
Computer-Aided Screening of Phytoconstituents from Ocimum tenuiflorum against Diabetes Mellitus Targeting DPP4 Inhibition: A Combination of Molecular Docking, Molecular Dynamics, and Pharmacokinetics Approaches

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Docking Simulation
2.2. Molecular Dynamics Simulations
2.3. Binding Free Energy Calculations
2.4. Druglikeliness and Pharmacokinetics Analyses
3. Results
3.1. Molecular Docking Simulations
3.2. Molecular Dynamics Simulation
3.3. Binding Free Energy Calculations
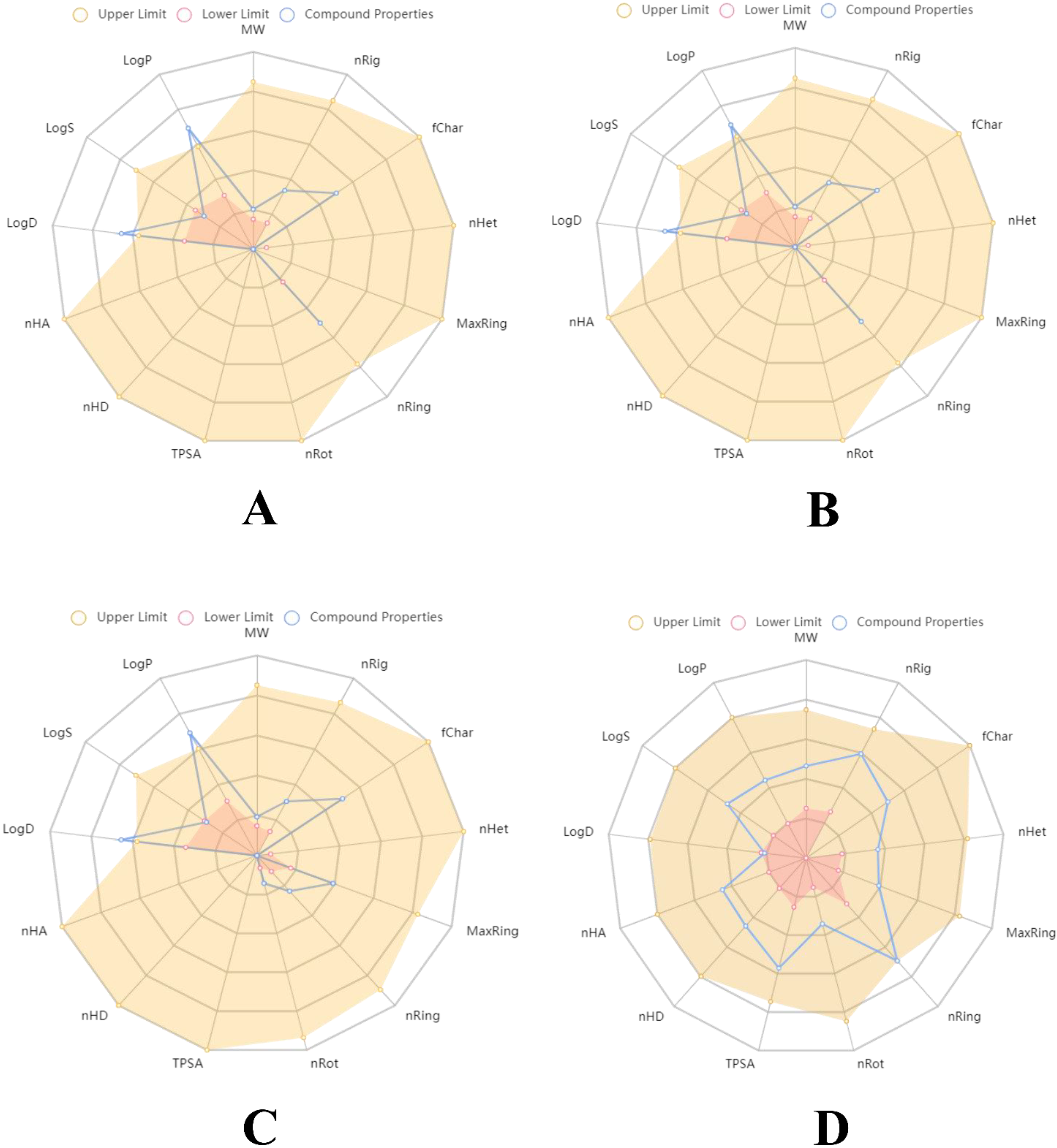
3.4. Druglikeliness and Pharmacokinetics Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Patil, S.M.; Shirahatti, P.S.; Ramu, R. Azadirachta indica A. Juss (neem) against diabetes mellitus: A critical review on its phytochemistry, pharmacology, and toxicology. J. Pharm. Pharmacol. 2021, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F. Physiology and pharmacology of DPP-4 in glucose homeostasis and the treatment of type 2 diabetes. Front. Endocrinol. 2019, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.H.; Kim, S.W. Soluble dipeptidyl peptidase-4 levels are associated with decreased renal function in patients with type 2 diabetes mellitus. Diabetes Metab. J. 2019, 43, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef]
- Röhrborn, D.; Wronkowitz, N.; Eckel, J. DPP4 in diabetes. Front. Immunol. 2015, 6, 386. [Google Scholar] [CrossRef]
- El, K.; Gray, S.M.; Capozzi, M.E.; Knuth, E.R.; Jin, E.; Svendsen, B.; Clifford, A.; Brown, J.L.; Encisco, S.E.; Chazotte, B.M.; et al. GIP mediates the incretin effect and glucose tolerance by dual actions on α cells and β cells. Sci. Adv. 2021, 7, eabf1948. [Google Scholar] [CrossRef]
- Sesti, G.; Avogaro, A.; Belcastro, S.; Bonora, B.M.; Croci, M.; Daniele, G.; Dauriz, M.; Dotta, F.; Formichi, C.; Frontoni, S.; et al. Ten years of experience with DPP-4 inhibitors for the treatment of type 2 diabetes mellitus. Acta Diabetol. 2019, 56, 605–617. [Google Scholar] [CrossRef]
- Salvo, F.; Moore, N.; Arnaud, M.; Robinson, P.; Raschi, E.; De Ponti, F.; Bégaud, B.; Pariente, A. Addition of dipeptidyl peptidase-4 inhibitors to sulphonylureas and risk of hypoglycaemia: Systematic review and meta-analysis. BMJ 2016, 353, i2231. [Google Scholar] [CrossRef]
- Pathak, R.; Bridgeman, M.B. Dipeptidyl peptidase-4 (DPP-4) inhibitors in the management of diabetes. P T 2010, 35, 509–513. [Google Scholar]
- Karagiannis, T.; Boura, P.; Tsapas, A. Safety of dipeptidyl peptidase 4 inhibitors: A perspective review. Ther. Adv. Drug Saf. 2014, 5, 138–146. [Google Scholar] [CrossRef]
- Gallwitz, B. Clinical Use of DPP-4 Inhibitors. Front. Endocrinol. 2019, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chaudhuri, P.K. A review on phytochemical and pharmacological properties of Holy basil (Ocimum sanctum L.). Ind. Crops. Prod. 2018, 118, 367–382. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Kumar, B.P.; Kumar, N. Evaluation of flavonoids from banana pseudostem and flower (quercetin and catechin) as potent inhibitors of α-glucosidase: An in silico perspective. J. Biomol. Struct. Dyn. 2021, 7, 1–5. [Google Scholar] [CrossRef]
- Sutton, J.M.; Clark, D.E.; Dunsdon, S.J.; Fenton, G.; Fillmore, A.; Harris, N.V.; Higgs, C.; Hurley, C.A.; Krintel, S.L.; MacKenzie, R.E.; et al. Novel heterocyclic DPP-4 inhibitors for the treatment of type 2 diabetes. Bioorg. Med. Chem. Lett. 2012, 22, 1464–1468. [Google Scholar] [CrossRef]
- Maradesha, T.; Patil, S.M.; Al-Mutairi, K.A.; Ramu, R.; Madhunapantula, S.V.; Alqadi, T. Inhibitory effect of polyphenols from the whole green jackfruit flour against α-glucosidase, α-amylase, aldose reductase and glycation at multiple stages and their interaction: Inhibition kinetics and molecular simulations. Molecules 2022, 27, 1888. [Google Scholar] [CrossRef]
- Jyothi, M.; Khamees, H.A.; Patil, S.M.; Ramu, R.; Khanum, S.A. Microwave-assisted synthesis, characterization, docking studies and molecular dynamic of some novel phenyl thiazole analogs as xanthine oxidase inhibitor. J. Iran. Chem. Soc. 2022, 10, 1–5. [Google Scholar] [CrossRef]
- Martiz, R.M.; Patil, S.M.; Abdulaziz, M.; Babalghith, A.; Al-Areefi, M.; Al-Ghorbani, M.; Mallappa Kumar, J.; Prasad, A.; Mysore Nagalingaswamy, N.P.; Ramu, R. Defining the role of isoeugenol from Ocimum tenuiflorum against diabetes mellitus-linked Alzheimer’s disease through network pharmacology and computational methods. Molecules 2022, 27, 2398. [Google Scholar] [CrossRef]
- Patil, S.M.; Al-Mutairi, K.A.; Firdose, N.; Ramu, R.; Martiz, R.M.; Ashwini, P. Pharmacoinformatics based screening discovers swertianolin from Lavandula angustifolia as a novel neuromodulator targeting epilepsy, depression, and anxiety. South. Afr. J. Bot. 2022, 149, 712–730. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Satish, A.M.; Shbeer, A.M.; Ageel, M.; Al-Ghorbani, M.; Parameswaran, S.; Ramu, R. Discovery of novel coumarin derivatives as potential dual inhibitors against α-glucosidase and α-amylase for the management of post-prandial hyperglycemia via molecular modelling approaches. Molecules 2022, 27, 3888. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Chandra, J.S.; Ranganatha, L.V. In silico identification of novel benzophenone-coumarin derivatives as SARS-CoV-2 RNAdependent RNA polymerase (RdRp) inhibitors. J. Biomol. Struct. Dyn. 2021, 11, 1–17. [Google Scholar]
- Kumar, V.; Shetty, P.; Arunodaya, H.S.; Chandra, K.S.; Ramu, R.; Patil, S.M.; Baliga, A.; Rai, V.M.; Shalini Shenoy, M.; Udupi, V.; et al. Potential fluorinated anti-MRSA thiazolidinone derivatives with antibacterial, antitubercular activity and molecular docking studies. Chem. Biodivers. 2022, 19, e202100532. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Ramu, R.; Shirahatti, P.S.; Kumari, V.C.; Sushma, P.; Mandal, S.P.; Patil, S.M. α-glucosidase; α-amylase inhibition; kinetics and docking studies of novel (2-chloro-6-(trifluoromethyl) benzyloxy) arylidene) based rhodanine and rhodanine acetic acid derivatives. Chem. Select 2021, 6, 9637–9644. [Google Scholar] [CrossRef]
- Martiz, R.M.; Patil, S.M.; Ramu, R.; Jayanthi, M.K.; Ranganatha, L.V.; Khanum, S.A.; Silina, E.; Stupin, V.; Achar, R.R. Discovery of novel benzophenone integrated derivatives as anti-Alzheimer’s agents targeting presenilin-1 and presenilin-2 inhibition: A computational approach. PLoS ONE 2022, 17, e0265022. [Google Scholar] [CrossRef] [PubMed]
- Gurupadaswamy, H.D.; Ranganatha, V.L.; Ramu, R.; Patil, S.M.; Khanum, S.A. Competent synthesis of biaryl analogs via asymmetric Suzuki–Miyaura cross-coupling for the development of anti-inflammatory and analgesic agents. J. Iran. Chem. Soc. 2022, 19, 2421–2436. [Google Scholar] [CrossRef]
- Ganavi, D.; Ramu, R.; Kumar, V.; Patil, S.M.; Martiz, R.M.; Shirahatti, P.S.; Sathyanarayana, R.; Poojary, B.; Holla, B.S.; Poojary, V. In vitro and in silico studies of fluorinated 2,3-disubstituted thiazolidinone-pyrazoles as potential α-amylase inhibitors and antioxidant agents. Arch. Pharm. 2021, 12, e2100342. [Google Scholar] [CrossRef]
- Patil, S.M.; Maruthi, K.R.; Bajpe, N.S.; Vyshali, V.M.; Sushmitha, S.; Chagalamari, A.; Ramith, R. Comparative molecular docking and simulation analysis of molnupiravir and remdesivir with SARS-CoV-2 RNA dependent RNA polymerase (RdRp). Bioinformation 2021, 7, 932–939. [Google Scholar]
- Ramu, R.; Shirahatti, P.S.; Deepika, T.H.; Bajpe, S.N.; Sreepathi, N.; Patil, S.M.; Prasad, N. Investigating Musa paradisiaca (Var. Nanjangud rasa bale) pseudostem in preventing hyperglycemia along with improvement of diabetic complications. J. Appl. Biol. Biotechnol. 2022, 10, 56–65. [Google Scholar] [CrossRef]
- Patil, S.M.; Sujay, S.; Tejaswini, M.; Sushma, P.; Prithvi, S.; Ramu, R. Bioactive peptides: Its production and potential role on health. Innov. Food. Sci. Emerg. Technol. 2020, 7, 167–182. [Google Scholar]
- Ramu, R.; Patil, S.M. A perspective on the effective conduction of functional-based coaching program on diabetic Indonesian communities. Oman Med. J. 2021, 36, e281. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Shirahatti, P.S.; Kumari, V.B.C.; Ramu, R.; Prasad, N. Azadirachta indica A. Juss (neem) as a contraceptive: An evidence-based review on its pharmacological efficiency. Phytomedicine 2021, 88, 153596. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.M.; Ramu, R.; Shirahatti, P.S.; Shivamallu, C.; Amachawadi, R.G. A systematic review on ethnopharmacology; phytochemistry and pharmacological aspects of Thymus vulgaris Linn. Heliyon 2021, 7, e07054. [Google Scholar] [CrossRef] [PubMed]
- Parasuraman, S.; Balamurugan, S.; Christapher, P.V.; Petchi, R.R.; Yeng, W.Y.; Sujithra, J.; Vijaya, C. Evaluation of antidiabetic and antihyperlipidemic effects of hydroalcoholic extract of leaves of Ocimum tenuiflorum (Lamiaceae) and prediction of biological activity of its phytoconstituents. Pharmacogn. Res. 2015, 7, 156–165. [Google Scholar] [CrossRef]
- Suanarunsawat, T.; Anantasomboon, G.; Piewbang, C. Anti-diabetic and anti-oxidative activity of fixed oil extracted from Ocimum sanctum L. leaves in diabetic rats. Exp. Ther. Med. 2016, 11, 832–840. [Google Scholar] [CrossRef]
- Jamshidi, N.; Cohen, M.M. The Clinical Efficacy and Safety of Tulsi in Humans: A systematic review of the literature. Evid.-Based Complement. Altern. Med. 2017, 2017, 9217567. [Google Scholar] [CrossRef]
- Kumari, V.C.; Patil, S.M.; Shirahatti, P.S.; Sujay, S.; Tejaswini, M.; Ranganatha, L.V.; Jayanthi, M.K.; Ramu, R. The current status and perspectives for the emerging pandemic: Covid-19. Int. J. Pharm. Pharm. Sci. 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Abu Khalaf, R.; Awad, M.; Al-Essa, L.; Mefleh, S.; Sabbah, D.; Al-Shalabi, E.; Shabeeb, I. Discovery, synthesis and in combo studies of Schiff’s bases as promising dipeptidyl peptidase-IV inhibitors. Mol. Divers. 2021, 26, 1213–1225. [Google Scholar] [CrossRef]
- Deng, X.; Liang, Y.; Hu, J.; Yang, Y. Studies on the Mechanism of Gegen Qinlian decoction in treating diabetes mellitus based on network pharmacology. Nat. Prod. Commun. 2021, 16, 1–11. [Google Scholar] [CrossRef]
- Sharma, P.; Joshi, T.; Mathpal, S.; Chandra, S.; Tamta, S. In silico identification of antidiabetic target for phytochemicals of A. Marmelos and mechanistic insights by molecular dynamics simulations. J. Biomol. Struct. Dyn. 2021, 1–18. [Google Scholar] [CrossRef]
- Kim, B.R.; Kim, H.Y.; Choi, I.; Kim, J.B.; Jin, C.H.; Han, A.R. DPP-IV inhibitory potentials of flavonol glycosides isolated from the seeds of Lens culinaris: In vitro and molecular docking analyses. Molecules 2018, 23, 1998. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Mishra, A. Molecular dynamics simulation and free energy calculation studies of Coagulin L as dipeptidyl peptidase-4 inhibitor. J. Biomol. Struct. Dyn. 2022, 40, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, C.Y.; Xie, J.; Dai, J.H.; He, S.L.; Tian, Y. Identification of potential dipeptidyl peptidase (DPP)-IV inhibitors among Moringa oleifera phytochemicals by virtual screening, molecular docking analysis, adme/t-based prediction, and in vitro analyses. Molecules 2020, 25, 189. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Fonseca, V. Saxagliptin overview: Special focus on safety and adverse effects. Expert Opin. Drug. Saf. 2013, 12, 103–109. [Google Scholar] [CrossRef]
- Dave, D.J. Saxagliptin: A dipeptidyl peptidase-4 inhibitor in the treatment of type 2 diabetes mellitus. J. Pharmacol. Pharmacother. 2011, 2, 230–235. [Google Scholar] [CrossRef]
- Raz, I.; Bhatt, D.L.; Hirshberg, B.; Mosenzon, O.; Scirica, B.M.; Umez-Eronini, A.; Im, K.; Stahre, C.; Buskila, A.; Iqbal, N.; et al. Incidence of pancreatitis and pancreatic cancer in a randomized controlled multicenter trial (SAVOR-TIMI 53) of the dipeptidyl peptidase-4 inhibitor saxagliptin. Diabetes Care. 2014, 37, 2435–2441. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.; Polyak, A.; Mathe, D.; Kiraly, R.; Thuroczy, J.; Terez, M.; Janoki, G.; Ting, Y.; Bucci, L.R.; Schauss, A.G. Absorption, uptake and tissue affinity of high-molecular-weight hyaluronan after oral administration in rats and dogs. J. Agric. Food Chem. 2008, 56, 10582–10593. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.Y.; Jang, M.K.; Nah, J.W. Influence of molecular weight on oral absorption of water soluble chitosans. J. Control. Release 2005, 102, 383–394. [Google Scholar] [CrossRef]
- Al Kury, L.T.; Abdoh, A.; Ikbariah, K.; Sadek, B.; Mahgoub, M. In vitro and in vivo antidiabetic potential of monoterpenoids: An update. Molecules 2021, 27, 182. [Google Scholar] [CrossRef]
- Salehi, B.; Upadhyay, S.; Erdogan Orhan, I.; Kumar Jugran, A.; LD Jayaweera, S.; ADias, D.; Sharopov, F.; Taheri, Y.; Martins, N.; Baghalpour, N.; et al. Therapeutic potential of α- and β-pinene: A miracle gift of nature. Biomolecules 2019, 9, 738. [Google Scholar] [CrossRef]
- Özbek, H.; Yılmaz, B.S. Anti-inflammatory and hypoglycemic activities of alpha-pinene. Acta Pharm. Sci. 2017, 55, 7–14. [Google Scholar] [CrossRef]
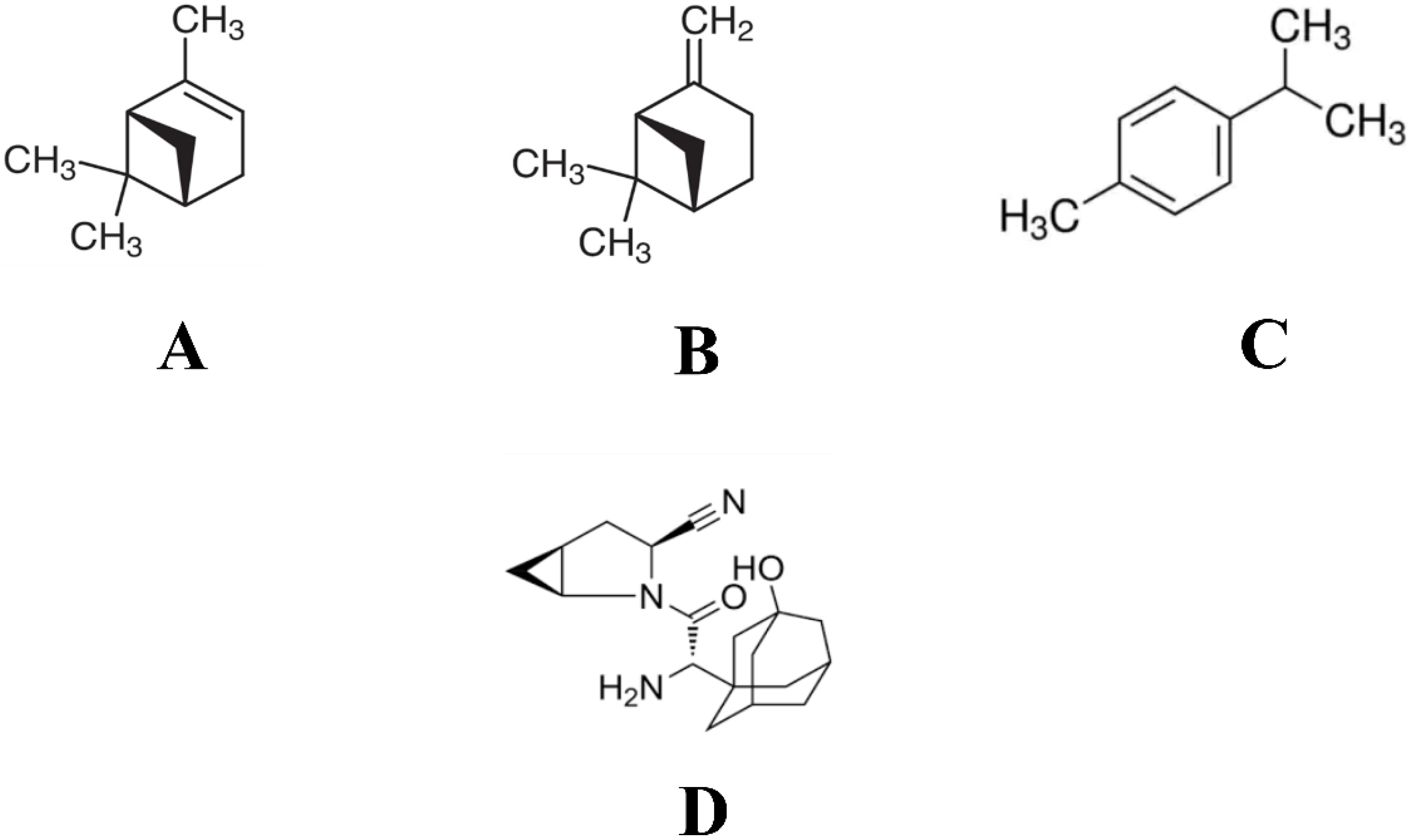
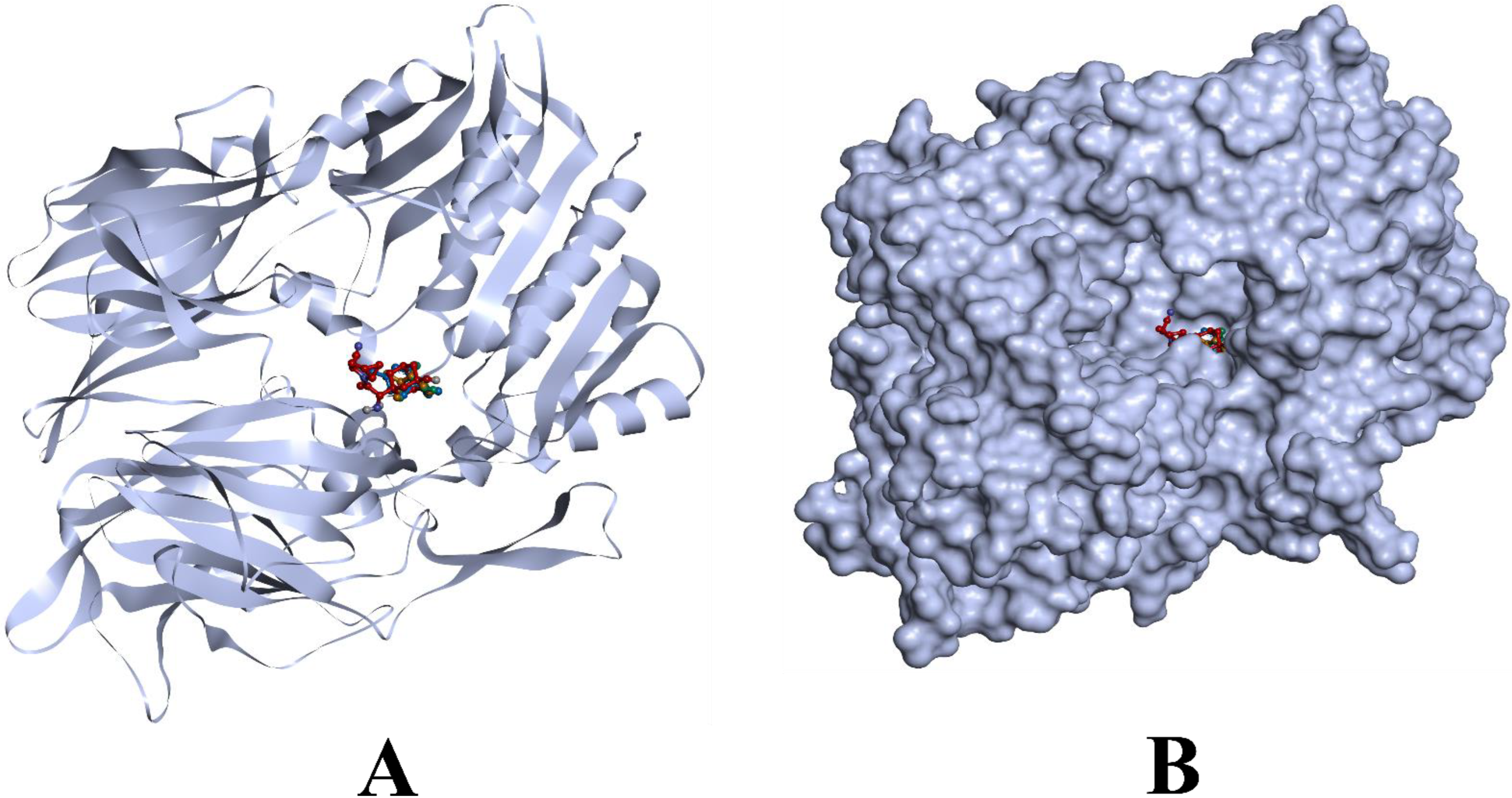
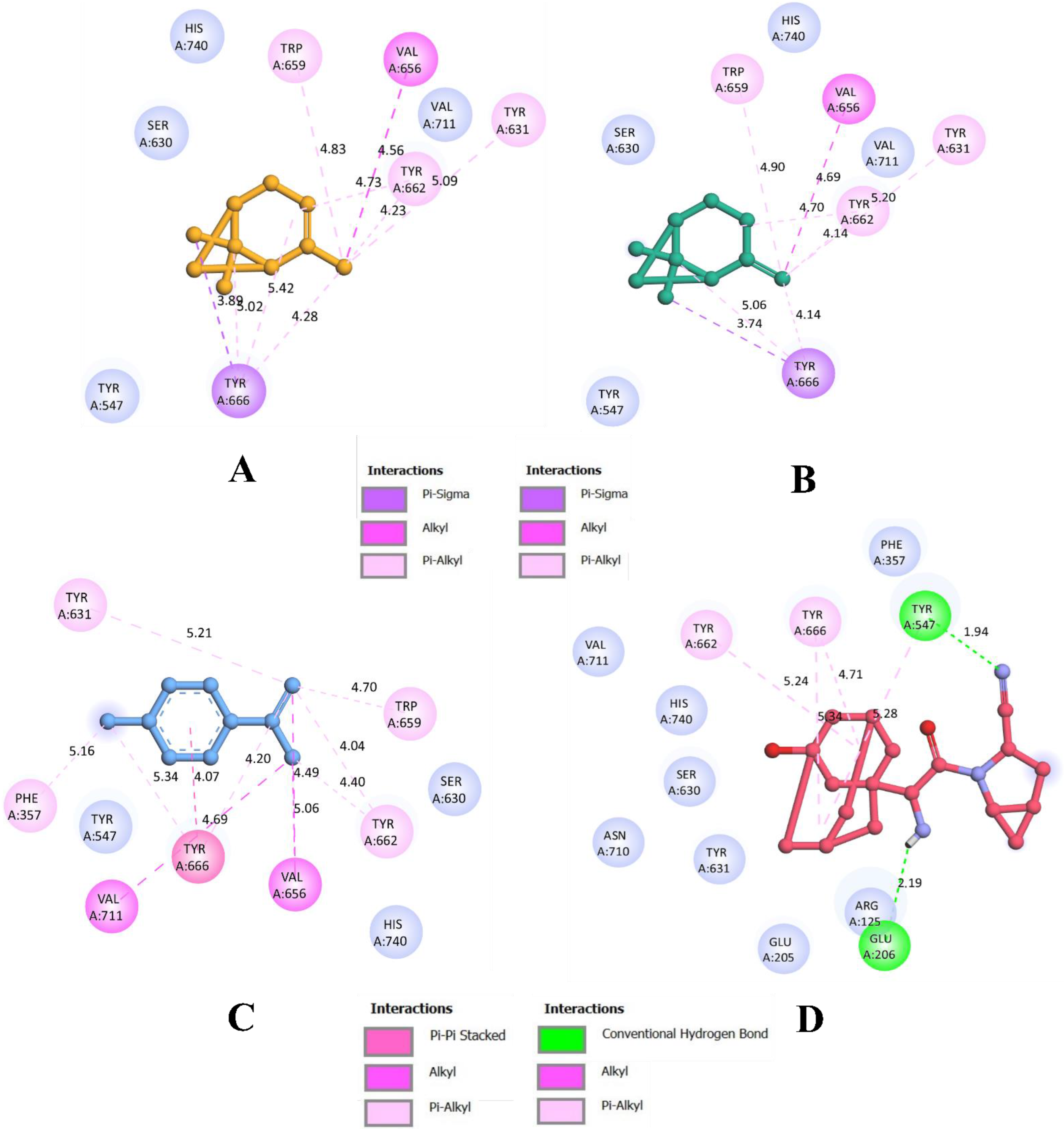
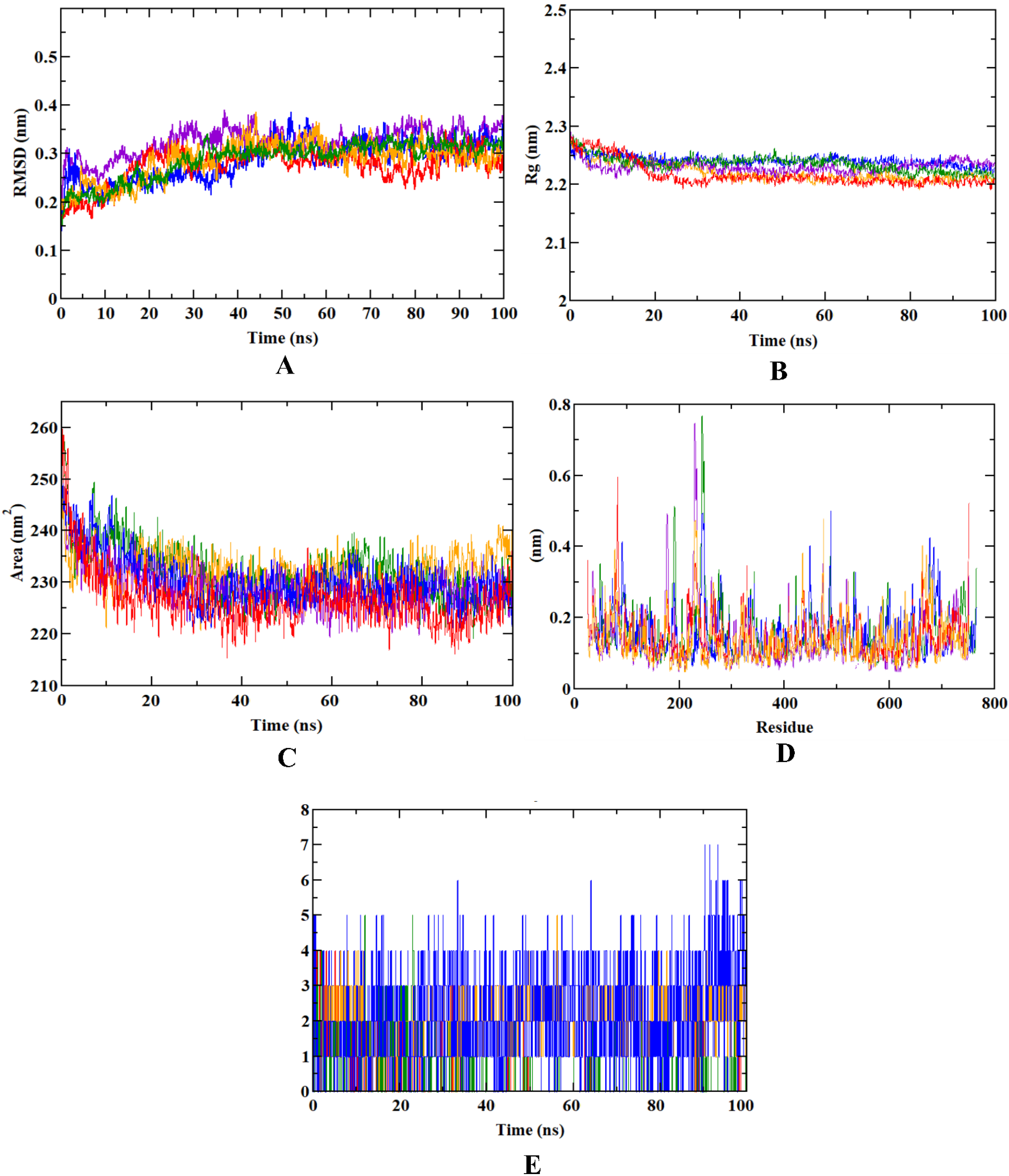

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Name of the Compound | Binding Affinity (kcal/mol) | Total No. of Intermolecular Interactions | Total No. of Hydrophobic Bonds |
|---|---|---|---|---|
| 1 | (−)-Alloaromadendrene | −6.3 | 4 | 4 |
| 2 | (−)-Camphene | −5.4 | 2 | 2 |
| 3 | (−)-Linalool | −5.7 | 11 | 10 |
| 4 | (+)-α-Phellandrene | −5.9 | 6 | 6 |
| 5 | (+)-Endo-β-bergamotene | −6.5 | 6 | 6 |
| 6 | (1S)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-one | −5.7 | 2 | 2 |
| 7 | (1S, 2R, 4S)-(−)-Bornyl acetate | −6.2 | 1 | 0 |
| 8 | (E)-β-ocimene | −5.4 | 9 | 9 |
| 9 | 1S-α-pinene | −6.2 | 9 | 9 |
| 10 | 2,3-Dimethylaniline | −5.5 | 2 | 2 |
| 11 | 3-Carene | −6.1 | 6 | 6 |
| 12 | 4-Terpineol | −6.1 | 3 | 3 |
| 13 | Acetyleugenol | −6.2 | 6 | 6 |
| 14 | α-Fenchene | −5.8 | 7 | 7 |
| 15 | α-Terpineol | −5.8 | 8 | 7 |
| 16 | β-Caryophyllene | −6.0 | 2 | 2 |
| 17 | β-Pinene | −6.2 | 8 | 8 |
| 18 | Cis-Anethole | −5.8 | 6 | 6 |
| 19 | Cyclo-(L-Val-L-Leu) | −6.6 | 6 | 6 |
| 20 | Dehydro-p-cymene | −6.4 | 11 | 11 |
| 21 | Eucalyptol | −6.1 | 1 | 1 |
| 22 | γ-Selinene | −6.6 | 3 | 3 |
| 23 | Geranyl Acetate | −6.1 | 5 | 5 |
| 24 | Isoeugenol | −5.6 | 7 | 7 |
| 25 | Myrcene | −5.5 | 10 | 10 |
| 26 | Phytosterols | −8.3 | 3 | 2 |
| Control | Saxagliptin | −7.4 | 6 | 4 |
| MD Trajectory Plots | RMSD (nm) | RMSF (nm) | Rg (nm) | SASA (nm2) | Number of Ligand H-Bonds |
|---|---|---|---|---|---|
| Apoprotein | 0.321 | 0.262 | 2.341 | 236.25 | |
| DPP4-1S-α-pinene complex | 0.287 | 0.291 | 2.349 | 237.25 | 5 |
| DPP4-β-pinene complex | 0.285 | 0.245 | 2.296 | 239.60 | 5 |
| DPP4-dehydro-p-cymene | 0.291 | 0.259 | 2.340 | 235.10 | 7 |
| DPP4-saxagliptin complex | 0.232 | 0.250 | 2.310 | 231.12 | 4 |
| Types of Binding Free Energies | DPP4-1S-α-Pinene Complex (kJ/mol) | DPP4-β-Pinene Complex (kJ/mol) | DPP4-Dehydro-p-Cymene (kJ/mol) | DPP4-Saxagliptin Complex (kJ/mol) |
|---|---|---|---|---|
| Van der Waal energy | −51.461 ± 10.125 | −49.871 ± 9.012 | −52.918 ± 5.670 | −39.920 ± 24.183 |
| Electrostatic energy | −1.028 ± 9.716 | −0.389 ± 12.133 | −1.209 ± 6.999 | −0.214 ± 1.305 |
| Polar solvation energy | 32.514 ± 12.871 | 28.574 ± 20.445 | 21.841 ± 10.001 | 22.912 ± 35.181 |
| SASA energy | −3.898 ± 2.999 | −2.901 ± 2.101 | −4.996 ± 7.120 | −4.614 ± 3.652 |
| Binding energy | −34.807 ± 11.099 | −27.569 ± 13.122 | −33.124 ± 14.909 | −29.752 ± 48.193 |
| Categories | Types of Parameters | 1S-α-Pinene | β-Pinene | Dehydro-p-Cymene |
|---|---|---|---|---|
| Druglikeliness based on Lipinski’s rule of five | Molecular weight | 136.13 g/mol | 136.13 g/mol | 315.19 g/mol |
| Topological polar surface area (Å) | 0.0 | 0.0 | 90.35 | |
| No. of hydrogen bond donors | 0 | 0 | 3 | |
| No. of hydrogen bond acceptors | 0 | 0 | 5 | |
| No. of rotatable bonds | 0 | 0 | 3 | |
| Adsorption | Caco-2 Permeability | −4.303 | −4.46 | −5.212 |
| MDCK Permeability | 1.8e − 05 | 2e − 05 | 9.4e − 05 | |
| Distribution | Volume distribution | 1.73 | 1.091 | 1.426 |
| Plasma Protein Binding | 86.33% | 64.33% | 8.648% | |
| Metabolism | CYP1A2 inhibition | 0.469 | 0.296 | 0.006 |
| CYP2C19 inhibition | 0.267 | 0.163 | 0.029 | |
| CYP2C9 inhibition | 0.312 | 0.321 | 0.039 | |
| CYP2D6 inhibition | 0.012 | 0.009 | 0.019 | |
| CYP3A4 inhibition | 0.034 | 0.026 | 0.338 | |
| Excretion | Clearance | 15.022 | 10.097 | 8.981 |
| Half-life (T 1/2) | 0.114 | 0.107 | 0.187 | |
| Toxicity | Human Hepatotoxicity | 0.196 | 0.109 | 0.912 |
| hERG inhibition | 0.006 | 0.005 | 0.013 | |
| Carcinogenicity | 0.056 | 0.042 | 0.919 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sajal, H.; Patil, S.M.; Raj, R.; Shbeer, A.M.; Ageel, M.; Ramu, R. Computer-Aided Screening of Phytoconstituents from Ocimum tenuiflorum against Diabetes Mellitus Targeting DPP4 Inhibition: A Combination of Molecular Docking, Molecular Dynamics, and Pharmacokinetics Approaches. Molecules 2022, 27, 5133. https://doi.org/10.3390/molecules27165133
Sajal H, Patil SM, Raj R, Shbeer AM, Ageel M, Ramu R. Computer-Aided Screening of Phytoconstituents from Ocimum tenuiflorum against Diabetes Mellitus Targeting DPP4 Inhibition: A Combination of Molecular Docking, Molecular Dynamics, and Pharmacokinetics Approaches. Molecules. 2022; 27(16):5133. https://doi.org/10.3390/molecules27165133
Chicago/Turabian StyleSajal, Harshit, Shashank M. Patil, Ranjith Raj, Abdullah M. Shbeer, Mohammed Ageel, and Ramith Ramu. 2022. "Computer-Aided Screening of Phytoconstituents from Ocimum tenuiflorum against Diabetes Mellitus Targeting DPP4 Inhibition: A Combination of Molecular Docking, Molecular Dynamics, and Pharmacokinetics Approaches" Molecules 27, no. 16: 5133. https://doi.org/10.3390/molecules27165133
APA StyleSajal, H., Patil, S. M., Raj, R., Shbeer, A. M., Ageel, M., & Ramu, R. (2022). Computer-Aided Screening of Phytoconstituents from Ocimum tenuiflorum against Diabetes Mellitus Targeting DPP4 Inhibition: A Combination of Molecular Docking, Molecular Dynamics, and Pharmacokinetics Approaches. Molecules, 27(16), 5133. https://doi.org/10.3390/molecules27165133