L.; Wang, Q.L.; Liao, Z.J.; Zhang, N.Y.; Guo, Y.J.; Xiang, Z.H. Hierarchically porous metal-free carbon with record high mass activity for oxygen reduction and Zn-air batteries. J. Mater. Chem. A 2019, 7, 9831–9836. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, L.Z.; Du, A.J.; Gao, G.P.; Chen, J.; Yan, X.C.; Brown, C.L.; Yao, X.D. Defect Graphene as a Trifunctional Catalyst for Electrochemical Reactions. Adv. Mater. 2016, 28, 9532–9538. [Google Scholar] [CrossRef]
- Makimizu, Y.; Yoo, J.; Poornajar, M.; Nguyen, N.T.; Ahn, H.J.; Hwang, I.; Kment, S.; Schmuki, P. Effects of low oxygen annealing on the photoelectrochemical water splitting properties of alpha-Fe2O3. J. Mater. Chem. A 2020, 8, 1315–1325. [Google Scholar] [CrossRef]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.T.; Qiao, S.Z. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, X.Y.; Qin, J.Q.; Liu, R.P. Revealing Ni-based layered double hydroxides as high-efficiency electrocatalysts for the oxygen evolution reaction: A DFT study. J. Mater. Chem. A 2019, 7, 23091–23097. [Google Scholar] [CrossRef]
- Zhang, H.C.; Li, Y.J.; Zhang, G.X.; Xu, T.H.; Wan, P.B.; Sun, X.M. A metallic CoS2 nanopyramid array grown on 3D carbon fiber paper as an excellent electrocatalyst for hydrogen evolution. J. Mater. Chem. A 2015, 3, 6306–6310. [Google Scholar] [CrossRef]
- Cui, Z.T.; Sa, R.J.; Du, W.; Xiao, C.W.; Li, Q.H.; Ma, Z.J. Theoretical screening of group IIIA-VIIA elements doping to promote hydrogen evolution of MoS2 basal plane. Appl. Surf. Sci. 2021, 542, 148535. [Google Scholar] [CrossRef]
- Geng, X.M.; Sun, W.W.; Wu, W.; Chen, B.; Al-Hilo, A.; Benamara, M.; Zhu, H.L.; Watanabe, F.; Cui, J.B.; Chen, T.P. Pure and stable metallic phase molybdenum disulfide nanosheets for hydrogen evolution reaction. Nat. Commun. 2016, 7, 10672. [Google Scholar] [CrossRef] [PubMed]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jorgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Norskov, J.K. Biornimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, M.A.; Daniel, A.S.; Meng, F.; Forticaux, A.; Li, L.S.; Jin, S. Enhanced Hydrogen Evolution Catalysis from Chemically Exfoliated Metallic MoS2 Nanosheets. J. Am. Chem. Soc. 2013, 135, 10274–10277. [Google Scholar] [CrossRef]
- Tsai, C.; Chan, K.R.; Abild-Pedersen, F.; Norskov, J.K. Active edge sites in MoSe2 and WSe2 catalysts for the hydrogen evolution reaction: A density functional study. Phys. Chem. Chem. Phys. 2014, 16, 13156–13164. [Google Scholar] [CrossRef]
- Zhang, K.L.; Li, Y.H.; Deng, S.J.; Shen, S.H.; Zhang, Y.; Pan, G.X.; Xiong, Q.Q.; Liu, Q.; Xia, X.H.; Wang, X.L.; et al. Molybdenum Selenide Electrocatalysts for Electrochemical Hydrogen Evolution Reaction. Chemelectrochem 2019, 6, 3530–3548. [Google Scholar] [CrossRef]
- Jaramillo, T.F.; Jorgensen, K.P.; Bonde, J.; Nielsen, J.H.; Horch, S.; Chorkendorff, I. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 2007, 317, 100–102. [Google Scholar] [CrossRef]
- Li, H.; Tsai, C.; Koh, A.L.; Cai, L.L.; Contryman, A.W.; Fragapane, A.H.; Zhao, J.H.; Han, H.S.; Manoharan, H.C.; Abild-Pedersen, F.; et al. Activating and optimizing MoS2 basal planes for hydrogen evolution through the formation of strained sulphur vacancies. Nat. Mater. 2016, 15, 48. [Google Scholar] [CrossRef]
- Wang, H.T.; Tsai, C.; Kong, D.S.; Chan, K.R.; Abild-Pedersen, F.; Norskov, J.; Cui, Y. Transition-metal doped edge sites in vertically aligned MoS2 catalysts for enhanced hydrogen evolution. Nano Res. 2015, 8, 566–575. [Google Scholar] [CrossRef]
- Ye, G.L.; Gong, Y.J.; Lin, J.H.; Li, B.; He, Y.M.; Pantelides, S.T.; Zhou, W.; Vajtai, R.; Ajayan, P.M. Defects Engineered Monolayer MoS2 for Improved Hydrogen Evolution Reaction. Nano Lett. 2016, 16, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.-Y.; Zhu, H.; Xiao, J.; Chuu, C.-P.; Han, Y.; Chiu, M.-H.; Cheng, C.-C.; Yang, C.-W.; Wei, K.-H.; Yang, Y.; et al. Janus monolayers of transition metal dichalcogenides. Nat. Nanotechnol. 2017, 12, 744–749. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, S.; Kholmanov, I.; Dong, L.; Er, D.; Chen, W.; Guo, H.; Jin, Z.; Shenoy, V.B.; Shi, L.; et al. Janus Monolayer Transition-Metal Dichalcogenides. ACS Nano 2017, 11, 8192–8198. [Google Scholar] [CrossRef]
- Dong, L.; Lou, J.; Shenoy, V.B. Large In-Plane and Vertical Piezoelectricity in Janus Transition Metal Dichalchogenides. ACS Nano 2017, 11, 8242–8248. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Guo, Y.; Gao, H.; Guo, W. Tribo-piezoelectricity in Janus transition metal dichalcogenide bilayers: A first-principles study. Nano Energy 2019, 56, 33–39. [Google Scholar] [CrossRef]
- Li, L.; Li, B.; Guo, Q.; Li, B. Theoretical Screening of Single-Atom-Embedded MoSSe Nanosheets for Electrocatalytic N2 Fixation. J. Phys. Chem. C 2019, 123, 14501–14507. [Google Scholar] [CrossRef]
- Shi, W.; Li, G.; Wang, Z. Triggering Catalytic Active Sites for Hydrogen Evolution Reaction by Intrinsic Defects in Janus Monolayer MoSSe. J. Phys. Chem. C 2019, 123, 12261–12267. [Google Scholar] [CrossRef]
- Er, D.; Ye, H.; Frey, N.C.; Kumar, H.; Lou, J.; Shenoy, V.B. Prediction of Enhanced Catalytic Activity for Hydrogen Evolution Reaction in Janus Transition Metal Dichalcogenides. Nano Lett. 2018, 18, 3943–3949. [Google Scholar] [CrossRef]
- Ma, X.; Wu, X.; Wang, H.; Wang, Y. A Janus MoSSe monolayer: A potential wide solar-spectrum water-splitting photocatalyst with a low carrier recombination rate. J. Mater. Chem. A 2018, 6, 2295–2301. [Google Scholar] [CrossRef]
- Zhao, F.; Li, J.; Chen, Y.; Zhang, M.; Zhang, H. Photocatalytic activity of co-doped Janus monolayer MoSSe for solar water splitting: A computational investigation. Appl. Surf. Sci. 2021, 544, 148741. [Google Scholar] [CrossRef]
- Jain, A.; Bar Sadan, M.; Ramasubramaniam, A. Promoting Active Sites for Hydrogen Evolution in MoSe2 via Transition-Metal Doping. J. Phys. Chem. C 2020, 124, 12324–12336. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Hongzhiwei Technology, Device Studio, Version 2021A, China. 2021. Available online: https://iresearch.net.cn/cloud-software (accessed on 15 September 2022).
- Guo, X.; Lin, S.; Gu, J.; Zhang, S.; Chen, Z.; Huang, S. Simultaneously Achieving High Activity and Selectivity toward Two-Electron O2 Electroreduction: The Power of Single-Atom Catalysts. ACS Catal. 2019, 9, 11042–11054. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Norskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Valdes, A.; Qu, Z.W.; Kroes, G.J.; Rossmeisl, J.; Norskov, J.K. Oxidation and photo-oxidation of water on TiO2 surface. J. Phys. Chem. C 2008, 112, 9872–9879. [Google Scholar] [CrossRef]
- Li, J.J.; Jiang, Y.F.; Wang, Q.; Xu, C.Q.; Wu, D.J.; Banis, M.N.; Adair, K.R.; Doyle-Davis, K.; Meira, D.M.; Finfrock, Y.Z.; et al. A general strategy for preparing pyrrolic-N4 type single-atom catalysts via pre-located isolated atoms. Nat. Commun. 2021, 12, 6806. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Cao, L.; Zhang, W.; Chen, T.; Lin, Y.; Wang, H.; Wang, Y.; Yao, T. Active Sites of Single-Atom Iron Catalyst for Electrochemical Hydrogen Evolution. J. Phys. Chem. Lett. 2020, 11, 6691–6696. [Google Scholar] [CrossRef] [PubMed]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TM@MoSSe | q (Mo) | q (S) | q (Se) | q (TM) |
|---|---|---|---|---|
| Fe | 1.063 | −0.670 | −0.462 | 0.609 |
| Co | 1.074 | −0.662 | −0.463 | 0.466 |
| Ni | 1.077 | −0.654 | −0.464 | 0.379 |
| Ru | 1.078 | −0.651 | −0.467 | 0.368 |
| Rh | 1.076 | −0.636 | −0.466 | 0.227 |
| Pd | 1.080 | −0.637 | −0.466 | 0.206 |
| Ir | 1.065 | −0.620 | −0.461 | 0.144 |
| Pt | 1.068 | −0.614 | −0.460 | 0.044 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Wang, X.; Zheng, M.; Zhou, X. Improving Catalytic Activity of “Janus” MoSSe Based on Surface Interface Regulation. Molecules 2022, 27, 6038. https://doi.org/10.3390/molecules27186038
Wang M, Wang X, Zheng M, Zhou X. Improving Catalytic Activity of “Janus” MoSSe Based on Surface Interface Regulation. Molecules. 2022; 27(18):6038. https://doi.org/10.3390/molecules27186038
Chicago/Turabian StyleWang, Mingqian, Xin Wang, Ming Zheng, and Xin Zhou. 2022. "Improving Catalytic Activity of “Janus” MoSSe Based on Surface Interface Regulation" Molecules 27, no. 18: 6038. https://doi.org/10.3390/molecules27186038
APA StyleWang, M., Wang, X., Zheng, M., & Zhou, X. (2022). Improving Catalytic Activity of “Janus” MoSSe Based on Surface Interface Regulation. Molecules, 27(18), 6038. https://doi.org/10.3390/molecules27186038