
Alteration of Biomolecular Conformation by Aluminum-Implications for Protein Misfolding Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Aluminum (Al3+) and Adenosine Triphosphate (ATP)
3. Al3+, DNA and Linker Histone H1°
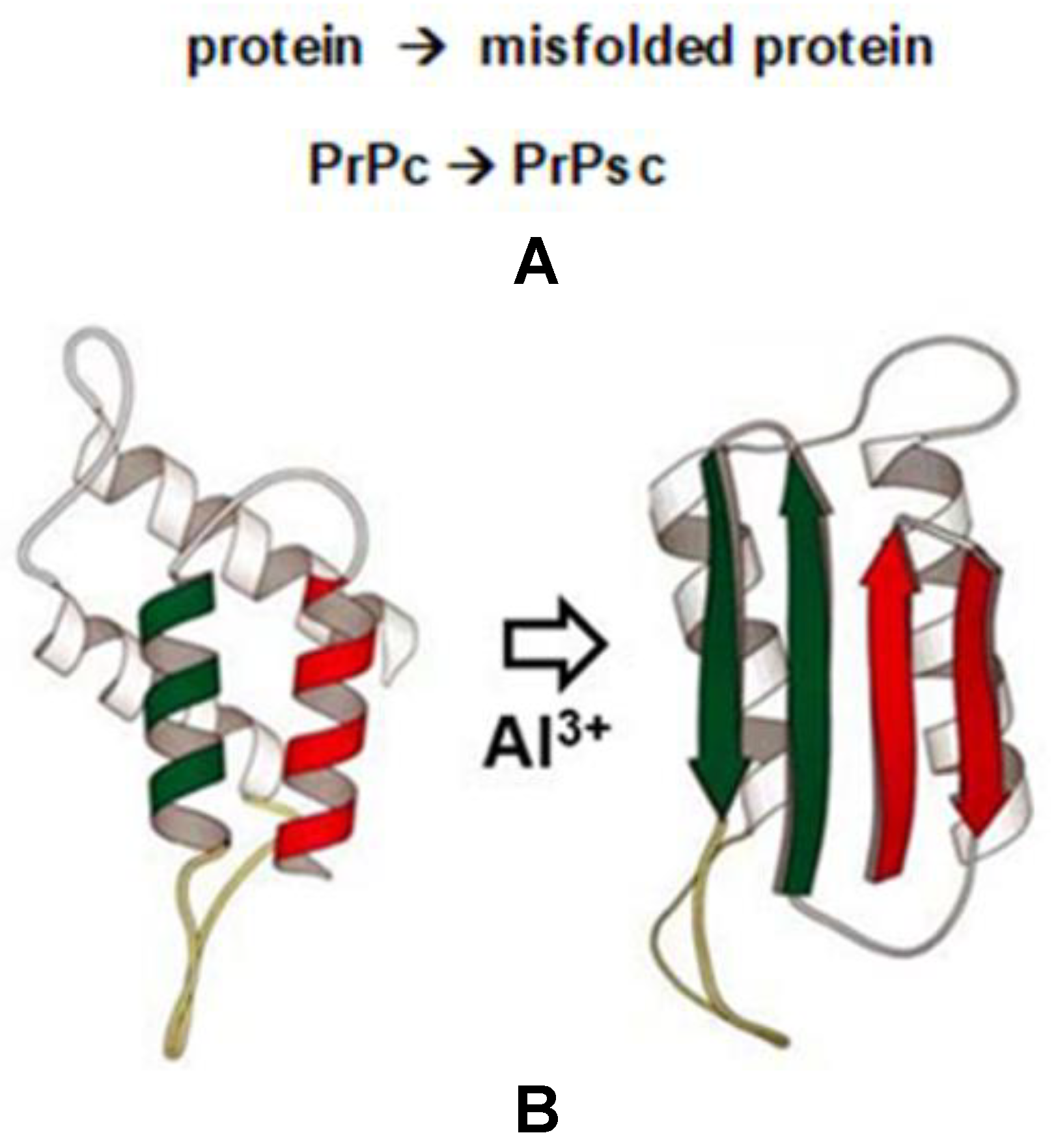
4. Al3+, Protein Misfolding and the PrPc to PrPsc Transition in Neurodegenerative Disease
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Aluminum Institute. Aluminum for Future Generations. Available online: https://primary.world-aluminium.org/aluminium-facts/industry-structure/ (accessed on 13 June 2022).
- Pogue, A.I.; Lukiw, W.J. The mobilization of aluminum into the biosphere. Front. Neurol. 2014, 5, 262. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.B. Aluminum speciation in biology. In Ciba Foundation Symposium 169—Aluminium in Biology and Medicine: Aluminium in Biology and Medicine: Ciba Foundation Symposium 169; Ciba Foundation: London, UK, 1992; pp. 5–18. [Google Scholar] [CrossRef]
- Atkári, K.; Kiss, T.; Bertani, R.; Martin, R.B. Interactions of aluminum (III) with phosphates. Inorg. Chem. 1996, 35, 7089–7094. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, D.R.C.; Lukiw, W.J.; Kruck, T.P.A. New evidence for an active role of aluminum in Alzheimer’s disease. Can. J. Neurol. Sci. 1989, 16, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Drago, D.; Bolognin, S.; Zatta, P. Role of metal ions in the Abeta oligomerization in Alzheimer’s disease and in other neurological disorders. Curr. Alzheimer Res. 2008, 5, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Bondy, S.C. Prolonged exposure to low levels of aluminum leads to changes associated with brain aging and neurodegeneration. Toxicology 2014, 315, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Krishnan, B.; Wong, L.; Kruck, T.P.A.; Bergeron, C.; McLachlan, D.R.C. Nuclear compartmentalization of aluminum in Alzheimer’s disease (AD). Neurobiol. Aging 1992, 13, 115–121. [Google Scholar] [CrossRef]
- Bharathi; Rao, K.S.J.; Stein, R. First evidence on induced topological changes in supercoiled DNA by an aluminum D-aspartate complex. J. Biol. Inorg. Chem. 2003, 8, 823–830. [Google Scholar] [CrossRef]
- Pogue, A.I.; Lukiw, W.J. Aluminum, the genetic apparatus of the human CNS and Alzheimer’s disease (AD). Morphologie 2016, 100, 56–64. [Google Scholar] [CrossRef]
- Niu, Q. Overview of the relationship between aluminum exposure and health of human being. Adv. Exp. Med. Biol. 2018, 1091, 1–31. [Google Scholar] [CrossRef]
- Krupińska, I. Aluminium drinking water treatment residuals and their toxic impact on human health. Molecules 2020, 25, 641. [Google Scholar] [CrossRef]
- Zhao, H.Z.; Yang, H.W.; Guan, Y.T.; Jiang, Z.P. The transformation of aluminum species in the processes of coagulation, sedimentation and filtration. Water Sci. Technol. 2006, 53, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Nidheesh, P.V.; Khatri, J.; Anantha Singh, T.S.; Gandhimathi, R.; Ramesh, S.T. Review of zero-valent aluminium based water and wastewater treatment methods. Chemosphere 2018, 200, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.J.; Zhang, X.; Wang, W.; Li, D.; Ren, Y.; Kang, J.; Liu, D. Evaluation of carboxymethylpullulan-AlCl3 as a coagulant for water treatment: A case study with kaolin. Water Environ. Res. 2020, 92, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.R.; LeBlanc, J.; Sikorska, M. Effects of aluminum and other cations on the structure of brain and liver chromatin. Biochemistry 1989, 28, 3911–3915. [Google Scholar] [CrossRef]
- Lukiw, W.J. Evidence supporting a biological role for aluminum in brain chromatin compaction and epigenetics. J. Inorg. Biochem. 2010, 104, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Kruck, T.P.A.; Percy, M.E.; Pogue, A.I.; Alexandrov, P.N.; Walsh, W.J.; Sharfman, N.M.; Jaber, V.R.; Zhao, Y.; Li, W.; et al. Aluminum in neurological disease—A 36 year multicenter study. J. Alzheimer’s Dis. Parkinsonism 2019, 8, 457. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Kanbe, T.; Yoshikawa, K. Compaction and multiple chain assembly of DNA with the cationic polymer poly (aluminum chloride) (PAC). Langmuir 2004, 20, 6439–6442. [Google Scholar] [CrossRef]
- Ohyama, T. New aspects of magnesium function: A key regulator in nucleosome self-assembly, chromatin folding and phase separation. Int. J. Mol. Sci. 2019, 20, 4232. [Google Scholar] [CrossRef]
- Liu, W.; Zhong, W.; Chen, J.; Huang, B.; Hu, M.; Li, Y. Understanding regulatory mechanisms of brain function and disease through 3D genome organization. Genes 2022, 13, 586. [Google Scholar] [CrossRef]
- Sigismondo, G.; Papageorgiou, D.N.; Krijgsveld, J. Cracking chromatin with proteomics: From chromatome to histone modifications. Proteomics 2022, 2100206. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Kruck, T.P.; McLachlan, D.R.C. Alterations in human linker histone-DNA binding in the presence of aluminum salts in vitro and in Alzheimer’s disease. Neurotoxicology 1987, 8, 291–301. [Google Scholar] [PubMed]
- Karlik, S.J.; Chong, A.A.; Eichhorn, G.L.; De Boni, U. Reversible toroidal compaction of DNA by aluminum. Neurotoxicology 1989, 10, 167–176. [Google Scholar] [PubMed]
- Strong, M.J.; Garruto, R.M.; Joshi, J.G.; Mundy, W.R.; Shafer, T.J. Can the mechanisms of aluminum neurotoxicity be integrated into a unified scheme? J. Toxicol. Environ. Health 1996, 48, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Uryu, K.; Sung, S.; Tang, S.; Trojanowski, J.Q.; Lee, V.M. Aluminum-modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 2002, 16, 1138–1140. [Google Scholar] [CrossRef]
- McLachlan, D.R.C.; Lewis, P.N.; Lukiw, W.J.; Sima, A.; Bergeron, C.; De Boni, U. Chromatin structure in dementia. Ann. Neurol. 1984, 15, 329–334. [Google Scholar] [CrossRef]
- McLachlan, D.R.C.; Lukiw, W.J.; Kruck, T.P. Aluminum, altered transcription, and the pathogenesis of Alzheimer’s disease. Environ. Geochem. Health 1990, 12, 103–114. [Google Scholar] [CrossRef]
- Pogue, A.I.; Dua, P.; Hill, J.M.; Lukiw, W.J. Progressive inflammatory pathology in the retina of aluminum-fed 5xFAD transgenic mice. J. Inorg. Biochem. 2015, 152, 206–209. [Google Scholar] [CrossRef]
- Saha, A.; Dalal, Y. A glitch in the snitch: The role of linker histone H1 in shaping the epigenome in normal and diseased cells. Open Biol. 2021, 11, 210124. [Google Scholar] [CrossRef]
- McLachlan, D.R.C.; Lukiw, W.J.; Wong, L.; Bergeron, C.; Bech-Hansen, N.T. Selective messenger RNA reduction in Alzheimer’s disease. Brain Res. 1988, 427, 255–261. [Google Scholar] [CrossRef]
- Lukiw, W.J. Chromatin Structure, Gene Expression and Nuclear Aluminum in Alzheimer’s Disease. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 1992. ISBN 0315739398, 9780315739390. [Google Scholar]
- Bryant, P.L.; Lukiw, W.J.; Gan, Z.; Hall, R.W.; Butler, L.G. High-field 19.6T 27Al solid-state MAS NMR of in vitro aluminated brain tissue. J. Magn. Reson. 2004, 170, 257–262. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.R.; DeArmond, S.J.; Cohen, F.E. Prion protein biology. Cell 1998, 93, 337–348. [Google Scholar] [CrossRef]
- Horwich, A.L.; Weissman, J.S. Deadly conformations—Protein misfolding in prion disease. Cell 1997, 89, 499–510. [Google Scholar] [CrossRef]
- Ayers, J.I.; Paras, N.A.; Prusiner, S.B. Expanding spectrum of prion diseases. Emerg. Top Life Sci. 2020, 4, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Prusiner, S.B. Prion protein—Mediator of toxicity in multiple proteinopathies. Nat. Rev. Neurol. 2020, 16, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Carlson, G.A.; Prusiner, S.B. How an infection of sheep revealed prion mechanisms in Alzheimer’s disease and other neurodegenerative disorders. Int. J. Mol. Sci. 2021, 22, 4861. [Google Scholar] [CrossRef] [PubMed]
- Ruttkay-Nedecky, B.; Sedlackova, E.; Chudobova, D.; Cihalova, K.; Jimenez, A.M.J.; Krizkova, S.; Richtera, L.; Adam, V.; Kizek, R. Prion protein and its interactions with metal ions (Cu2+, Zn2+, and Cd2+) and metallothionein. ADMET DMPK 2015, 3, 287–295. [Google Scholar] [CrossRef][Green Version]
- Kawahara, M. Conformational change, pore formation and neurotoxicity of amyloidogenic proteins. Rinsho Byori. 2008, 56, 130–136. [Google Scholar]
- Lukiw, W.J. Recent advances in our molecular and mechanistic understanding of misfolded cellular proteins in Alzheimer’s disease (AD) and prion disease (PrD). Biomolecules 2022, 12, 166. [Google Scholar] [CrossRef]
- Di Paolo, C.; Reverte, I.; Colomina, M.T.; Domingo, J.L.; Gómez, M. Chronic exposure to aluminum and melatonin through the diet: Neurobehavioral effects in a transgenic mouse model of Alzheimer disease. Food Chem. Toxicol. 2014, 69, 320–329. [Google Scholar] [CrossRef]
- Garcia, T.; Esparza, J.L.; Nogués, M.R.; Romeu, M.; Domingo, J.L.; Gómez, M. Oxidative stress status and RNA expression in hippocampus of an animal model of Alzheimer’s disease after chronic exposure to aluminum. Hippocampus 2010, 20, 218–225. [Google Scholar] [CrossRef]
- Ricchelli, F.; Fusi, P.; Tortora, P.; Valtorta, M.; Riva, M.; Tognon, G.; Chieregato, K.; Bolognin, S.; Zatta, P. Destabilization of non-pathological variants of ataxin-3 by metal ions results in aggregation/fibrillogenesis. Int. J. Biochem. Cell Biol. 2007, 39, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Ventura, S. What makes a protein sequence a prion? PLoS Comput. Biol. 2015, 11, e1004013. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.J.; Phillips, K.E.; Schramm, P.T.; McKenzie, D.; Aiken, J.M.; Pedersen, J.A. Prions adhere to soil minerals and remain infectious. PLoS Pathog. 2006, 2, e32. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.H.; Kuech, T.R.; Pedersen, J.A. Attachment of pathogenic prion protein to model oxide surfaces. Environ. Sci. Technol. 2013, 47, 6925–6934. [Google Scholar] [CrossRef] [PubMed]
- Pritzko, S.; Morales, R.; Lyon, A.; Concha-Marambio, L.; Urayama, A.; Soto, C. Efficient prion disease transmission through common environmental materials. J. Biol. Chem. 2018, 293, 3363–3373. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, W.; Luo, Q.; Luo, K.; Huang, L.; Wang, W.; Huang, T.; Chen, R.; Lin, Y.; Pang, D.; et al. Copper (II) promotes the formation of soluble neurotoxic PrP oligomers in acidic environment. J. Cell Biochem. 2010, 111, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.K.; Yin, J.; Aguzzi, A.; James, M.N.G. Transition of the prion protein from a structured cellular form (PrPc) to the infectious scrapie agent (PrPsc). Protein Sci. 2019, 28, 2055–2063. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Pogue, A.; Bhattacharjee, S.; Lukiw, W.J. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer’s disease. NeuroReport 2011, 22, 623–627. [Google Scholar] [CrossRef]
- Pogue, A.I.; Jaber, V.; Zhao, Y.; Lukiw, W.J. Systemic Inflammation in C57BL/6J mice receiving dietary aluminum sulfate; up-regulation of the pro-inflammatory cytokines IL-6 and TNFα, C-reactive protein (CRP) and miRNA-146a in blood serum. J. Alzheimer’s Dis. Parkinsonism 2017, 7, 403. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Zhao, Y.; Jones, B.M.; Bhattacharjee, S.; Lukiw, W.J. Expression of the phagocytosis-essential protein TREM2 is down-regulated by an aluminum-induced miRNA-34a in a murine microglial cell line. J. Inorg. Biochem. 2013, 128, 267–269. [Google Scholar] [CrossRef]
- He, C.; Zhao, X.; Lei, Y.; Nie, J.; Lu, X.; Song, J.; Wang, L.; Li, H.; Liu, F.; Zhang, Y.; et al. Whole-transcriptome analysis of aluminum-exposed rat hippocampus and identification of ceRNA networks to investigate neurotoxicity of Al. Mol. Ther. Nucleic Acids 2021, 26, 1401–1417. [Google Scholar] [CrossRef] [PubMed]
- Esparza, J.L.; Garcia, T.; Gómez, M.; Nogués, M.R.; Giralt, M.; Domingo, J.L. Role of deferroxamine on enzymatic stress markers in an animal model of Alzheimer’s disease after chronic aluminum exposure. Biol. Trace Elem. Res. 2011, 141, 232–245. [Google Scholar] [CrossRef] [PubMed]
- García, T.; Esparza, J.L.; Giralt, M.; Romeu, M.; Domingo, J.L.; Gómez, M. Protective role of melatonin on oxidative stress status and RNA expression in cerebral cortex of APP transgenic mice after chronic exposure to aluminum. Biol. Trace Elem. Res. 2010, 135, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.L.; Jia, L.; Jiao, X.; Guo, W.L.; Ji, J.W.; Yang, H.L.; Niu, Q. APP/PS1 transgenic mice treated with aluminum: An update of an Alzheimer’s disease model. Int. J. Immunopathol. Pharmacol. 2012, 25, 49–58. [Google Scholar] [CrossRef]
- Oshima, E.; Ishihara, T.; Yokota, O.; Nakashima-Yasuda, H.; Nagao, S.; Ikeda, C.; Naohara, J.; Terada, S.; Uchitomi, Y. Accelerated tau aggregation, apoptosis and neurological dysfunction caused by chronic oral administration of aluminum in a mouse model of tauopathies. Brain Pathol. 2013, 23, 633–644. [Google Scholar] [CrossRef]
- Ribes, D.; Colomina, M.T.; Vicens, P.; Domingo, J.L. Impaired spatial learning and unaltered neurogenesis in a transgenic model of Alzheimer’s disease after oral aluminum exposure. Curr. Alzheimer Res. 2010, 7, 401–408. [Google Scholar] [CrossRef]
- Sanajou, S.; Şahin, G.; Baydar, T. Aluminum in cosmetics and personal care products. J. Appl. Toxicol. 2021, 41, 1704–1718. [Google Scholar] [CrossRef]
- Coulson, J.M.; Hughes, B.W. Dose-response relationships in aluminum toxicity in humans. Clin. Toxicol. 2022, 60, 415–428. [Google Scholar] [CrossRef]
- Dórea, J.G. Neurotoxic effects of combined exposures to aluminum and mercury in early life. Environ. Res. 2020, 188, 109734. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Pogue, A.I.; Lukiw, W.J. Synergism in aluminum and mercury neurotoxicity. Integr. Food Nutr. Metab. 2018, 5, 1–7. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, R.; Liu, R.; Ba, Y.; Huang, H. The roles of histone modifications in metal-induced neurological disorders. Biol. Trace Elem. Res. 2022. preprint. [Google Scholar] [CrossRef] [PubMed]
- Crestini, A.; Santilli, F.; Martellucci, S.; Carbone, E.; Sorice, M.; Piscopo, P.; Mattei, V. Prions and neurodegenerative diseases: A focus on Alzheimer’s disease. J. Alzheimer’s Dis. 2022, 85, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Kruck, T.P.; Cui, J.G.; Percy, M.E.; Lukiw, W.J. Molecular shuttle chelation: The use of ascorbate, desferrioxamine and Feralex-G in combination to remove nuclear bound aluminum. Cell Mol. Neurobiol. 2004, 24, 443–459. [Google Scholar] [CrossRef] [PubMed]
- ELBini-Dhouib, I.; Doghri, R.; Ellefi, A.; Degrach, I.; Srairi-Abid, N.; Gati, A. Curcumin attenuated neurotoxicity in sporadic animal model of Alzheimer’s disease. Molecules 2021, 26, 3011. [Google Scholar] [CrossRef] [PubMed]
- Deokar, R.G.; Barooah, N.; Barik, A. Interaction of esculetin with aluminium ion by spectroscopic studies and isothermal titration calorimetry: A probable molecule for chelation therapy. J. Biomol. Struct. Dyn. 2021, 40, 6163–6170. [Google Scholar] [CrossRef] [PubMed]
- Rahimzadeh, M.R.; Rahimzadeh, M.R.; Kazemi, S.; Amiri, R.J.; Pirzadeh, M.; Moghadamnia, A.A. Aluminum poisoning with emphasis on its mechanism and treatment of intoxication. Emerg. Med. Int. 2022, 2022, 1480553. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Pogue, A.I.; Alexandrov, P.N.; Butler, L.G.; Li, W.; Jaber, V.R.; Lukiw, W.J. Alteration of Biomolecular Conformation by Aluminum-Implications for Protein Misfolding Disease. Molecules 2022, 27, 5123. https://doi.org/10.3390/molecules27165123
Zhao Y, Pogue AI, Alexandrov PN, Butler LG, Li W, Jaber VR, Lukiw WJ. Alteration of Biomolecular Conformation by Aluminum-Implications for Protein Misfolding Disease. Molecules. 2022; 27(16):5123. https://doi.org/10.3390/molecules27165123
Chicago/Turabian StyleZhao, Yuhai, Aileen I. Pogue, Peter N. Alexandrov, Leslie G. Butler, Wenhong Li, Vivian R. Jaber, and Walter J. Lukiw. 2022. "Alteration of Biomolecular Conformation by Aluminum-Implications for Protein Misfolding Disease" Molecules 27, no. 16: 5123. https://doi.org/10.3390/molecules27165123
APA StyleZhao, Y., Pogue, A. I., Alexandrov, P. N., Butler, L. G., Li, W., Jaber, V. R., & Lukiw, W. J. (2022). Alteration of Biomolecular Conformation by Aluminum-Implications for Protein Misfolding Disease. Molecules, 27(16), 5123. https://doi.org/10.3390/molecules27165123