
SARS-CoV-2 and MERS-CoV Spike Protein Binding Studies Support Stable Mimic of Bound 9-O-Acetylated Sialic Acids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
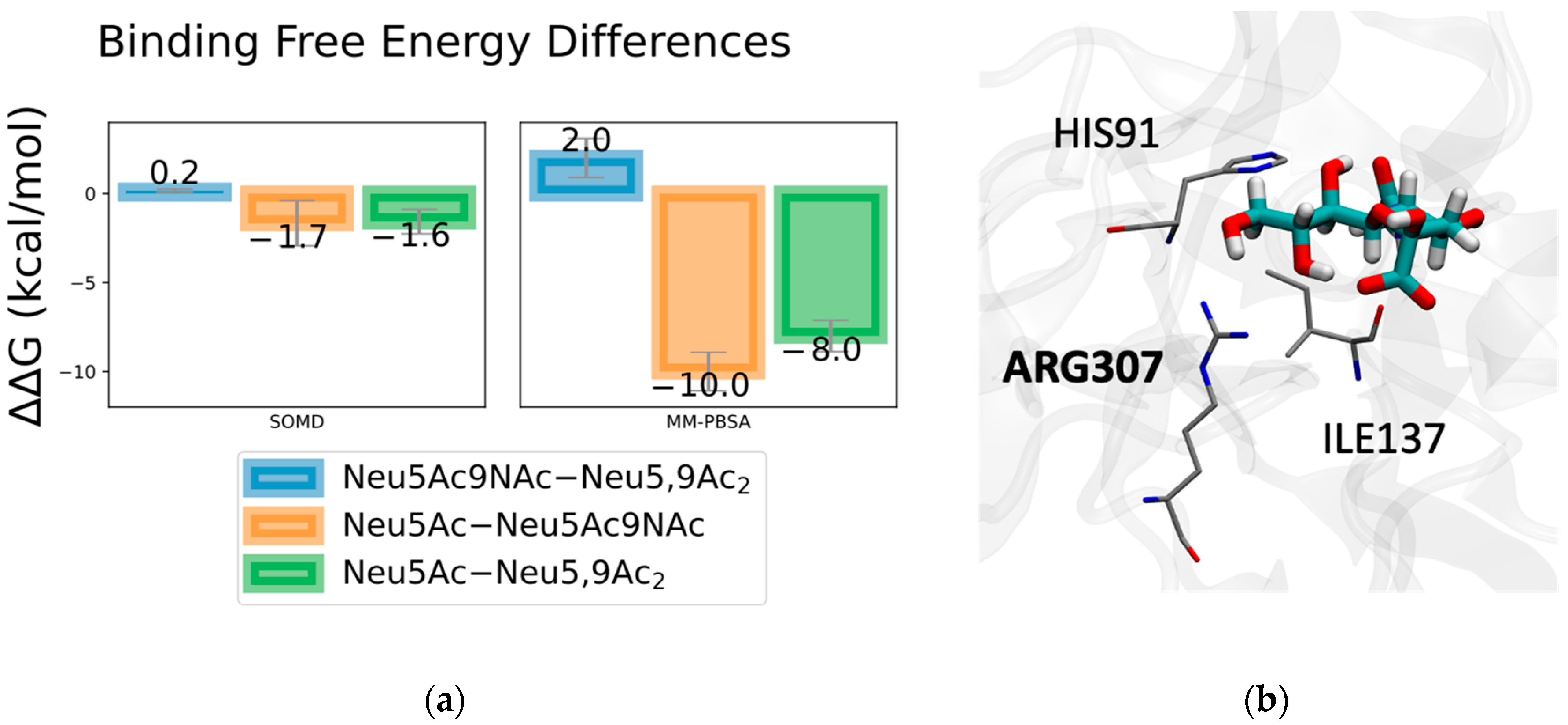
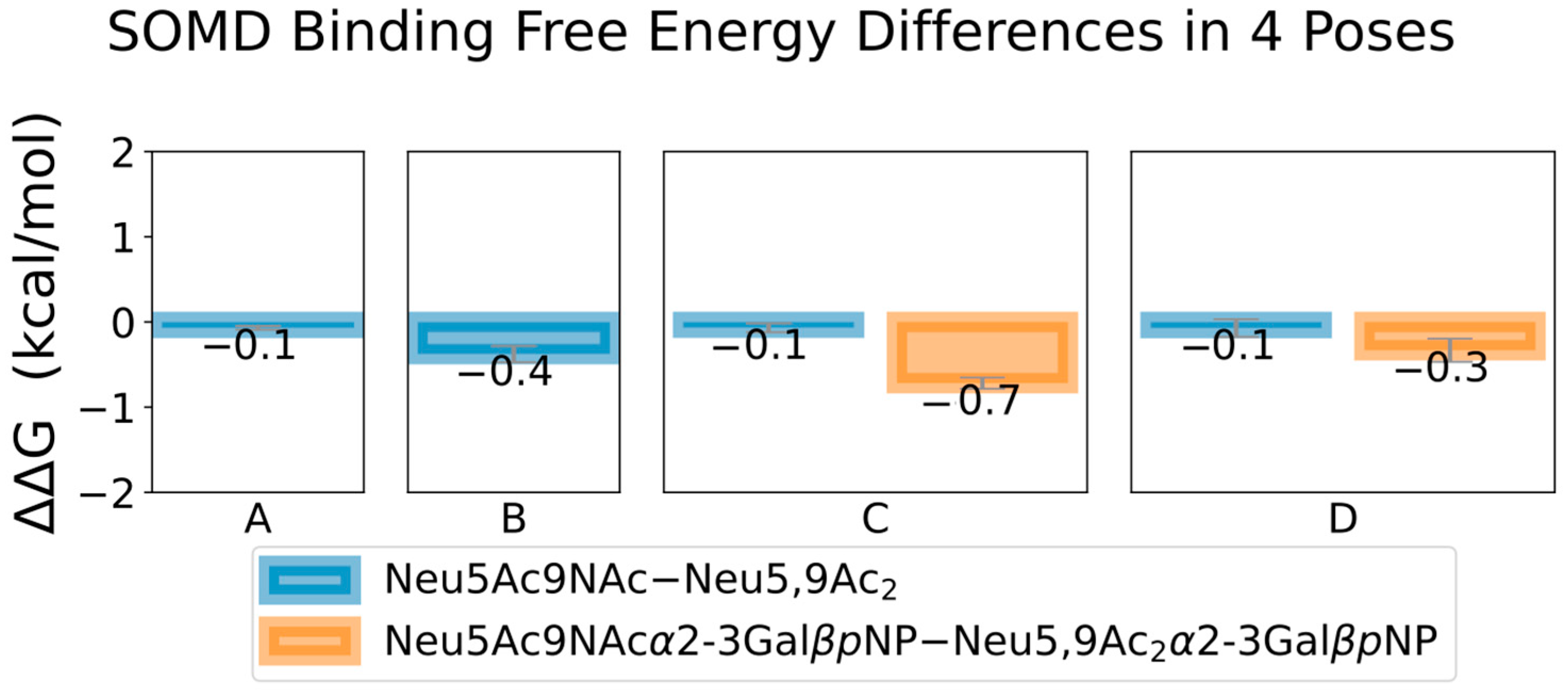
2. Results and Discussion
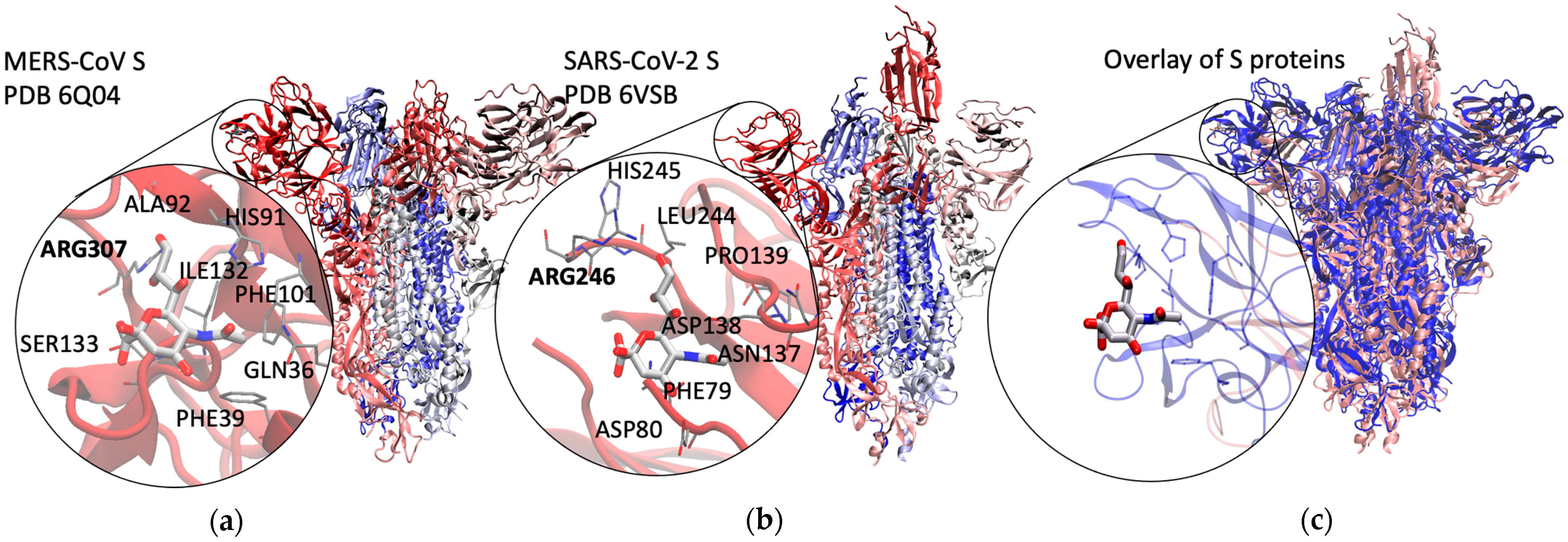
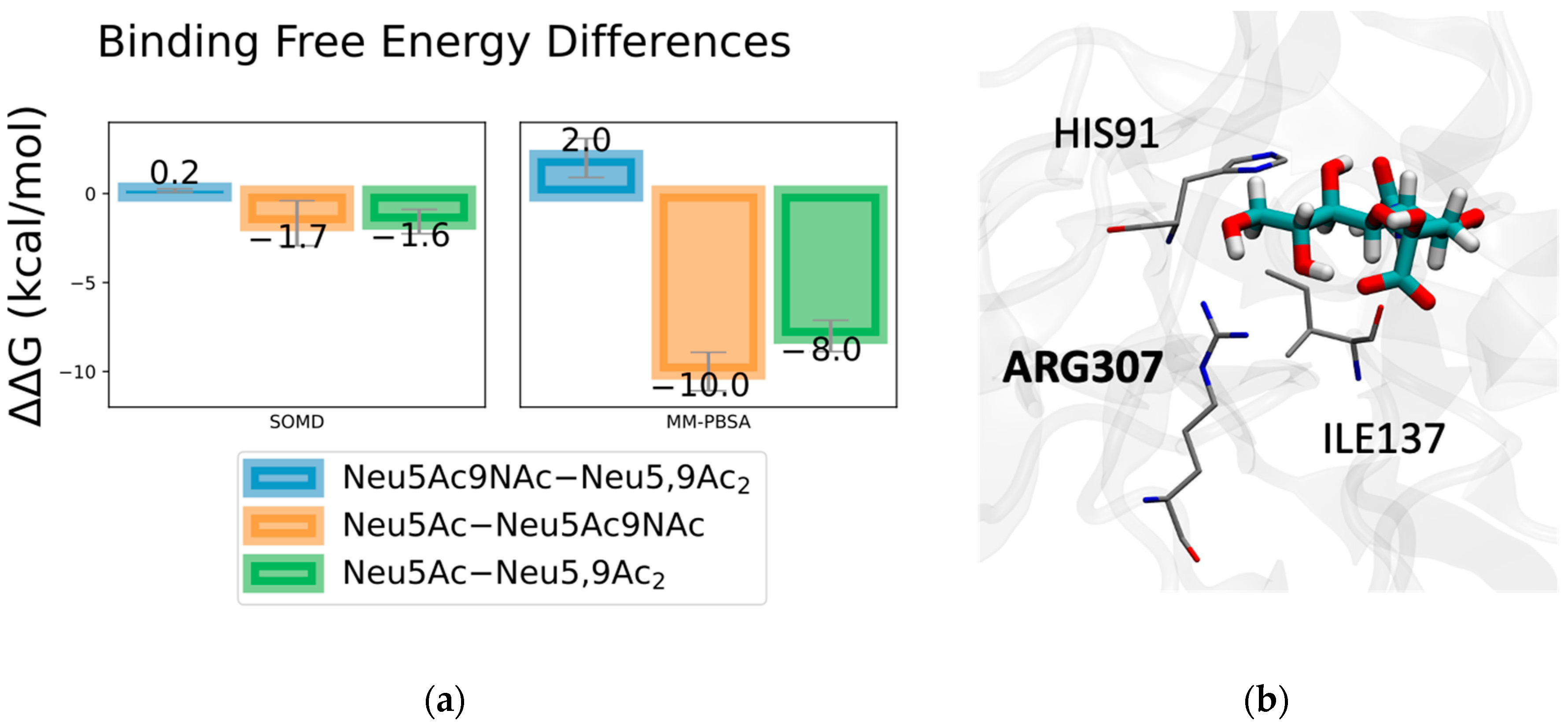
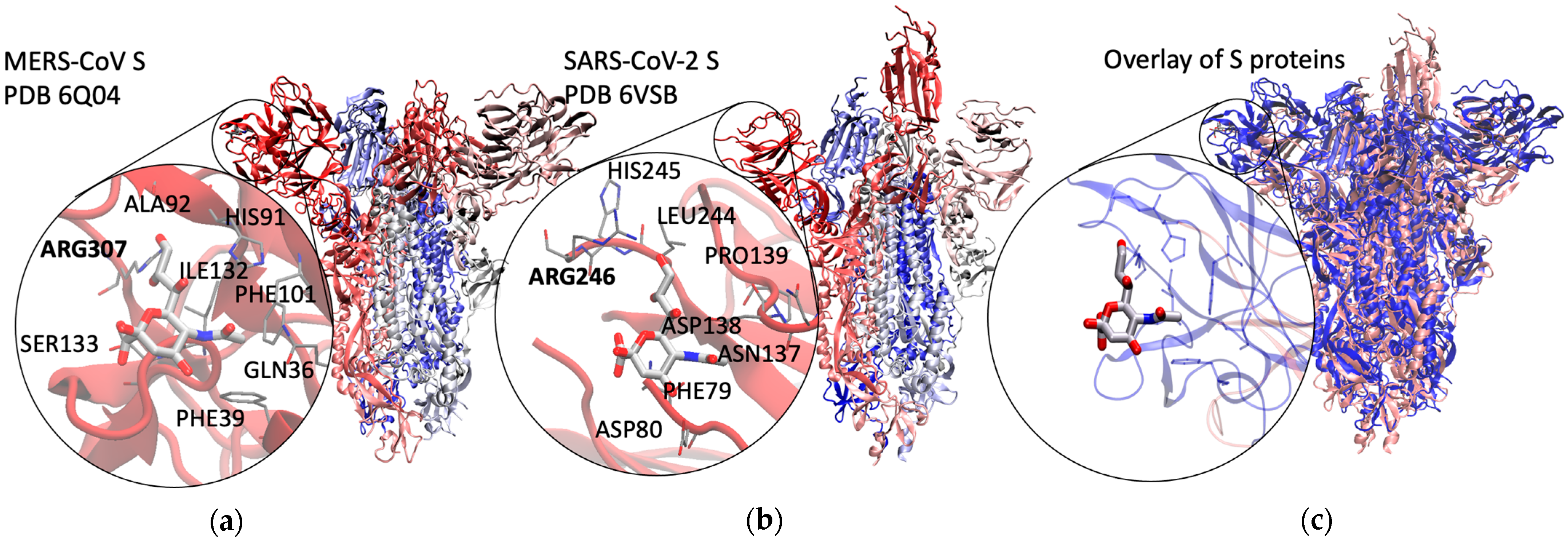
2.1. Method Validation with Sia-MERS-CoV S Protein Binding
2.2. Discovery and Analysis of Sias-SARS-CoV-2 S Protein Binding
3. Methods
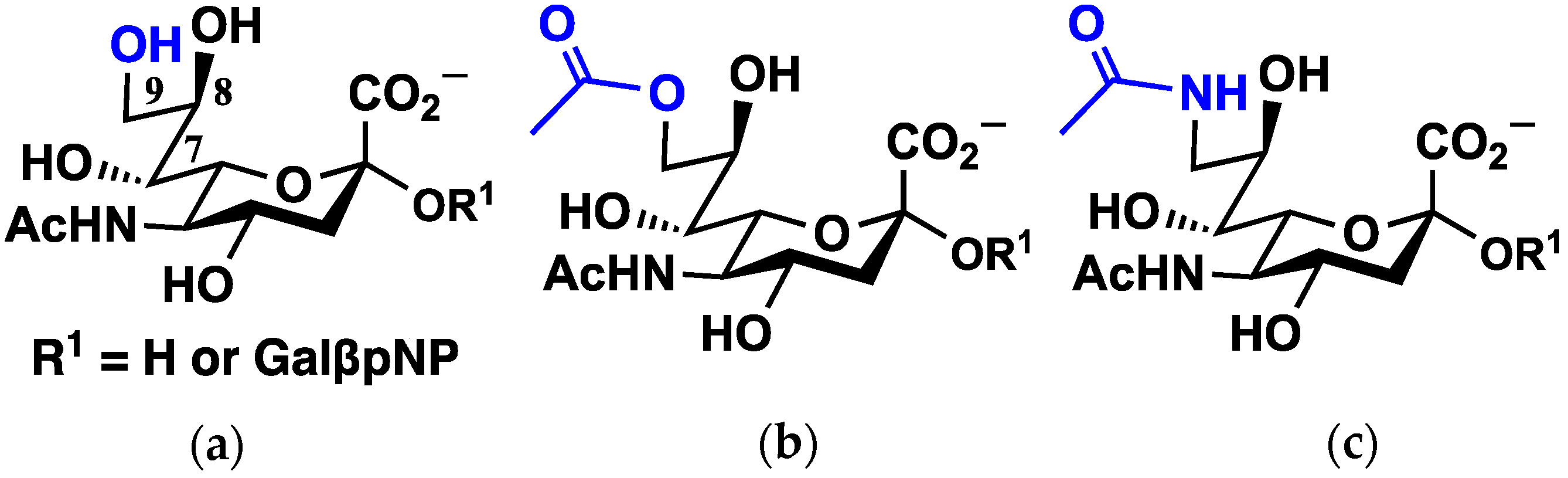
3.1. Choice of Protein Structures, Sialic Acids, and Binding Poses
3.2. Molecular Dynamics Simulation Setup
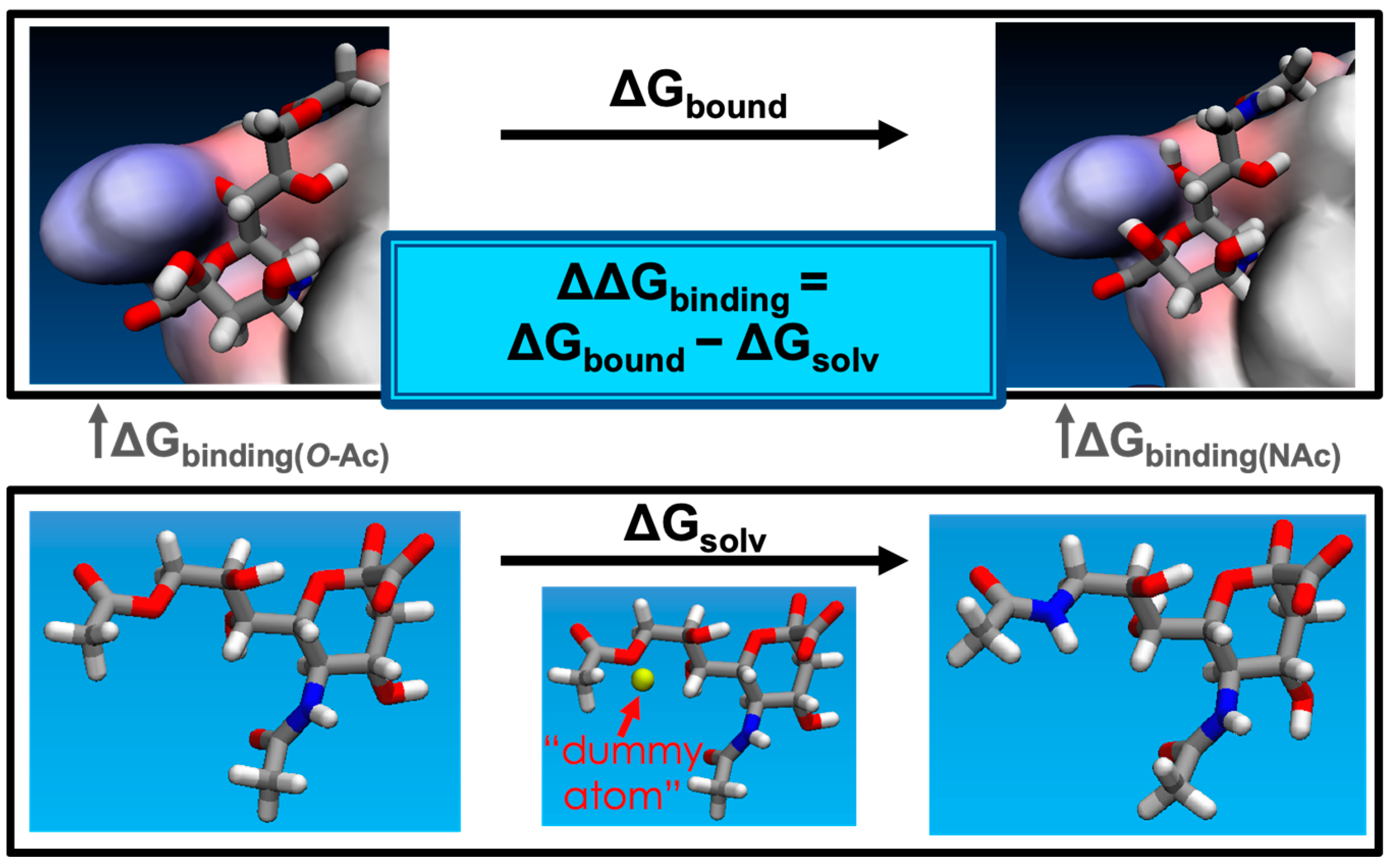
3.3. Computation of Binding Free Energies Using MM-PBSA and Alchemical Simulation Approaches
3.4. Alchemical Free Energy Simulations: Setup and Procedure
3.5. Sialic Acid Parametrization Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tortorici, M.A.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.-J.; Bosch, B.-J.; Rey, F.A.; de Groot, R.J.; et al. Structural Basis for Human Coronavirus Attachment to Sialic Acid Receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A.; Schauer, R. Sialic Acids. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; ISBN 978-0-87969-770-9. [Google Scholar]
- Park, Y.-J.; Walls, A.C.; Wang, Z.; Sauer, M.M.; Li, W.; Tortorici, M.A.; Bosch, B.-J.; DiMaio, F.; Veesler, D. Structures of MERS-CoV Spike Glycoprotein in Complex with Sialoside Attachment Receptors. Nat. Struct. Mol. Biol. 2019, 26, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulswit, R.J.G.; Lang, Y.; Bakkers, M.J.G.; Li, W.; Li, Z.; Schouten, A.; Ophorst, B.; van Kuppeveld, F.J.M.; Boons, G.-J.; Bosch, B.-J.; et al. Human Coronaviruses OC43 and HKU1 Bind to 9-O-Acetylated Sialic Acids via a Conserved Receptor-Binding Site in Spike Protein Domain A. Proc. Natl. Acad. Sci. USA 2019, 116, 2681–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Lang, Y.; Liu, L.; Bunyatov, M.I.; Sarmiento, A.I.; de Groot, R.J.; Boons, G.-J. Synthetic O-Acetylated Sialosides Facilitate Functional Receptor Identification for Human Respiratory Viruses. Nat. Chem. 2021, 13, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; McCord, K.A.; Bui, D.T.; Bouwman, K.M.; Kitova, E.N.; Elaish, M.; Kumawat, D.; Daskhan, G.C.; Tomris, I.; Han, L.; et al. Sialic Acid-Containing Glycolipids Mediate Binding and Viral Entry of SARS-CoV-2. Nat. Chem. Biol. 2022, 18, 81–90. [Google Scholar] [CrossRef]
- Nguyen, K.; Chakraborty, S.; Mansbach, R.A.; Korber, B.; Gnanakaran, S. Exploring the Role of Glycans in the Interaction of SARS-CoV-2 RBD and Human Receptor ACE2. Viruses 2021, 13, 927. [Google Scholar] [CrossRef]
- Baker, A.N.; Richards, S.-J.; Guy, C.S.; Congdon, T.R.; Hasan, M.; Zwetsloot, A.J.; Gallo, A.; Lewandowski, J.R.; Stansfeld, P.J.; Straube, A.; et al. The SARS-CoV-2 Spike Protein Binds Sialic Acids and Enables Rapid Detection in a Lateral Flow Point of Care Diagnostic Device. ACS Cent. Sci. 2020, 6, 2046–2052. [Google Scholar] [CrossRef]
- Dhar, C.; Sasmal, A.; Diaz, S.; Verhagen, A.; Yu, H.; Li, W.; Chen, X.; Varki, A. Are Sialic Acids Involved in COVID-19 Pathogenesis? Glycobiology 2021, 31, 1068–1071. [Google Scholar] [CrossRef]
- Li, B.; Wang, L.; Ge, H.; Zhang, X.; Ren, P.; Guo, Y.; Chen, W.; Li, J.; Zhu, W.; Chen, W.; et al. Identification of Potential Binding Sites of Sialic Acids on the RBD Domain of SARS-CoV-2 Spike Protein. Front. Chem. 2021, 9, 659764. [Google Scholar] [CrossRef]
- Wang, C.-W.; Lee, O.K.; Fischer, W.B.; Wang, C.-W.; Lee, O.K.; Fischer, W.B. Screening Coronavirus and Human Proteins for Sialic Acid Binding Sites Using a Docking Approach. AIMS Biophys. 2021, 8, 248–263. [Google Scholar] [CrossRef]
- Milanetti, E.; Miotto, M.; Di Rienzo, L.; Nagaraj, M.; Monti, M.; Golbek, T.W.; Gosti, G.; Roeters, S.J.; Weidner, T.; Otzen, D.E.; et al. In-Silico Evidence for a Two Receptor Based Strategy of SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 509. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Di Scala, C.; Chahinian, H.; Yahi, N. Structural and Molecular Modelling Studies Reveal a New Mechanism of Action of Chloroquine and Hydroxychloroquine against SARS-CoV-2 Infection. Int. J. Antimicrob. Agents 2020, 55, 105960. [Google Scholar] [CrossRef] [PubMed]
- Zamorano Cuervo, N.; Grandvaux, N. ACE2: Evidence of Role as Entry Receptor for SARS-CoV-2 and Implications in Comorbidities. eLife 2020, 9, e61390. [Google Scholar] [CrossRef] [PubMed]
- Vandelli, A.; Monti, M.; Milanetti, E.; Armaos, A.; Rupert, J.; Zacco, E.; Bechara, E.; Delli Ponti, R.; Tartaglia, G.G. Structural Analysis of SARS-CoV-2 Genome and Predictions of the Human Interactome. Nucleic Acids Res. 2020, 48, 11270–11283. [Google Scholar] [CrossRef]
- Seyran, M.; Takayama, K.; Uversky, V.N.; Lundstrom, K.; Palù, G.; Sherchan, S.P.; Attrish, D.; Rezaei, N.; Aljabali, A.A.A.; Ghosh, S.; et al. The Structural Basis of Accelerated Host Cell Entry by SARS-CoV-2. FEBS J. 2021, 288, 5010–5020. [Google Scholar] [CrossRef]
- Awasthi, M.; Gulati, S.; Sarkar, D.P.; Tiwari, S.; Kateriya, S.; Ranjan, P.; Verma, S.K. The Sialoside-Binding Pocket of SARS-CoV-2 Spike Glycoprotein Structurally Resembles MERS-CoV. Viruses 2020, 12, 909. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Sun, X.-L. The Role of Cell Surface Sialic Acids for SARS-CoV-2 Infection. Glycobiology 2021, 31, 1245–1253. [Google Scholar] [CrossRef]
- Khedri, Z.; Xiao, A.; Yu, H.; Landig, C.S.; Li, W.; Diaz, S.; Wasik, B.R.; Parrish, C.R.; Wang, L.-P.; Varki, A.; et al. A Chemical Biology Solution to Problems with Studying Biologically Important but UnsTable 9-O-Acetyl Sialic Acids. ACS Chem. Biol. 2017, 12, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Battistel, M.D.; Reeves, H.; Oh, L.; Yu, H.; Chen, X.; Wang, L.-P.; Freedberg, D.I. A Combined NMR, MD and DFT Conformational Analysis of 9-O-Acetyl Sialic Acid-Containing GM3 Ganglioside Glycan and Its 9-N-Acetyl Mimic. Glycobiology 2020, 30, 787–801. [Google Scholar] [CrossRef]
- Ji, Y.; Sasmal, A.; Li, W.; Oh, L.; Srivastava, S.; Hargett, A.A.; Wasik, B.R.; Yu, H.; Diaz, S.; Choudhury, B.; et al. Reversible O-Acetyl Migration within the Sialic Acid Side Chain and Its Influence on Protein Recognition. ACS Chem. Biol. 2021, 16, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Oh, L.; Ji, Y.; Li, W.; Varki, A.; Chen, X.; Wang, L.-P. O-Acetyl Migration within the Sialic Acid Side Chain: A Mechanistic Study by the Ab Initio Nanoreactor. Department of Chemistry, University of California, Davis, CA, USA. 2022; submitted. [Google Scholar]
- Varki, A.; Angata, T. Siglecs—The Major Subfamily of I-Type Lectins. Glycobiology 2006, 16, 1R–27R. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, L. Strong Ligand-Protein Interactions Derived from Diffuse Ligand Interactions with Loose Binding Sites. BioMed Res. Int. 2015, 2015, e746980. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Rizzi, A.; Murkli, S.; McNeill, J.N.; Yao, W.; Sullivan, M.; Gilson, M.K.; Chiu, M.W.; Isaacs, L.; Gibb, B.C.; Mobley, D.L.; et al. Overview of the SAMPL6 Host–Guest Binding Affinity Prediction Challenge. J. Comput. Aided Mol. Des. 2018, 32, 937–963. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef]
- Woods, C.J.; Mey, A.S.; Calabro, G.; Julien, M. Sire Molecular Simulation Framework. Version 2020.1.0. Available online: Http://Siremol.Org.2020.1.0 (accessed on 19 May 2020).
- Li, W.; Hulswit, R.J.G.; Widjaja, I.; Raj, V.S.; McBride, R.; Peng, W.; Widagdo, W.; Tortorici, M.A.; van Dieren, B.; Lang, Y.; et al. Identification of Sialic Acid-Binding Function for the Middle East Respiratory Syndrome Coronavirus Spike Glycoprotein. Proc. Natl. Acad. Sci. USA 2017, 114, E8508–E8517. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.K.; Penkler, D.L.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: A Software Suite for Analyzing Molecular Dynamics Trajectories. Bioinformatics 2017, 33, 2768–2771. [Google Scholar] [CrossRef] [Green Version]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemost. 2020, 120, 1700–1715. [Google Scholar] [CrossRef]
- Kim, S.H.; Kearns, F.L.; Rosenfeld, M.A.; Casalino, L.; Papanikolas, M.J.; Simmerling, C.; Amaro, R.E.; Freeman, R. GlycoGrip: Cell Surface-Inspired Universal Sensor for Betacoronaviruses. ACS Cent. Sci. 2022, 8, 22–42. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.C.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 Spike Glycoprotein Conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.; Eargle, J.; Wright, D.; Luthey-Schulten, Z. MultiSeq: Unifying Sequence and Structure Data for Evolutionary Analysis. BMC Bioinform. 2006, 7, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible Shape-Guided Docking For Pose Prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef]
- OEDOCKING 3.3.1: OpenEye Scientific Software, Inc., Santa Fe, NM. Available online: http://www.eyesopen.com (accessed on 28 May 2019).
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without Compromise—A Mixed Precision Model for GPU Accelerated Molecular Dynamics Simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Nguyen, P.H.; Pham, K.; Huynh, D.; Le, T.-B.N.; Wang, H.; Ren, P.; Luo, R. Calculating Protein-Ligand Binding Affinities with MMPBSA: Method and Error Analysis: Calculating Protein-Ligand Binding Affinities. J. Comput. Chem. 2016, 37, 2436–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Srinivasan, J.; Miller, J.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of RNA Hairpin Loops and Helices. J. Biomol. Struct. Dyn. 1998, 16, 671–682. [Google Scholar] [CrossRef]
- Woods, C. FESetup: Automating Setup for Alchemical Free Energy Simulations. J. Chem. Inf. Modeling 2015, 55, 2485–2490. [Google Scholar]
- Mey, A.S.J.S.; Jiménez, J.J.; Michel, J. Impact of Domain Knowledge on Blinded Predictions of Binding Energies by Alchemical Free Energy Calculations. J. Comput. Aided Mol. Des. 2018, 32, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, H.H.; Bosisio, S.; Duarte Ramos Matos, G.; Suh, D.; Roux, B.; Mobley, D.L.; Michel, J. Reproducibility of Free Energy Calculations across Different Molecular Simulation Software Packages. J. Chem. Theory Comput. 2018, 14, 5567–5582. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.; Firth-Clark, S.; Tosco, P.; Mey, A.S.J.S.; Mackey, M.; Michel, J. Automated Assessment of Binding Affinity via Alchemical Free Energy Calculations. J. Chem. Inf. Model. 2020, 60, 3120–3130. [Google Scholar] [CrossRef] [PubMed]
- Mey, A.S.J.S.; Allen, B.K.; Macdonald, H.E.B.; Chodera, J.D.; Hahn, D.F.; Kuhn, M.; Michel, J.; Mobley, D.L.; Naden, L.N.; Prasad, S.; et al. Best Practices for Alchemical Free Energy Calculations [Article v1.0]. Living J. Comput. Mol. Sci. 2020, 2, 18378. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Chodera, J.D. Statistically Optimal Analysis of Samples from Multiple Equilibrium States. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Merz, K.M.; Kollman, P.A. Free Energy Perturbation Simulations of the Inhibition of Thermolysin: Prediction of the Free Energy of Binding of a New Inhibitor. J. Am. Chem. Soc. 1989, 111, 5649–5658. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A Generalizable Biomolecular Force Field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ufimtsev, I.S.; Martinez, T.J. Quantum Chemistry on Graphical Processing Units. 3. Analytical Energy Gradients, Geometry Optimization, and First Principles Molecular Dynamics. J. Chem. Theory Comput. 2009, 5, 2619–2628. [Google Scholar] [CrossRef]
- Ufimtsev, I.S.; Martínez, T.J. Quantum Chemistry on Graphical Processing Units. 1. Strategies for Two-Electron Integral Evaluation. J. Chem. Theory Comput. 2008, 4, 222–231. [Google Scholar] [CrossRef]
- Ufimtsev, I.S.; Martinez, T.J. Quantum Chemistry on Graphical Processing Units. 2. Direct Self-Consistent-Field Implementation. J. Chem. Theory Comput. 2009, 5, 1004–1015. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-P.; Song, C. Geometry Optimization Made Simple with Translation and Rotation Coordinates. J. Chem. Phys. 2016, 144, 214108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-P.; McKiernan, K.A.; Gomes, J.; Beauchamp, K.A.; Head-Gordon, T.; Rice, J.E.; Swope, W.C.; Martínez, T.J.; Pande, V.S. Building a More Predictive Protein Force Field: A Systematic and Reproducible Route to AMBER-FB15. J. Phys. Chem. B 2017, 121, 4023–4039. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, M.K.; Karton, A.; Martin, J.M.L. Benchmark Ab Initio Conformational Energies for the Proteinogenic Amino Acids through Explicitly Correlated Methods. Assessment of Density Functional Methods. J. Chem. Theory Comput. 2016, 12, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-P.; Martinez, T.J.; Pande, V.S. Building Force Fields: An Automatic, Systematic, and Reproducible Approach. J. Phys. Chem. Lett. 2014, 5, 1885–1891. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, L.; Varki, A.; Chen, X.; Wang, L.-P. SARS-CoV-2 and MERS-CoV Spike Protein Binding Studies Support Stable Mimic of Bound 9-O-Acetylated Sialic Acids. Molecules 2022, 27, 5322. https://doi.org/10.3390/molecules27165322
Oh L, Varki A, Chen X, Wang L-P. SARS-CoV-2 and MERS-CoV Spike Protein Binding Studies Support Stable Mimic of Bound 9-O-Acetylated Sialic Acids. Molecules. 2022; 27(16):5322. https://doi.org/10.3390/molecules27165322
Chicago/Turabian StyleOh, Lisa, Ajit Varki, Xi Chen, and Lee-Ping Wang. 2022. "SARS-CoV-2 and MERS-CoV Spike Protein Binding Studies Support Stable Mimic of Bound 9-O-Acetylated Sialic Acids" Molecules 27, no. 16: 5322. https://doi.org/10.3390/molecules27165322
APA StyleOh, L., Varki, A., Chen, X., & Wang, L.-P. (2022). SARS-CoV-2 and MERS-CoV Spike Protein Binding Studies Support Stable Mimic of Bound 9-O-Acetylated Sialic Acids. Molecules, 27(16), 5322. https://doi.org/10.3390/molecules27165322