
A Concise Synthesis of Sacidumlignan B




Abstract
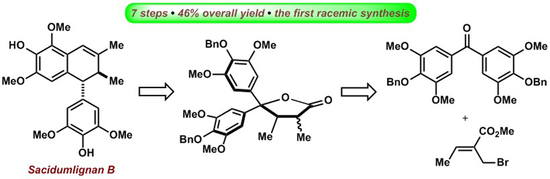
:
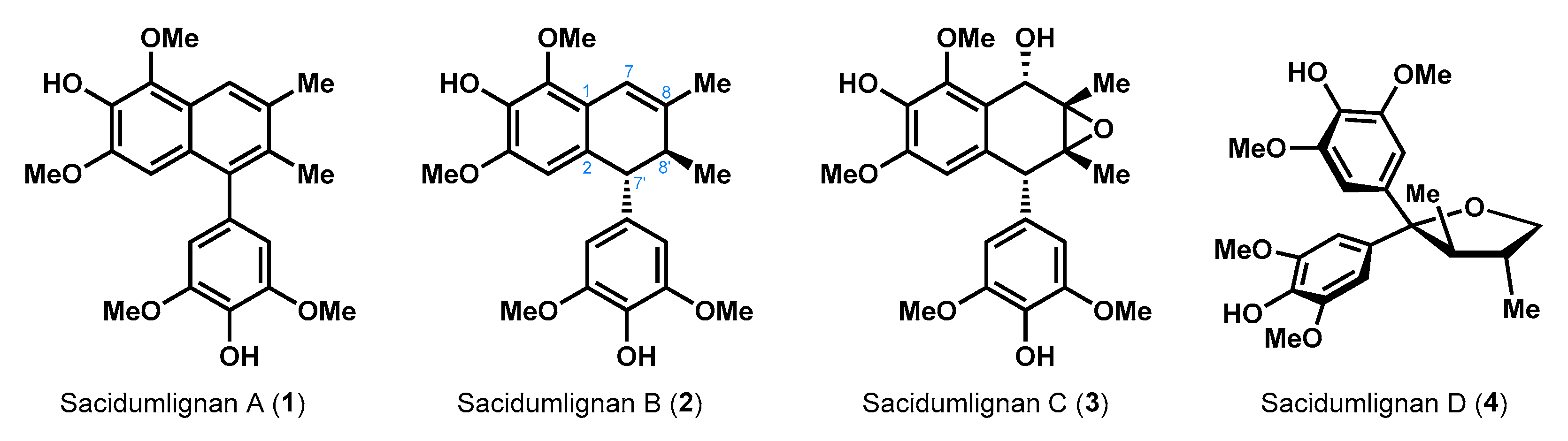
1. Introduction
2. Results and Discussion
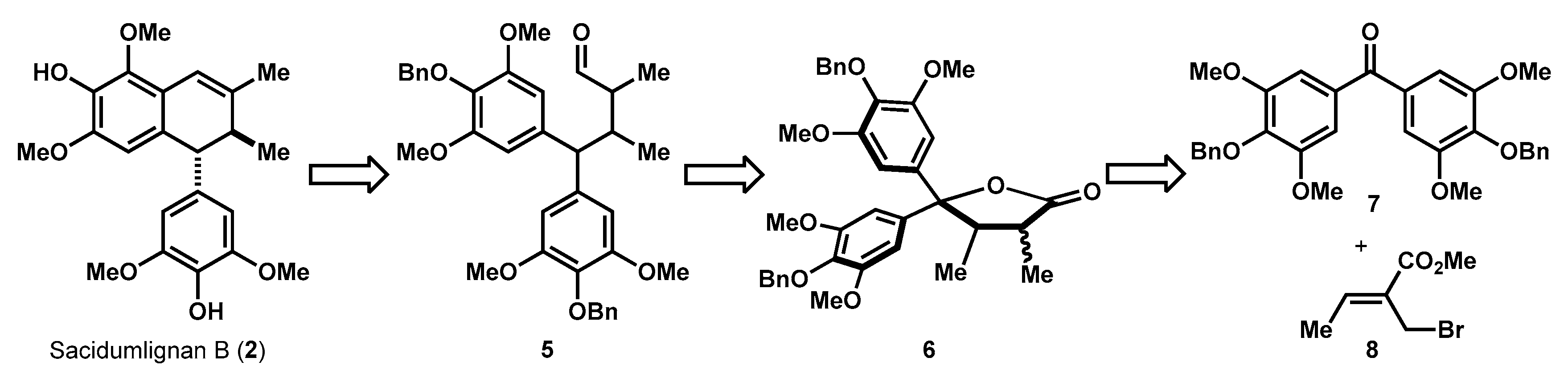
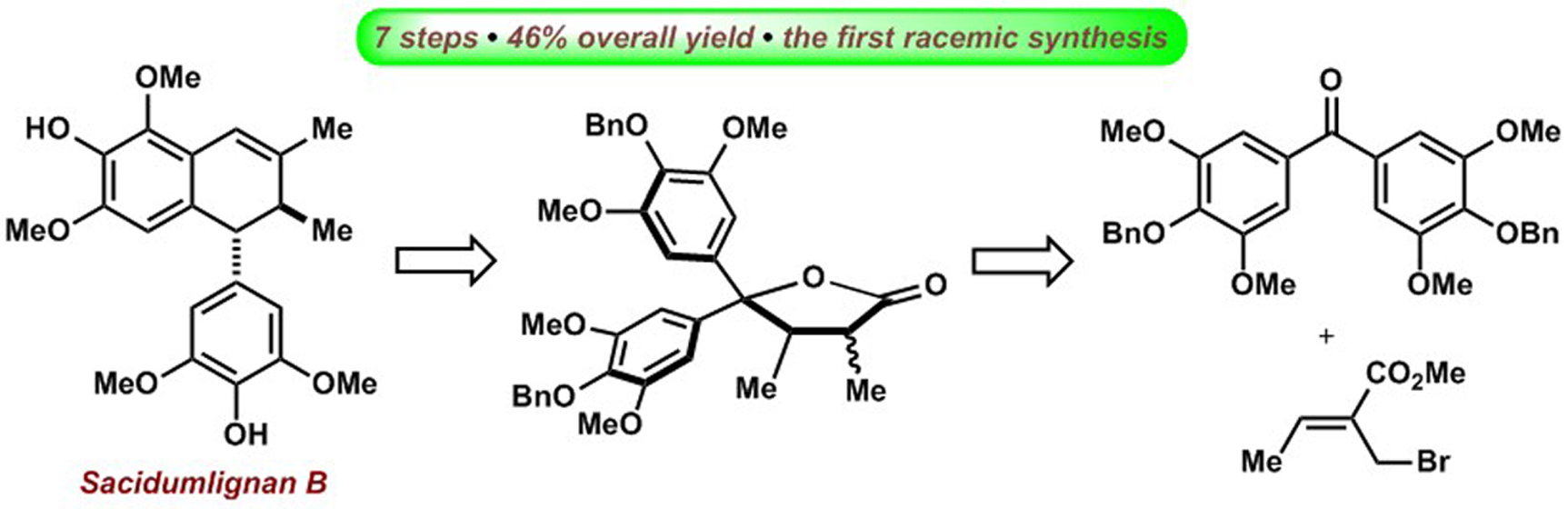
2.1. Retrosynthetic Analysis of Sacidumlignan B
2.2. Facile Construction of Sacidumlignan B Skeleton
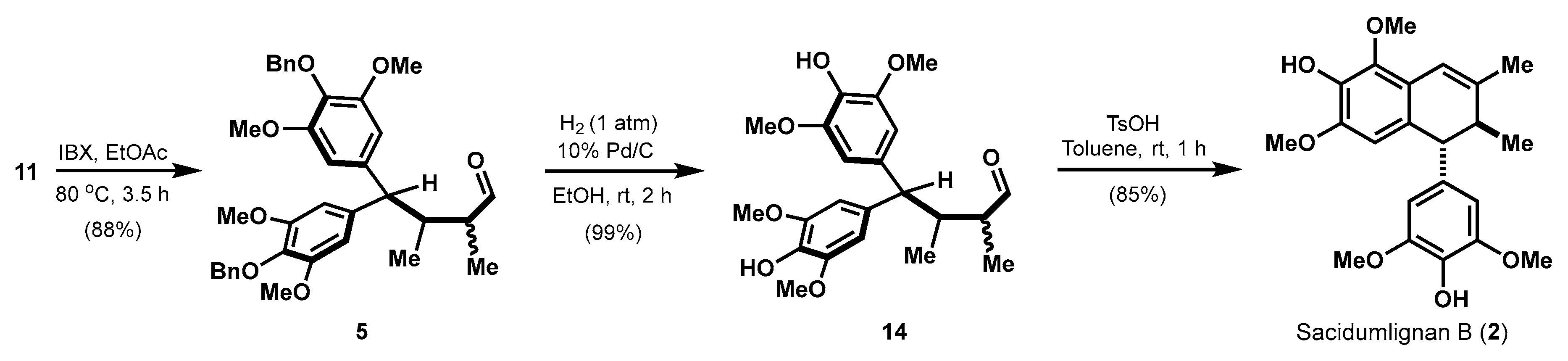
2.3. Investigation to Debenzylation
2.4. Efficient Synthesis of Sacidumlignan B
3. Materials and Methods
3.1. General Procedure
3.2. Synthesis of Compound 9
3.3. Synthesis of Compound 6
3.4. Synthesis of Compound 10
3.5. Synthesis of Compound 11
3.6. Synthesis of Compound 12
3.7. Synthesis of Compound 13 and Sacidumlignan B (2)
3.8. Alternative Synthesis of Sacidumlignan B (2)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gan, L.-S.; Yang, S.-P.; Fan, C.-Q.; Yue, J.-M. Lignans and Their Degraded Derivatives from Sarcostemma acidum. J. Nat. Prod. 2005, 68, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Q.; Yan, C.-X.; Xiao, J.; Wang, Y.-W.; Peng, Y. Recent Advances in the Total Synthesis of 2,7′-cyclolignans. Org. Biomol. Chem. 2022, 20, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Hu, X. Advances in the Synthesis of Lignan Natural Products. Molecules 2018, 23, 3385. [Google Scholar] [CrossRef] [PubMed]
- Rout, J.K.; Ramana, C.V. Total Synthesis of (−)-Sacidumlignans B and D. J. Org. Chem. 2012, 77, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Yan, C.-S.; Peng, Y.; Luo, Z.-B.; Xu, X.-B.; Wang, Y.-W. Total Synthesis of (±)-Sacidumlignans D and A through Ueno−Stork Radical Cyclization Reaction. Org. Biomol. Chem. 2013, 11, 2498–2513. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.-B.; Wang, Y.-W.; Peng, Y. Base-Promoted Addition of DMA with 1,1-Diarylethylenes: Application to a Total Synthesis of (−)-Sacidumlignan B. Org. Biomol. Chem. 2020, 18, 2054–2057. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Bai, D.; Huang, S.-H.; Jia, X.; Hong, R. Kinetic Resolution of Diols via Etherification Catalyzed by a Chiral Phosphoric Acid: Concise Synthesis of (+)-Sacidumlignan D. Asian J. Org. Chem. 2014, 3, 277–280. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, L.; Zhang, Y.; Hong, R. Bioinspired and Concise Synthesis of (±)-Stemoamide. Angew. Chem. Int. Ed. 2011, 50, 2787–2790. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Wooten, C.M.; Park, Y.; Hong, J. Stereoselective Synthesis of Tetrahydrofuran Lignans via BF3·OEt2-Promoted Reductive Deoxygenation/Epimerization of Cyclic Hemiketal: Synthesis of (−)-Odoratisol C, (−)-Futokadsurin A, (−)-Veraguensin, (+)-Fragransin A2, (+)-Galbelgin, and (+)-Talaumidin. Org. Lett. 2007, 9, 3965–3968. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Xiao, J.; Wang, Y.-W.; Peng, Y. Advances on Asymmetric Construction of Diarylmethine Stereocenters. Acta Chim. Sinica 2021, 79, 1303–1319. [Google Scholar] [CrossRef]
- Akiyama, T.; Hirofuji, H.; Ozaki, S. AlCl3-N,N-Dimethylaniline: A New Benzyl and Allyl Ether Cleavage Reagent. Tetrahedron Lett. 1991, 32, 1321–1324. [Google Scholar] [CrossRef]
- Jempty, T.; Gogins, K.A.Z.; Mazur, Y.; Miller, L. FeCl3/SiO2 Reacts as Oxidant or Lewis Acid with Phenol Ethers. J. Org. Chem. 1981, 46, 4545–4551. [Google Scholar] [CrossRef]
- Hori, H.; Nishida, Y.; Ohrui, H.; Meguro, H. Regioselective De-O-benzylation with Lewis Acid. J. Org. Chem. 1989, 54, 1346–1353. [Google Scholar] [CrossRef]
- Loh, T.-P.; Cao, G.-Q.; Pei, J. Studies Towards Total Synthesis of Antillatoxin: Synthesis of C1-C11 Fragment. Tetrahedron Lett. 1998, 39, 1457–1460. [Google Scholar] [CrossRef]
- Xiao, J.; Cong, X.-W.; Yang, G.-Z.; Wang, Y.-W.; Peng, Y. Divergent Asymmetric Syntheses of Podophyllotoxin and Related Family Members via Stereoselective Reductive Ni-catalysis. Org. Lett. 2018, 9, 3965–3968. [Google Scholar]
- Xiao, J.; Nan, G.; Wang, Y.-W.; Peng, Y. Concise Syntheses of (+)-β- and γ-Apopicropodophyllins, and Dehydrodesoxypodophyllotoxin. Molecules 2018, 23, 3037. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Shen, L.; Hu, X. Asymmetric Total Synthesis of (+)-Ovafolinins A and B. Chem. Commun. 2018, 54, 7539–7541. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.J.; Barker, D. Total Synthesis of Ovafolinins A and B: Unique Polycyclic Benzoxepin Lignans through a Cascade Cyclization. Angew. Chem. Int. Ed. 2017, 56, 9483–9486. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Reagents | Solvent | T/°C | 2 Yield (%) | 13 Yield (%) |
| 1 | Pd-C/H2 | EtOH | 20 | 0 | 98 |
| 2 | AlCl3 | Me2S | 22 | 0 | 0 |
| 3 | FeCl3 | DCM | 22 | 0 | 0 |
| 4 | TiCl4 | DCM | 0 | 15 | 0 |
| 5 | TiCl4 | DCM | −45 | 47 | 0 |
| 6 | TiCl4 | DCM | −78 | 39 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuang, Z.; Luo, Z.; Yao, S.; Wang, Y.; Peng, Y. A Concise Synthesis of Sacidumlignan B. Molecules 2022, 27, 5775. https://doi.org/10.3390/molecules27185775
Zhuang Z, Luo Z, Yao S, Wang Y, Peng Y. A Concise Synthesis of Sacidumlignan B. Molecules. 2022; 27(18):5775. https://doi.org/10.3390/molecules27185775
Chicago/Turabian StyleZhuang, Zhiyuan, Zhenbiao Luo, Sichen Yao, Yawen Wang, and Yu Peng. 2022. "A Concise Synthesis of Sacidumlignan B" Molecules 27, no. 18: 5775. https://doi.org/10.3390/molecules27185775
APA StyleZhuang, Z., Luo, Z., Yao, S., Wang, Y., & Peng, Y. (2022). A Concise Synthesis of Sacidumlignan B. Molecules, 27(18), 5775. https://doi.org/10.3390/molecules27185775