

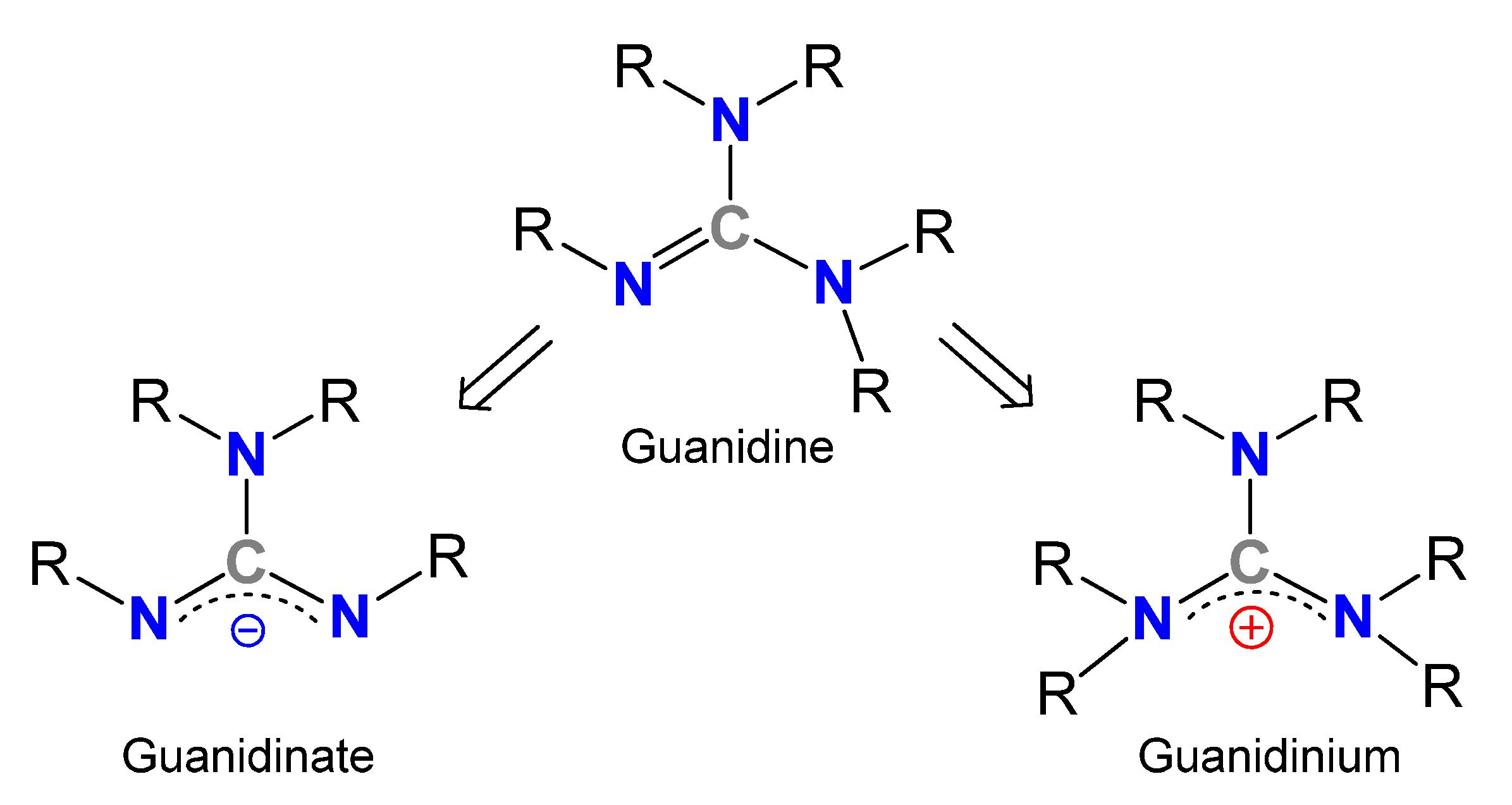
Guanidinates as Alternative Ligands for Organometallic Complexes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
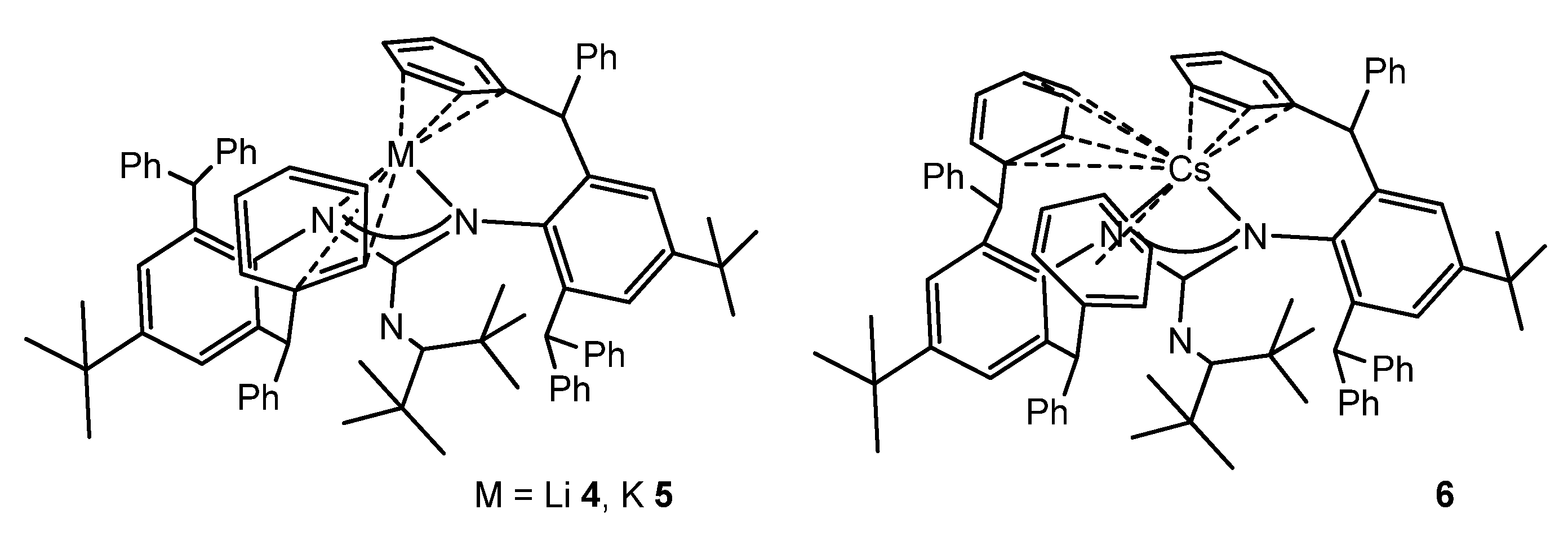
{kind=link}
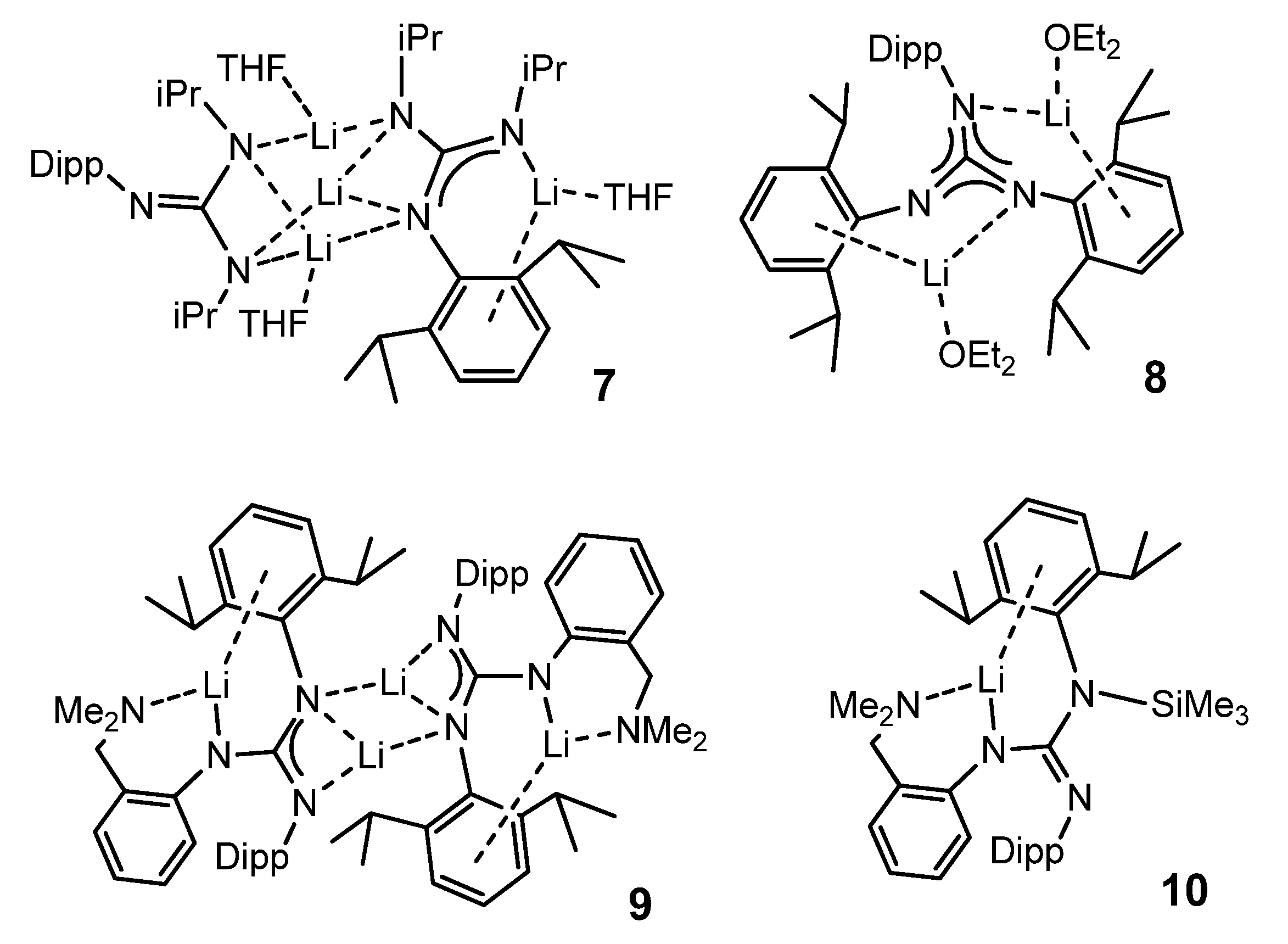
{kind=link}
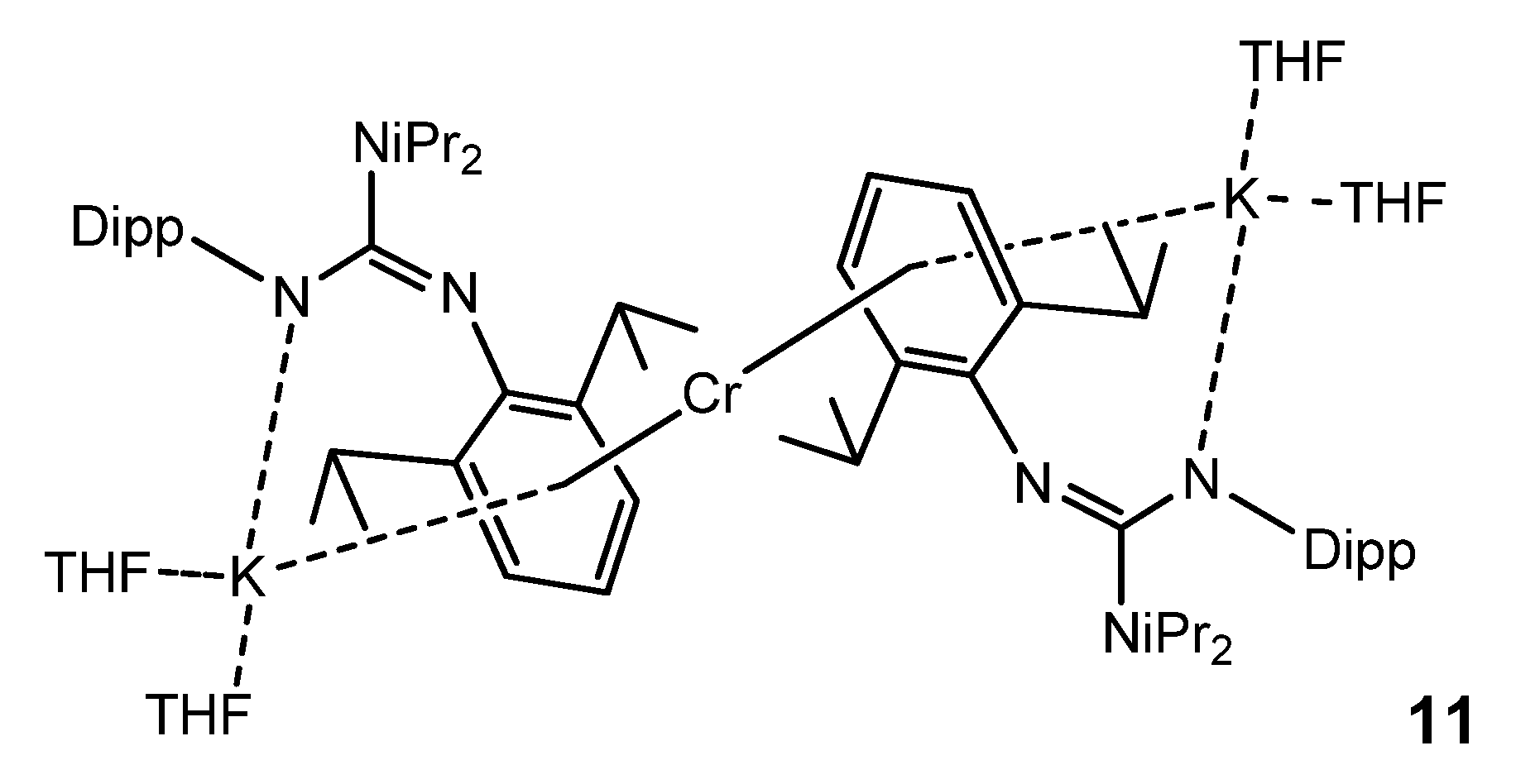
{kind=link}
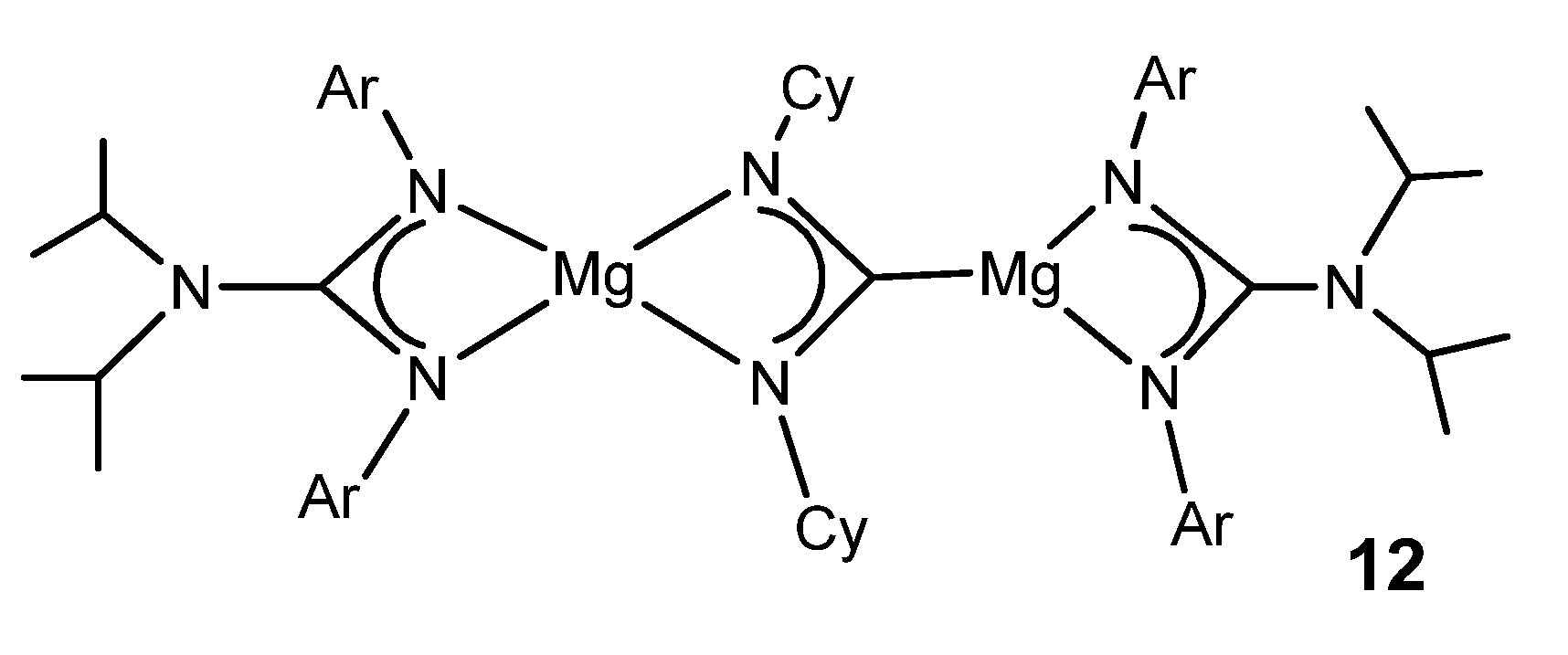
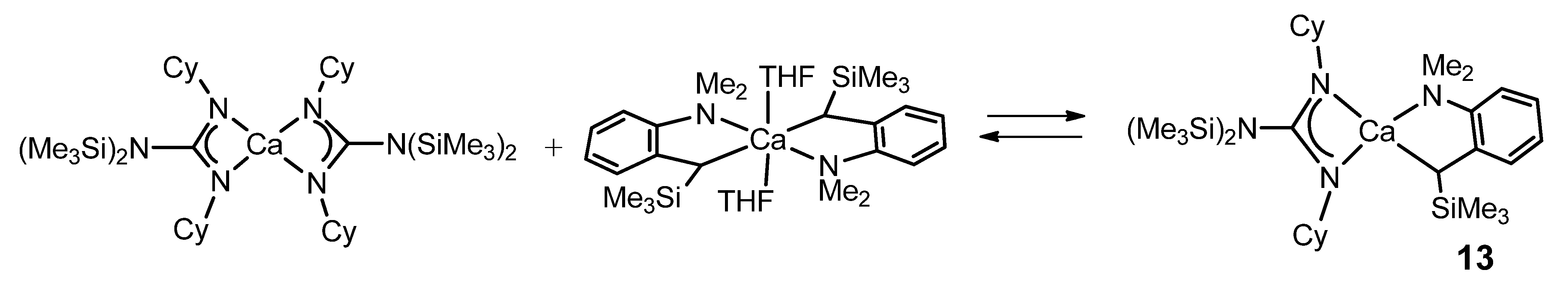{kind=link}
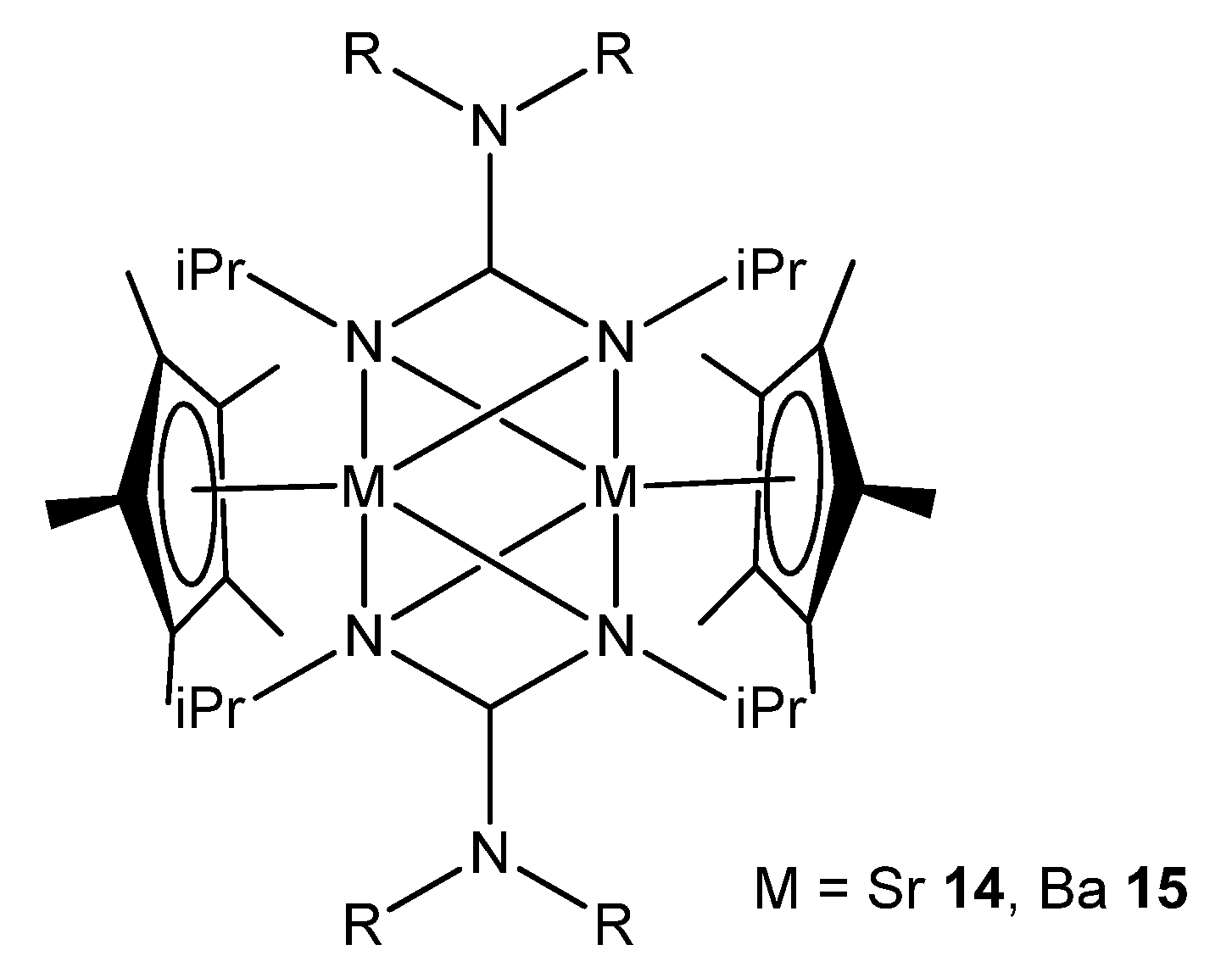{kind=link}
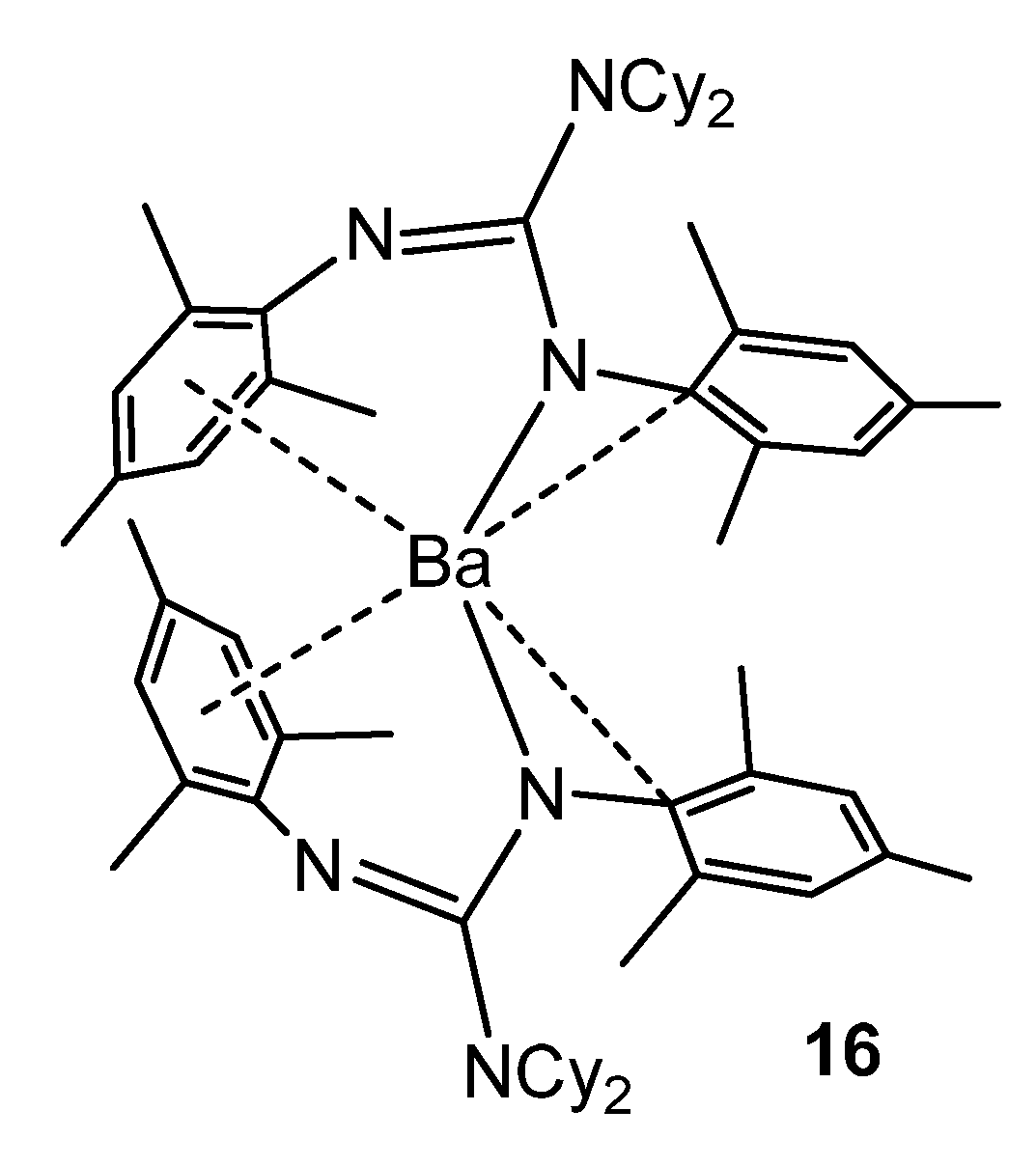{kind=link}
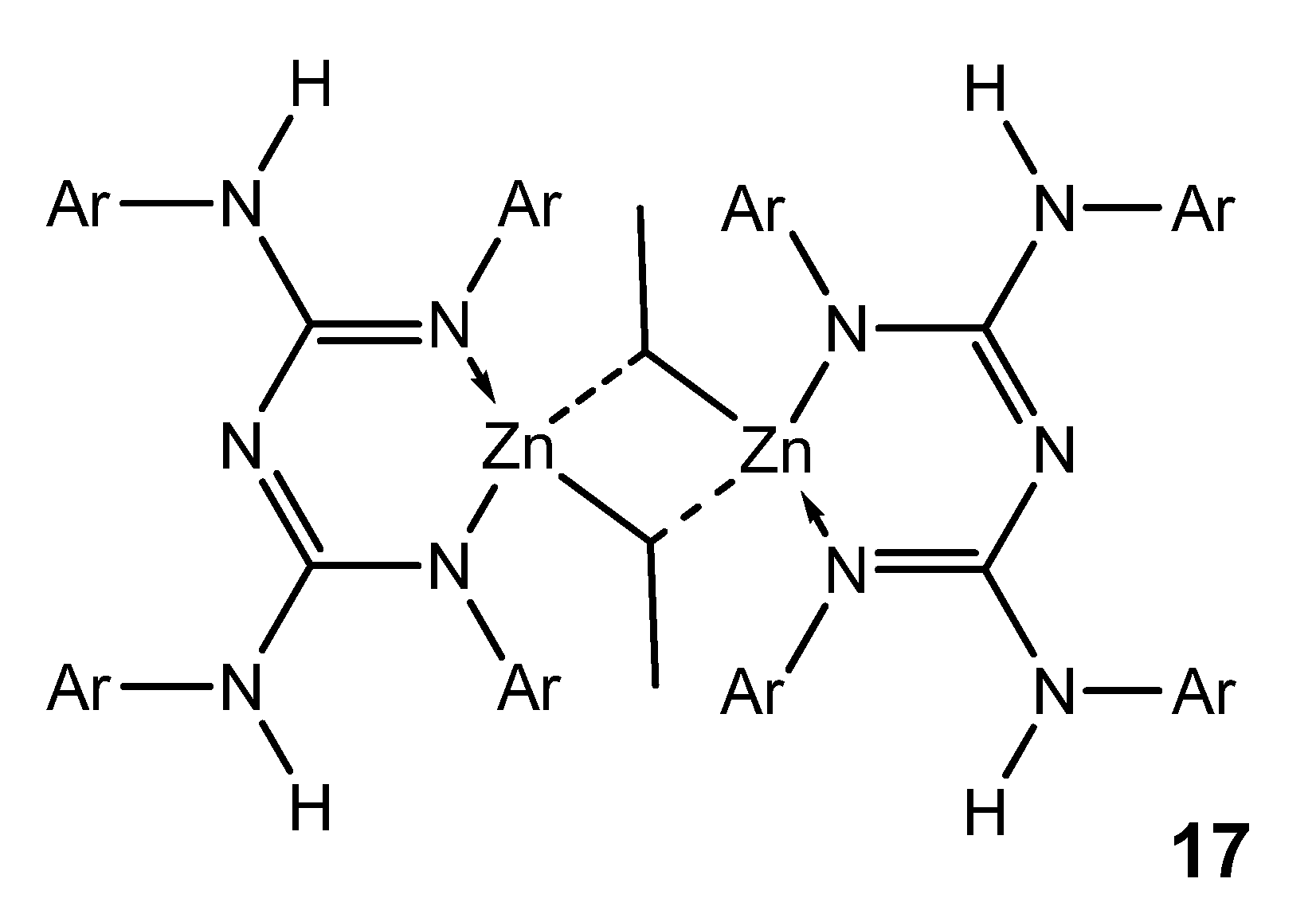{kind=link}
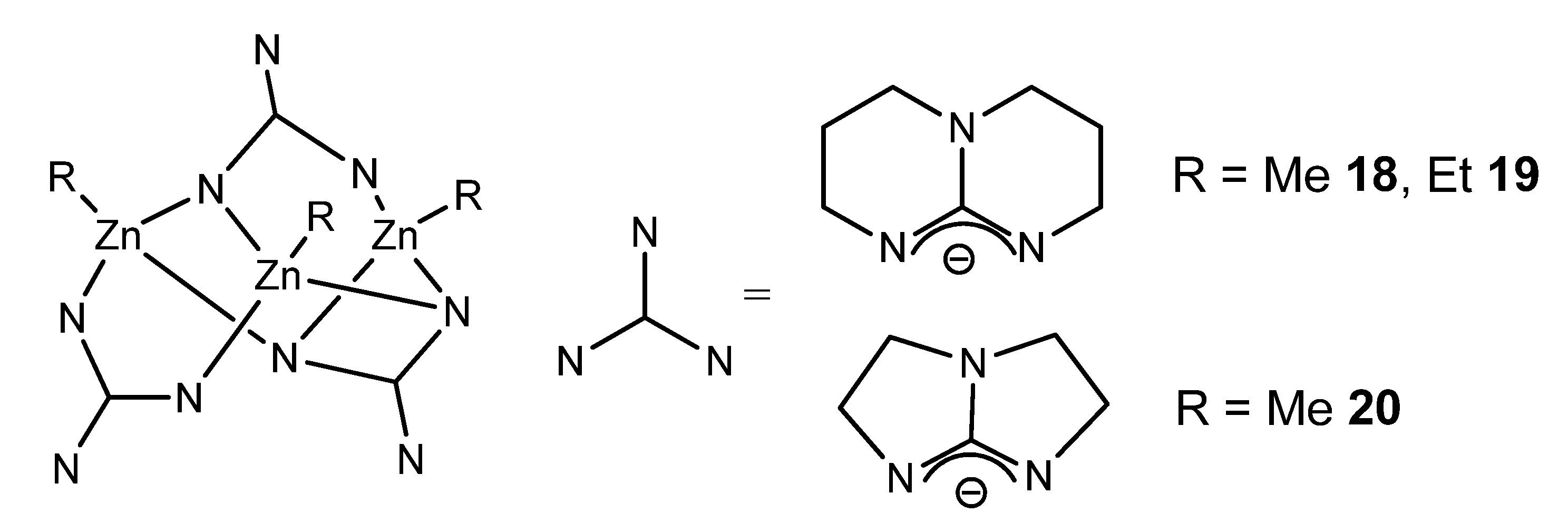{kind=link}
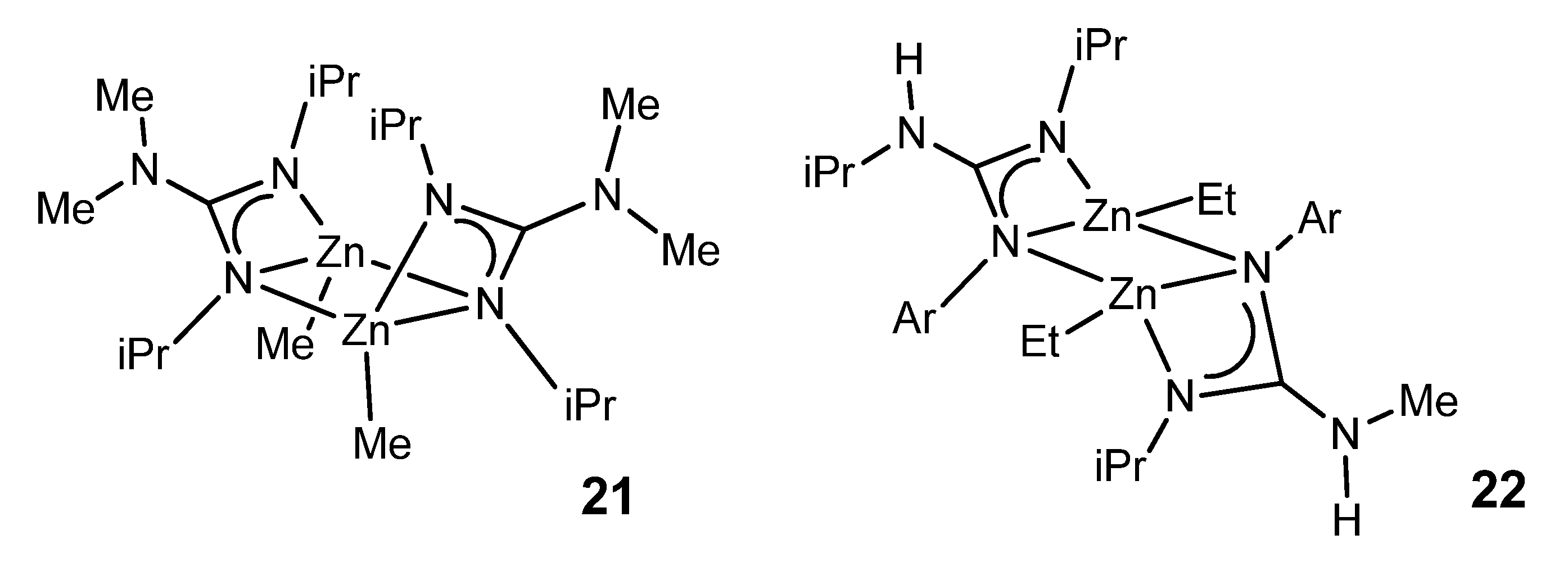{kind=link}
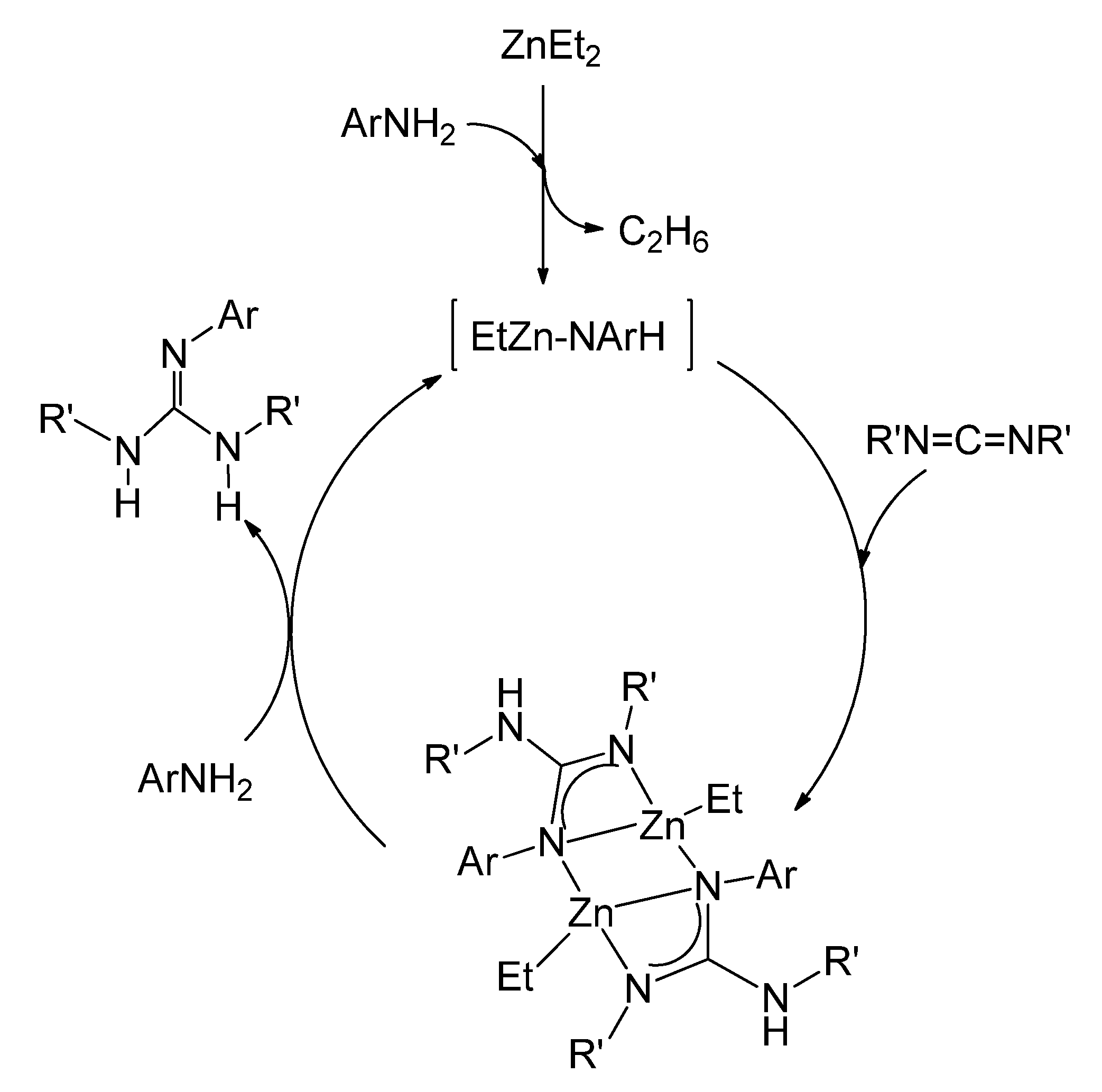{kind=link}
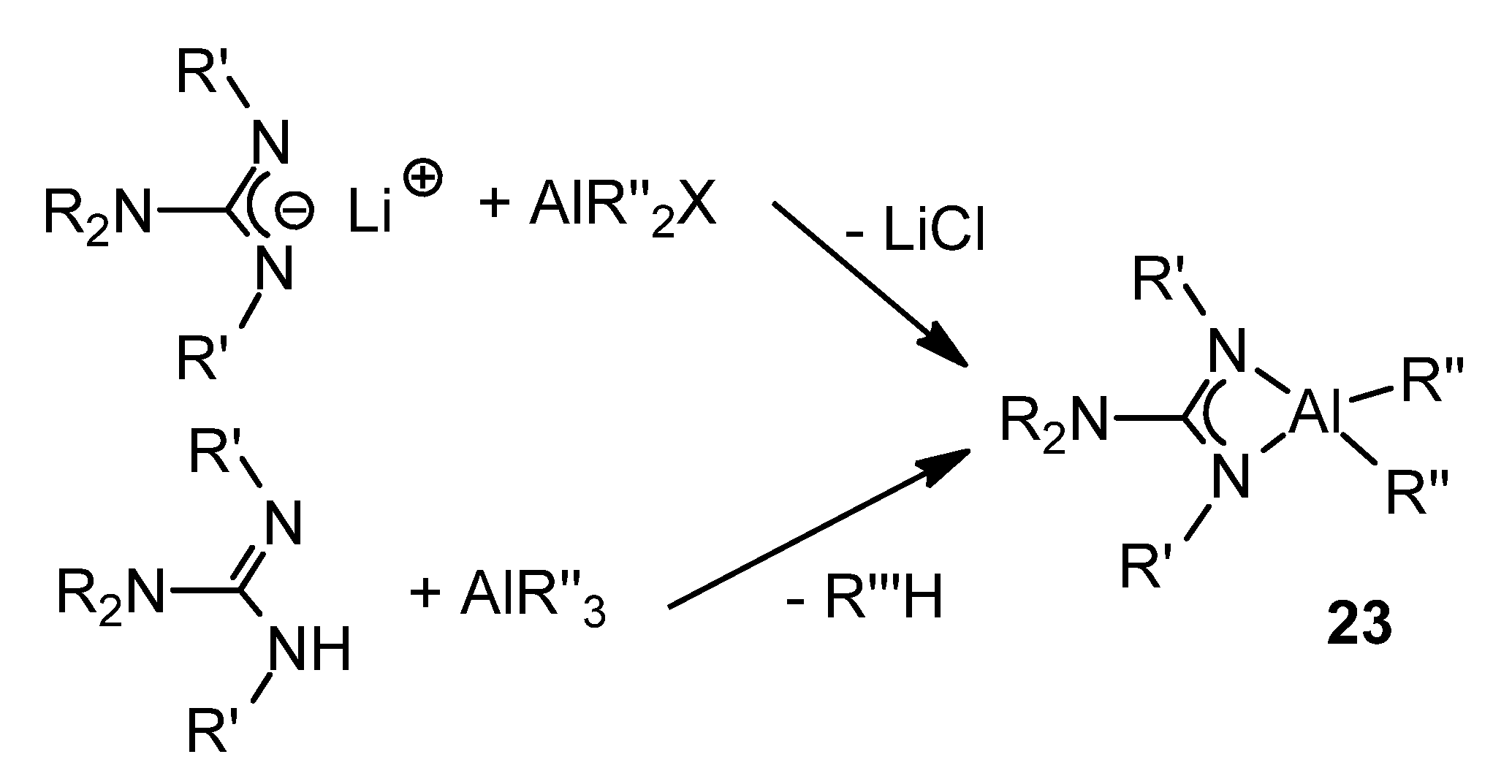{kind=link}
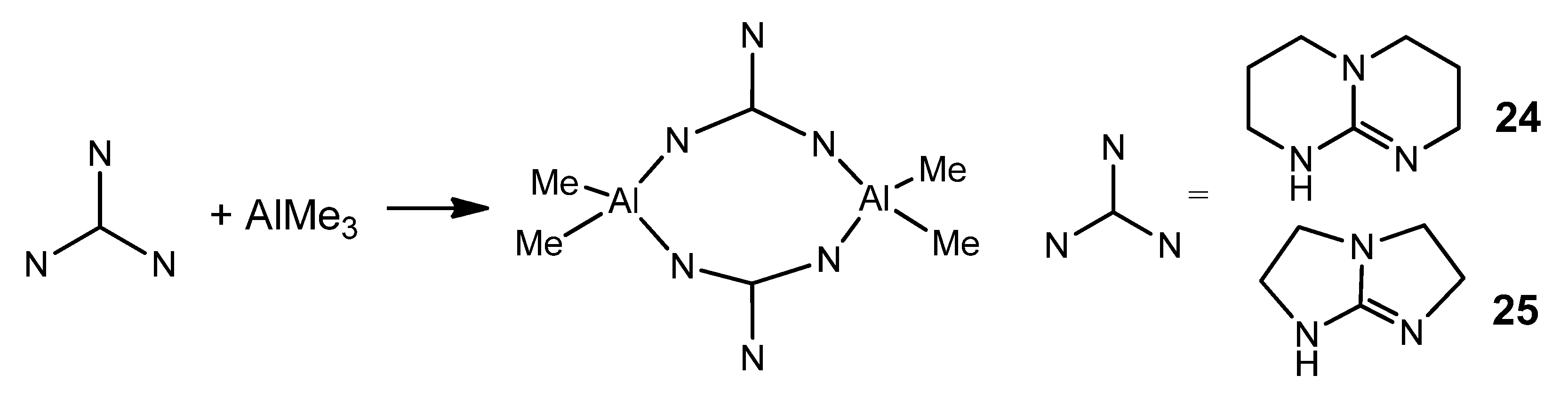{kind=link}
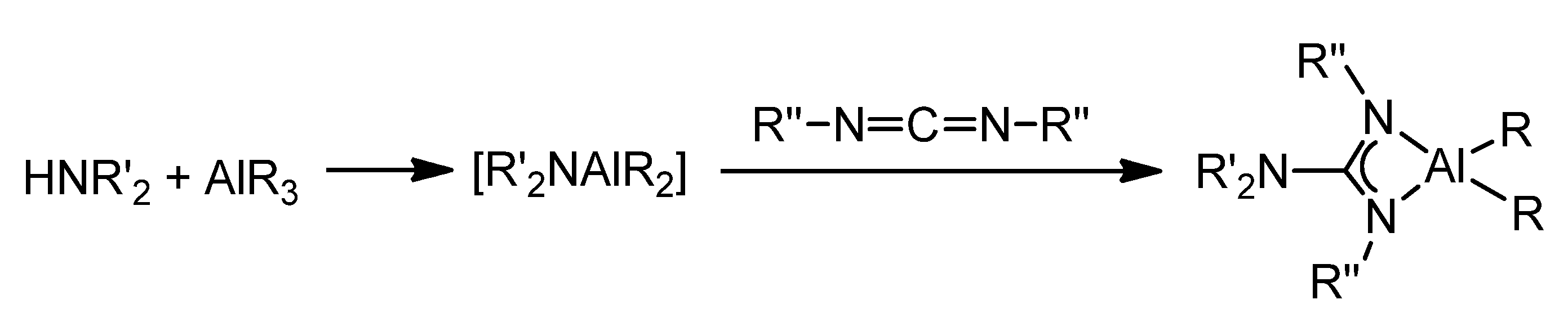{kind=link}
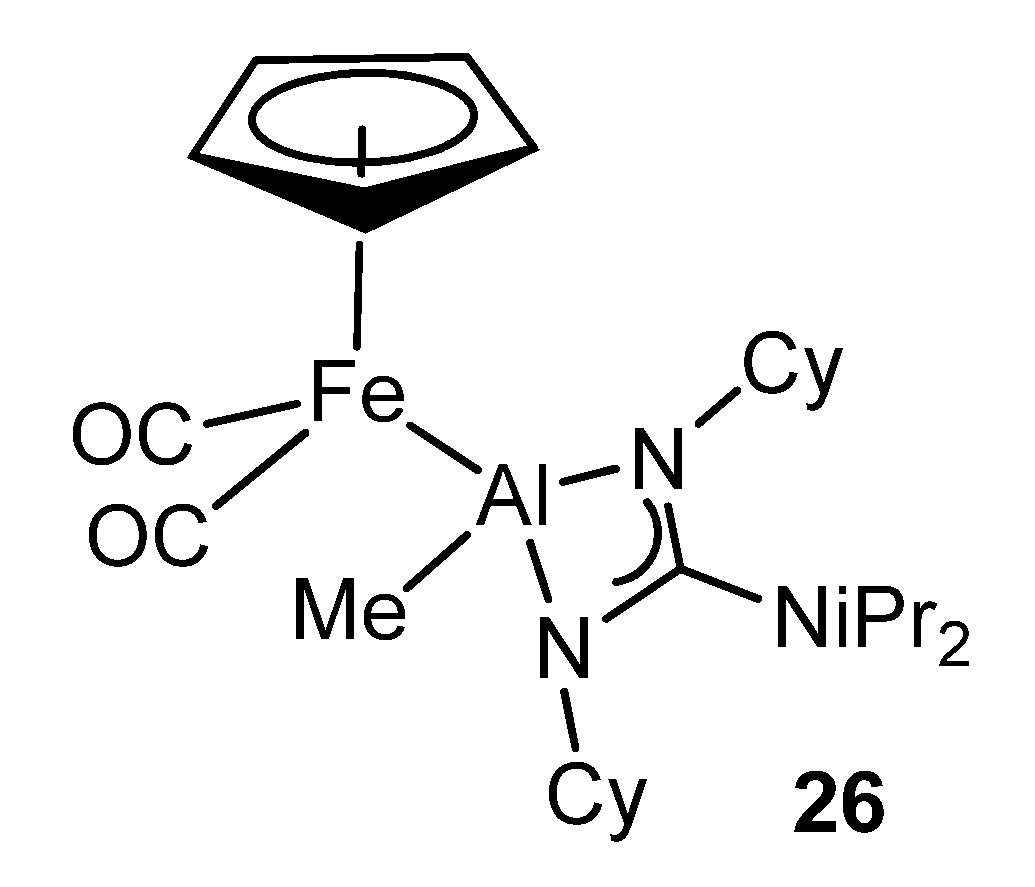{kind=link}
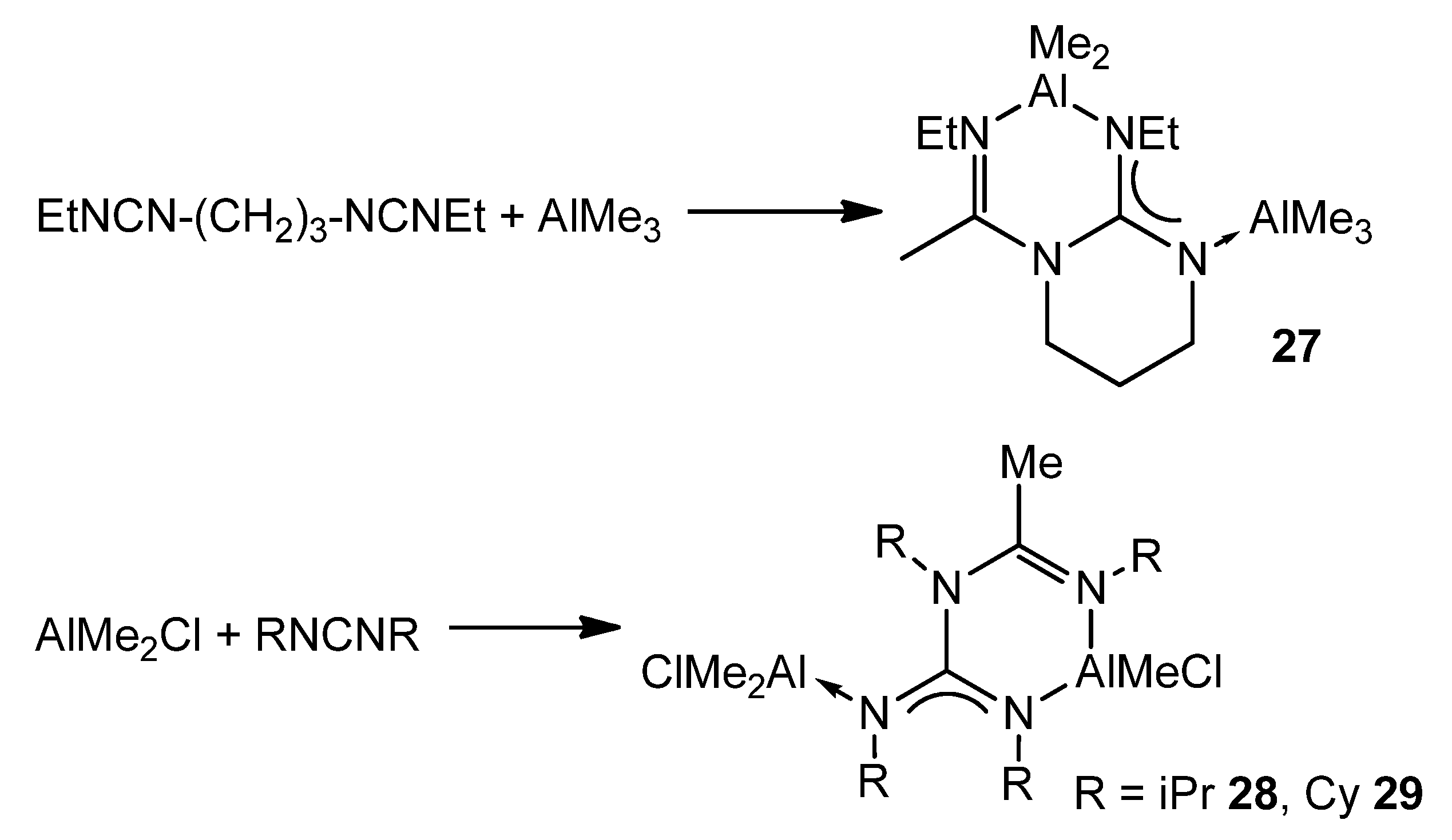{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
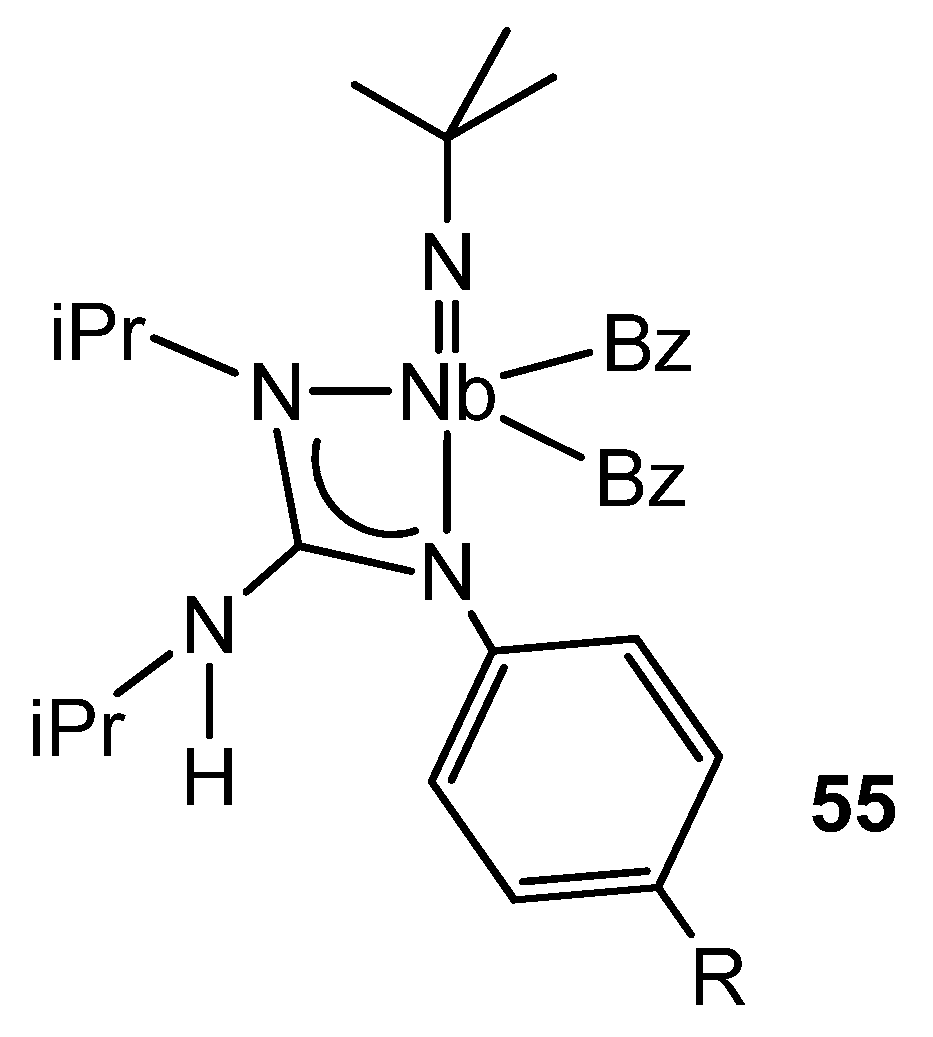{kind=link}
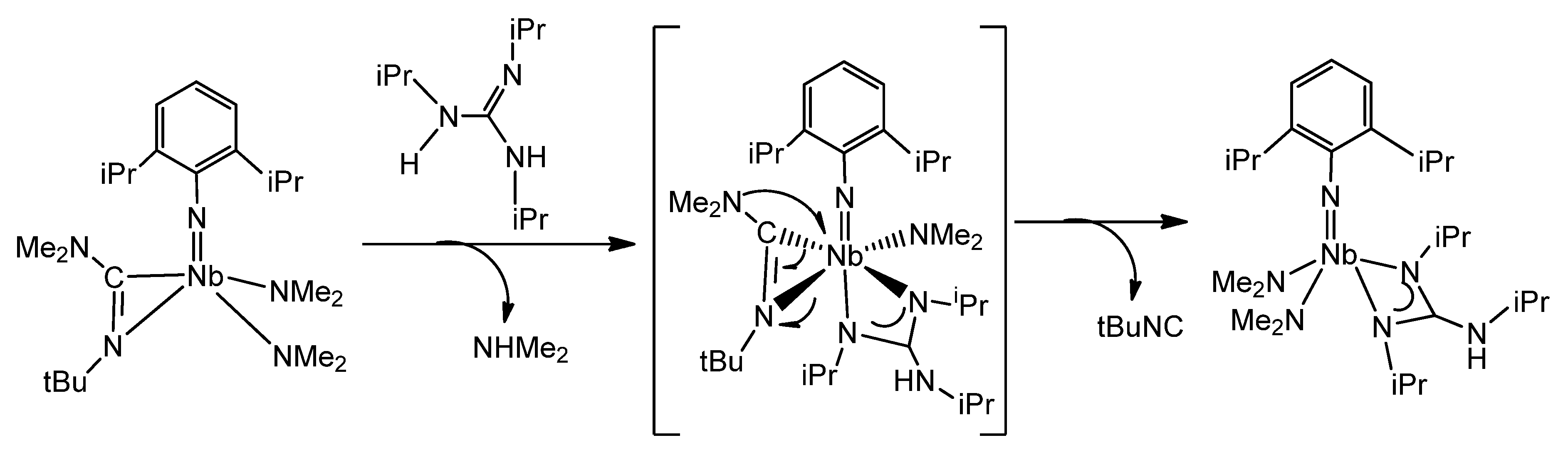{kind=link}
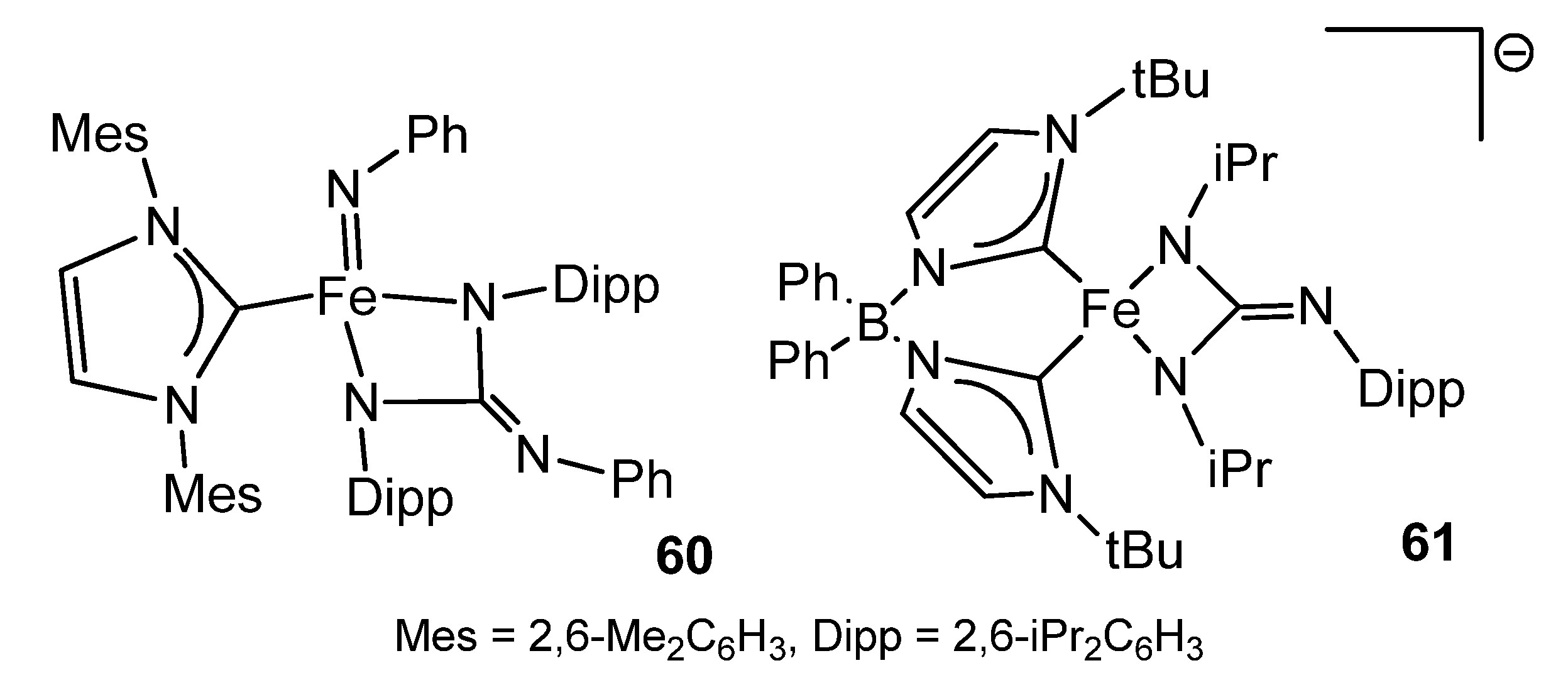{kind=link}
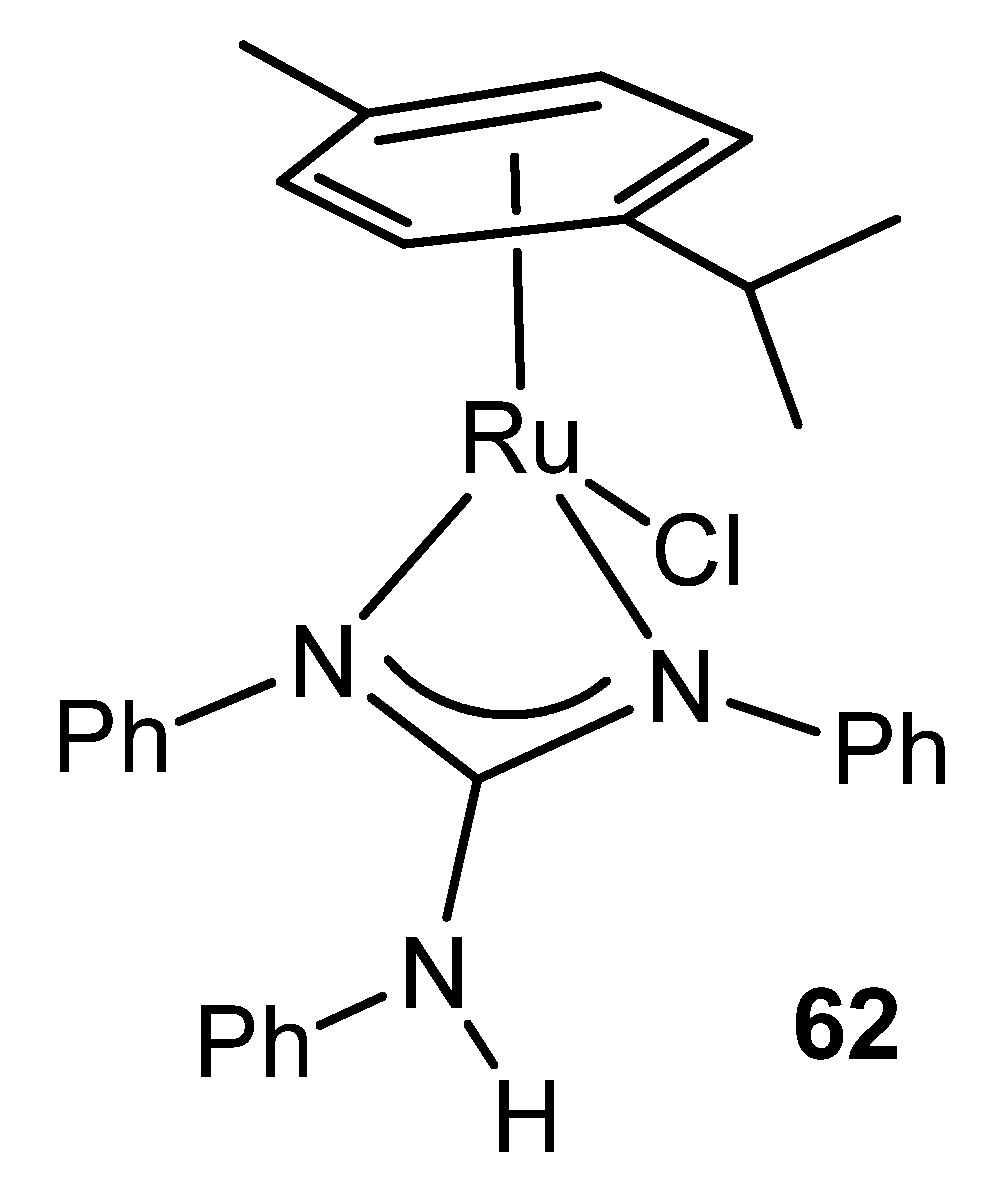{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
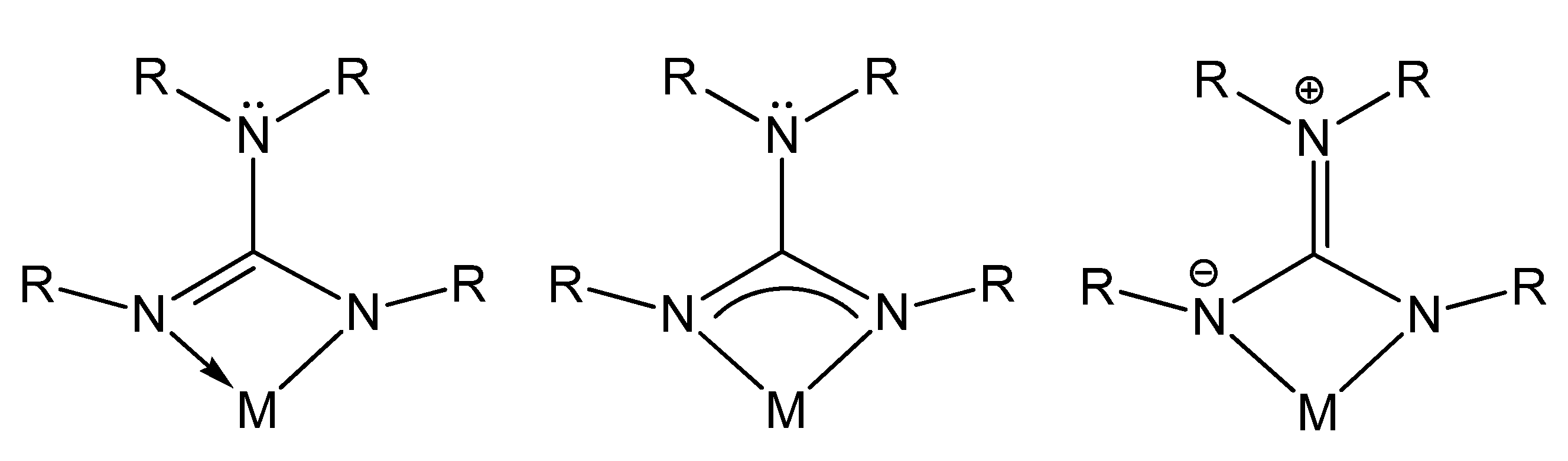
1. Introduction
2. Organometallic Chemistry of Guanidines
2.1. Main Group Complexes
2.2. Transition Metal Complexes
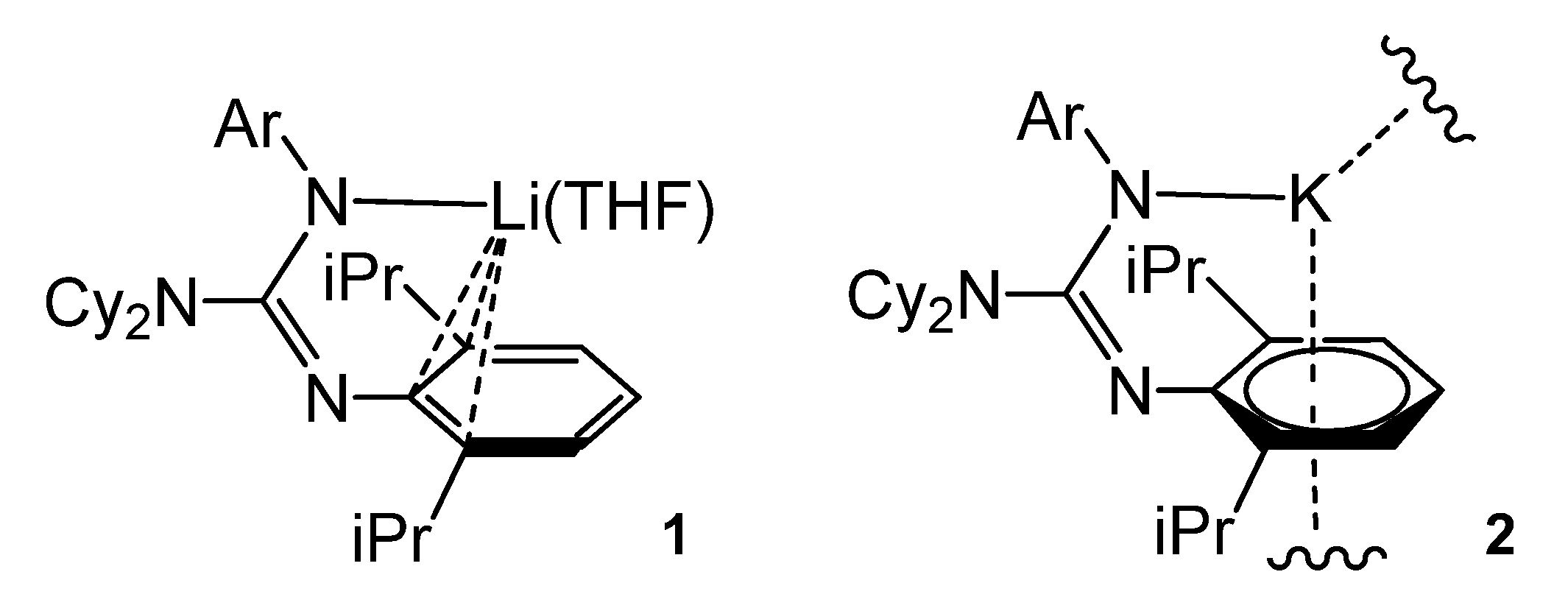
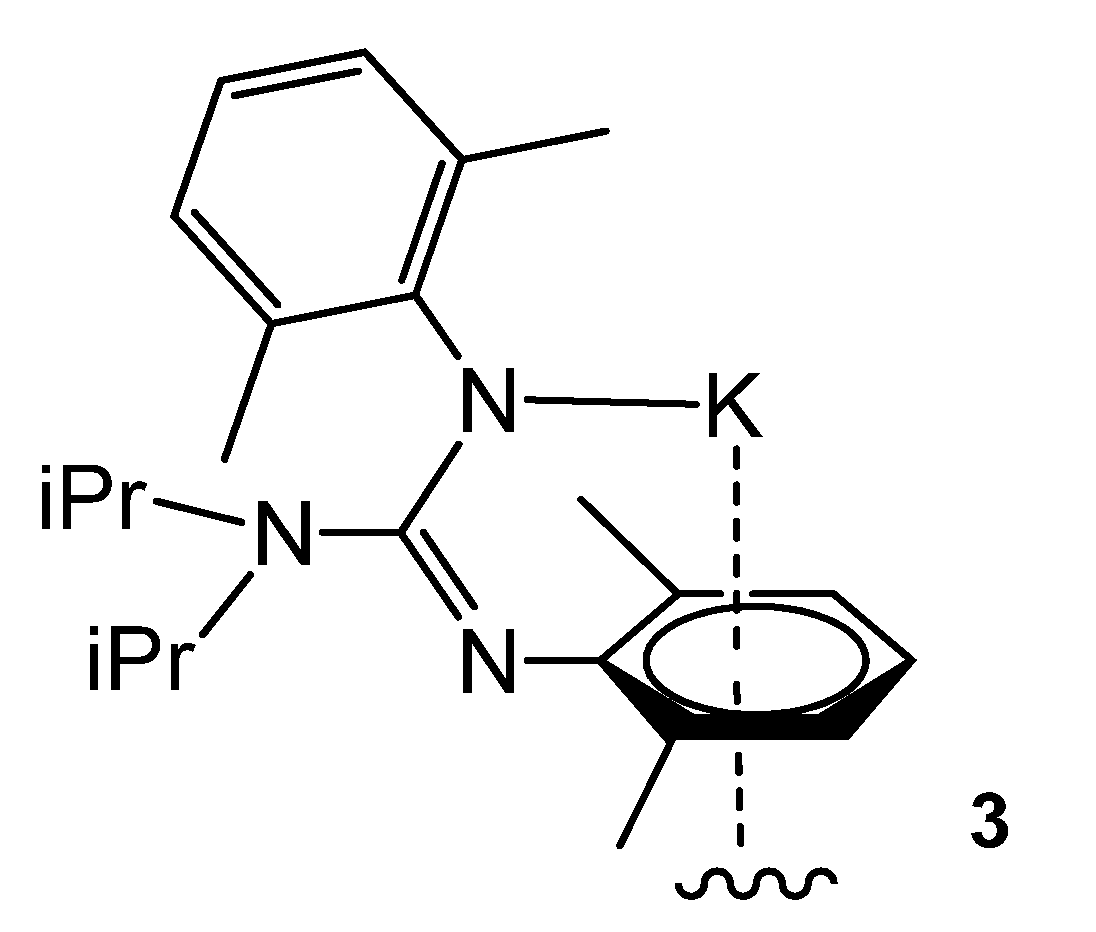
2.3. Group 3 and the Lanthanoid Complexes
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ishikawa, T.; Kumamoto, T. Guanidines in Organic Synthesis. Synthesis 2006, 2006, 737–752. [Google Scholar] [CrossRef]
- Ishikawa, T. Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Berlinck, R.G.S. Natural guanidine derivatives. Nat. Prod. Rep. 1999, 16, 339–365. [Google Scholar] [CrossRef]
- Berlinck, R.G.; Trindade-Silva, A.E.; Santos, M.F. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2012, 29, 1382–1406. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Kossuga, M.H. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2008, 25, 919–954. [Google Scholar] [CrossRef]
- Alegre-Requena, J.V.; Marqués-López, E.; Herrera, R.P. Guanidine Motif in Biologically Active Peptides. Aust. J. Chem. 2014, 67, 965–971. [Google Scholar] [CrossRef]
- Berlinck, R.G.S.; Romminger, S. The chemistry and biology of guanidine natural products. Nat. Prod. Rep. 2016, 33, 456–490. [Google Scholar] [CrossRef]
- Kim, S.-H.; Semenya, D.; Castagnolo, D. Antimicrobial drugs bearing guanidine moieties: A review. Eur. J. Med. Chem. 2021, 216, 113293. [Google Scholar] [CrossRef]
- Oliver, D.W.; Dormehl, I.C.; Wikberg, J.E.S.; Dambrova, M. Guanidines: From molecule to primate. Med. Chem. Res. 2004, 13, 427–438. [Google Scholar] [CrossRef]
- Ishikawa, T. Guanidine Chemistry. Chem. Pharm. Bull. 2010, 58, 1555–1564. [Google Scholar] [CrossRef]
- Fu, X.; Tan, C.-H. Mechanistic considerations of guanidine-catalyzed reactions. Chem. Commun. 2011, 47, 8210–8222. [Google Scholar] [CrossRef]
- Corey, E.J.; Grogan, M. Enantioselective synthesis of alpha-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. J. Org. Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef]
- Ishikawa, T.; Araki, Y.; Kumamoto, T.; Seki, H.; Fukuda, K.; Isobe, T. Modified guanidines as chiral superbases: Application to asymmetric Michael reaction of glycine imine with acrylate or its related compounds. Chem. Commun. 2001, 245–246. [Google Scholar] [CrossRef]
- Shen, J.; Nguyen, T.T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. Chiral Bicyclic Guanidine-Catalyzed Enantioselective Reactions of Anthrones. J. Am. Chem. Soc. 2006, 128, 13692–13693. [Google Scholar] [CrossRef]
- Allingham, M.T.; Howard-Jones, A.; Murphy, P.J.; Thomas, D.A.; Caulkett, P.W.R. Synthesis and applications of C2 Symmetric Guanidine Bases. Tetrahedron Lett. 2003, 44, 8677–8680. [Google Scholar] [CrossRef]
- Sohtome, Y.; Hashimoto, Y.; Nagasawa, K. Guanidine-Thiourea Bifunctional Organocatalyst for the Asymmetric Henry (Nitroaldol) Reaction. Adv. Synth. Catal. 2005, 347, 1643–1648. [Google Scholar] [CrossRef]
- Terada, M.; Ube, H.; Yaguchi, Y. Axially Chiral Guanidine as Enantioselective Base Catalyst for 1,4-Addition Reaction of 1,3-Dicarbonyl Compounds with Conjugated Nitroalkenes. J. Am. Chem. Soc. 2006, 128, 1454–1455. [Google Scholar] [CrossRef]
- Fu, X.; Jiang, Z.; Tan, C.-H. Bicyclic guanidine-catalyzed enantioselective phospha-Michael reaction: Synthesis of chiral β-aminophosphine oxides and β-aminophosphines. Chem. Commun. 2007, 5058–5060. [Google Scholar] [CrossRef]
- Ye, W.; Jiang, Z.; Zhao, Y.; Goh, S.L.M.; Leow, D.; Soh, Y.-T.; Tan, C.-H. Chiral Bicyclic Guanidine as a Versatile Brønsted Base Catalyst for the Enantioselective Michael Reactions of Dithiomalonates and β-Keto Thioesters. Adv. Synth. Catal. 2007, 349, 2454–2458. [Google Scholar] [CrossRef]
- Leow, D.; Lin, S.; Chittimalla, S.K.; Fu, X.; Tan, C.-H. Enantioselective Protonation Catalyzed by a Chiral Bicyclic Guanidine Derivative. Angew. Chem. Int. Ed. 2008, 47, 5641–5645. [Google Scholar] [CrossRef]
- Terada, M.; Ikehara, T.; Ube, H. Enantioselective 1, 4-addition reactions of diphenyl phosphite to nitroalkenes catalyzed by an axially chiral guanidine. J. Am. Chem. Soc. 2007, 129, 14112–14113. [Google Scholar] [CrossRef]
- Ye, W.; Xu, J.; Tan, C.T.; Tan, C.-H. 1, 5, 7-Triazabicyclo[4.4.0]dec-5-ene (TBD) catalyzed Michael reactions. Tetrahedron Lett. 2005, 46, 6875–6878. [Google Scholar] [CrossRef]
- Kita, T.; Georgieva, A.; Hashimoto, Y.; Nakata, T.; Nagasawa, K. C2-Symmetric Chiral Pentacyclic Guanidine: A Phase-Transfer Catalyst for the Asymmetric Alkylation of tert-Butyl Glycinate Schiff Base. Angew. Chem. Int. Ed. 2002, 41, 2832–2834. [Google Scholar] [CrossRef]
- Ishikawa, T.; Isobe, T. Modified guanidines as chiral auxiliaries. Chem. Eur. J. 2002, 8, 553–557. [Google Scholar] [CrossRef]
- Terada, M.; Nakano, M.; Ube, H. Axially chiral guanidine as highly active and enantioselective catalyst for electrophilic amination of unsymmetrically substituted 1, 3-dicarbonyl compounds. J. Am. Chem. Soc. 2006, 128, 16044–16045. [Google Scholar] [CrossRef]
- Pratt, R.C.; Lohmeijer, B.G.G.; Long, D.A.; Waymouth, R.M.; Zhang, S.; He, L.-N. Capture and fixation of CO2 promoted by guanidine derivatives. Aust. J. Chem. 2014, 67, 980–988. [Google Scholar]
- Mesías-Salazar, Á.; Martínez, J.; Rojas, R.S.; Carrillo-Hermosilla, F.; Ramos, A.; Fernández-Galán, R.; Antiñolo, A. Aromatic guanidines as highly active binary catalytic systems for the fixation of CO2 into cyclic carbonates under mild conditions. Catal. Sci. Technol. 2019, 9, 3879–3886. [Google Scholar] [CrossRef]
- Claver, C.; Yeamin, M.B.; Reguero, M.; Masdeu-Bultó, A.M. Recent advances in the use of catalysts based on natural products for the conversion of CO2 into cyclic carbonates. Green Chem. 2020, 22, 7665–7706. [Google Scholar] [CrossRef]
- Das Neves Gomes, C.; Blondiaux, E.; Thuéry, P.; Cantat, T. Metal-Free Reduction of CO2 with Hydroboranes: Two Efficient Pathways at Play for the Reduction of CO2 to Methanol. Chem. Eur. J. 2014, 20, 7098–7106. [Google Scholar] [CrossRef] [PubMed]
- von Wolff, N.; Lefèvre, G.; Berthet, J.-C.; Thuéry, P.; Cantat, T. Implications of CO2 Activation by Frustrated Lewis Pairs in the Catalytic Hydroboration of CO2: A View Using N/Si+ Frustrated Lewis Pairs. ACS Catal. 2016, 6, 4526–4535. [Google Scholar] [CrossRef]
- Simon, L.; Goodman, J.M. The mechanism of TBD-catalyzed ring-opening polymerization of cyclic esters. J. Org. Chem. 2007, 72, 9656–9662. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Feng, X.; Liu, X. Chiral guanidines and their derivatives in asymmetric synthesis. Chem. Soc. Rev. 2018, 47, 8525–8540. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Rogovoy, B.V. Recent developments in guanylating agents. Arkivoc 2005, 4, 49–87. [Google Scholar] [CrossRef]
- Ishikawa, T. Roads to new guanidine chemistry from 2-imidazolidinones through 2-chloroamidinium derivatives. Arkivoc 2006, 7, 148–168. [Google Scholar] [CrossRef]
- Rauws, T.R.M.; Maes, B.U.W. Transition metal-catalyzed N-arylations of amidines and guanidines. Chem. Soc. Rev. 2012, 41, 2463–2497. [Google Scholar] [CrossRef]
- Alonso-Moreno, C.; Antiñolo, A.; Carrillo-Hermosilla, F.; Otero, A. Guanidines: From classical approaches to efficient catalytic syntheses. Chem. Soc. Rev. 2014, 43, 3406–3425. [Google Scholar] [CrossRef]
- Tahir, S.; Badshah, A.; Hussain, R.A. Guanidines from ‘toxic substances’ to compounds with multiple biological applications—Detailed outlook on synthetic procedures employed for the synthesis of guanidines. Bioorg. Chem. 2015, 59, 39–79. [Google Scholar] [CrossRef]
- Chandra, G.; Jenkins, A.D.; Lappert, M.F.; Srivastava, R.C. Amido-derivatives of metals and metalloids. Part X. Reactions of titanium(IV), zirconium(IV), and hafnium(IV) amides with unsaturated substrates, and some related experiments with amides of boron, silicon, germanium, and tin(IV). J. Chem. Soc. A 1970, 2550–2558. [Google Scholar]
- Bailey, P.J.; Pace, S. The coordination chemistry of guanidines and guanidinates. Coord. Chem. Rev. 2001, 214, 91–141. [Google Scholar] [CrossRef]
- Coles, M.P. Application of neutral amidines and guanidines in coordination chemistry. Dalton Trans. 2006, 985–1001. [Google Scholar] [CrossRef]
- Edelmann, F.T. Advances in the Coordination Chemistry of Amidinate and Guanidinate Ligands. Organomet. Chem. 2008, 57, 183–352. [Google Scholar]
- Edelmann, F.T. Recent progress in the chemistry of metal amidinates and guanidinates: Syntheses, catalysis and materials. Adv. Organomet. Chem. 2013, 61, 55–374. [Google Scholar]
- Edelmann, F.T. Lanthanide amidinates and guanidinates: From laboratory curiosities to efficient homogeneous catalysts and precursors for rare-earth oxide thin films. Chem. Soc. Rev. 2009, 38, 2253–2268. [Google Scholar] [CrossRef]
- Edelmann, F.T. Lanthanide amidinates and guanidinates in catalysis and materials science: A continuing success story. Chem. Soc. Rev. 2012, 41, 7657–7672. [Google Scholar] [CrossRef]
- Trifonov, A.A. Guanidinate and amidopyridinate rare-earth complexes: Towards highly reactive alkyl and hydrido species. Coord. Chem. Rev. 2010, 254, 1327–1347. [Google Scholar] [CrossRef]
- Sengupta, D.; Gómez-Torres, A.; Fortier, S. Amidinates, Formamidinates, and Guanidinates. In Comprehensive Coordination Chemistry III; Constable, E.C., Parkin, G., Que, L., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2021; pp. 366–405. [Google Scholar]
- Jin, G.; Jones, C.; Junk, P.C.; Lippert, K.-A.; Rose, R.P.; Stasch, A. Synthesis and characterisation of bulky guanidines and phosphaguanidines: Precursors for low oxidation state metallacycles. N. J. Chem. 2009, 33, 64–75. [Google Scholar] [CrossRef]
- Barman, M.K.; Baishya, A.; Nembenna, S.J. Bulky guanidinate stabilized homoleptic magnesium, calcium and zinc complexes and their catalytic activity in the Tishchenko reaction. J. Organomet. Chem. 2015, 785, 52–60. [Google Scholar] [CrossRef]
- Maity, A.K.; Fortier, S.; Griego, L.; Metta-Magana, A. Synthesis of a “Super Bulky” Guanidinate Possessing an Expandable Coordination Pocket. J. Inorg. Chem. 2014, 53, 8155–8164. [Google Scholar] [CrossRef]
- Chlupaty, T.; Nevoralova, J.; Ruzickova, Z.; Ruzicka, A. Lithium and Dilithium Guanidinates, a Starter Kit for Metal Complexes Containing Various Mono-and Dianionic Ligands. Inorg. Chem. 2020, 59, 10854–10865. [Google Scholar] [CrossRef]
- Noor, A.; Bauer, T.; Todorova, T.K.; Weber, B.; Gagliardi, L.; Kempe, R. The Ligand-Based Quintuple Bond-Shortening Concept and Some of Its Limitations. Chem. Eur. J. 2013, 19, 9825–9832. [Google Scholar] [CrossRef]
- Bonyhady, S.J.; Green, S.P.; Jones, C.; Nembenna, S.; Stasch, A. A Dimeric Magnesium(I) Compound as a Facile Two-Center/Two-Electron Reductant. Angew. Chem. Int. Ed. 2009, 48, 2973–2977. [Google Scholar] [CrossRef]
- Feil, F.; Harder, S. Guanidinate Complexes of Heavier Alkaline-Earth Metals (Ca, Sr): Syntheses, Structures, Styrene Polymerization and Unexpected Reaction Behaviour. Eur. J. Inorg. Chem. 2005, 4438–4443. [Google Scholar] [CrossRef]
- Cameron, T.M.; Xu, C.; Dipasquale, A.G.; Rheingold, A.L. Synthesis and structure of strontium and barium guanidinates and mixed-ligand guanidinate pentamethylcyclopentadienyl complexes. Organometallics 2008, 27, 1596–1604. [Google Scholar] [CrossRef]
- Moxey, G.J.; Blake, A.J.; Lewis, W.; Kays, D.L. Alkaline Earth Complexes of a Sterically Demanding Guanidinate Ligand. Eur. J. Inorg. Chem. 2015, 5892–5902. [Google Scholar] [CrossRef]
- Sahoo, R.K.; Mahato, M.; Jana, A.; Nembenna, S. Zinc hydride-catalyzed hydrofuntionalization of ketones. J. Org. Chem. 2020, 85, 11200–11210. [Google Scholar] [CrossRef] [PubMed]
- Birch, S.J.; Boss, S.R.; Cole, S.C.; Coles, M.P.; Haigh, R.; Hitchcock, P.B.; Wheatley, A.E.H. The structural characteristics of organozinc complexes incorporating N,N′-bidentate ligands. Dalton Trans. 2004, 3568–3574. [Google Scholar] [CrossRef]
- Khalaf, M.S.; Coles, M.P.; Hitchcock, P.B. A structural, theoretical and coordinative evaluation of the bicyclic guanidinate derived from 1,4,6-triazabicyclo[3.3.0]oct-4-ene. Dalton Trans. 2008, 4288–4295. [Google Scholar] [CrossRef]
- Zelga, K.; Leszczyński, M.; Justyniak, I.; Kornowicz, A.; Cabaj, M.; Wheatley, A.E.H.; Lewiński, J. Synthesis, structure and unique reactivity of the ethylzinc derivative of a bicyclic guanidine. Dalton Trans. 2012, 41, 5934–5938. [Google Scholar] [CrossRef]
- Coles, M.P.; Hitchcock, P.B. Bicyclic guanidinate compounds of magnesium and their activity as pre-catalysts in the Tishchenko reaction. Eur. J. Inorg. Chem. 2004, 2662–2672. [Google Scholar] [CrossRef]
- Alonso-Moreno, C.; Carrillo-Hermosilla, F.; Garcés, A.; Otero, A.; López-Solera, I.; Rodríguez, A.M.; Antiñolo, A. Simple, versatile, and efficient catalysts for guanylation of amines. Organometallics 2010, 29, 2789–2795. [Google Scholar] [CrossRef]
- Harder, S. Early Main Group Metal. Catalysis: Concepts and Rections; Wiley-VCH Verlag GmbH & Co. KGaAL: Weinheim, Germany, 2020; Volume 12, p. 69469. [Google Scholar]
- Aeilts, S.L.; Coles, M.P.; Swenson, D.C.; Jordan, R.F.; Young, V.G. Aluminum alkyl complexes containing guanidinate ligands. Organometallics 1998, 17, 3265–3270. [Google Scholar] [CrossRef]
- Koller, J.; Bergman, R.G. Synthesis, characterization, and reactivity of aluminum alkyl/amide complexes supported by guanidinate and monoanionic OCO-pincer ligands. Organometallic 2010, 29, 3350–3356. [Google Scholar] [CrossRef]
- Koller, J.; Bergman, R.G. Highly efficient aluminum-catalyzed hydro-amination/-hydrazination of carbodiimides. Organometallics 2010, 29, 5946–5952. [Google Scholar] [CrossRef]
- Cole, M.L.; Davies, A.J.; Jones, C.; Junk, P.C.; McKay, A.I.; Stasch, A.Z. Aluminum and indium complexes derived from guanidines, triazenes, and amidines. Anorg. Allg. Chem. 2015, 641, 2233–2244. [Google Scholar] [CrossRef]
- Han, H.-F.; Zhang, S.-F.; Guo, Z.-Q.; Tong, H.-B.; Wei, X.-H. Three asymmetric guanidinato metal complexes: Synthesis, crystal structures and their use as pre-catalysts in the Meerwein–Ponndorf–Verley reduction. Polyhedron 2015, 99, 71–76. [Google Scholar] [CrossRef]
- Chang, C.-C.; Hsiung, C.-S.; Su, H.-L.; Srinivas, B.; Chiang, M.Y.; Lee, G.-H.; Wang, Y. Carbodiimide insertion into organoaluminum compounds and thermal rearrangement of the products. Organometallics 1998, 17, 1595–1601. [Google Scholar] [CrossRef]
- Zhang, W.-X.; Li, D.; Wang, Z.; Xi, Z. Alkyl aluminum-catalyzed addition of amines to carbodiimides: A highly efficient route to substituted guanidines. Organometallics 2009, 28, 882–887. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, S.; Zhou, S.; Feng, Z.; Guo, L.; Zhu, X.; Mu, X.; Yao, F. Aluminum Alkyl Complexes Supported by Bidentate N,N Ligands: Synthesis, Structure, and Catalytic Activity for Guanylation of Amines. Organometallics 2015, 34, 1882–1889. [Google Scholar] [CrossRef]
- Riddlestone, I.M.; Urbano, J.; Phillips, N.; Kelly, M.J.; Vidovic, D.; Bates, J.I.; Taylor, R.; Aldridge, S. Salt metathesis for the synthesis of M–Al and M–H–Al bonds. Dalton Trans. 2013, 42, 249–258. [Google Scholar] [CrossRef]
- Bayram, M.; Bläser, D.; Wölper, C.; Schulz, S. Syntheses and Structures of Bis-Amidinate–Alane Complexes. Organometallics 2014, 33, 2080–2087. [Google Scholar] [CrossRef]
- Chlupatý, T.; Bílek, M.; Moncol’, J.; Ruzicková, Z.; Ruzicka, A. Addition of dimethylaluminium chloride to N,N′-Disubstituted carbodiimides. J. Organomet. Chem. 2015, 786, 48–54. [Google Scholar] [CrossRef]
- Han, H.-F.; Guo, Z.-Q.; Zhang, S.-F.; Li, J.; Wei, X.-H. Guanidinato aluminum complexes: Synthesis, crystal structures and reactivities. RSC Adv. 2016, 6, 101437–101446. [Google Scholar] [CrossRef]
- Han, H.; Guo, Z.; Zhang, S.; Hua, Y.; Wei, X. Synthesis and crystal structures of guanidinatoaluminum complexes and catalytic study for MPV reduction. Polyhedron 2017, 126, 214–219. [Google Scholar] [CrossRef]
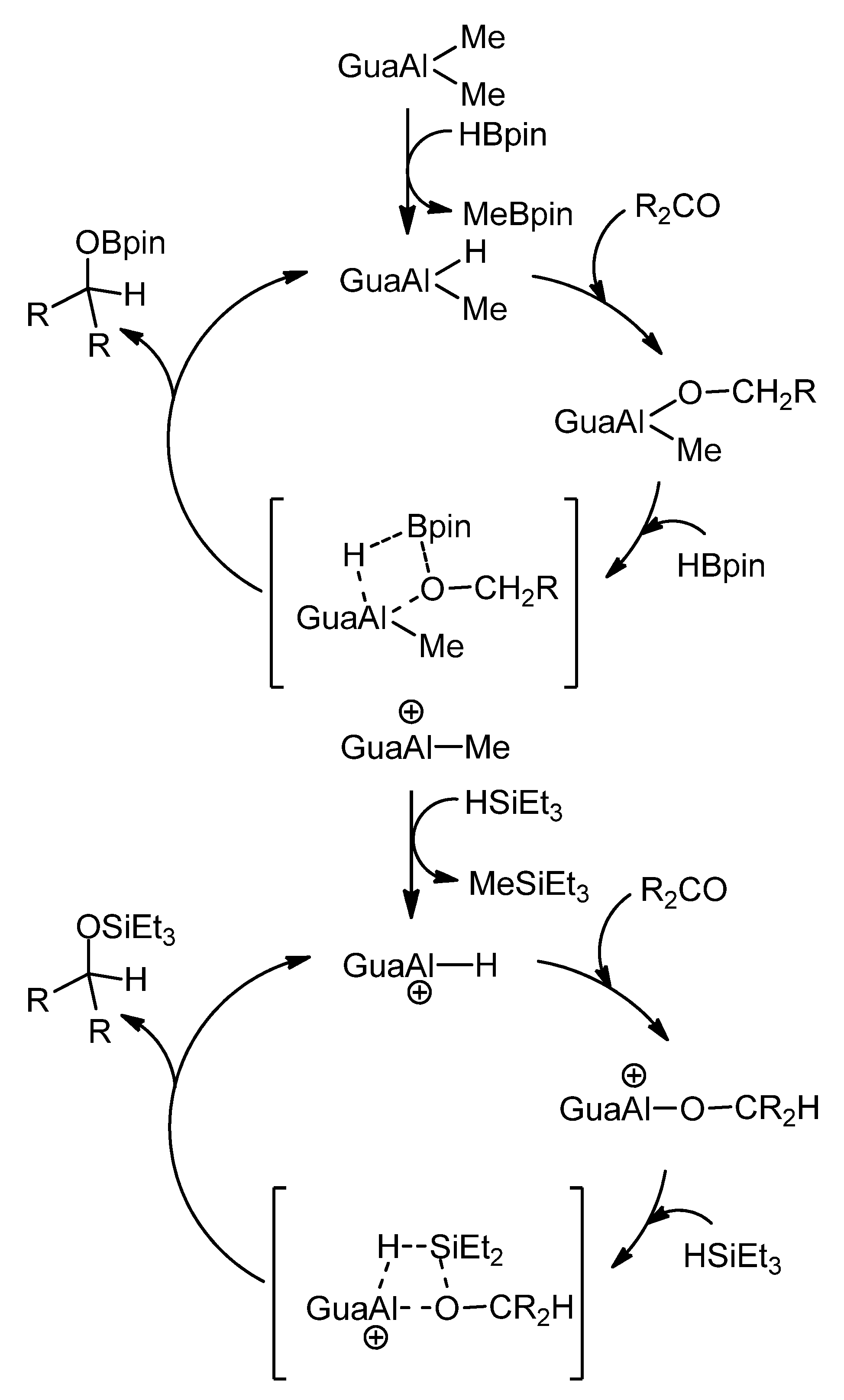
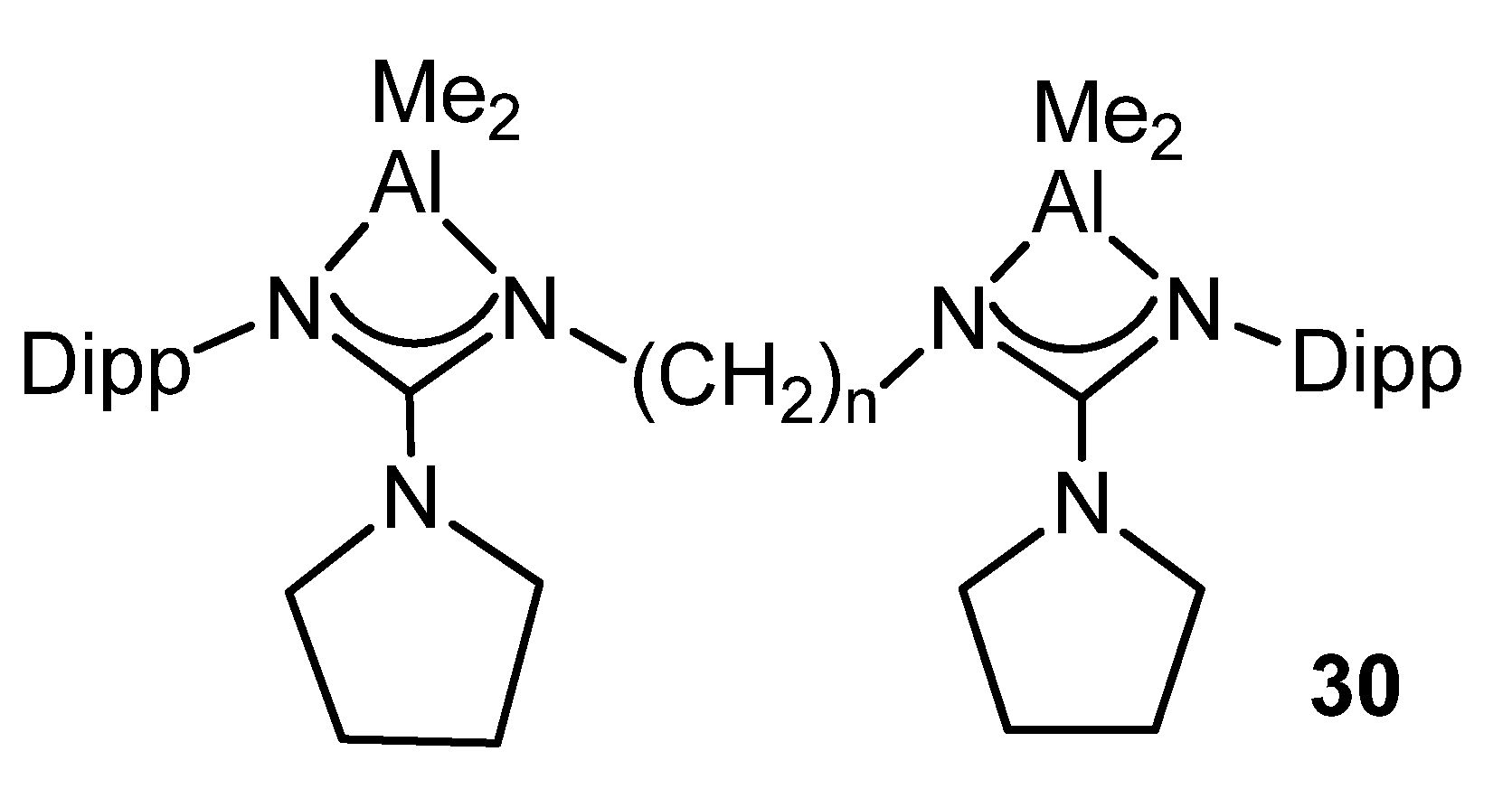
- Peddarao, T.; Sarkar, N.; Nembenna, S. Mono- and Bimetallic Aluminum Alkyl, Alkoxide, Halide and Hydride Complexes of a Bulky Conjugated Bis-Guanidinate (CBG) Ligand and Aluminum Alkyls as Precatalysts for Carbonyl Hydroboration. Inorg. Chem. 2020, 59, 4693–4702. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, N.; Sahoo, R.K.; Mukhopadhyay, S.; Nembenna, S. Organoaluminum Cation Catalyzed Selective Hydrosilylation of Carbonyls, Alkenes, and Alkynes. Eur. J. Inorg. Chem. 2022, e202101030. [Google Scholar] [CrossRef]
- Rösch, A.; Seifert, F.; Vass, V.; Görls, H.; Kretschmer, R. Synthesis, structure, and catalytic activity of dinuclear aluminium bis (amidinate) and bis (guanidinate) complexes. New J. Chem. 2021, 45, 972–981. [Google Scholar] [CrossRef]
- Rios Yepes, Y.; Mesías-Salazar, Á.; Becerra, A.; Daniliuc, C.G.; Ramos, A.; Fernández-Galán, R.; Rodríguez-Diéguez, A.; Antiñolo, A.; Carrillo-Hermosilla, F.; Rojas, R.S. Mono- and Dinuclear Asymmetric Aluminum Guanidinates for the Catalytic CO2 Fixation into Cyclic Carbonates. Organometallics 2021, 40, 2859–2869. [Google Scholar] [CrossRef]
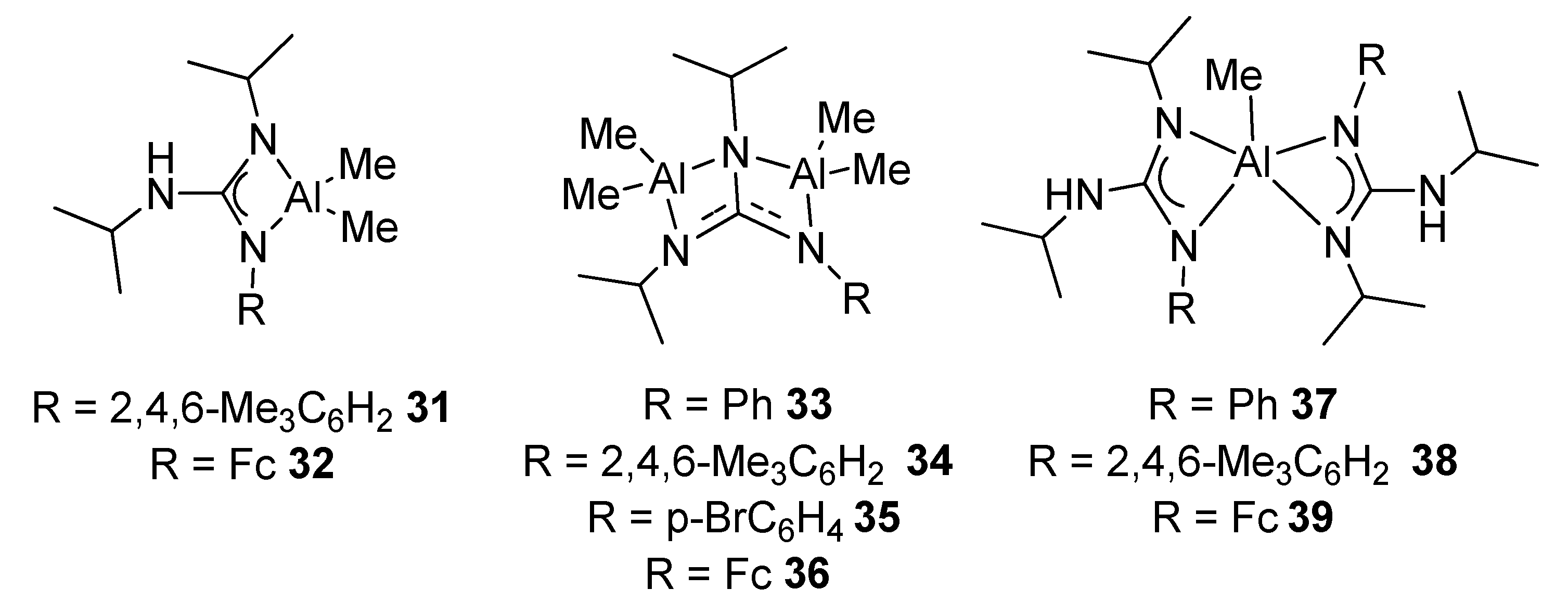
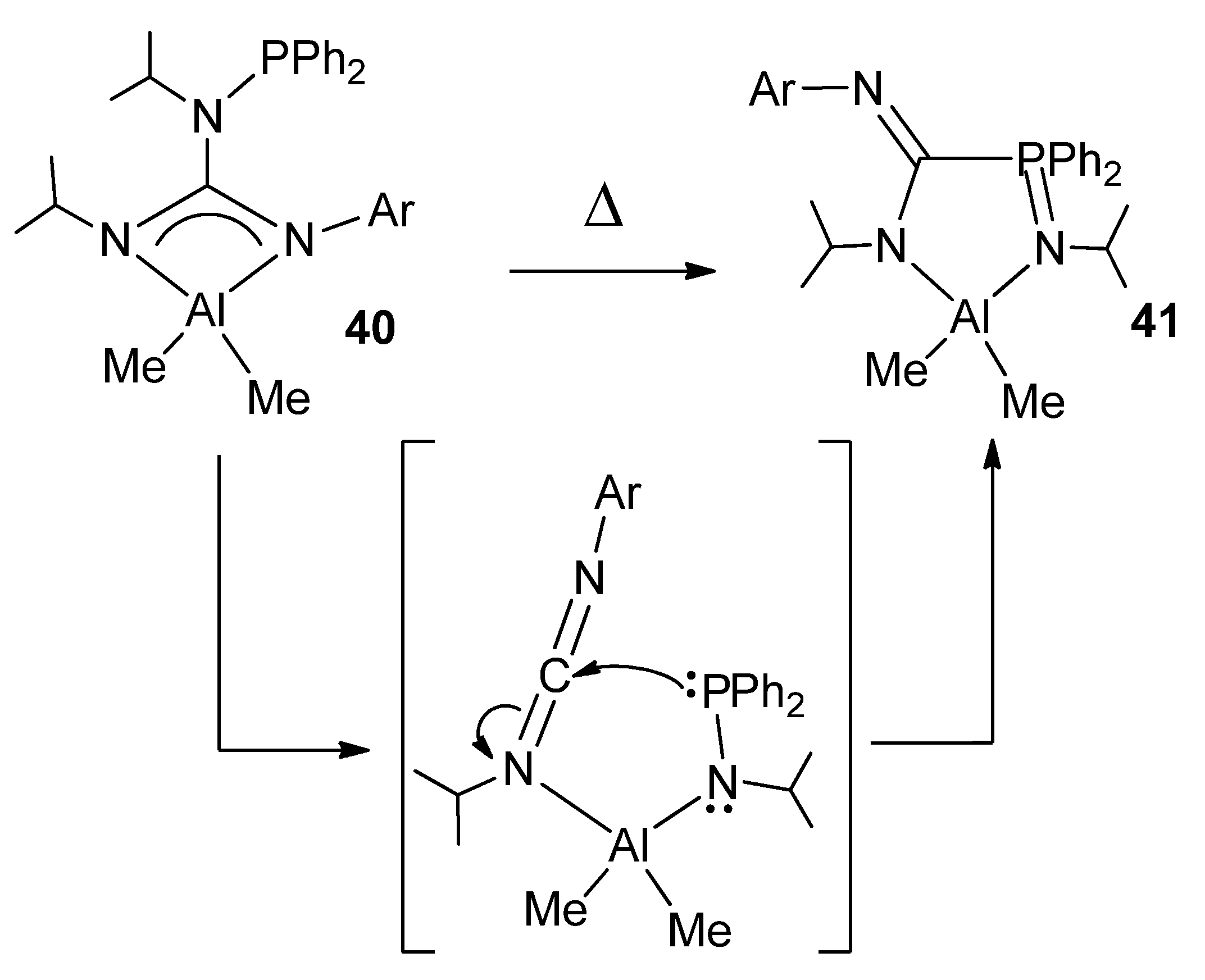
- Fernández-Galán, R.; Ramos, A.; Huergo, E.; Antiñolo, A.; Carrillo-Hermosilla, F.; Rodríguez-Diéguez, A.; García-Vivó, D. Unusual ligand rearrangement: From N-phosphinoguanidinato to phosphinimine-amidinato compounds. Chem. Commun. 2019, 55, 2809–2812. [Google Scholar] [CrossRef] [PubMed]
- Huergo, E.; Fernández-Galán, R.; Ramos, A.; Antiñolo, A.; Carrillo-Hermosilla, F.; Rodríguez-Diéguez, A.; García-Vivó, D. Reactivity of N-Phosphinoguanidines of the Formula (HNR)(Ph2PNR)C(NAr) toward Main Group MetalAlkyls: Facile Ligand Rearrangement from N-Phosphinoguanidinates to Phosphinimine-Amidinates. Inorg. Chem. 2020, 59, 15262–15275. [Google Scholar] [CrossRef] [PubMed]
- Brazeau, A.L.; Barry, S.T. Atomic layer deposition of aluminum oxide thin films from a heteroleptic, amidinate-containing precursor. Chem. Mat. 2008, 20, 7287–7291. [Google Scholar] [CrossRef]
- Dodonov, V.A.; Xiao, L.; Kushnerova, O.A.; Baranov, E.V.; Zhao, Y.; Yang, X.J.; Fedushkin, I.L. Transformation of carbodiimides to guanidine derivatives facilitated by gallylenes. Chem Commun. 2020, 56, 7475–7478. [Google Scholar] [CrossRef]
- Horeglad, P.; Litwińska, A.; Zukowska, G.Z.; Kubicki, D.; Szczepaniak, G.; Dranka, M.; Zachara, J. The influence of organosuperbases on the structure and activity of dialkylgallium alkoxides in the polymerization of rac -lactide: The road to stereo diblock PLA copolymers. App. Organomet. Chem. 2013, 27, 328–336. [Google Scholar] [CrossRef]
- Kassymbek, A.; Britten, J.F.; Spasyuk, D.; Gabidullin, B.; Nikonov, G.I. Interaction of multiple bonds with NacNacGa: Oxidative cleavage vs coupling and cyclization. Inorg. Chem. 2019, 58, 8665–8672. [Google Scholar] [CrossRef]
- Jin, G.; Jones, C.; Junk, P.C.; Stasch, A.; Woodul, W.D. Group 13 metal (I) and (II) guanidinate complexes: Effect of ligand backbone on metal oxidation state and coordination sphere. New J. Chem. 2008, 32, 835–842. [Google Scholar] [CrossRef]
- Jones, C.; Junk, P.C.; Platts, J.A.; Stasch, A. Four-membered group 13 metal(I) N-heterocyclic carbene analogues: Synthesis, characterization, and theoretical studies. J. Am. Chem. Soc. 2006, 128, 2206–2207. [Google Scholar] [CrossRef]
- Matsuda, I.; Itoh, K.; Ishii, Y. Reactions of CO2 and CO2 Analogs (CXY with X, Y = O, S, NR) with Reagents Containing Si–H and Si–N Units. J. Organomet. Chem. 1974, 69, 353–359. [Google Scholar] [CrossRef]
- Das, S.; Pati, S.K. Computational Exploration of Intramolecular Sn/N Frustrated Lewis Pairs for Hydrogen Activation and Catalytic Hydrogenation. Organometallics 2021, 40, 194–202. [Google Scholar] [CrossRef]
- George, T.A.; Jones, K.; Lappert, M.F. Amino-derivatives of metals and metalloids. Part II. Ainostannylation of unsaturated substrates, and the infrared spectra and structures of carbamato- and dithiocarbamato-trimethylstannanes and related compounds. J. Chem. Soc. 1965, 2157–2165. [Google Scholar]
- Hänssgen, D.; Odenhausen, E. Organotin heterocycles. J. Organomet. Chem. 1977, 124, 143–150. [Google Scholar] [CrossRef]
- Hänssgen, D.; Pohl, I. Ringerweiterungs-und-spaltungsreaktionen der Cyclodistannazane. Chem. Ber. 1979, 112, 2798–2803. [Google Scholar] [CrossRef]
- Kupchik, E.J.; Hanke, H.E.; DiMarco, J.P.; Chessari, R.J. Synthesis of N,N,N′-trisubstituted N′′-cyanoguanidines and N-aryl-N′-(triorganostannyl)-N′N′′-dicyanoguanidines. J. Chem. Eng. Data 1981, 26, 105–106. [Google Scholar] [CrossRef]
- Andrade-López, N.; Ariza-Castolo, A.; Contreras, R.; Vázquez-Olmos, A.; Barba-Behrens, N.; Tlahuext, H. Versatile behavior of 2-guanidinobenzimidazole nitrogen atoms toward protonation, coordination and methylation. Heteroat. Chem. 1997, 8, 397–410. [Google Scholar] [CrossRef]
- Fialon, M.-P.; Andrade-Lopez, N.; Barba-Behrens, N.; Contreras, R. Organometallic tin complexes derived from 2-guanidinobenzimidazole. Heteroat. Chem. 1998, 9, 637–641. [Google Scholar] [CrossRef]
- Wood, D.; Yap, G.P.A.; Richeson, D.S. N-Substituted Guanidinate Anions as Ancillary Ligands in Group 4 Chemistry. Syntheses and Characterization of M{RNC[N(SiMe3)2]NR}2Cl2, [M{CyNC[N(SiMe3)2]NCy}Cl3]− (M = Zr, Hf; R = iPr, Cy), and Zr{CyNC[N(SiMe3)2]NCy}(CH2Ph)3. Inorg. Chem. 1999, 38, 5788–5794. [Google Scholar] [CrossRef]
- Giesbrecht, G.R.; Whitener, G.D.; Arnold, J. Crystal Packing Forces Dictate η1- versus η2-Coordination of Benzyl Groups in [Guanidinate]Zr(CH2Ph)3. Organometallics 2000, 19, 2809–2812. [Google Scholar] [CrossRef]
- Coles, M.P.; Hitchcock, P.B. Exploration of the suitability of bicyclic guanidinates as ligands in catalytic chemistry mediated by titanium. Organometallics 2003, 22, 5201–5211. [Google Scholar] [CrossRef]
- Fandos, R.; Otero, A.; Rodríguez, A.; Terreros, P. Syntheses of new titanium(IV) and rhodium(I) guanidinido complexes. Collect. Czech. Chem. Commun. 2007, 72, 579–588. [Google Scholar] [CrossRef]
- Rodriguez, G.; Sperry, C.K.; Bazan, G.C. Boratabenzene complexes of zirconium, hafnium and chromium. J. Mol. Catal. A 1998, 128, 5–28. [Google Scholar] [CrossRef]
- Fernández-Galán, R.; Antiñolo, A.; Carrillo-Hermosilla, F.; López-Solera, I.; Otero, A.; Serrano-Laguna, A.; Villaseñor, E. New zirconium and zirconocene guanidinate complexes. J. Organomet. Chem. 2012, 711, s35–s42. [Google Scholar] [CrossRef]
- Zuckerman, R.L.; Bergman, R.G. Structural factors that influence the course of overall [2 + 2] cycloaddition reactions between imidozirconocene complexes and heterocumulenes. Organometallics 2000, 19, 4795–4809. [Google Scholar] [CrossRef]
- Zuckerman, R.L.; Bergman, R.G. Mechanistic investigation of cycloreversion/cycloaddition reactions between zirconocene metallacycle complexes and unsaturated organic substrates. Organometallics 2001, 20, 1792–1807. [Google Scholar] [CrossRef][Green Version]
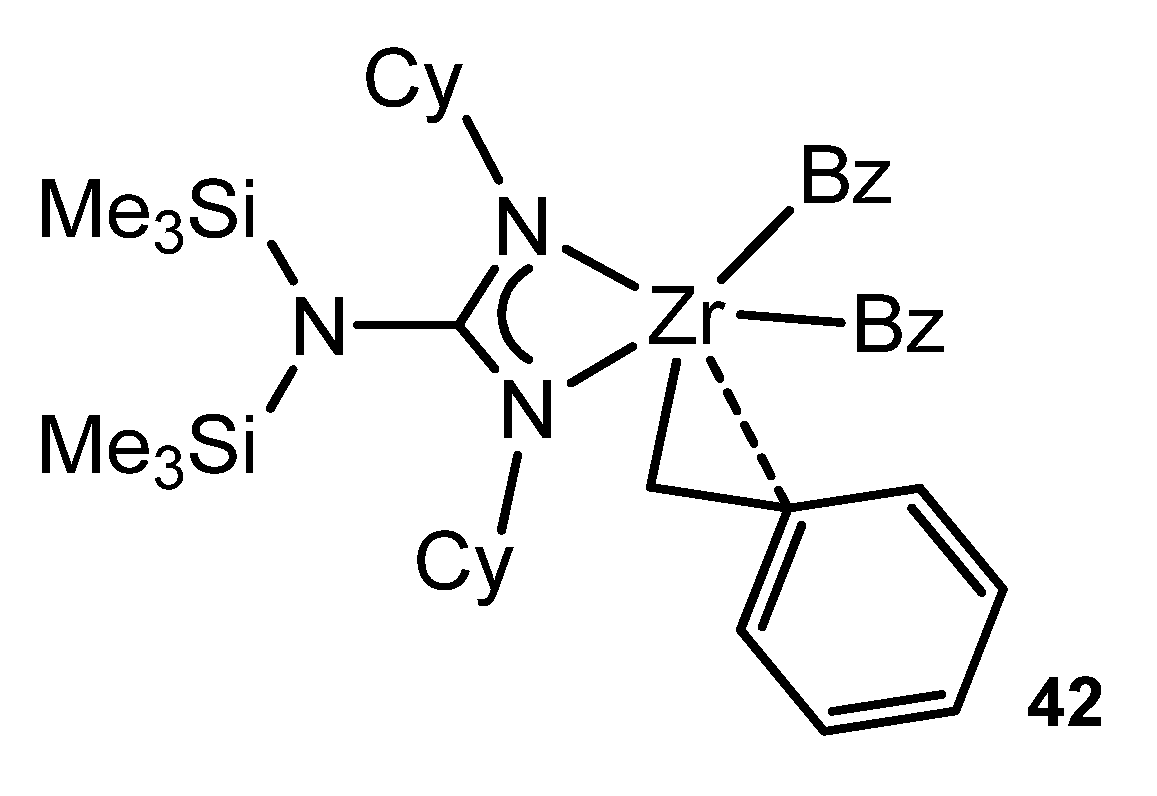
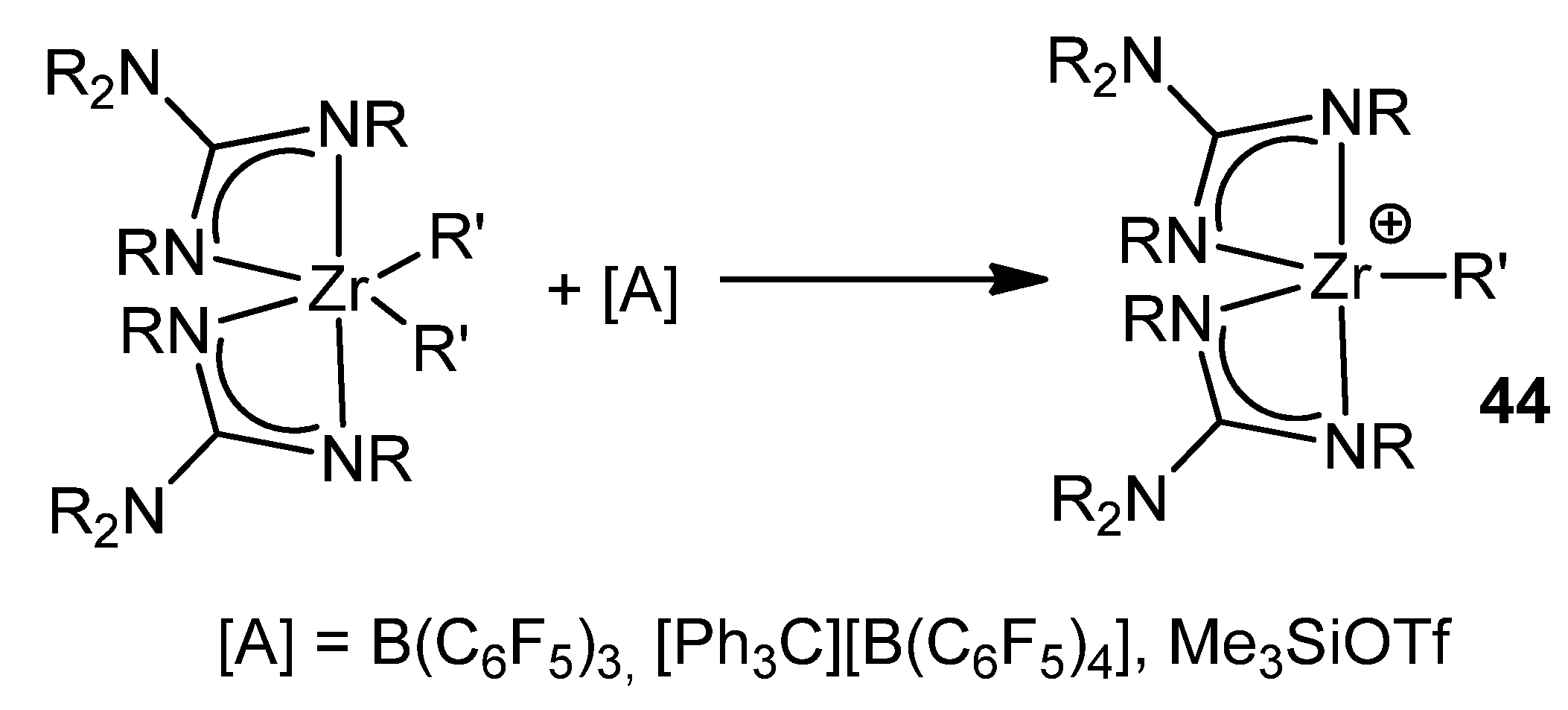
- Duncan, A.P.; Mullins, S.M.; Arnold, J.; Bergman, R.G. Synthesis, structural investigation, and reactivity of neutral and cationic bis (guanidinato) zirconium(IV) complexes. Organometallics 2001, 20, 1808–1819. [Google Scholar] [CrossRef]
- Bazinet, P.; Wood, D.; Yap, G.P.A.; Richeson, D.S. Synthesis and Structural Investigation of N,N′,N′′-Trialkylguanidinato-Supported Zirconium(IV) Complexes. Inorg. Chem. 2003, 42, 6225–6229. [Google Scholar] [CrossRef]
- Ong, T.-G.; Yap, G.P.A.; Richeson, D.S. Redefining the Coordination Geometry and Reactivity of Guanidinate Complexes by Covalently Linking the Guanidinate Ligands. Synthesis and Reactivity of [RN{NH(R)}CN(CH2)2NC{NH(R)}NR]M(CH2Ph)2 (R = iPr; M = Ti, Zr). Organometallics 2003, 22, 387–389. [Google Scholar] [CrossRef]
- Ong, T.-G.; Yap, G.P.A.; Richeson, D.S. Formation of a guanidinate-supported titanium imido complex: A catalyst for alkyne hydroamination. Organometallics 2002, 21, 2839–2841. [Google Scholar] [CrossRef]
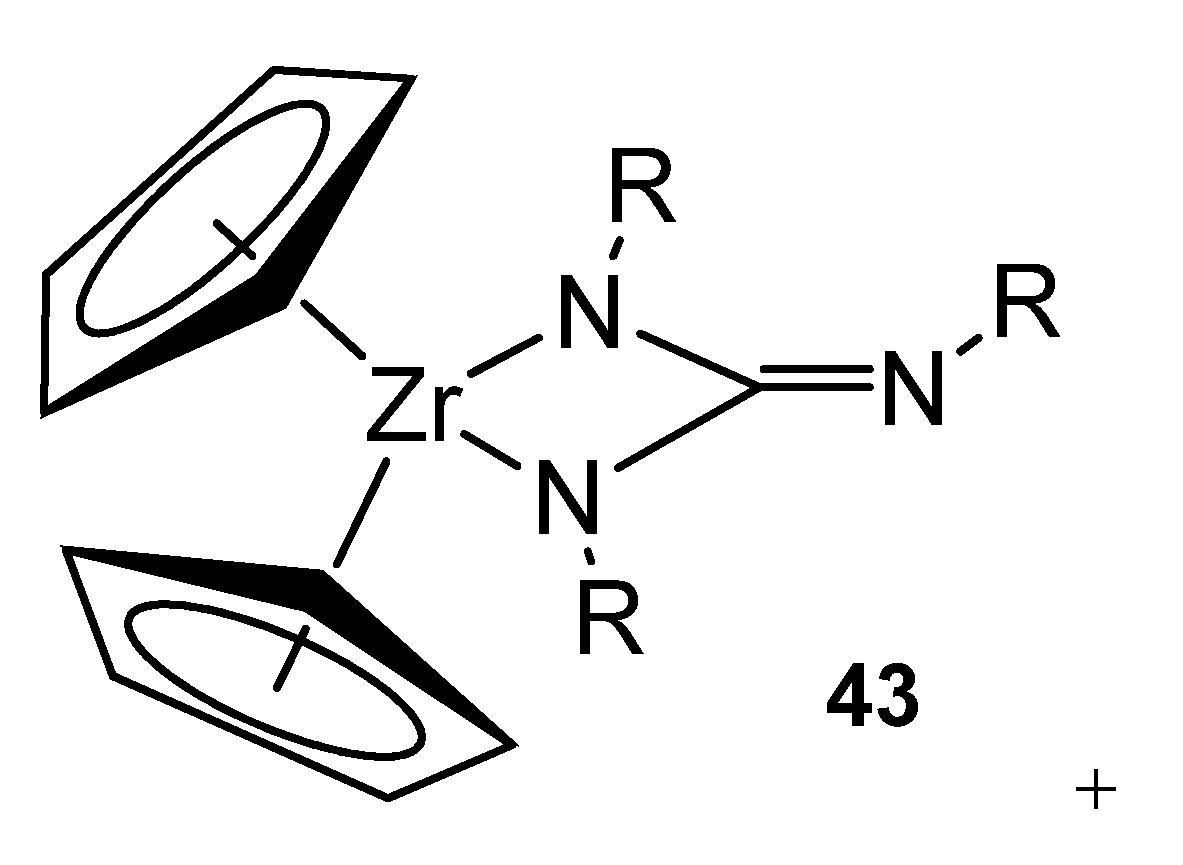
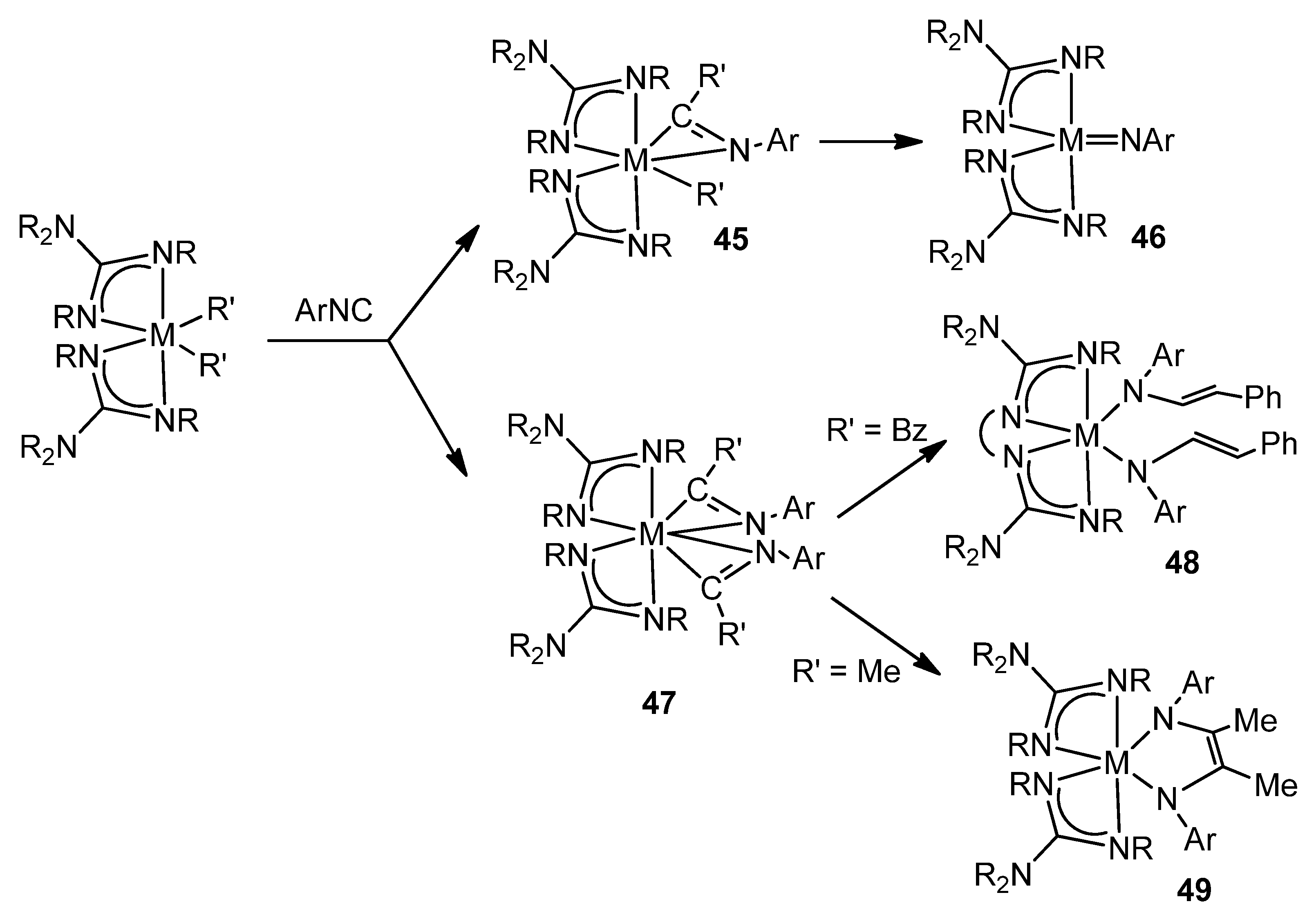
- Ong, T.-G.; Wood, D.; Yap, G.P.A.; Richeson, D.S. Transformations of aryl isocyanide on guanidinate-supported organozirconium complexes to yield terminal imido, iminoacyl, and enediamido ligands. Organometallics 2002, 21, 1–3. [Google Scholar] [CrossRef]
- Fernández-Galán, R.; Antiñolo, A.; Carrillo-Hermosilla, F.; López-Solera, I.; Otero, A.; Serrano-Laguna, A.; Villaseñor, E. Migratory Insertion Reactions in Asymmetrical Guanidinate-Supported Zirconium Complexes. Organometallics 2012, 31, 8360–8369. [Google Scholar] [CrossRef]
- Zhang, J.; Han, F.; Han, Y.; Chen, Z.; Zhou, X. Synthesis and structures of titanium and yttrium complexes with N,N′-tetramethylguanidinate ligands: Different reactivity of the M–N bonds toward phenyl isocyanate. Dalton Trans. 2009, 1806–1811. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, Q.; Tong, H.; Yan, L.; Wang, X. Organoamido zirconium(IV) and titanium(IV) complexes and their catalysis towards ethylene polymerization. RSC Adv. 2015, 5, 105292–105298. [Google Scholar] [CrossRef]
- Shen, H.; Chan, H.-S.; Xie, Z. Guanylation of amines catalyzed by a half-sandwich titanacarborane amide complex. Organometallics 2006, 25, 5515–5517. [Google Scholar] [CrossRef]
- Shen, H.; Chan, H.-S.; Xie, Z. Reaction of [σ:η5-(C9H6)C2B9H10]Zr(NMe2)(DME) with Guanidines: Metallacarborane-Mediated C−N Bond Cleavage and 1,5-Sigmatropic Rearrangement. J. Am. Chem. Soc. 2007, 129, 12934–12935. [Google Scholar] [CrossRef]
- Gott, A.L.; Coles, S.R.; Clarke, A.J.; Clarkson, G.J.; Scott, P. Chiral alkoxide-functionalized guanidinates from ring-opening rearrangement of aminooxazolinate complexes. Organometallics 2007, 26, 136–142. [Google Scholar] [CrossRef]
- Gott, A.L.G.; Clarkson, J.; Deeth, R.J.; Hammond, M.L.; Morton, C.; Scott, P. Constrained geometry aminooxazolinate ligands giving chiral zirconium guanidinates; catalytic cyclohydroamination. Dalton Trans. 2008, 2983–2990. [Google Scholar] [CrossRef]
- Potts, S.E.; Carmalt, C.J.; Blackman, C.S.; Abou-Chahine, F.; Pugh, D.; Davies, H.O. Synthesis of zirconium guanidinate complexes and the formation of zirconium carbonitride via low pressure CVD. Organometallics 2009, 28, 1838–1844. [Google Scholar] [CrossRef]
- Wasslen, Y.A.; Tois, E.; Haukka, S.; Kreisel, K.A.; Yap, G.P.A.; Halls, M.D.; Barry, S.T. A family of heteroleptic titanium guanidinates: Synthesis, thermolysis, and surface reactivity. Inorg. Chem. 2010, 49, 1976–1982. [Google Scholar] [CrossRef]
- Xu, K.; Milanov, A.P.; Winter, M.; Barreca, D.; Gasparotto, A.; Becker, H.-W.; Devi, A. Heteroleptic Guanidinate- and Amidinate-Based Complexes of Hafnium as New Precursors for MOCVD of HfO2. Eur. J. Inorg. Chem. 2010, 1679–1688. [Google Scholar] [CrossRef]
- Kurek, A.; Gordon, P.G.; Karle, S.; Devi, A.; Barry, S.T. Recent advances using guanidinate ligands for chemical vapour deposition (CVD) and atomic layer deposition (ALD) applications. Aust. J. Chem. 2014, 67, 989–996. [Google Scholar] [CrossRef]
- Hirotsu, M.; Fontaine, P.P.; Zavalij, P.Y.; Sita, L.R. Extreme N⋮N Bond Elongation and Facile N-Atom Functionalization Reactions within Two Structurally Versatile New Families of Group 4 Bimetallic “Side-on-Bridged” Dinitrogen Complexes for Zirconium and Hafnium. J. Am. Chem. Soc. 2007, 129, 12690–12692. [Google Scholar] [CrossRef]
- Aguilar-Calderón, J.R.; Metta-Magaña, A.J.; Noll, B.; Fortier, S. Reversible oxidative-addition and reductive-elimination of thiophene from a titanium complex and its thermally-induced hydrodesulphurization chemistry. Angew. Chem. Int. Ed. 2016, 55, 1–6. [Google Scholar]
- Giménez-Torres, A.; Aguilar-Calderón, J.R.; Encerrado-Manríquez, A.M.; Pink, M.; Metta-Magaça, A.J.; Lee, W.-Y.; Fortier, S. Titanium-Mediated Catalytic Hydrogenation of Monocyclic and Polycyclic Arenes. Chem. Eur. J. 2020, 26, 2803–2807. [Google Scholar] [CrossRef]
- Aguilar-Calderón, J.R.; Murillo, J.; Gómez-Torres, A.; Saucedo, C.; Jordan, A.; Metta-Magaña, A.J.; Pink, M.; Fortier, S. Redox character and small molecule reactivity of a masked titanium(II) synthon. Organometallics 2020, 39, 295–311. [Google Scholar] [CrossRef]
- Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; López-Solera, I.; Menot, B.; Fernández-Galán, R.; Villaseñor, E.; Otero, A. New alkylimido niobium complexes supported by guanidinate ligands: Synthesis, characterization, and migratory insertion reactions. Organometallics 2012, 31, 1840–1848. [Google Scholar] [CrossRef]
- Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; López-Solera, I.; Fernández-Galán, R.; Serrano, A.; Villaseñor, E. Synthesis, characterization and reactivity of new dinuclear guanidinate diimidoniobium complexes. Eur. J. Inorg. Chem. 2013, 2940–2946. [Google Scholar] [CrossRef]
- Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; Suárez, F.J.; López-Solera, I.; Fernández-Galán, R.; Villaseñor, E. Asymmetric niobium guanidinates as intermediates in the catalytic guanylation of amines. Dalton Trans. 2013, 42, 8223–8230. [Google Scholar] [CrossRef]
- Elorriaga, D.; Carrillo-Hermosilla, F.; Antiñolo, A.; López-Solera, I.; Fernández-Galán, R.; Villaseñor, E. Unexpected mild C–N bond cleavage mediated by guanidine coordination to a niobium iminocarbamoyl complex. Chem. Commun. 2013, 49, 8701–8703. [Google Scholar] [CrossRef]
- Thirupathi, N.; Yap, G.P.A.; Richeson, D.S. Mono- and Dianionic Guanidinate Ligands. Reactivity of [iPrNC(NiPr)2]Ta(NMe2)3 and [(iPrNH)C(NiPr)2]TaCl(NMe2)3 with Me3SiCl and ArNC (Ar = 2,6-Me2C6H4). Organometallics 2000, 19, 2573–2579. [Google Scholar] [CrossRef]
- Mohammad, A.; Olson, J.R.; Rotsch, D.A.; Bemowski, R.D.; Swenson, D.C.; Messerle, L. High-and Mid-Valent Tantalum and Mono (peralkylcyclopentadienyl) tantalum Complexes of the Bicyclic Guanidinate Hexahydropyrimidopyrimidinate. Organometallics 2013, 32, 6232–6239. [Google Scholar] [CrossRef]
- Yonke, B.L.; Keane, A.J.; Zavalij, P.Y.; Sita, L.R. Mononuclear Tantalum(IV, d1) Imido Complexes Supported by the Monocyclopentadienyl, Amidinate and Guanidinate Ligand Sets As Models to Explore Dinitrogen Fixation by “End-On-Bridged” Dinuclear {[Ta(IV, d1)]}2(μ-η1:η1-N2) Complexes. Organometallics 2012, 31, 345–355. [Google Scholar] [CrossRef]
- Keane, A.J.; Yonke, B.L.; Hirotsu, M.; Zavalij, P.Y.; Sita, L.R. Fine-Tuning the Energy Barrier for Metal-Mediated Dinitrogen N≡N Bond Cleavage. J. Am. Chem. Soc. 2014, 136, 9906–9909. [Google Scholar] [CrossRef]
- Saadati, F.; Griffin, S.E.; Schafer, L.L. Guanidinate Early-Transition-Metal Complexes: Efficient and Selective Hydroaminoalkylation of Alkenes. Organometallics 2022, 41, 1816–1822. [Google Scholar] [CrossRef]
- Hao, H.; Cui, C.; Bai, G.; Roesky, H.W.; Noltemeyer, M.; Schmidt, H.-G.; Ding, Y.Z. Bis(arylimido) Molybdenum(VI) Amidinate and Guanidinate Complexes; Molecular Structures of [(ArN)2MoMe{N(Cy)C[N(i -Pr)2]N(Cy)}] (Ar = 2,6-i -Pr2C6H3; Cy = Cyclohexyl) and [(2,6-i -Pr2C6H3N)2MoCl2] · [NH=C(C6H5)CH(SiMe3)2]. Anorg. Allg. Chem. 2000, 626, 1660–1664. [Google Scholar] [CrossRef]
- Wang, L.; Hu, L.; Zhang, H.; Chen, H.; Deng, L. Three-coordinate iron(IV) bisimido complexes with aminocarbene ligation: Synthesis, structure, and reactivity. J. Am. Chem. Soc. 2015, 137, 14196–14207. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Carta, V.; Pink, M.; Smith, J.M. Catalytic Carbodiimide Guanylation by a Nucleophilic, High Spin Iron(II) Imido Complex. J. Am. Chem. Soc. 2021, 143, 5324–5329. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.J.; Mitchell, L.A.; Parsons, S. Guanidine anions as chelating ligands; syntheses and crystal structures of [Rh(η-C5Me5){η2-(NPh)2CNHPh}Cl] and [Ru(η-MeC6H4Pri-p)-{η2-(NPh)2CNHPh}Cl]. J. Chem. Soc. Dalton Trans. 1996, 2839–2841. [Google Scholar] [CrossRef]
- Dinger, M.B.; Henderson, W.; Nicholson, B.K. Organometallic complexes of platinum-group metals incorporating substituted guanidine dianion (triazatrimethylenemethane) ligands. J. Organomet. Chem. 1998, 556, 75–88. [Google Scholar] [CrossRef]
- Singh, T.; Kishan, R.; Nethaji, M.; Thirupathi, N. Synthesis, Reactivity Studies, Structural Aspects, and Solution Behavior of Half Sandwich Ruthenium(II) N,N′,N″-Triarylguanidinate Complexes. Inorg. Chem. 2012, 51, 157–169. [Google Scholar] [CrossRef]
- Kishan, R.; Kumar, R.; Baskaran, S.; Sivasankar, C.; Thirupathi, N. Ionic and Neutral Half-Sandwich Guanidinatoruthenium(II) Complexes and Their Solution Behavior. Eur. J. Inorg. Chem. 2015, 3182–3194. [Google Scholar] [CrossRef]
- Bailey, P.J.; Grant, K.J.; Mitchell, L.A.; Pace, S.; Parkin, A.; Parsons, S. Guanidinates as chelating anionic ligands for early, middle and late transition metals: Syntheses and crystal structures of [Ti{η2-(NPh)2CNEt2}2Cl2], [Ru{η2-(NPh)2CNHPh}3] and [Pt{η2-(NPh)2CNHPh}2]. J. Chem. Soc. Dalton Trans. 2000, 1887–1891. [Google Scholar] [CrossRef]
- Robin Kumar, R.; Ujjval, R.; Thirupathi, N. Half Sandwich Electron Deficient N,N′,N′′-Triarylguanidinato-ruthenium(II) Complexes: Syntheses, Reactivity Studies, and Structural Aspects. Eur. J. Inorg. Chem. 2019, 31, 3619–3628. [Google Scholar] [CrossRef]
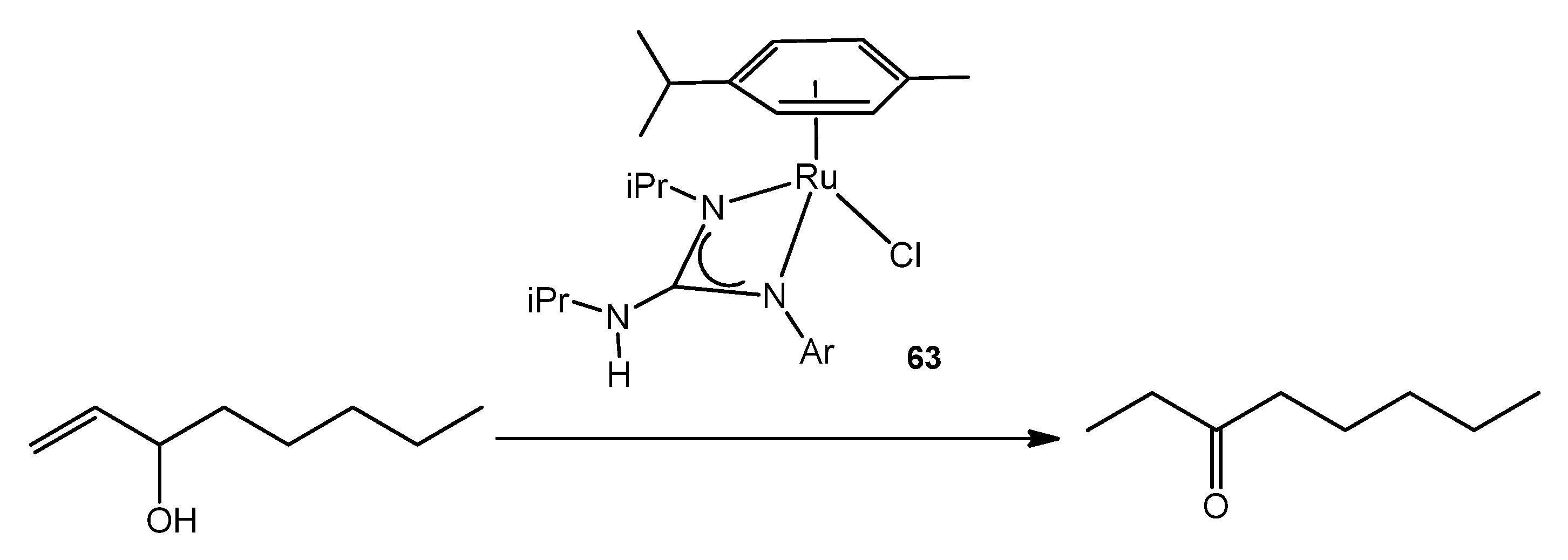
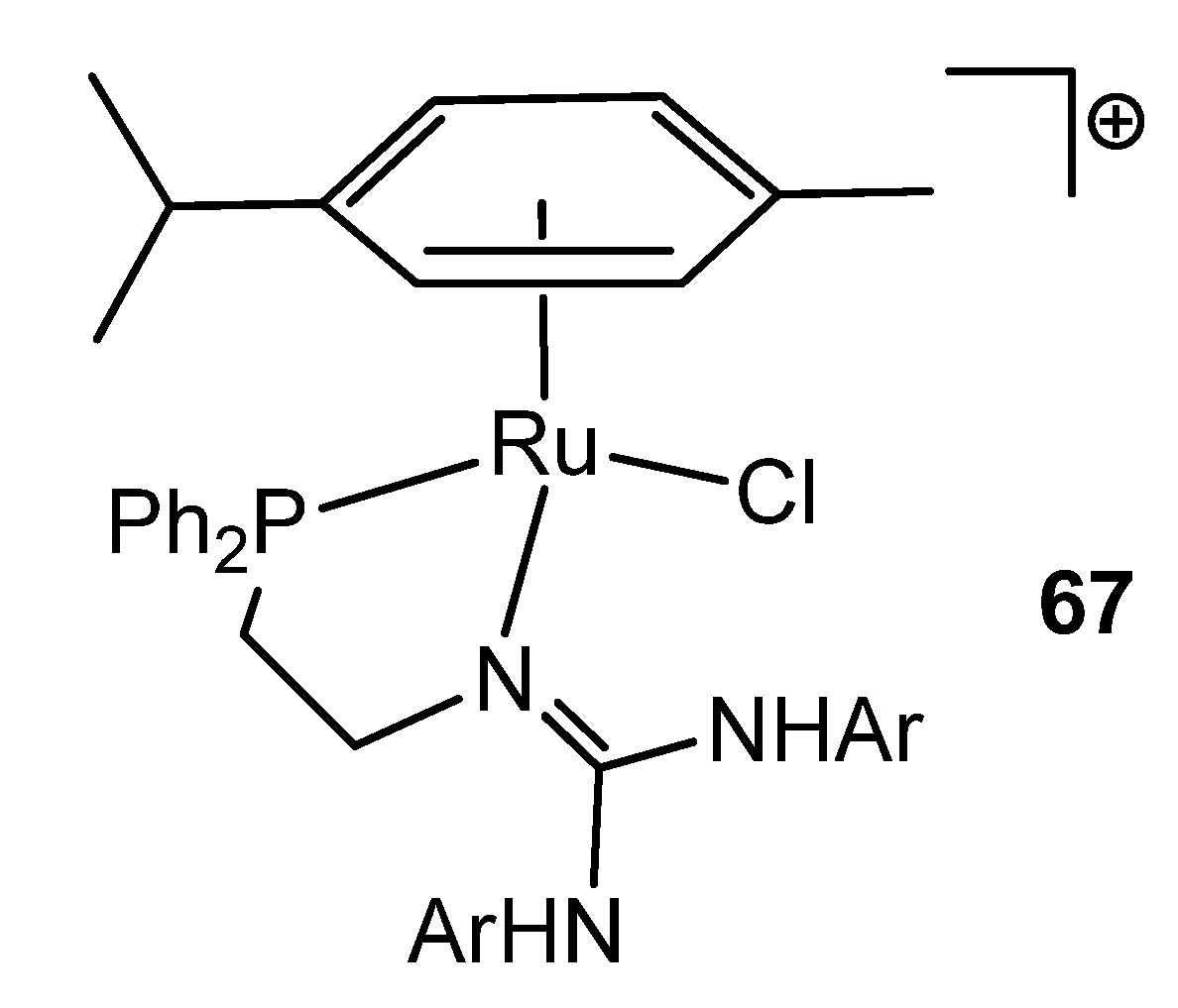
- García-Álvarez, R.; Suárez, F.J.; Dıéz, J.; Crochet, P.; Cadierno, V.; Antiñolo AFernández-Galán, R.; Carrillo-Hermosilla, F. Ruthenium(II) Arene Complexes with Asymmetrical Guanidinate Ligands: Synthesis, Characterization, and Application in the Base-Free Catalytic Isomerization of Allylic Alcohols. Organometallics 2012, 31, 8301–8311. [Google Scholar] [CrossRef]
- Menéndez-Rodríguez, L.; Tomás-Mendivil, E.; Francos, J.; Crochet, P.; Cadierno, V.; Antiñolo, A.; Fernández-Galán, R.; Carrillo-Hermosilla, F. Reactivity of the Dimer [{RuCl(μ-Cl)(η3:η3-C10H16)}2] (C10H16 = 2,7-Dimethylocta-2,6-diene-1,8-diyl) toward Guanidines: Access to Ruthenium(IV) and Ruthenium(II) Guanidinate Complexes. Organometallics 2015, 34, 2796–2809. [Google Scholar] [CrossRef]
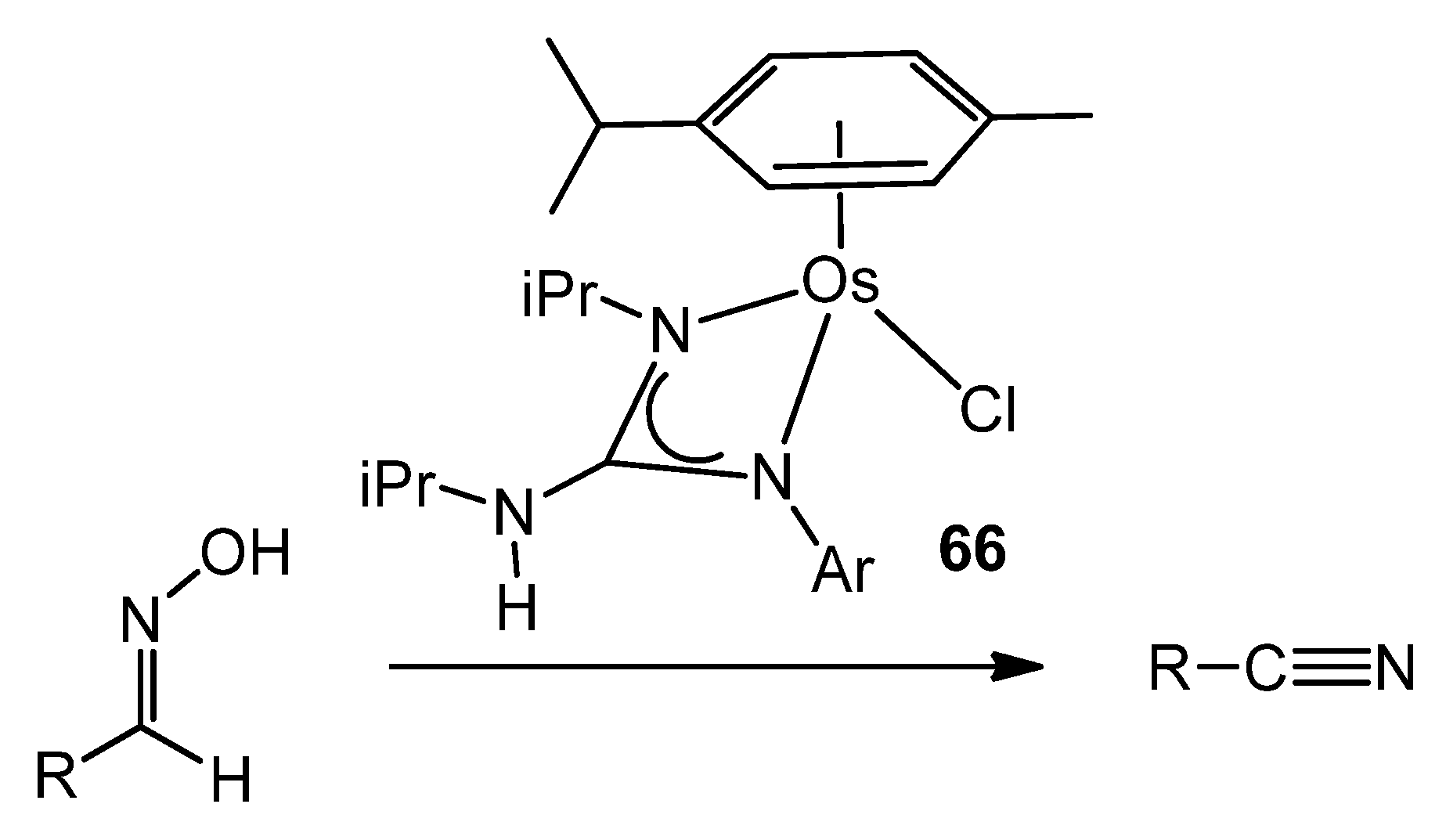
- Francos, J.; Gonzalez-Liste, P.J.; Menéndez-Rodríguez, L.; Crochet, P.; Cadierno, V.; Borge, J.; Antinolo, A.; Fernández-Galán, R.; Carrillo-Hermosilla, F. Half-Sandwich Guanidinate–Osmium(II) Complexes: Synthesis and Application in the Selective Dehydration of Aldoximes. Eur. J. Inorg. Chem. 2016, 393–402. [Google Scholar] [CrossRef]
- Wilkinson, E.-T.; Viguri, F.; Rodríguez, R.; López, J.A.; García-Orduña, P.; Lahoz, F.J.; Lamata, P.; Carmona, D. Strained Ruthenium Complexes Bearing Tridentate Guanidine-Derived Ligands. Helv. Chim. Acta 2021, 104, e2100044. [Google Scholar] [CrossRef]
- Parker, A.; Lamata, P.; Viguri, F.; Rodríguez, R.; López, J.A.; Lahoz, F.J.; García-Orduña, P.; Carmona, D. Half-sandwich complexes of osmium containing guanidine-derived ligands. Dalton Trans. 2020, 49, 13601–13617. [Google Scholar] [CrossRef] [PubMed]
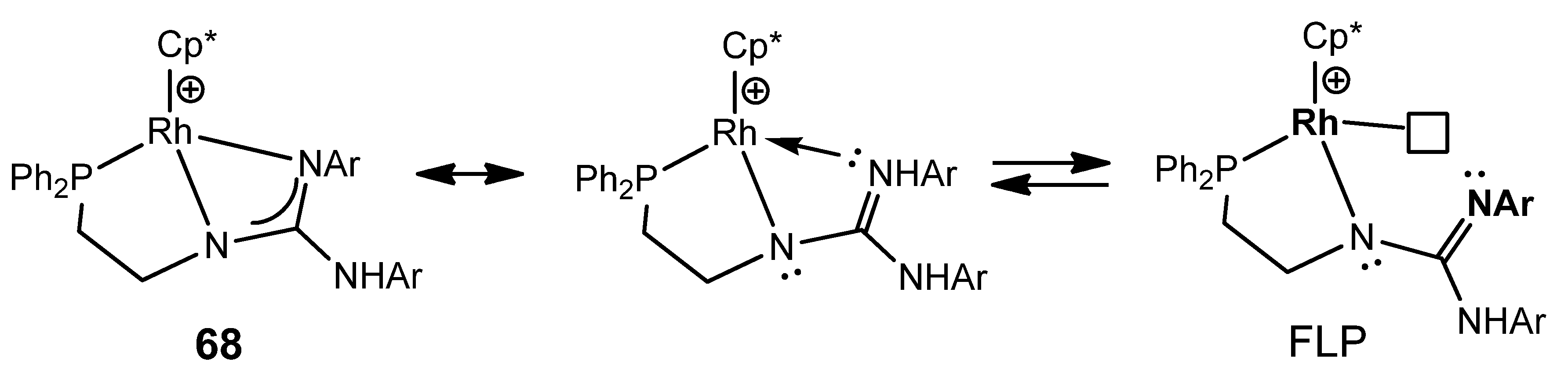
- Carmona, M.; Ferrer, J.; Rodríguez, R.; Passarelli, V.; Lahoz, F.J.; García-Orduña, P.; Cañadillas-Delgado, L.; Carmona, D. Reversible Activation of Water by an Air- and Moisture-Stable Frustrated Rhodium Nitrogen Lewis Pair. Chem. Eur. J. 2019, 25, 13665–13670. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, C.; Ferrer, J.; Passarelli, V.; Lahoz, F.J.; García-Orduña, P.; Carmona, D. Well-Stabilized but Strained Frustrated Lewis Pairs Based on Rh/N and Ir/N Couples. Organometallics 2022, 41, 1445–1453. [Google Scholar] [CrossRef]
- Kumar, R.; Kishan, R.; Thomas, J.M.; Chinnappan, S.; Thirupathi, N. Probing the factors that influence the conformation of a guanidinato ligand in [(η5-C5Me5)M(NN)X] (NN = chelating N,N′,N′′-tri(o-substituted aryl)guanidinate(1−); X = chloro, azido and triazolato). New J. Chem. 2018, 42, 1853–1866. [Google Scholar] [CrossRef]
- Kumar, R.; Thirupathi, N. Syntheses, characterisation, and catalytic role of (η⁵-C₅Me₅)Rh(III) guanidinato complexes in transfer hydrogenation (TH) and TH–etherification. RSC Adv. 2017, 7, 33890–33904. [Google Scholar] [CrossRef]
- Jones, C.; Mills, D.P.; Stasch, A. Flexible coordination of bulky amidinates and guanidinates towards rhodium(I): Conversion of kinetic to thermodymanic isomers. Dalton Trans. 2008, 4799–4804. [Google Scholar] [CrossRef]
- Rohde, J.-U.; Kelley, M.R.; Lee, W.-T. Synthesis, Characterization, and O2 Reactivity of Iridium(I) Complexes Supported by Guanidinato Ligands. Inorg. Chem. 2008, 47, 11461–11463. [Google Scholar] [CrossRef]
- Rohde, J.-U.; Lee, W.-T. Stabilization of Iridium(IV) by Monoanionic Dialkyldiarylguanidinato Ligands. J. Am. Chem. Soc. 2009, 131, 9162–9163. [Google Scholar] [CrossRef]
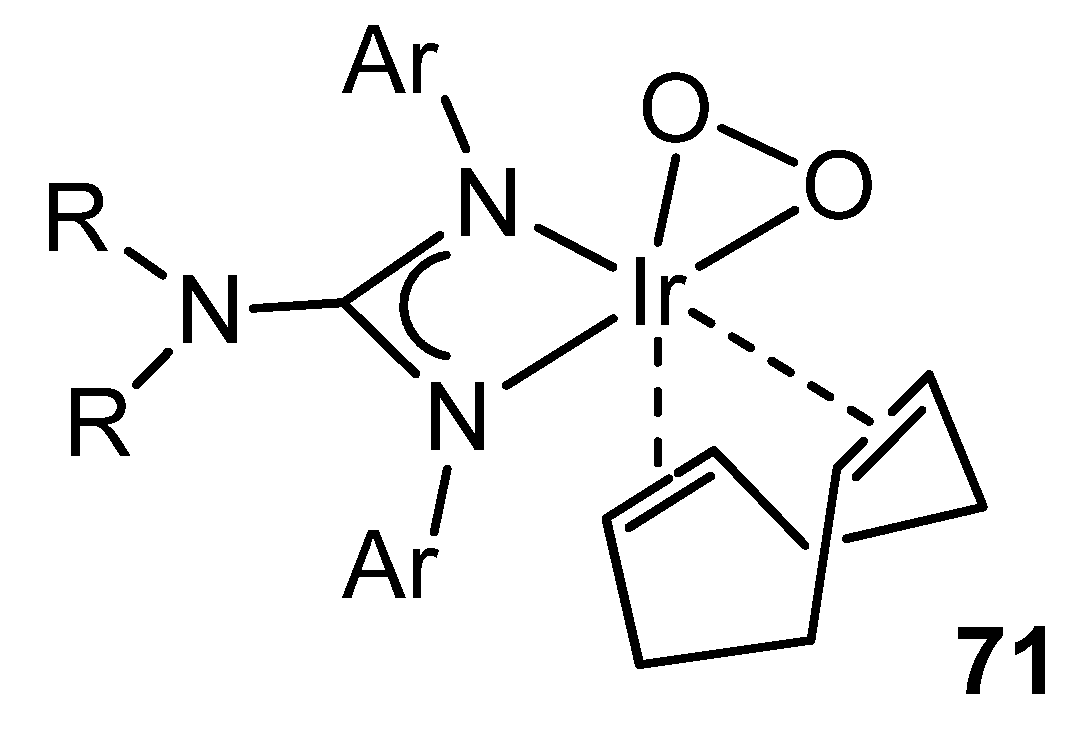
- Kelley, M.R.; Rohde, J.-U. Evidence for a reactive (alkene)peroxoiridium(III) intermediate in the oxidation of an alkene complex with O2. Chem. Commun. 2012, 48, 2876–2878. [Google Scholar] [CrossRef]
- Kelley, M.R.; Rohde, J.-U. Guanidinato Complexes of Iridium: Ligand-Donor Strength, O2 Reactivity, and (Alkene)peroxoiridium(III) Intermediates. Inorg. Chem. 2013, 52, 2564–2580. [Google Scholar] [CrossRef]
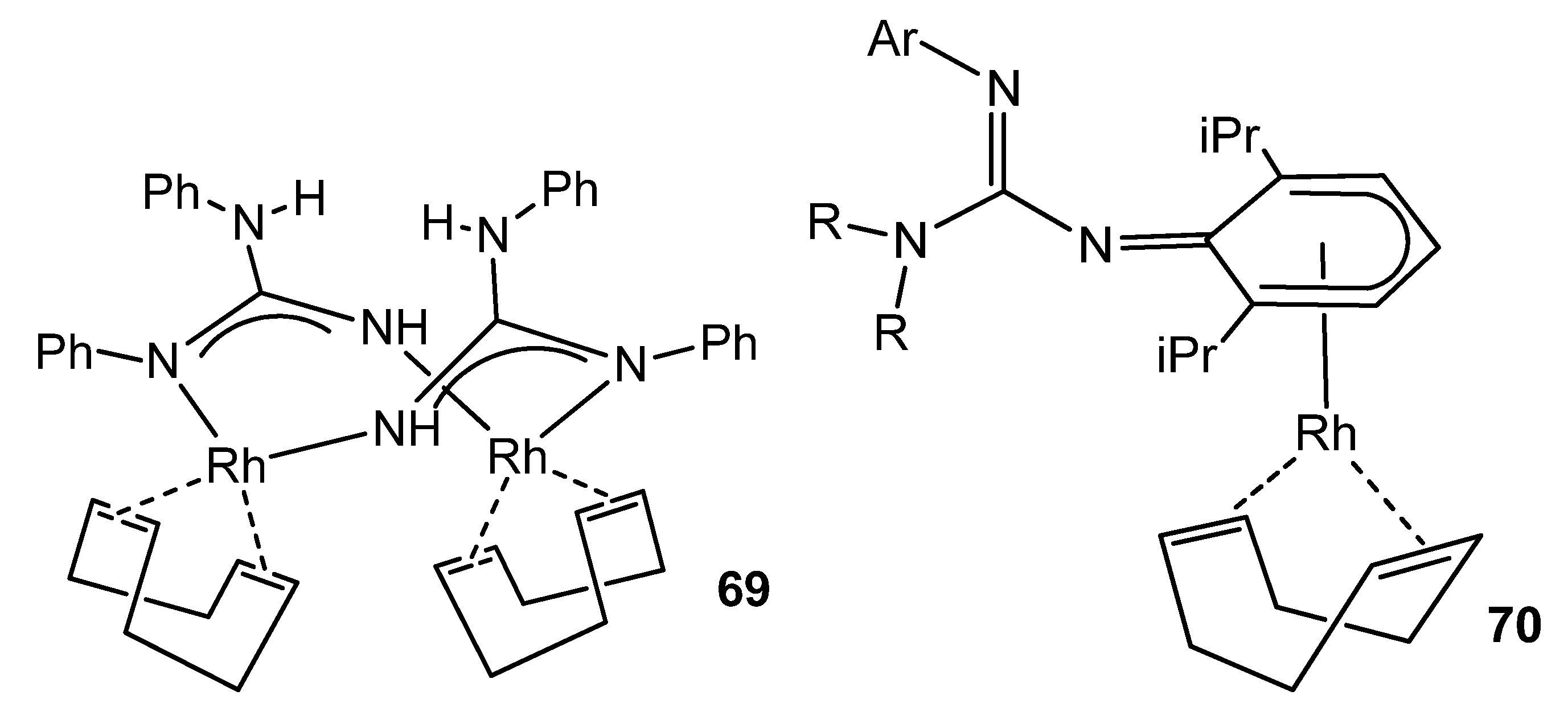
- Holland, A.W.; Bergman, R.G. Heterocumulene Metathesis by Iridium Guanidinate and Ureylene Complexes: Catalysis Involving Reversible Insertion to Form Six-Membered Metallacycles. J. Am. Chem. Soc. 2002, 124, 9010–9011. [Google Scholar] [CrossRef]
- Holland, A.W.; Bergman, R.G. Thermal reactivity of Group 9 transition metal pinacolate complexes: Heterocumulene cleavage and CO bond formation. Inorg. Chim. Acta 2002, 341, 99–106. [Google Scholar] [CrossRef]
- Ogata, K.; Oka, O.; Toyota, A.; Suzuki, N.; Fukuzawa, S. Phosphine-Dependent Selective Cross-Dimerization between Terminal Alkylacetylene and Silylacetylene by Iridium(I) Guanidinate Complex-Phosphine System. Synlett 2008, 2663–2666. [Google Scholar] [CrossRef]
- Xu, H.; Chen, R.; Sun, Q.; Lai, W.; Su, Q.; Huang, W.; Liu, X. Recent progress in metal–organic complexes for optoelectronic applications. Chem. Soc. Rev. 2014, 43, 3259–3302. [Google Scholar] [CrossRef]
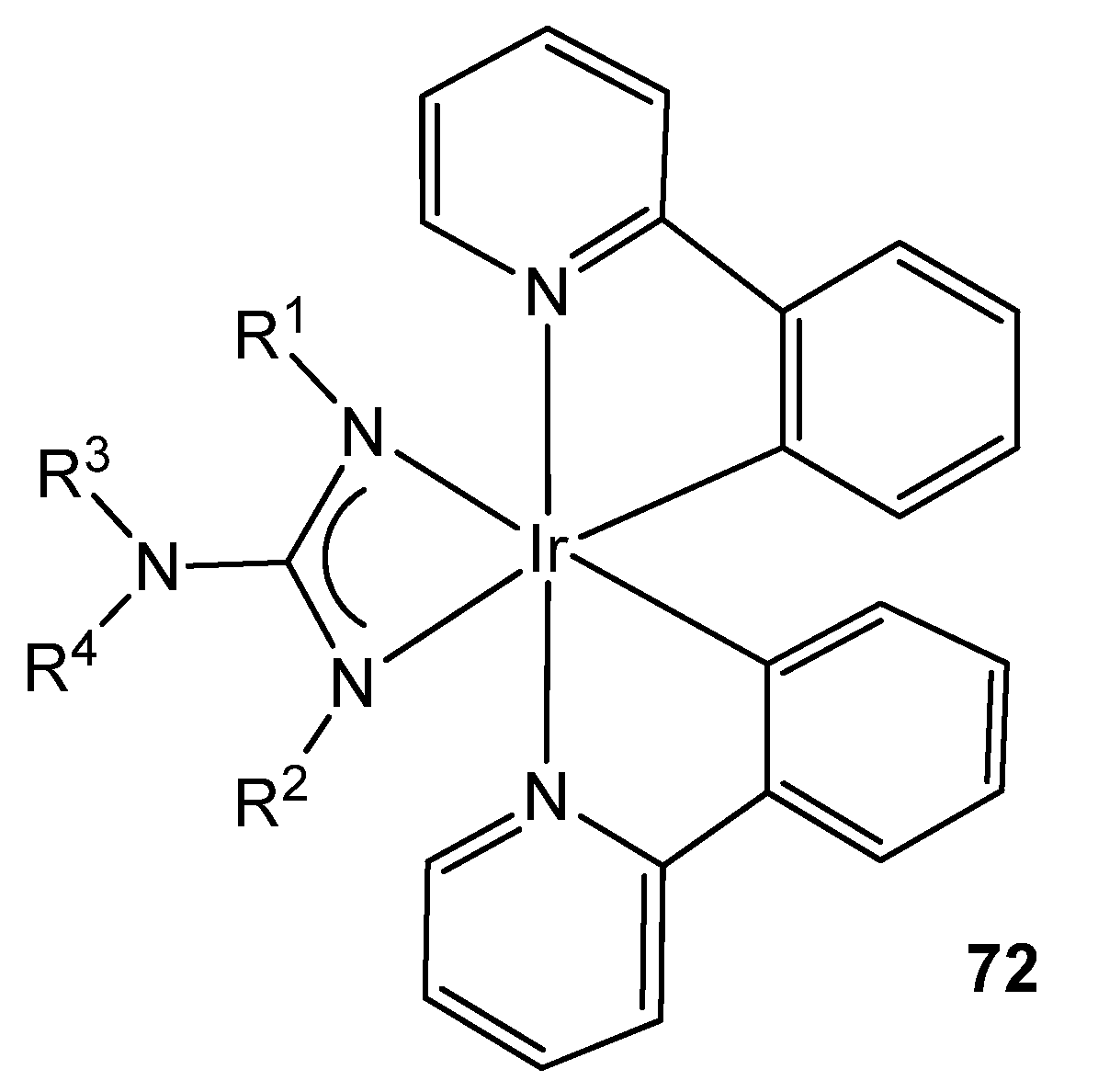
- Rai, V.K.; Nishiura, M.; Takimoto, M.; Zhao, S.; Liu, Y.; Hou, Z. Bis-Cyclometalated Iridium(III) Complexes Bearing Ancillary Guanidinate Ligands. Synthesis, Structure, and Highly Efficient Electroluminescence. Inorg. Chem. 2012, 51, 822–835. [Google Scholar] [CrossRef]
- Feng, Y.; Li, P.; Zhuang, X.; Ye, K.; Peng, T.; Liu, Y.; Wang, Y. A novel bipolar phosphorescent host for highly efficient deep-red OLEDs at a wide luminance range of 1000–10000 cdm−2. Chem. Commun. 2015, 51, 12544–12547. [Google Scholar] [CrossRef]
- Hasan, K.; Pal, A.K.; Auvray, T.; Zysman-Colman, E.; Hanan, G.S. Blue-green emissive cationic iridium(III) complexes using partially saturated strongly-donating guanidyl-pyridine/-pyrazine ancillary ligands. Chem. Commun. 2015, 51, 14060–14063. [Google Scholar] [CrossRef]
- Kabir, E.; Wu, Y.; Sittel, S.; Nguyen, B.-L.; Teets, T.S. Improved deep-red phosphorescence in cyclometalated iridium complexes via ancillary ligand modification. Inorg. Chem. Front. 2020, 7, 1362–1373. [Google Scholar] [CrossRef]
- Jones, C.; Schulten, C.; Rose, R.P.; Stasch, A.; Aldridge, S.; Woodul, W.D.; Murray, K.S.; Moubaraki, B.; Brynda, M.; La Macchia, G.; et al. Amidinato– and Guanidinato–Cobalt(I) Complexes: Characterization of Exceptionally Short Co–Co Interactions. Angew. Chem. Int. Ed. 2009, 48, 7406–7410. [Google Scholar] [CrossRef]
- Gopi, K.; Thirupathi, N. Synthesis, Reactivity, Structural Aspects, and Solution Dynamics of Cyclopalladated Compounds of N,N′,N′′-Tris(2-anisyl)guanidine. Organometallics 2011, 30, 572–583. [Google Scholar] [CrossRef]
- Elumalai, P.; Thirupathi, N.; Nethaj, M. Six-membered [C,N] cyclopalladated sym N,N′,N″-tri(4-tolyl)guanidines: Synthesis, reactivity studies and structural aspects. J. Organomet. Chem. 2013, 741–742, 141–147. [Google Scholar] [CrossRef]
- Gopi, K.; Saxena, P.; Nethaji, M.; Thirupathi, N. Influence of steric effect on the structural aspects of N,N′,N″-triarylguanidine derived six-membered [C,N] palladacycles. Polyhedron 2013, 52, 1041–1052. [Google Scholar] [CrossRef]
- Elumalai, P.; Thirupathi, N.; Nethaji, M. Dual Role of Acetate as a Nucleophile and as an Internal Base in Cycloplatination Reaction of sym-N,N′,N″-Triarylguanidines. Inorg. Chem. 2013, 52, 1883–1894. [Google Scholar] [CrossRef]
- Mishra, V.; Thirupathi, N. Critical Role of Anions in Platinum(II) Precursors upon the Structural Motifs of Six-Membered Cycloplatinated N,N′,N″-Triarylguanidines. ACS Omega 2018, 3, 6075–6090. [Google Scholar] [CrossRef]
- Ujjval, R.; Deepa, M.; Thomas, J.M.; Sivasankar, C.; Thirupathi, N. Unusual [Pt{κ2(C,N)}]+ → [Pt{κ2(N,N)}]+ Coordination Mode Flip of the Guanidinate(1−) Ligand in Cationic N,N′,N″-Tris(3,5-xylyl)guanidinatoplatinum(II) Bis(phosphine) Complexes. Syntheses, Structural and Theoretical Studies. Organometallics 2020, 39, 3663–3678. [Google Scholar] [CrossRef]
- Thakur, V.; Thirupathi, N. Syntheses and structural aspects of dinuclear cycloplatinated N,N′,N″-triarylguanidinate(2−) complexes with a novel tridentate μ2-κ2(C,N):κ1N coordination mode. J. Organomet. Chem. 2020, 911, 121138–121143. [Google Scholar] [CrossRef]
- Mishra, V.; Sinha, N.K.; Thirupathi, N. Reactions of Cycloplatinated Guanidine Complexes with Hg(OC(O)CF3)2: Formation of a One-Dimensional Coordination Polymer Containing a Pt2Hg(μ2-S(O)Me2-S,O) Repeating Unit versus a Discrete Pt2Hg2 Complex. Inorg. Chem. 2021, 60, 3879–3892. [Google Scholar] [CrossRef]
- Sinha, N.K.; Thirupathi, N. [6,5] CNN Palladium(II) Pincer Complexes Containing N-Substituted Monoanionic and Dianionic Guanidinate Ligands: Syntheses, Structural Aspects, and Their Utility in Suzuki–Miyaura Coupling Reactions. Organometallics 2021, 40, 3535–3549. [Google Scholar] [CrossRef]
- Thakur, V.; Thirupathi, N. Reactivity studies of cycloplatinated triarylguanidinato complexes, [Pt(TAG-κC,κN)(X)(S(O)Me2)] (X = Cl and OC(O)CF3) towards Lewis bases, and structural aspects of the products. J. Organomet. Chem. 2022, 959, 122200–122210. [Google Scholar] [CrossRef]
- Saxena, P.; Thomas, J.M.; Sivasankarb, C.; Thirupathi, N. Syntheses and structural aspects of six-membered palladacyclic complexes derived from N,N′,N′′-triarylguanidines with N- or S-thiocyanate ligands. New J. Chem. 2019, 43, 2307–2327. [Google Scholar] [CrossRef]
- Romanov, A.S.; Chotard, F.; Rashid, J.; Bochmann, M. Synthesis of copper(I) cyclic (alkyl)(amino)carbene complexes with potentially bidentate N^N, N^S and S^S ligands for efficient white photoluminescence. Dalton Trans. 2019, 48, 15445–15454. [Google Scholar] [CrossRef] [PubMed]
- Day, B.M.; Guo, F.-S.; Layfield, R.A. Cyclopentadienyl Ligands in Lanthanide Single-Molecule Magnets: One Ring to Rule Them All? Acc. Chem. Res. 2018, 51, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yap, G.P.A.; Richeson, D.S. N-Substituted Guanidinate Anions as Ancillary Ligands in Organolanthanide Chemistry. Synthesis and Characterization of {CyNC[N(SiMe3)2]NCy}2SmCH(SiMe3)2. Organometallics 1998, 17, 4387–4391. [Google Scholar] [CrossRef]
- Lu, Z.; Yap, G.P.A.; Richeson, D.S. Tetrasubstituted Guanidinate Anions as Supporting Ligands in Organoyttrium Chemistry. Organometallics 2001, 20, 706–712. [Google Scholar] [CrossRef]
- Lyubov, D.M.; Fukin, G.K.A.; Trifonov, A. N,N′-Diisopropyl-N߱-bis(trimethylsilyl)guanidinate Ligand as a Supporting Coordination Environment in Yttrium Chemistry. Synthesis, Structure, and Properties of Complexes [(Me3Si)2NC(Ni-Pr)2]YCl2(THF)2, [(Me3Si)2NC(Ni-Pr)2]Y(CH2SiMe3)2(THF)2, and [(Me3Si)2NC(Ni-Pr)2]Y[(μ-H)(μ-Et)2BEt]2(THF). Inorg. Chem. 2007, 46, 11450–11456. [Google Scholar]
- Trifonov, A.A.; Lyubov, D.M.; Fedorova, E.A.; Fukin, G.K.; Schumann, H.; Mühle, S.; Hummert, M.; Bochkarev, M.N. Chloro, Alkyl and Aryl Complexes of Rare Earth Metals Supported by Bulky Tetrasubstituted Guanidinate Ligands. Eur. J. Inorg. Chem. 2006, 747–756. [Google Scholar] [CrossRef]
- Trifonov, A.A.; Lyubov, D.M.; Fukin, G.K.; Baranov, E.V.; Kurskii, Y.A. Alkylyttrium Complexes Supported by N,N′-Dicyclohexyl-N′-bis(trimethylsilyl)guanidinate Ligands. Organometallics 2006, 25, 3935–3942. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, R.; Weng, L.; Zhou, X. Insertion of a Carbodiimide into the Ln−N σ-Bond of Organolanthanide Complexes. Isomerization and Rearrangement of Organolanthanides Containing Guanidinate Ligands. Organometallics 2004, 23, 3303–3308. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, J.; Cai, R.; Chen, Z.; Weng, L.; Zhou, X. Synthesis and reactivity of organolanthanide complexes containing phenothiazine ligand toward carbodiimide and isothiocyanate. J. Organomet. Chem. 2005, 690, 4926–4932. [Google Scholar] [CrossRef]
- Pi, C.; Zhang, Z.; Pang, Z.; Zhang, J.; Luo, J.; Chen, Z.; Weng, L.; Zhou, X. Multiple N−H Bond Activation: Synthesis and Reactivity of Functionalized Primary Amido Ytterbium Complexes. Organometallics 2007, 26, 1934–1946. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, L.; Zhang, L. Reactivity of a mixed methyl–aminobenzyl guanidinate lutetium complex towards iPrN=C=NiPr, CS2 and Ph2PH. Dalton Trans. 2022, 51, 12650–12660. [Google Scholar] [CrossRef]
- Pi, C.; Liu, R.; Zheng, P.; Chen, Z.; Zhou, X. Selective Reaction Based on the Linked Diamido Ligands of Dinuclear Lanthanide Complexes. Inorg. Chem. 2007, 46, 5252–5259. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, X.; Cai, R.; Weng, L. Reactivity of Organolanthanide and Organolithium Complexes Containing the Guanidinate Ligands toward Isocyanate or Carbodiimide: Synthesis and Crystal Structures. Inorg. Chem. 2005, 44, 716–722. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Li, Y.; Hong, L.; Chen, Z.; Zhou, X. Activation of Bis(guanidinate)lanthanide Alkyl and Aryl Complexes on Elemental Sulfur: Synthesis and Characterization of Bis(guanidinate)lanthanide Thiolates and Disulfides. Inorg. Chem. 2010, 49, 5715–5722. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Xue, M.; Zhang, Y.; Yao, Y.; Shen, Q. Synthesis and structure of samarium benzyl complex supported by bridged bis(guanidinate) ligand and its reactivity toward nitriles and phenyl isocyanate. J. Organomet. Chem. 2012, 716, 86–94. [Google Scholar] [CrossRef]
- Zheng, P.; Hong, J.; Liu, R.; Zhang, Z.; Pang, Z.; Weng, L.; Zhou, X. Synthesis and Reactivities of Guanidinate Dianion Complexes of Heterobimetallic Lanthanide−Lithium Cp2Ln[(CyN)2CNPh]Li(THF)3. Organometallics 2010, 29, 1284–1289. [Google Scholar] [CrossRef]
- Ge, S.; Meetsma, A.; Hessen, B. Highly Efficient Hydrosilylation of Alkenes by Organoyttrium Catalysts with Sterically Demanding Amidinate and Guanidinate Ligands. Organometallics 2008, 27, 3131–3135. [Google Scholar] [CrossRef]
- Trifonov, A.A.; Skvortsov, G.G.; Lyubov, D.M.; Skorodumova, N.A.; Fukin, G.K.; Baranov, E.V.; Glushakova, V.N. Postmetallocene Lanthanide–Hydrido Chemistry: A New Family of Complexes [{Ln{(Me3Si)2NC(Ni Pr)2}2(μ-H)}2] (Ln=Y, Nd, Sm, Gd, Yb) Supported by Guanidinate Ligands—Synthesis, Structure, and Catalytic Activity in Olefin Polymerization. Chem. Eur. J. 2006, 12, 5320–5327. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, Y.; Shen, Q. [(SiMe3)2NC(NiPr)2]2Ln(μ-Me)2Li(TMEDA) (Ln = Nd, Yb) as Effective Single-Component Initiators for Styrene Polymerization. Macromolecules 2002, 35, 8670–8671. [Google Scholar] [CrossRef]
- Trifonov, A.A.; Fedorova, E.A.; Fukin, G.K.; Bochkarev, M.N. Post-Metallocene Hydridolanthanide Chemistry: [Lu{(Me3Si)2NC(NiPr)2}2(μ-H)]2—A Novel Lanthanide Hydride in a Non-Cyclopentadienyl Coordination Environment; Synthesis, Structure and Catalytic Activity in Olefin Polymerization. Eur. J. Inorg. Chem. 2004, 4396–4401. [Google Scholar] [CrossRef]
- Lyubov, D.M.; Bubnov, A.M.; Fukin, G.K.; Dolgushin, F.M.; Antipin, M.Y.; Pelcé, O.; Schappacher, M.; Guillaume, S.M.; Trifonov, A.A. Hydrido Complexes of Yttrium and Lutetium Supported by Bulky Guanidinato Ligands [Ln(μ-H){(Me3Si)2NC(NCy)2}2]2 (Ln = Y, Lu): Synthesis, Structure, and Reactivity. Eur. J. Inorg. Chem. 2008, 2090–2098. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, Y.; Shen, Q.; Yu, K.; Weng, L. Synthesis and Characterization of Lanthanide(III) Bis(guanidinate) Derivatives and the Catalytic Activity of Methyllanthanide Bis(guanidinate) Complexes for the Polymerization of ϵ-Caprolactone and Methyl Methacrylate. Eur. J. Inorg. Chem. 2003, 318–323. [Google Scholar] [CrossRef]
- Beer, S.M.J.; Krusenbaum, A.; Winter, M.; Vahlas, C.; Devi, A. Study on Structural and Thermal Characteristics of Heteroleptic Yttrium Complexes as Potential Precursors for Vapor Phase Deposition. Eur. J. Inorg. Chem. 2020, 3587–3596. [Google Scholar] [CrossRef]



















































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrillo-Hermosilla, F.; Fernández-Galán, R.; Ramos, A.; Elorriaga, D. Guanidinates as Alternative Ligands for Organometallic Complexes. Molecules 2022, 27, 5962. https://doi.org/10.3390/molecules27185962
Carrillo-Hermosilla F, Fernández-Galán R, Ramos A, Elorriaga D. Guanidinates as Alternative Ligands for Organometallic Complexes. Molecules. 2022; 27(18):5962. https://doi.org/10.3390/molecules27185962
Chicago/Turabian StyleCarrillo-Hermosilla, Fernando, Rafael Fernández-Galán, Alberto Ramos, and David Elorriaga. 2022. "Guanidinates as Alternative Ligands for Organometallic Complexes" Molecules 27, no. 18: 5962. https://doi.org/10.3390/molecules27185962
APA StyleCarrillo-Hermosilla, F., Fernández-Galán, R., Ramos, A., & Elorriaga, D. (2022). Guanidinates as Alternative Ligands for Organometallic Complexes. Molecules, 27(18), 5962. https://doi.org/10.3390/molecules27185962