
2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization




Abstract
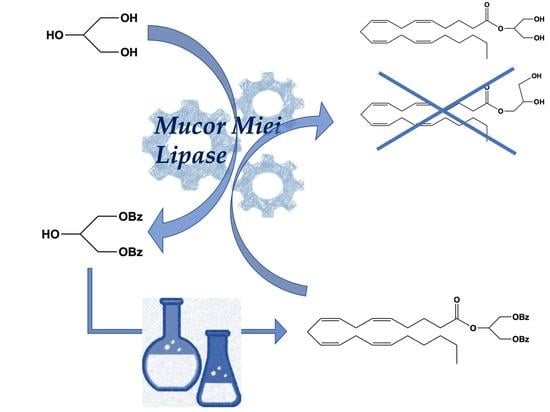
1. Introduction
2. Results and Discussion
3. Material and Methods
3.1. General Information

3.2. NMR Analysis
3.3. Synthesis and Purification of Glycerol Mono-, Di-, and Tri-Benzoate as Standard for Gas Chromatography Analyses
3.4. Gas Chromatography Separation of Glycerol Mono-, Di-, and Tri-Benzoate
3.5. Synthesis of 2-Hydroxypropane-1,3-diyl dibenzoate (1c)
3.6. Synthesis of 2-((5Z,8Z,11Z,14Z)-Icosa-5,8,11,14-tetraenoyloxy)propane-1,3-diyl dibenzoate (2)
3.7. Synthesis of (5Z,8Z,11Z,14Z)-1,3-Dihydroxypropan-2-yl Icosa-5,8,11,14-tetraenoate (3)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Pertwee, R.G. Ligands that target cannabinoid receptors in the brain: From THC to anandamide and beyond. Addict. Biol. 2008, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Vago, R.; Ravelli, A.; Bettiga, A.; Casati, S.; Lavorgna, G.; Benigni, F.; Salonia, A.; Montorsi, F.; Orioli, M.; Ciuffreda, P.; et al. Urine Endocannabinoids as Novel Non-Invasive Biomarkers for Bladder Cancer at Early Stage. Cancers 2020, 12, 870. [Google Scholar] [CrossRef] [PubMed]
- Pezzilli, R.; Ciuffreda, P.; Ottria, R.; Ravelli, A.; Melzi d’Eril, G.; Barassi, A. Serum endocannabinoids in assessing pain in patients with chronic pancreatitis and in those with pancreatic ductal adenocarcinoma. Scand. J. Gastroenterol. 2017, 52, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Bersani, G.; Pacitti, F.; Iannitelli, A.; Caroti, E.; Quartini, A.; Xenos, D.; Marconi, M.; Cuoco, V.; Bigio, B.; Bowles, N.P.; et al. Inverse correlation between plasma 2-arachidonoylglycerol levels and subjective severity of depression. Hum. Psychopharmacol. 2021, 36, e2779. [Google Scholar] [CrossRef]
- Marchioni, C.; de Souza, I.D.; Acquaro, V.R.; de Souza, C.J.A.; Tumas, V.; Queiroz, M.E.C. Recent advances in LC-MS/MS methods to determine endocannabinoids in biological samples: Application in neurodegenerative diseases. Anal. Chim. Acta 2018, 1044, 12–28. [Google Scholar] [CrossRef]
- Ottria, R.; Cappelletti, L.; Ravelli, A.; Mariotti, M.; Gigli, F.; Romagnosi, S.; Ciuffreda, P.; Banfi, G.; Drago, L. Plasma Endocannabinoids behavior in Total Knee and Hip Arthroplasty. J. Biol. Regul. Homeost. Agent 2016, 30, 1147–1152. [Google Scholar] [CrossRef]
- Lauria, S.; Perrotta, C.; Casati, S.; Di Renzo, I.; Ottria, R.; Eberini, I.; Palazzolo, L.; Parravicini, C.; Ciuffreda, P. Design, synthesis, molecular modelling and in vitro cytotoxicity analysis of novel carbamate derivatives as inhibitors of Monoacylglycerol lipase. Bioorg. Med. Chem. 2018, 26, 2561–2572. [Google Scholar] [CrossRef]
- Vago, R.; Bettiga, A.; Salonia, A.; Ciuffreda, P.; Ottria, R. Development of new inhibitors for N-acylethanolamine-hydrolyzing acid amidase as promising tool against bladder cancer. Bioorg. Med. Chem. 2017, 25, 1242–1249. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cordaro, M.; Siracusa, R.; Casili, G.; Evangelista, M.; Cuzzocrea, S. N-palmitoylethanolamide Prevents Parkinsonian Phenotypes in Aged Mice. Mol. Neurobiol. 2018, 55, 8455–8472. [Google Scholar] [CrossRef]
- Finn, D.P.; Haroutounian, S.; Hohmann, A.G.; Krane, E.; Soliman, N.; Rice, A.S.C. Cannabinoids, the endocannabinoid system, and pain: A review of preclinical studies. Pain 2021, 162, S5–S25. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.L.; Grenald, S.A.; Ciccone, H.A.; BassiriRad, N.; Niphakis, M.J.; Cravatt, B.F.; Largent-Milnes, T.M.; Vanderah, T.W. The endocannabinoid system alleviates pain in a murine model of cancer-induced bone pain. J. Pharmacol. Exp. Ther. 2020, 373, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Casati, S.; Giannasi, C.; Minoli, M.; Niada, S.; Ravelli, A.; Angeli, I.; Mergenthaler, V.; Ottria, R.; Ciuffreda, P.; Orioli, M.; et al. Quantitative lipidomic analysis of osteosarcoma cell-derived products by UHPLC-MS/MS. Biomolecules 2020, 10, 1302. [Google Scholar] [CrossRef]
- Compton, D.L.; Vermillion, K.E.; Laszlo, J.A. Acyl migration kinetics of 2-monoacylglycerols from soybean oil via 1H NMR. J. Amer. Oil Chem. Soc. 2007, 84, 343–348. [Google Scholar] [CrossRef]
- Boswinkel, G.; Derksen, J.T.P.; van’t Riet, K.; Cuperus, F.P. Kinetics of acyl migration in monoglycerides and dependence on acyl chainlength. J. Amer. Oil Chem. Soc. 1996, 73, 707–711. [Google Scholar] [CrossRef]
- Stamatov, S.D.; Stawinski, J. Novel, regioselective transformation of an oxirane system. An efficient approach to the synthesis of endocannabinoid 2-arachidonoylglycerol. Tetrahedron Lett. 2002, 43, 1759–1761. [Google Scholar] [CrossRef]
- Cartoni, A.; Margonelli, A.; Angelini, G.; Finazzi-Agrò, A.; Maccarrone, M. Simplified chemical and radiochemical synthesis of 2-arachidonoyl-glycerol, an endogenous ligand of cannabinoid receptors. Tetrahedron Lett. 2004, 45, 2723–2726. [Google Scholar] [CrossRef]
- Suhara, Y.; Takayama, H.; Nakane, S.; Miyashita, T.; Waku, K.; Sugiura, T. Synthesis and biological activities of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand, and its metabolically stable ether-linked analogues. Chem. Pharm. Bull. 2000, 48, 903–907. [Google Scholar] [CrossRef]
- Roche, M.J.; Madren, S.M.; Tallent, C.R.; Carroll, F.I.; Seltzman, H.H. Mild acetal cleavage using B-chlorocatecholborane in the synthesis of rearrangement-sensitive 2-arachidonoylglycerol. Tetrahedron Lett. 2012, 53, 3825–3827. [Google Scholar] [CrossRef]
- Whitten, K.M.; Makriyannis, A.; Vadivel, S.K. Application of chemoenzymatic hydrolysis in the synthesis of 2-monoacylglycerols. Tetrahedron 2012, 68, 5422–5428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vadivel, S.K.; Whitten, K.M.; Makriyannis, A. Chemoenzymatic synthesis of 2-arachidonoylglycerol, an endogenous ligand for cannabinoid receptors. Tetrahedron Lett. 2011, 52, 1149–1150. [Google Scholar] [CrossRef]
- Duclos, R.I.; Johnston, M.; Vadivel, S.K.; Makriyannis, A.; Glaser, S.T.; Gatley, S.J. A methodology for radiolabeling of the endocannabinoid 2-arachidonoylglycerol (2-AG). J. Org. Chem. 2011, 76, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, M.; Wang, T.; Jin, Q.; Wang, X. An improved method for the synthesis of 2-arachidonoylglycerol. Process Biochem. 2014, 49, 1415–1421. [Google Scholar] [CrossRef]
- Han, L.; Razdan, R.K. Total synthesis of 2-Arachidonylglycerol (2-Ara-Gl). Tetrahedron Lett. 1999, 40, 1631–1634. [Google Scholar] [CrossRef]
- Martin, J.B. Preparation of saturated and unsaturated symmetrical monoglycerides. J. Am. Chem. Soc. 1953, 75, 5482–5483. [Google Scholar] [CrossRef]
- Seltzman, H.H.; Fleming, D.N.; Hawkins, G.D.; Carroll, F. Facile synthesis and stabilization of 2-arachidonylglycerol via its 1,3-phenylboronate ester. Tetrahedron Lett. 2000, 41, 3589–3592. [Google Scholar] [CrossRef]
- Thangaraj, B.; Solomon, P.R. Immobilization of Lipases—A ReviewPart I: Enzyme Immobilization. ChemBioEng Rev. 2019, 6, 157–166. [Google Scholar] [CrossRef]
- Mulinari, J.; Oliveira, J.V.; Hotza, D. Lipase immobilization on ceramic supports: An overview on techniques and Materials. Biotechnol. Adv. 2020, 42, 107581. [Google Scholar] [CrossRef]
- Rafiee, F.; Rezaee, M. Different strategies for the lipase immobilization on the chitosan based supports and their applications. Int. J. Bio. Macromol. 2021, 179, 170–195. [Google Scholar] [CrossRef] [PubMed]
- Rodriguesa, R.C.; Virgen-Ortízb, J.J.; dos Santosc, J.C.; Berenguer-Murciad, Á.; Alcantarae, A.R.; Barbosaf, O.; Ortizg, C.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports: Immobilization mechanism, advantages, problems, and solutions. Biotechnol. Advan. 2019, 37, 746–770. [Google Scholar] [CrossRef] [PubMed]
- Casati, S.; Rota, P.; Allevi, P.; Mingione, A.; Ottria, R.; Ciuffreda, P. Clarifying the use of benzylidene protecting group for d-(+)-ribono-1,4-lactone, an essential building block in the synthesis of c-nucleosides. Molecules 2021, 26, 6447. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; Wiley: Hoboken, NJ, USA, 2006; ISBN 978-0-470-05348-5. [Google Scholar]
- Santaniello, E.; Casati, S.; Ciuffreda, P.; Gamberoni, L. Lipase-catalyzed alcoholysis of diol dibenzoates: Selective enzymatic access to the 2-benzoyl ester of 1,2-propanediol and preparation of the enantiomerically pure (R)-1-O-benzoyl-2-methylpropane-1,3-diol. Tetrahedron Asymmetry 2005, 16, 1705–1708. [Google Scholar] [CrossRef]
- Ciuffreda, P.; Casati, S.; Santaniello, E. Lipase-catalyzed monoprotection of 1,4-diols in an organic solvent using vinyl benzoate as acyl transfer agent. Tetrahedron Lett. 2003, 44, 3663–3665. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Jin, Q.; Wang, T. Improved synthesis of monopalmitin on a large scale by two enzymatic methods. J. Amer. Oil Chem. Soc. 2013, 90, 1455–1463. [Google Scholar] [CrossRef]
- Li, L.; Du, W.; Liu, D.; Wang, L.; Li, Z. Lipase-catalyzed transesterification of rapeseed oils for biodiesel production with a novel organic solvent as the reaction medium. J. Mol. Catal. B-Enzym 2006, 43, 58–62. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Derewenda, U.; Derewenda, Z.S.; Dodson, G.G.; Lawson, D.M.; Turkenburg, J.P.; Bjorkling, F.; Huge-Jensen, B.; Patkar, S.A.; Thim, L. A model for interfacial activation in lipases from the structure of a fungal lipase-inhibitor complex. Nature 1991, 351, 491–494. [Google Scholar] [CrossRef]
- Huge-Jensen, B.; Galluzzo, D.R.; Jensen, R.G. Partial purification and characterization of free and immobilized lipases from Mucor miehei. Lipids 1987, 22, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Handayani, N.; Loos, K.; Wahyuningrum, D.; Zulfikar, M.A. Immobilization of Mucor miehei lipase onto macroporous aminated polyethersulfone membrane for enzymatic reactions. Membranes 2012, 2, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Lazarević, J.; Šmelcerović, A.; Zvezdanović, J.; Yancheva, D.; Casati, S.; Ottria, R.; Ciuffreda, P. Lipid peroxidation inhibition study: A promising case of 1,3-di(1,1’-biphenyl-3-yl)urea. Chem. Biol. Interact. 2020, 326, 109137. [Google Scholar] [CrossRef]
- Ottria, R.; Casati, S.; Ciuffreda, P. Optimized synthesis and characterization of N-acylethanolamines and O-acylethanolamines, important family of lipid-signalling molecules. Chem. Phys. Lip. 2012, 165, 705–711. [Google Scholar] [CrossRef]
- Miceli, M.; Casati, S.; Ottria, R.; Di Leo, S.; Eberini, I.; Palazzolo, L.; Parravicini, C.; Ciuffreda, P. Set-Up and validation of a high throughput screening method for Human Monoacylglycerol Lipase (MAGL) Based on a new red fluorescent probe. Molecules 2019, 24, 2241. [Google Scholar] [CrossRef] [PubMed]
- Casati, S.; Ottria, R.; Ciuffreda, P. Simple Synthesis of 17-β-O-hemisuccinate of stanozolol for immunoanalytical methods. Molecules 2020, 25, 2019. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DIOX | DIOX-DCM | THF | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | 1a | 1b | 1c | 1d | 1a | 1b | 1c | 1d | |
| MML | 7 | 80 | 13 | - | 8 | 90 | 2 | - | 10 | 51 | 39 | - |
| CCL | 16 | 82 | 2 | - | 21 | 76 | 2 | - | 18 | 58 | 25 | - |
| CAL | 100 | - | - | - | 54 | 38 | 8 | - | 36 | 52 | 18 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottria, R.; Casati, S.; Rota, P.; Ciuffreda, P. 2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization. Molecules 2022, 27, 5190. https://doi.org/10.3390/molecules27165190
Ottria R, Casati S, Rota P, Ciuffreda P. 2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization. Molecules. 2022; 27(16):5190. https://doi.org/10.3390/molecules27165190
Chicago/Turabian StyleOttria, Roberta, Silvana Casati, Paola Rota, and Pierangela Ciuffreda. 2022. "2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization" Molecules 27, no. 16: 5190. https://doi.org/10.3390/molecules27165190
APA StyleOttria, R., Casati, S., Rota, P., & Ciuffreda, P. (2022). 2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization. Molecules, 27(16), 5190. https://doi.org/10.3390/molecules27165190