
An Efficient Method for Selective Syntheses of Sodium Selenide and Dialkyl Selenides

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
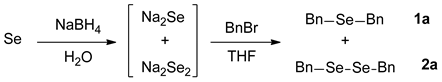
2.1. Optimization for the Reaction Conditions
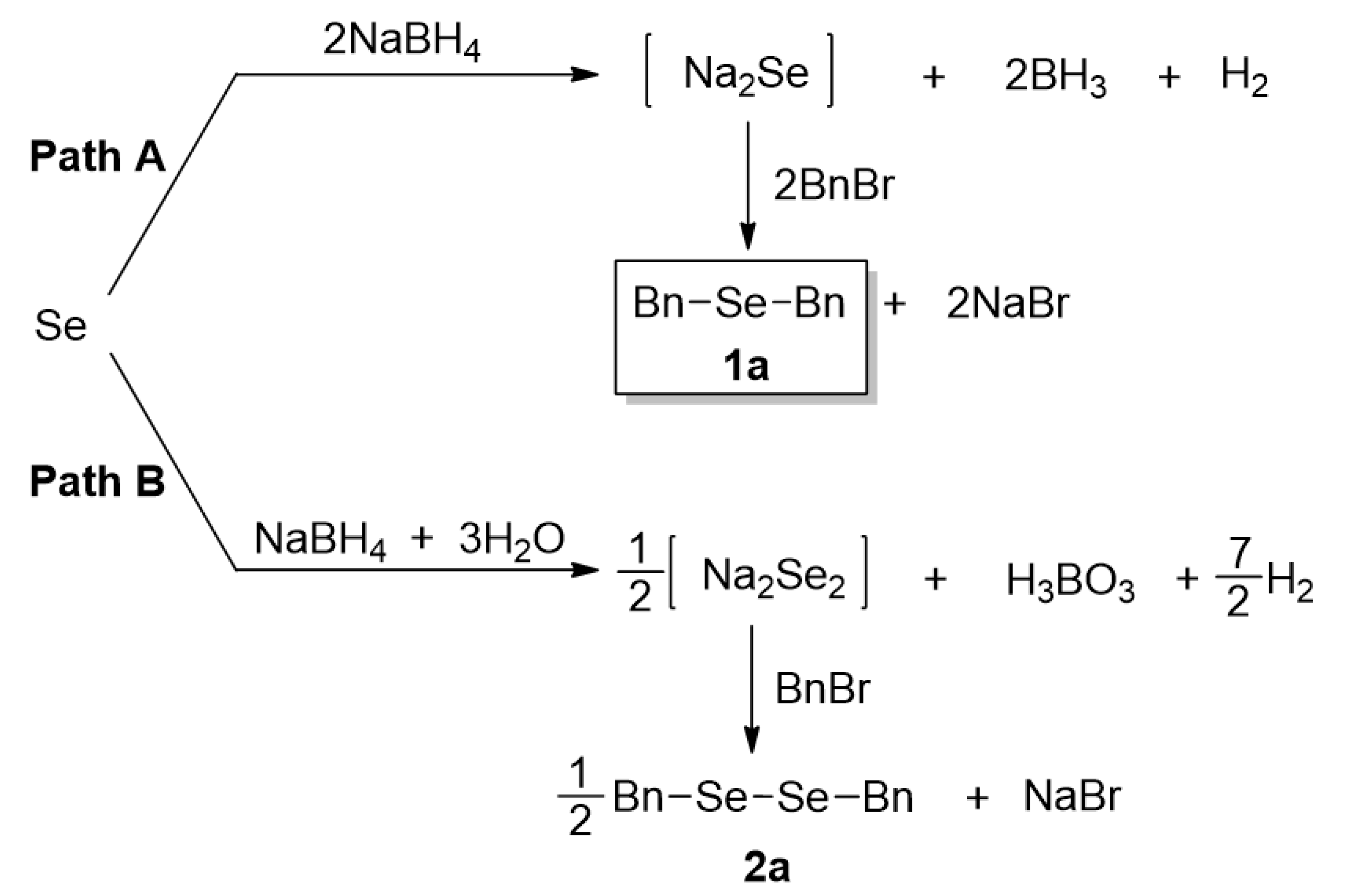
2.2. Reaction Pathways
2.3. Synthesis of Dialkyl Selenides 1
2.4. Solvent Studies
3. Experimental
3.1. General
3.2. General Procedure for the Synthesis of Dialkyl Monoselenides 1
3.2.1. 1,2-Dibenzyl Selenide (1a) [2]
3.2.2. 1,2-Bis(2-phenylethyl) Selenide (1b) [2]
3.2.3. 1,2-Diallyl Selenide (1c) [22]
3.2.4. 1,2-Di-n-butyl Selenide (1d) [19]
3.2.5. 1,2-Di-n-pentyl Selenide (1e) [2]
3.2.6. 1,2-Di-n-hexyl Selenide (1f) [19]
3.2.7. 1,2-Di-n-octyl Selenide (1g) [23]
3.2.8. 1,2-Di-c-pentyl Selenide (1h) [24]
3.2.9. 1,2-Di-c-hexyl Selenide (1i) [25]
3.2.10. 1,2-Bis(3-pentyl) Selenide (1j) [22]
3.2.11. 1,2-Bis(4-heptyl) Selenide (1k)
3.2.12. 1,3-Dihydrobenzo[c]Selenophene (1l) [26]
3.2.13. Tetrahydro-3,4-Selenophenediol (1m) [27]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Jain, V.K. An Overview of Organoselenium Chemistry: From Fundamentals to Synthesis. In Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Jain, V.K., Priyadarsini, K.I., Eds.; The Royal Society of Chemistry: London, UK, 2017; pp. 1–33. ISBN 978-1-78801-029-0. [Google Scholar]
- Gladysz, J.A.; Hornby, J.L.; Garbe, J.E. A Convenient One-Flask Synthesis of Dialkyl Selenides and Diselenides via Lithium Triethylborohydride Reduction of Sex. J. Org. Chem. 1978, 43, 1204–1208. [Google Scholar] [CrossRef]
- Ivanova, A.; Arsenyan, P. Rise of Diselenides: Recent Advances in the Synthesis of Heteroarylselenides. Coord. Chem. Rev. 2018, 370, 55–68. [Google Scholar] [CrossRef]
- Rayman, M.P. The Importance of Selenium to Human Health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef]
- Álvarez-Pérez, M.; Ali, W.; Marć, M.A.; Handzlik, J.; Domínguez-Álvarez, E. Selenides and Diselenides: A Review of their Anticancer and Chemopreventive Activity. Molecules 2018, 23, 628. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Speckmann, B.; Klotz, L.-O. Selenoproteins: Antioxidant selenoenzymes and beyond. Arch. Biochem. Biophys. 2016, 595, 113–119. [Google Scholar] [CrossRef]
- Wessjohann, L.A.; Schneider, A.; Abbas, M.; Brandt, W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol. Chem. 2007, 388, 997–1006. [Google Scholar] [CrossRef]
- Reich, H.J.; Hondal, R.J. Why Nature Chose Selenium. Acs. Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Sies, H. Protection against reactive oxygen species by selenoproteins. Biochim. Biophys. Acta 2009, 1790, 1478–1485. [Google Scholar] [CrossRef]
- Nascimento, V.; Alberto, E.E.; Tondo, D.W.; Dambrowski, D.; Detty, M.R.; Nome, F.; Braga, A.L. GPx-Like Activity of Selenides and Selenoxides: Experimental Evidence for the Involvement of Hydroxy Perhydroxy Selenane as the Active Species. J. Am. Chem. Soc. 2012, 134, 138–141. [Google Scholar] [CrossRef]
- Abdulah, R.; Miyazaki, K.; Nakazawa, M.; Koyama, H. Chemical forms of selenium for cancer prevention. J. Trace Elem. Med. Biol. 2005, 19, 141–150. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium in cancer prevention: A review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, H.; Dong, Y.; Park, Y.-M.; Ip, C. Endoplasmic Reticulum Stress Signal Mediators are Targets of Selenium Action. Cancer Res. 2005, 65, 9073–9079. [Google Scholar] [CrossRef] [PubMed]
- Sonn, G.A.; Aronson, W.; Litwin, M.S. Impact of diet on prostate cancer: A review. Prostate Cancer Prostatic Dis. 2005, 8, 304–310. [Google Scholar] [CrossRef]
- Hodage, A.S.; Prabhu, C.P.; Phadnis, P.P.; Wadawale, A.; Priyadarsini, K.I.; Jain, V.K. Synthesis, characterization, structures and GPx mimicking activity of pyridyl and pyrimidyl based organoselenium compounds. J. Organomet. Chem. 2012, 720, 19–25. [Google Scholar] [CrossRef]
- Milton, M.D.; Khan, S.; Singh, J.D.; Mishra, V.; Khandelwal, B.L. A facile access to chalcogen and dichalcogen bearing dialkylamines and diols. Tetrahedron Lett. 2005, 46, 755–758. [Google Scholar] [CrossRef]
- Andrade, L.H.; Silva, A.V.; Milani, P.; Koszelewski, D.; Kroutil, W. ω-Transaminases as efficient biocatalysts to obtain novel chiral selenium-amine ligands for Pd-catalysis. Org. Biomol. Chem. 2010, 8, 2043–2051. [Google Scholar] [CrossRef]
- Klayman, D.L.; Griffin, T.S. Reaction of Selenium with Sodium Borohydride in Protic Solvents. A Facile Method for the Introduction of Selenium into Organic Molecules. J. Am. Chem. Soc. 1973, 95, 197–199. [Google Scholar] [CrossRef]
- Krief, A.; Derock, M. Synthesis of diselenides and selenides from elemental selenium. Tetrahedron Lett. 2002, 43, 3083–3086. [Google Scholar] [CrossRef]
- Rossi, R.A.; Penenory, A.B. Direct (One Pot) Synthesis of Organoselenium and Organotellurium Compounds from the Metals. J. Org. Chem. 1981, 46, 4580–4582. [Google Scholar] [CrossRef]
- Lim, Y.J.; Shin, N.H.; Kim, C.; Kim, Y.E.; Cho, H.; Park, M.-S.; Lee, S.H. An efficient and practical method for the selective synthesis of sodium diselenide and diorganyl diselenides through selenium reduction. Tetrahedron 2020, 76, 131720. [Google Scholar] [CrossRef]
- Ouchi, A.; Liu, S.; Li, Z.; Kumar, S.A.; Suzuki, T.; Hyugano, T.; Kitahara, H. Factors Controlling Photochemical Cleavage of the Energetically Unfavorable Ph−Se Bond of Alkyl Phenyl Selenides. J. Org. Chem. 2007, 72, 8700–8706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-B.; Ruan, M.-D.; Fan, W.-Q. Reactions of Selenobenzamide and Alkyl Halides. Synthesis of Dialkyl Selenides and Diselenides. Synth. Commun. 1996, 26, 4665–4670. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Tanaka, H.; Takami, N.; Ogawa, A.; Ryu, I.; Sonoda, N. Reduction of Elemental Selenium by Samarium Diiodide: Selective Synthesis of Diorganyl Selenides and Diselenides. Heteroat. Chem. 1991, 2, 427–430. [Google Scholar] [CrossRef]
- Arase, A.; Masuda, Y. A New Route to Dialkyl Selenide from Olefin via Hydroboration. Chem. Lett. 1975, 4, 419–422. [Google Scholar] [CrossRef]
- Magdesieva, N.N.; Vdovin, V.A. Synthesis of 1,3-Dihydrobenzo[c]selenophene. Chem. Heterocycl. Compd. 1972, 8, 22–23. [Google Scholar] [CrossRef]
- Phadnis, P.P.; Wadawale, A.; Priyadarsini, K.I.; Jain, V.K.; Iwaoka, M. Synthesis, characterization, and structure of trans-3,4-dihydroxy-1-selenolane {DHS(OH)2} substituted derivatives. Tetrahedron Lett. 2015, 56, 2293–2296. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Na2Se Formation | 1a Formation (Reaction with BnBr) | Product | Side Product | |
| NaBH4 (eq) | Time (h) | Time (h) | 1a Yield b (%) | 2a Yield b (%) | |
| 1 | 1.0 | 1 | 2 | 25 | 40 |
| 2 | 1.5 | 1 | 2 | 27 | 40 |
| 3 | 2.0 | 0.5 | 1 | 59 | 8 |
| 4 | 1 | 2 | 44 | 34 | |
| 5 | 2 | 2 | 49 | 25 | |
| 6 | 4 | 2 | 28 | 56 | |
| 7 | 8 | 2 | c | 34 | |
| 8 | 3.0 | 0.5 | 1 | 80 | c |
| 9 | 1 | 2 | 87 | 2 | |
| 10 | 2 | 2 | 76 | c | |
| 11 | 4 | 2 | 79 | c | |
| 12 | 8 | 2 | 80 | c | |
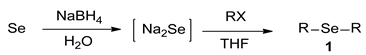 | ||||||
|---|---|---|---|---|---|---|
| Entry | Reaction with RX | Product | Yields (%) | |||
| RX | RX (eq) | Time (h) | Temp (°C) | |||
| 1 b | BnBr | 2.4 | 2 | rt | 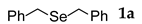 | 87 |
| 2 | PhCH2CHBr | 2.4 | 5 | rt | 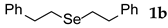 | 66 |
| 3 | allylBr | 2.4 | 25 | rt | 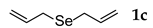 | 66 |
| 4 | n-BuBr | 2.4 | 3 | rt | n-Bu–Se-Bu-n1d | 93 |
| 5 | n-PenBr | 2.4 | 3 | rt | n-Pen–Se-Pen-n1e | 82 |
| 6 | n-HexBr | 2.4 | 5 | rt | n-Hex–Se-Hex-n1f | 85 |
| 7 | n-OctBr | 2.4 | 5 | rt | n-Oct–Se-Oct-n1g | 85 |
| 8 | 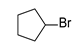 | 2.4 | 3 | 50 | 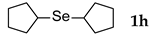 | 50 |
| 9 | 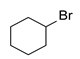 | 2.4 | 8 | 50 | 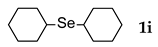 | 48 |
| 10 | 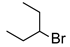 | 2.4 | 5 | 50 |  | 31 |
| 11 |  | 2.4 | 25 | 50 | 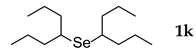 | 35 |
| 12 b | 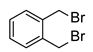 | 1.0 | 24 | rt | 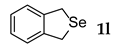 | 65 |
| 13 b | 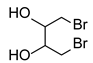 | 1.0 | 48 | rt | 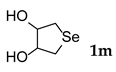 | 51 |
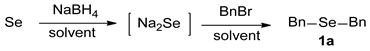 | ||||||
|---|---|---|---|---|---|---|
| Entry | Na2Se Formation | Reaction with BnBr | Product | Side Product | ||
| Solvent | Temp (°C) | Time (h) | Time (h) | 1a Yields b (%) | 2a Yields b (%) | |
| 1 | H2O-THF | 25 | 1 | 2 | 87 | 2 |
| 2 | EtOH | 25 | 1 | 2 | 81 | c |
| 3 | MeCN | 25 | 2 | 2 | 80 | c |
| 4 | THF | 50 | 1 | 48 | 50 | c |
| 5 | DME | 25 | 1 | 47 | 30 | 22 |
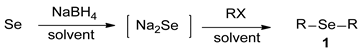 | ||||
|---|---|---|---|---|
| Entry | RX | Product | Yields (%) b | |
| Solvent (H2O-THF) | Solvent (MeCN) | |||
| 1 | BnBr | 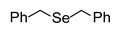 | 87% | 80% |
| 2 | BnCl | 79% | 70% | |
| 3 | PhCH2CH2Br | 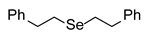 | 66% | 20% |
| 4 | PhCH2CH2Cl | 32% | 38% | |
| 5 | n-HexBr | n-Hex–Se-Hex-n | 85% | 63% |
| 6 | n-HexCl | 21% | 21% | |
| 7 | n-OctBr | n-Oct–Se-Oct-n | 85% | 56% |
| 8 | n-OctCl | 24% | 21% | |
| 9 | c-HexBr | 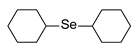 | 48% | 24% |
| 10 | c-HexCl | 3% | c % | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, N.H.; Lim, Y.J.; Kim, C.; Kim, Y.E.; Jeong, Y.R.; Cho, H.; Park, M.-S.; Lee, S.H. An Efficient Method for Selective Syntheses of Sodium Selenide and Dialkyl Selenides. Molecules 2022, 27, 5224. https://doi.org/10.3390/molecules27165224
Shin NH, Lim YJ, Kim C, Kim YE, Jeong YR, Cho H, Park M-S, Lee SH. An Efficient Method for Selective Syntheses of Sodium Selenide and Dialkyl Selenides. Molecules. 2022; 27(16):5224. https://doi.org/10.3390/molecules27165224
Chicago/Turabian StyleShin, Na Hye, Yoo Jin Lim, Chorong Kim, Ye Eun Kim, Yu Ra Jeong, Hyunsung Cho, Myung-Sook Park, and Sang Hyup Lee. 2022. "An Efficient Method for Selective Syntheses of Sodium Selenide and Dialkyl Selenides" Molecules 27, no. 16: 5224. https://doi.org/10.3390/molecules27165224
APA StyleShin, N. H., Lim, Y. J., Kim, C., Kim, Y. E., Jeong, Y. R., Cho, H., Park, M.-S., & Lee, S. H. (2022). An Efficient Method for Selective Syntheses of Sodium Selenide and Dialkyl Selenides. Molecules, 27(16), 5224. https://doi.org/10.3390/molecules27165224