
Inhibition of Endoplasmic Reticulum Stress Improves Acetylcholine-Mediated Relaxation in the Aorta of Type-2 Diabetic Rats

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
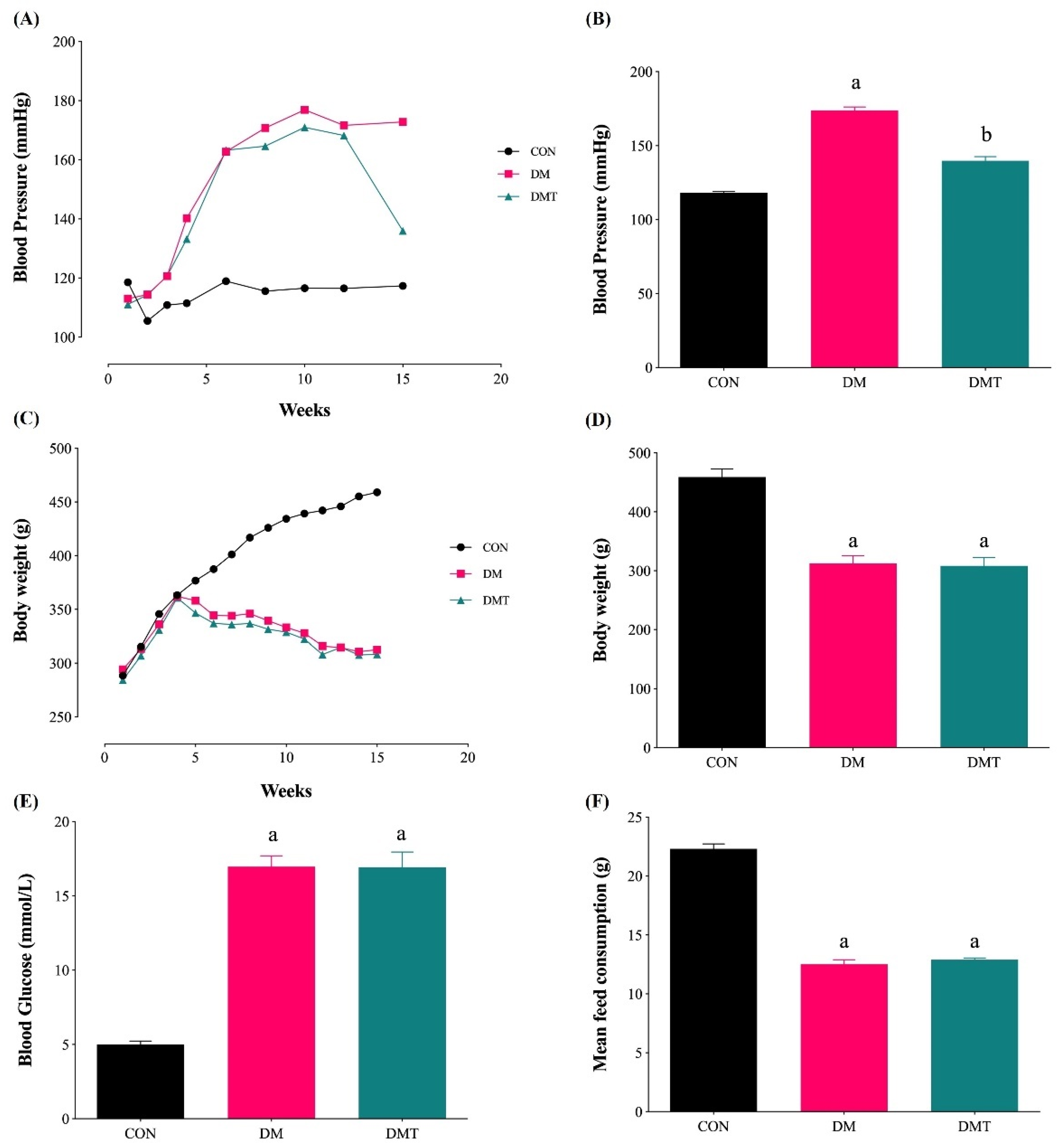
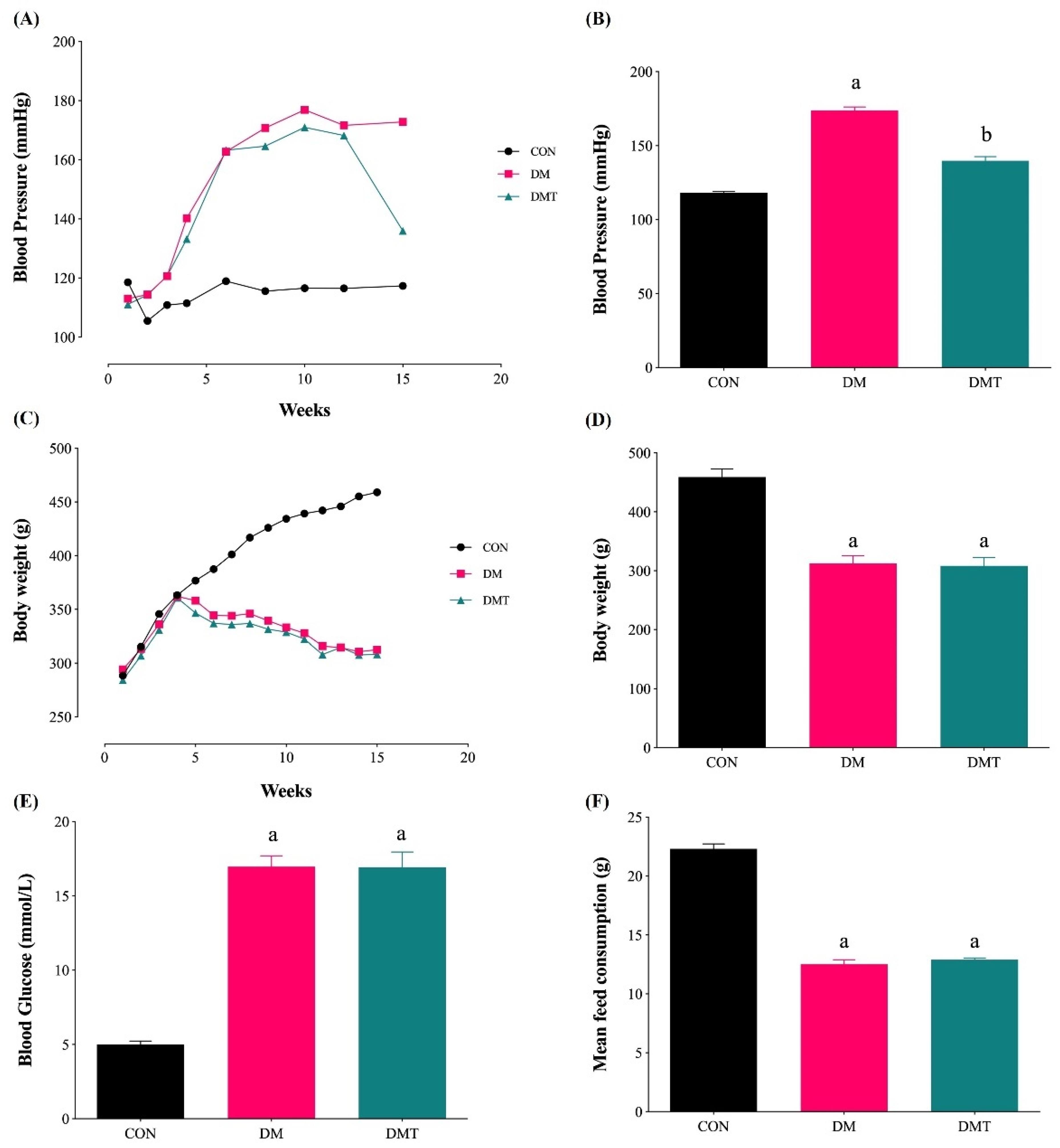
2.1. Effect of ER Stress Inhibition on Blood Pressure, Body Weight, Blood Glucose Level, and Food Consumption
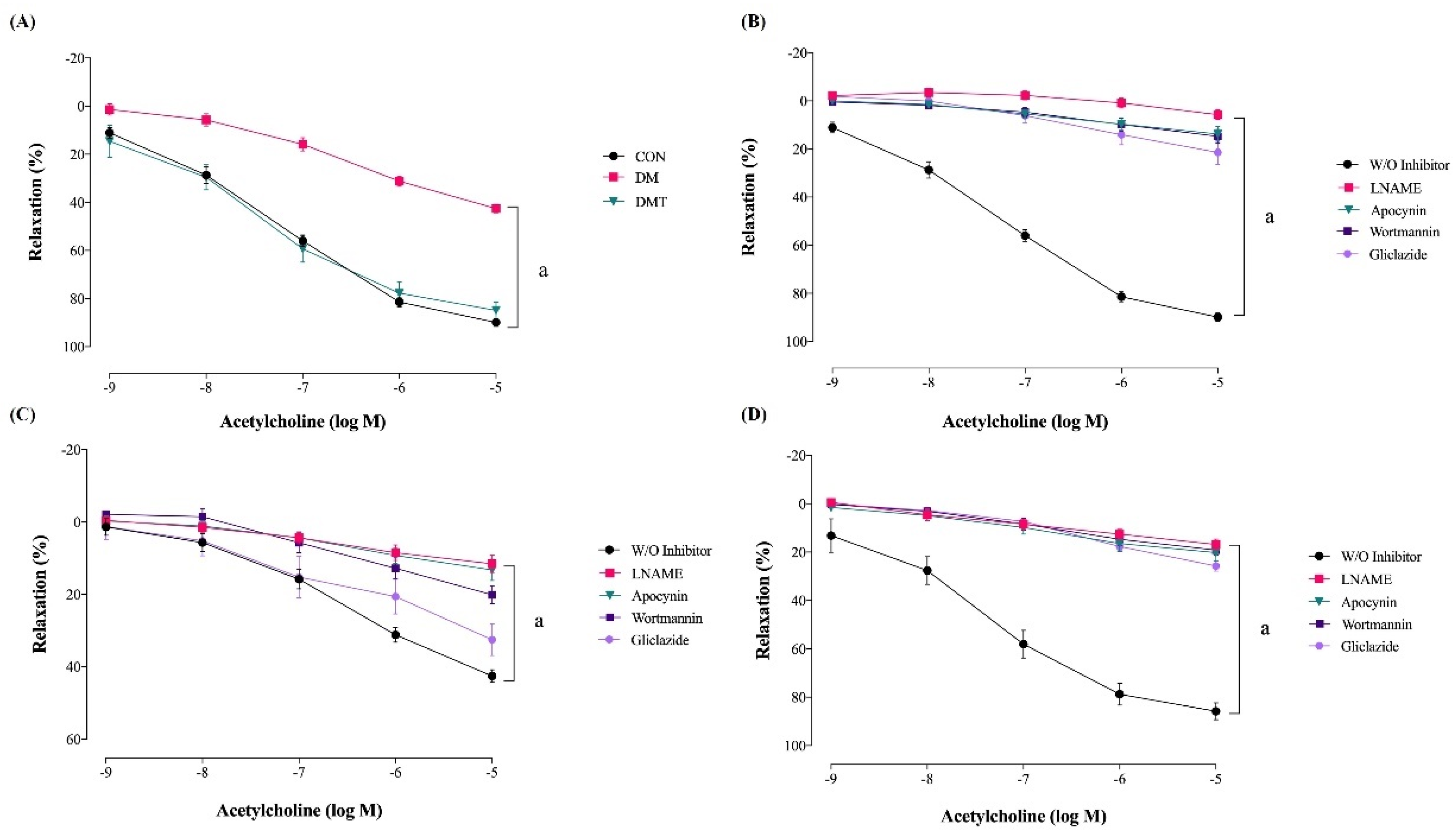
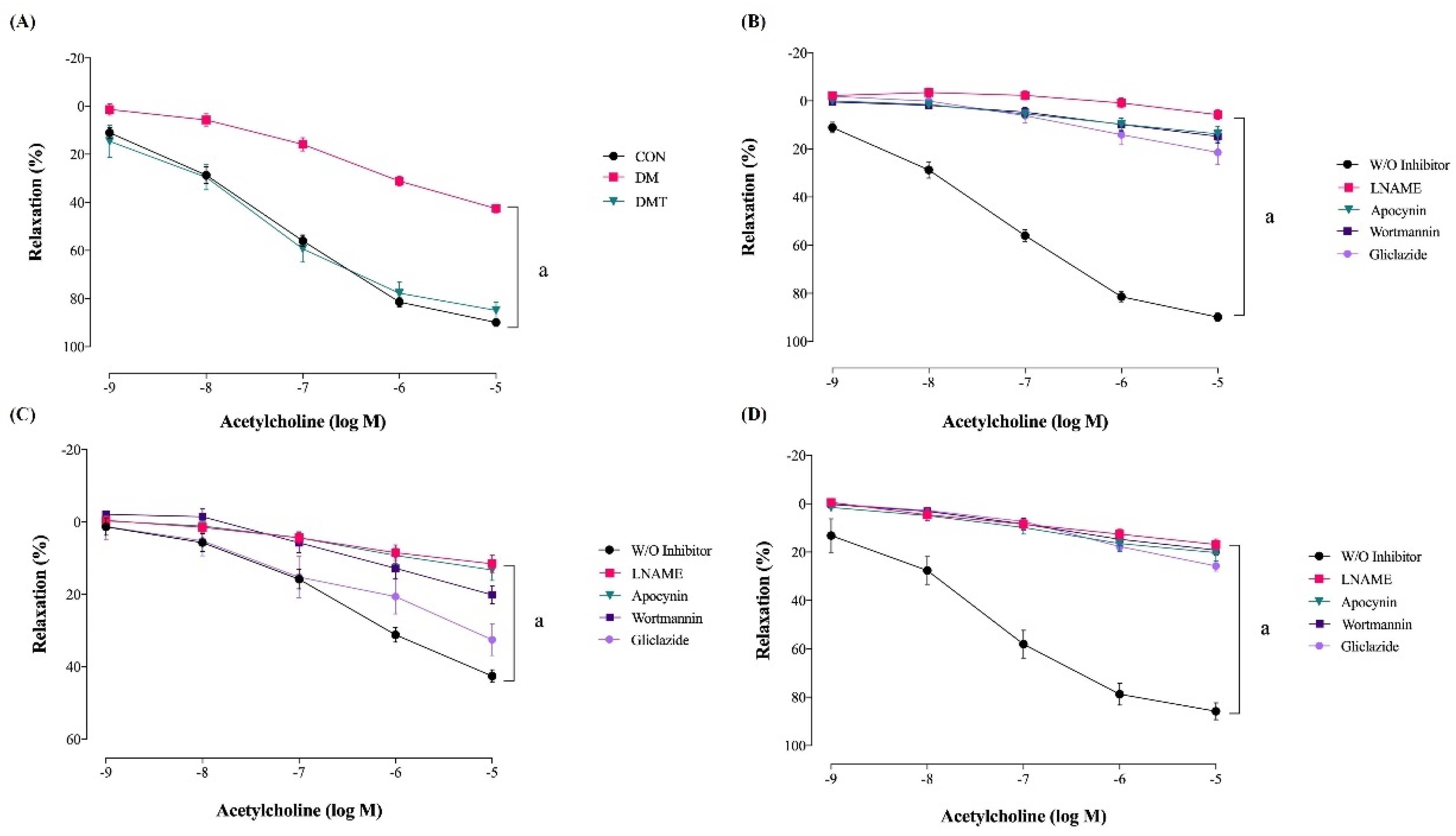
2.2. Effect of ER Stress Inhibition on Endothelium-Dependent Relaxation
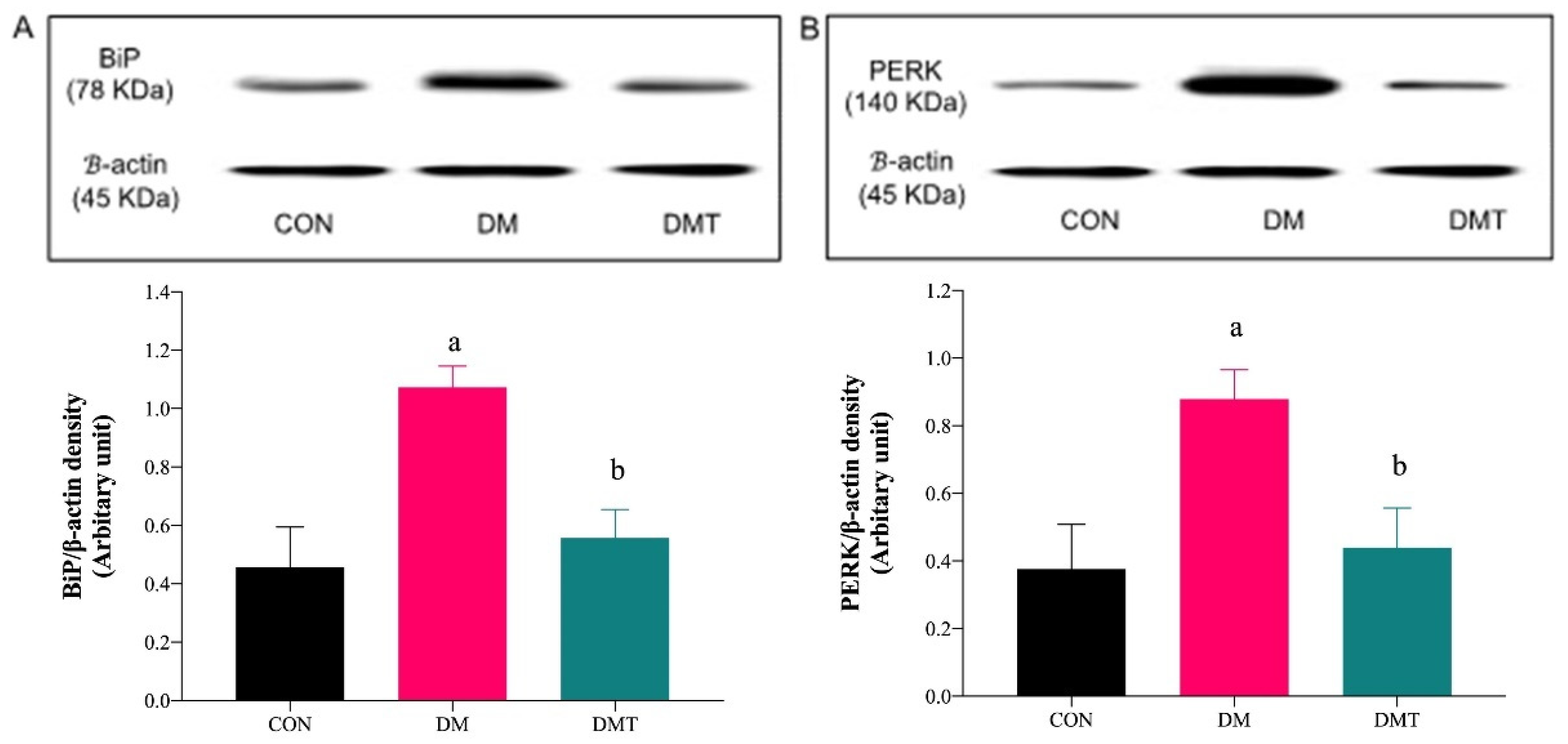
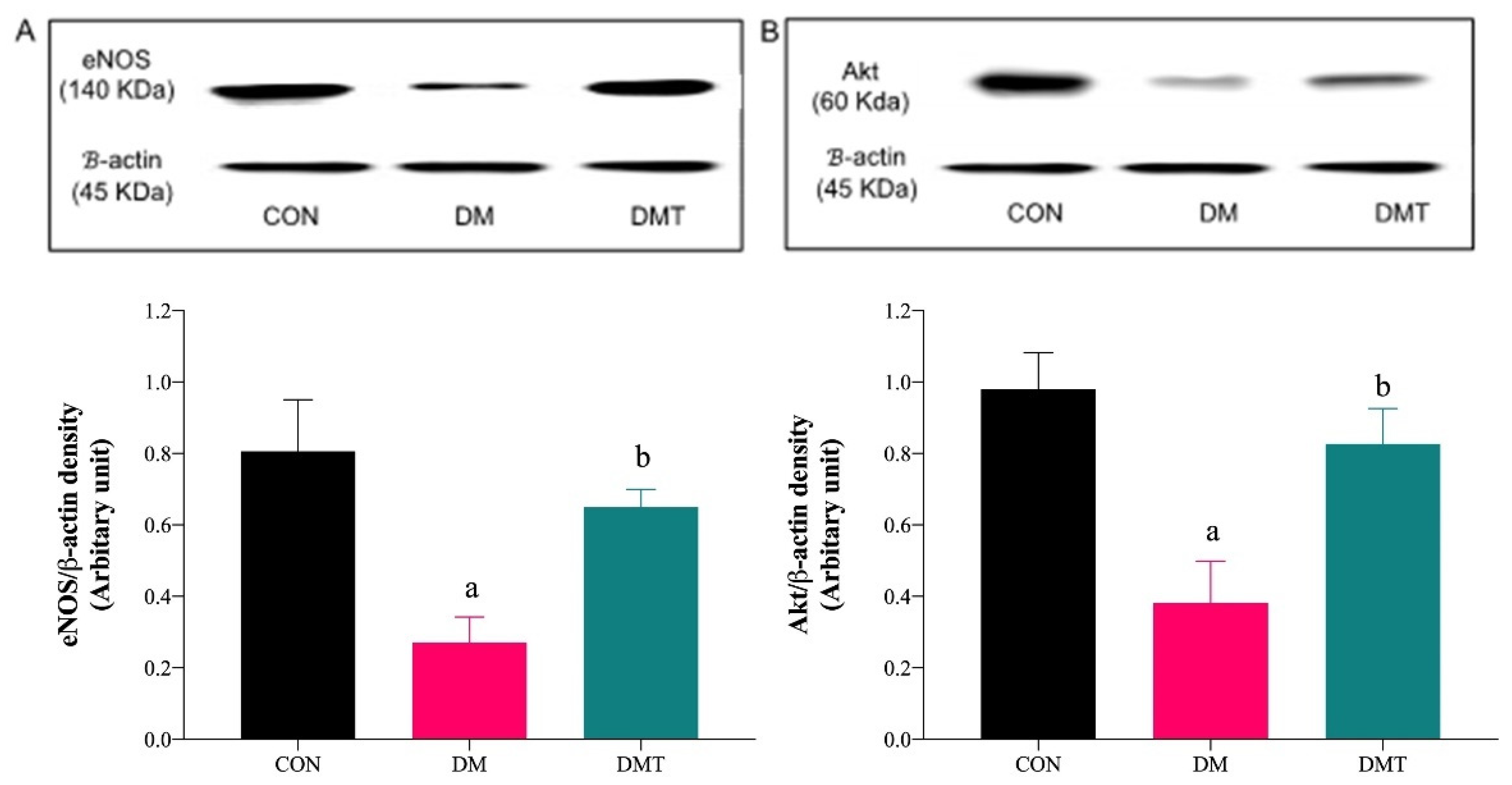
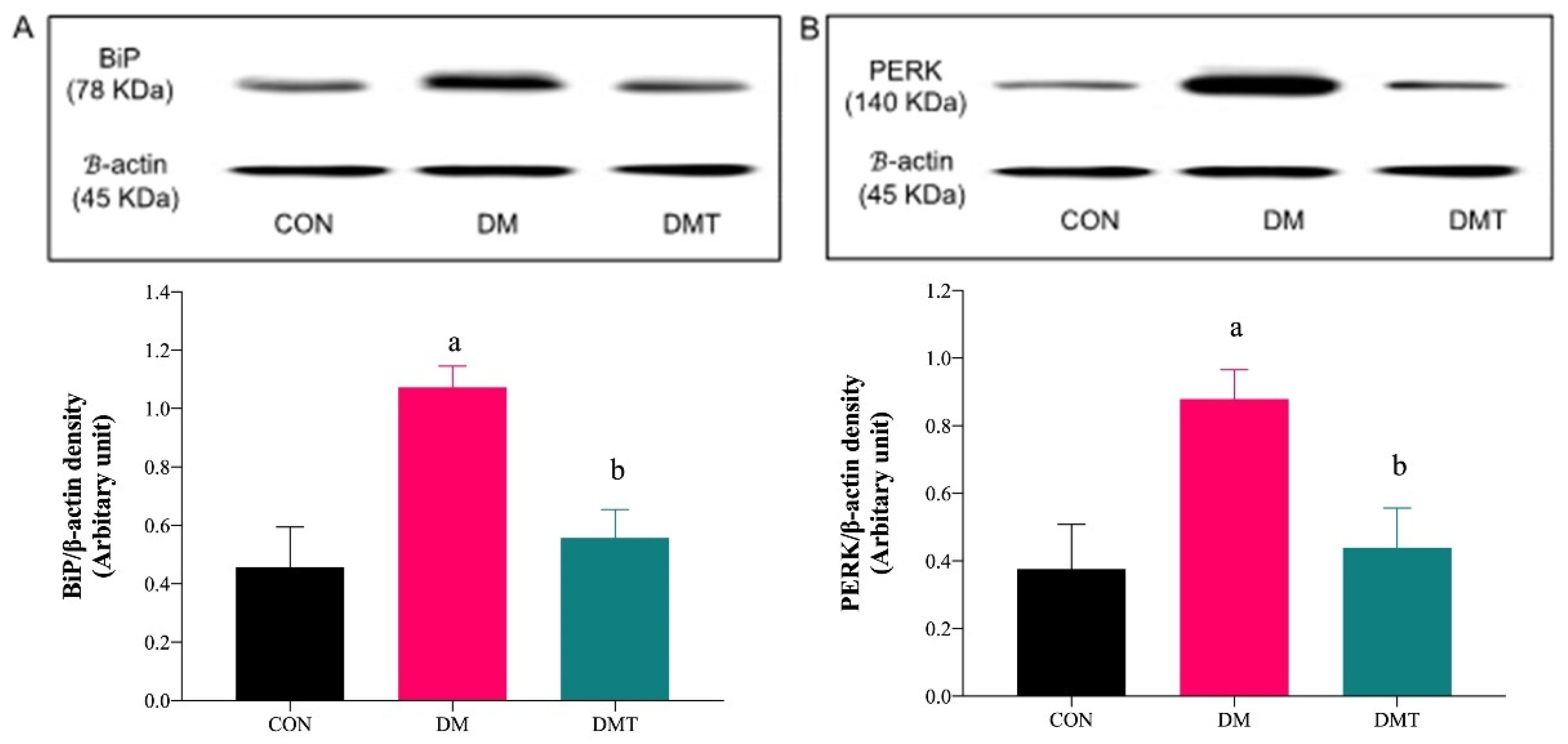
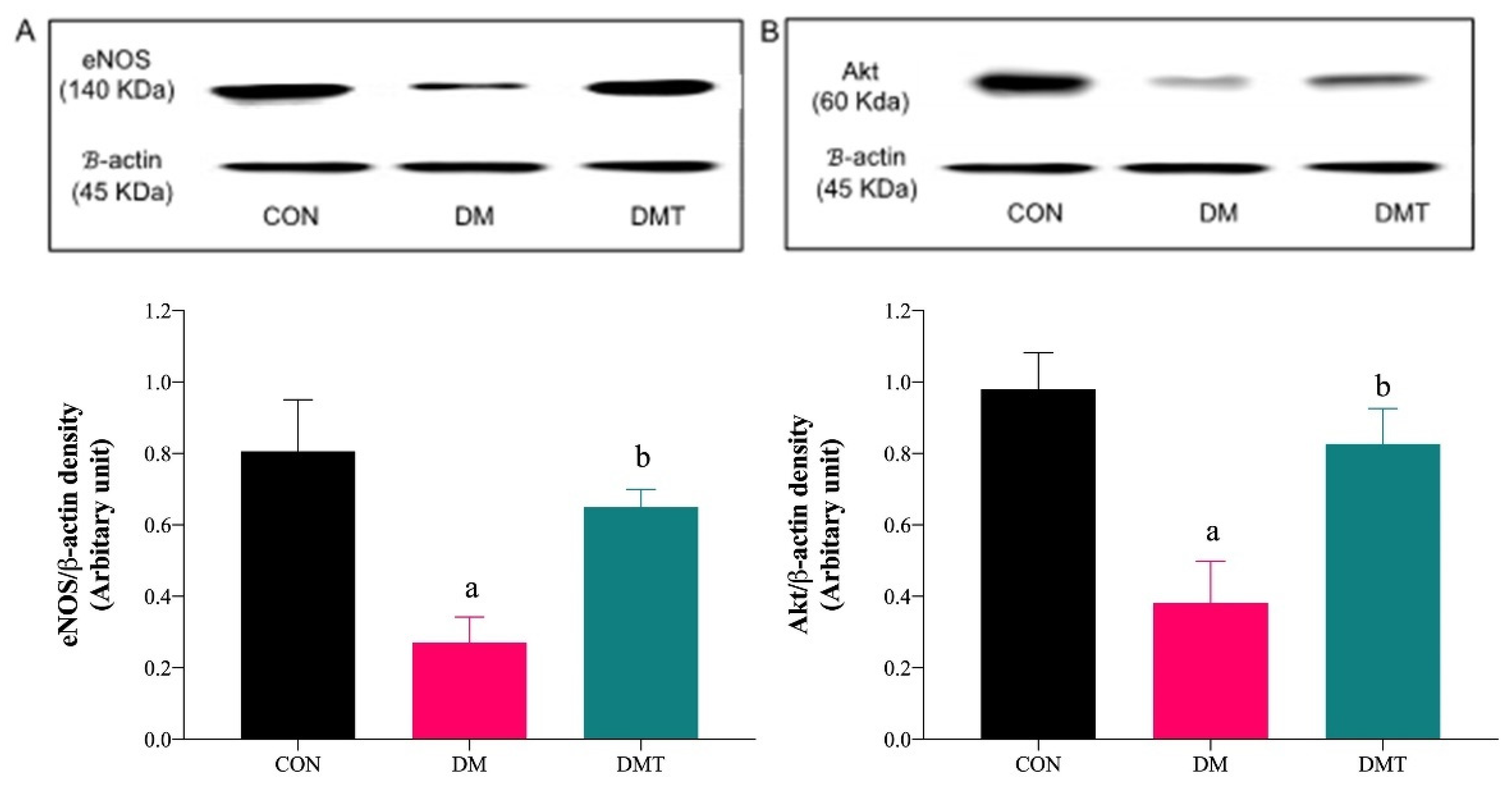
2.3. Effect of ER Stress Inhibition on the Protein Expression of BiP, PERK, eNOS, and Akt
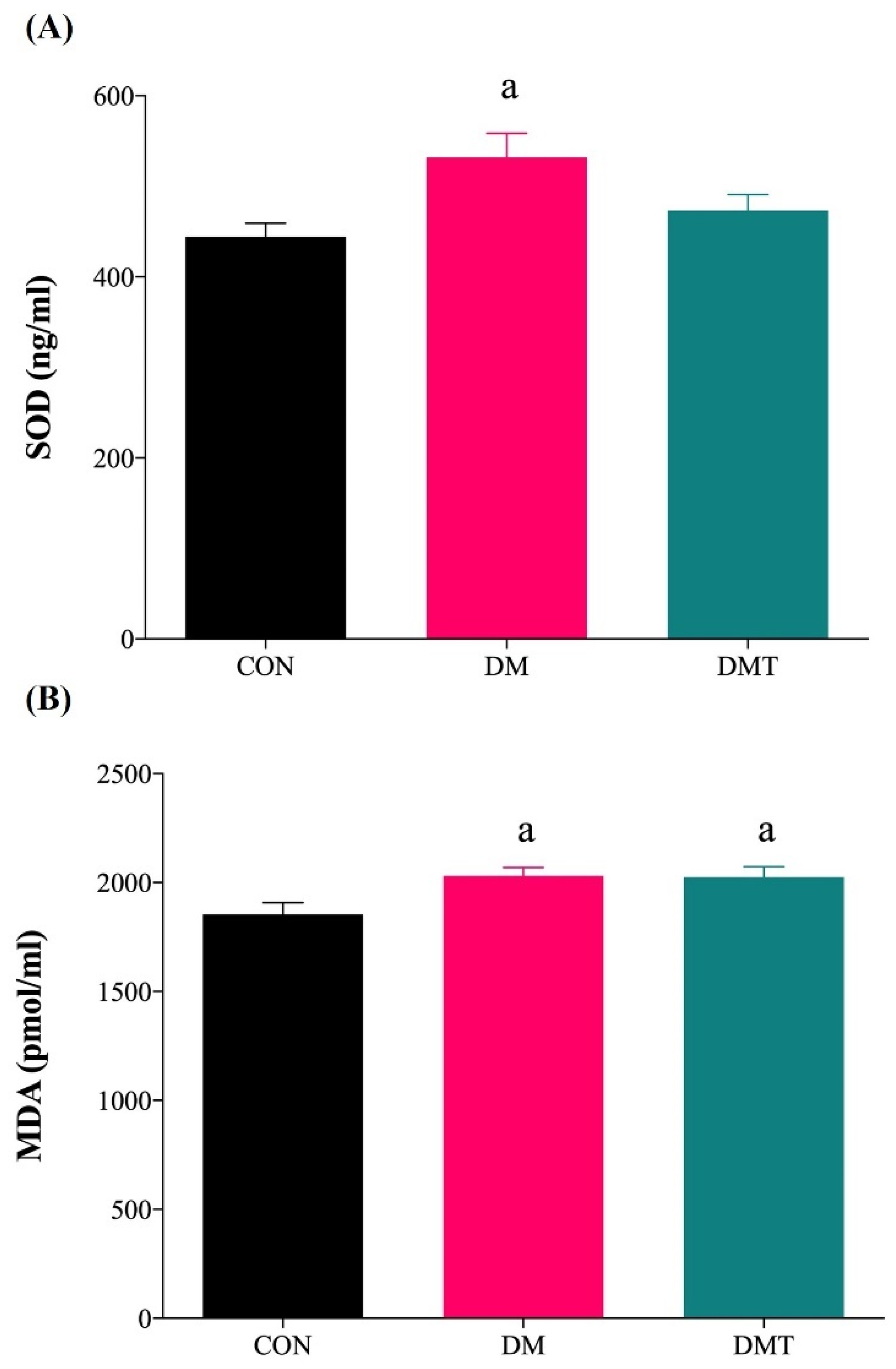
2.4. Effect of ER Stress Inhibition on SOD Activity and MDA Level
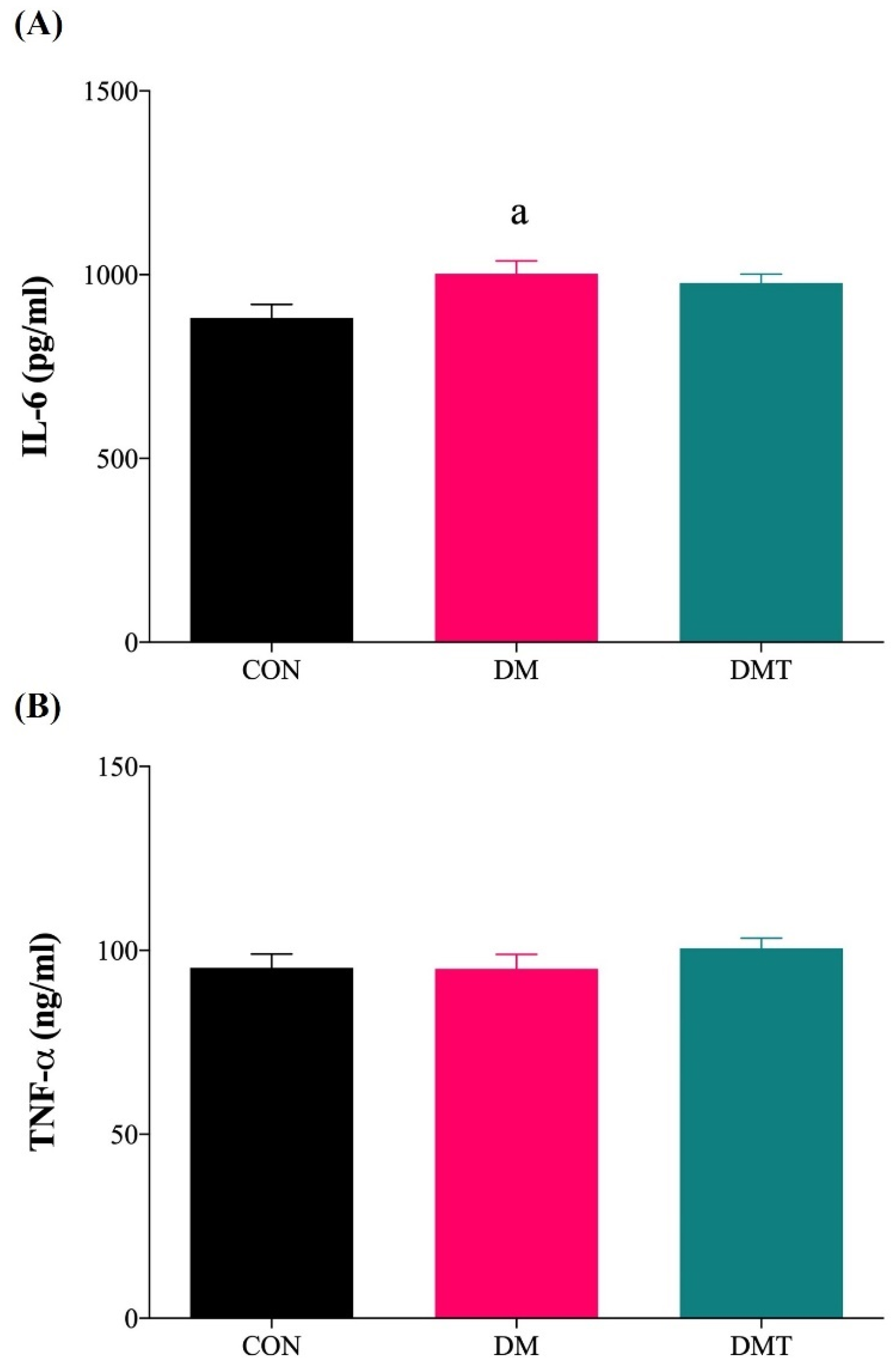
2.5. Effect of ER Stress Inhibition on IL-6 and TNF-α
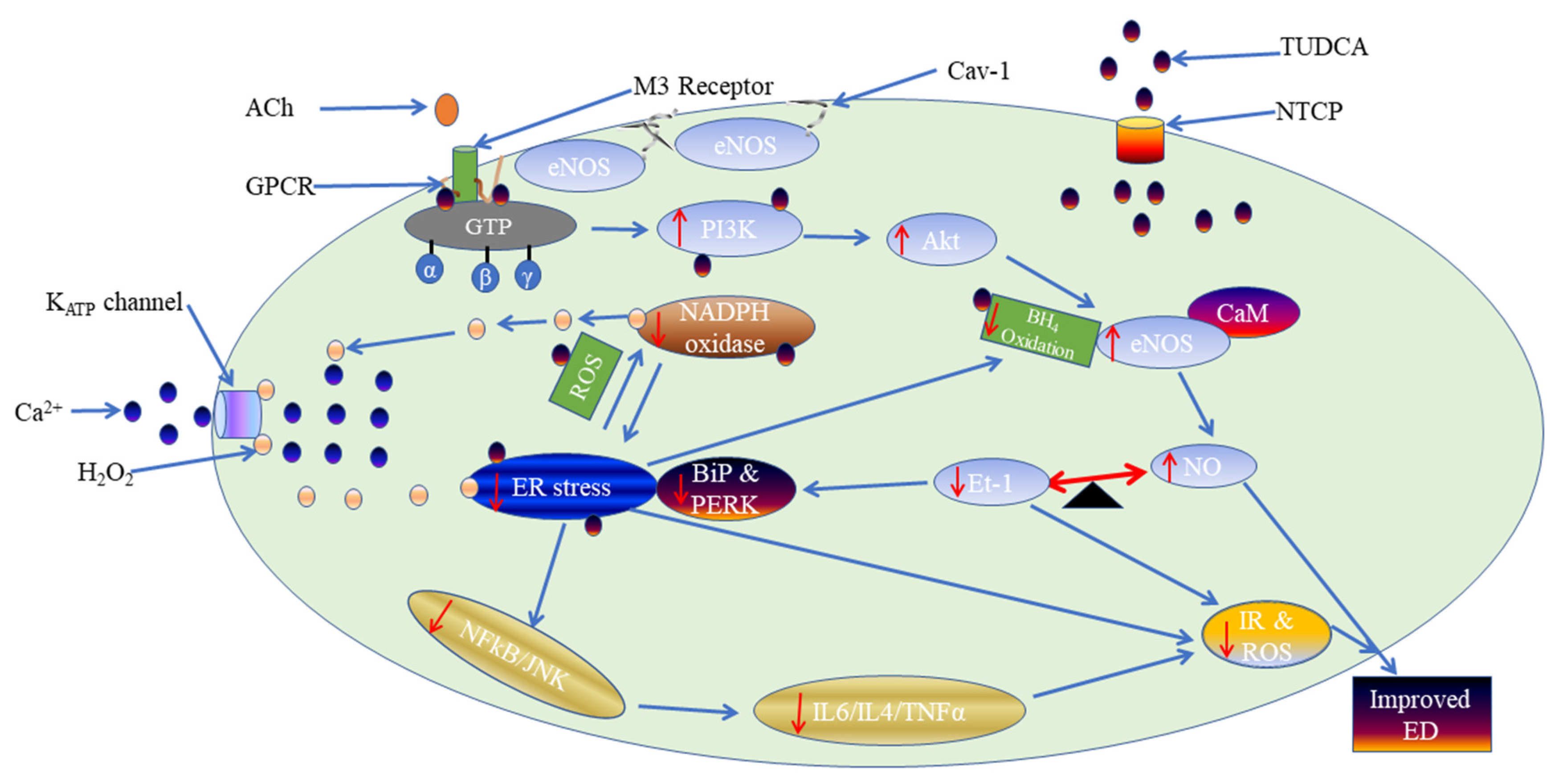
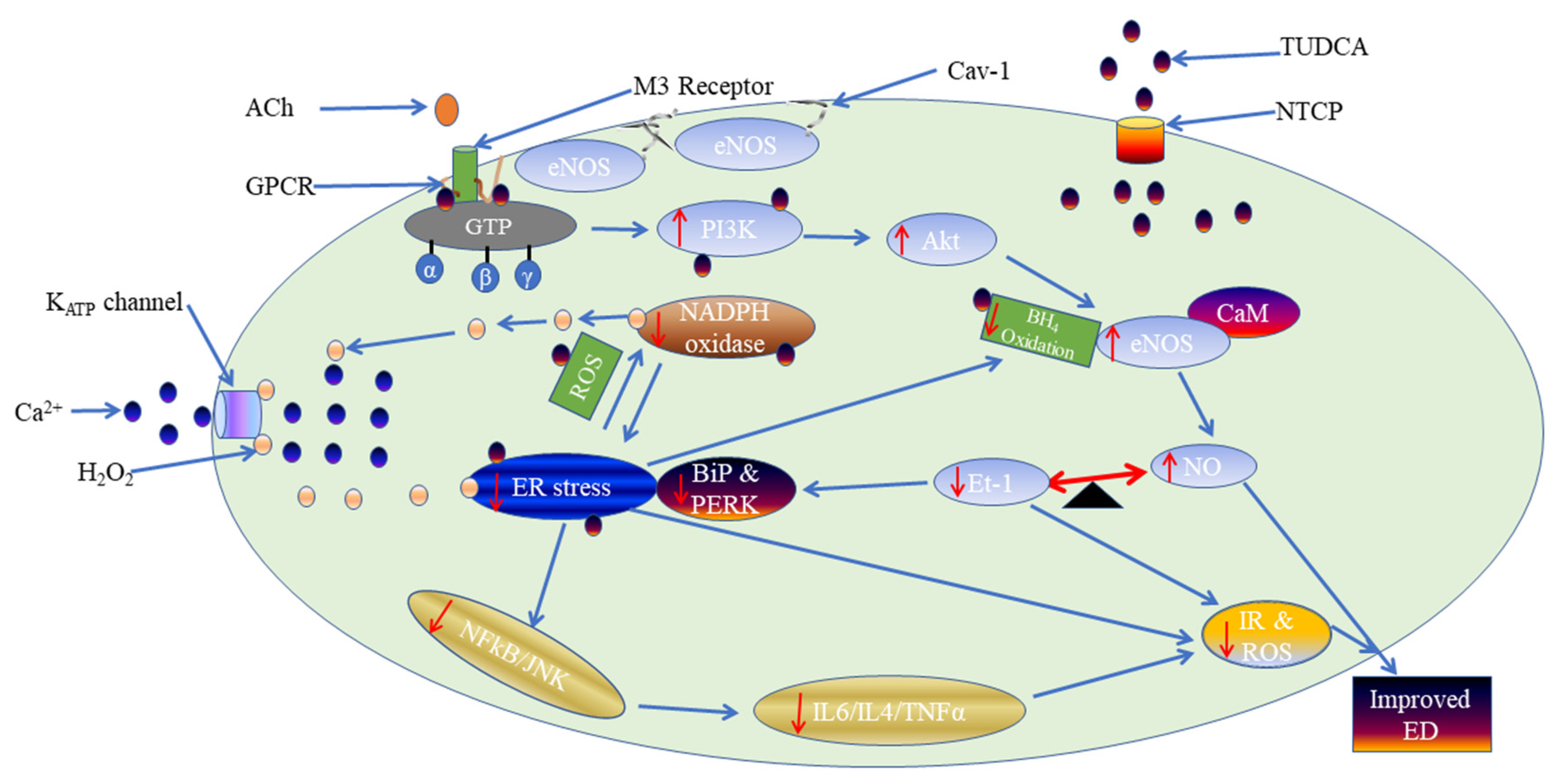
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Measurement of Blood Pressure
4.3. Functional Study
4.4. Western Blotting
4.5. Biochemical Analysis of Thoracic Aorta Tissue Lysate
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kosiborod, M.; Gomes, M.B.; Nicolucci, A.; Pocock, S.; Rathmann, W.; Shestakova, M.V.; Watada, H.; Shimomura, I.; Chen, H.; Ruzafa, J.C. Vascular complications in patients with type 2 diabetes: Prevalence and associated factors in 38 countries (the DISCOVER study program). Cardiovasc. Diabetol. 2018, 17, 150. [Google Scholar] [CrossRef]
- Mustapha, S.; Mohammed, M.; Yunusa, I.; Rasool, A.H.G.; Mokhtar, S.S. Potential risks of endoplasmic reticulum stress on vasculopathy in diabetes. Obes. Med. 2020, 19, 100274. [Google Scholar] [CrossRef]
- Mokhtar, S.S.; Rasool, A.H.G. Role of endothelium-dependent hyperpolarisation and prostacyclin in diabetes. Malays. J. Med. Sci. 2015, 22, 8–17. [Google Scholar] [PubMed]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the cardiac nitric oxide synthases: Tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Fernandes, C.; Liu, Y.; Wu, Y.; Wu, H.; Brophy, M.L.; Deng, L.; Song, K.; Wen, A.; Wong, S.; et al. Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diabetes Vasc. Dis. Res. 2016, 14, 14–23. [Google Scholar] [CrossRef]
- Hosford, P.S.; Christie, I.N.; Niranjan, A.; Aziz, Q.; Anderson, N.; Ang, R.; Lythgoe, M.F.; Wells, J.A.; Tinker, A.; Gourine, A.V. A critical role for the ATP-sensitive potassium channel subunit KIR6.1 in the control of cerebral blood flow. J. Cereb. Blood Flow Metab. 2018, 39, 2089–2095. [Google Scholar] [CrossRef]
- Tinker, A.; Aziz, Q.; Thomas, A.; Tinker, A.; Harvey, W. The role of ATP-sensitive potassium channels in cellular function and protection in the cardiovascular system. Br. J. Pharmacol. 2013, 171, 12–23. [Google Scholar] [CrossRef]
- Sharifi-Sanjani, M.; Zhou, X.; Asano, S.; Tilley, S.; Ledent, C.; Teng, B.; Dick, G.M.; Mustafa, S.J. Interactions between A2A adenosine receptors, hydrogen peroxide, and KATP channels in coronary reactive hyperemia. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, 1294–1301. [Google Scholar] [CrossRef]
- Aziz, Q.; Li, Y.; Anderson, N.; Ojake, L.; Tsisanova, E.; Tinker, A. Molecular and functional characterization of the endothelial ATP-sensitive potassium channel. J. Biol. Chem. 2017, 292, 17587–17597. [Google Scholar] [CrossRef]
- Nilius, B.; Droogmans, G.U.Y. Ion Channels and Their Functional Role in Vascular Endothelium. Physiol. Rev. 2001, 81, 1416–1447. [Google Scholar] [CrossRef]
- Mustapha, S.; Mohammed, M.; Azemi, A.K.; Yunusa, I.; Shehu, A.; Mustapha, L.; Wada, Y.; Ahmad, M.H.; Amir, W.; Wan, N.; et al. Potential Roles of Endoplasmic Reticulum Stress and Cellular Proteins Implicated in Diabesity. Oxid. Med. Cell. Longev. 2021, 2021, 8830880. [Google Scholar] [CrossRef]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Singh, P.; Rai, S.N. Factors a ff ecting obesity and its treatment. Obes. Med. 2019, 16, 100140. [Google Scholar] [CrossRef]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic reticulum stress: A critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef]
- Santos, C.X.C.; Tanaka, L.Y.; Wosniak, J.J.; Laurindo, F.R.M. Mechanisms and Implications of Reactive Oxygen Species Generation During the Unfolded Protein Response. Antioxid. Redox Signal 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol Heart. Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, S.; Mohammed, M.; Azemi, A.K.; Abubakar, I.J.; Shehu, A.; Mustapha, L.; Aliyu, M.; Danraka, N.; Amin, A.; Bala, A.A. Current status of endoplasmic reticulum stress in type II diabetes. Molecules 2021, 26, 4362. [Google Scholar] [CrossRef] [PubMed]
- Kassan, M.; Galán, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Jenkins, N.T. Induction of endoplasmic reticulum stress impairs insulin-stimulated vasomotor relaxation in rat aortic rings: Role of endothelin-1. J. Physiol. Pharmacol. 2013, 64, 557–564. [Google Scholar]
- Choi, S.K.; Lim, M.; Yeon, S.I.; Lee, Y.H. Inhibition of endoplasmic reticulum stress improves coronary artery function in type 2 diabetic mice. Exp. Physiol. 2016, 101, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Lim, M.; Byeon, S.H.; Lee, Y.H. Inhibition of endoplasmic reticulum stress improves coronary artery function in the spontaneously hypertensive rats. Sci. Rep. 2016, 6, 31925. [Google Scholar] [CrossRef]
- Spitler, K.M.; Matsumoto, T.; Webb, R.C. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am. J. Physiol Heart Circ. Physiol. 2013, 305, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Azemi, A.K.; Mokhtar, S.S.; Hanum, A.; Rasool, G. Clinacanthus nutans Leaves Extract Reverts Endothelial Dysfunction in Type 2 Diabetes Rats by Improving Protein Expression of eNOS. Oxid. Med. Cell. Longev. 2020, 2020, 7572892. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Taguchi, K.; Yasuhiro, T.; Matsumoto, T.; Kamata, K. Impairment of PI3-K/Akt Pathway Underlies Attenuated Endothelial Function in Aorta of Type 2 Diabetic Mouse Model. Hypertension 2004, 44, 956–962. [Google Scholar] [CrossRef]
- Mokhtar, S.S.; Vanhoutte, P.M.; Leung, S.W.S.; Suppian, R.; Yusof, M.I.; Rasool, A.H.G. Reduced nitric oxide-mediated relaxation and endothelial nitric oxide synthase expression in the tail arteries of streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2016, 773, 78–84. [Google Scholar] [CrossRef]
- Georgescu, A.; Popov, D.; Constantin, A.; Nemecz, M.; Alexandru, N.; Cochior, D.; Tudor, A. Dysfunction of human subcutaneous fat arterioles in obesity alone or obesity associated with Type 2 diabetes. Clin. Sci. 2011, 120, 463–472. [Google Scholar] [CrossRef]
- Akther, F.; Razan, R.M.; Shaligram, S.; Graham, J.L.; Stanhope, K.L.; Allen, K.N.; Vazquez-Medina, J.P.; Havel, P.J.; Rahimian, R. Potentiation of Acetylcholine-Induced Relaxation of Aorta in Male UC Davis Type 2 Diabetes Mellitus (UCD-T2DM) Rats: Sex-Specific Responses. Front. Pharmacol. 2021, 12, 616317. [Google Scholar] [CrossRef]
- Zhong, M.F.; Shen, W.L.; Tabuchi, M.; Nakamura, K.; Chen, Y.C.; Qiao, C.Z.; He, J.; Yang, J.; Zhang, C.; Kamenov, Z.; et al. Differential changes of aorta and carotid vasodilation in type 2 diabetic GK and OLETF rats: Paradoxical roles of hyperglycemia and insulin. Exp. Diabetes Res. 2012, 2012, 429020. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk between oxidative stress and endoplasmic reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: A focus on the protective roles of heme oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J., Jr.; Davies, P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Choy, K.W.; Lau, Y.S.; Murugan, D.; Mustafa, M.R. Chronic treatment with paeonol improves endothelial function in mice through inhibition of endoplasmic reticulum stress- mediated oxidative stress. PLoS ONE 2017, 12, e0178365. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.S.; Mustafa, M.R.; Choy, K.W.; Chan, S.M.H.; Potocnik, S.; Herbert, T.P.; Woodman, O.L. 3′,4′-Dihydroxyflavonol Ameliorates Endoplasmic Reticulum Stress-Induced Apoptosis and Endothelial Dysfunction in Mice. Sci. Rep. 2018, 8, 1818. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Scull, C.; Ozcan, L.; Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 2010, 191, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dellsperger, K.C.; Zhang, C. The link between metabolic abnormalities and endothelial dysfunction in type 2 diabetes: An update. Basic Res. Cardiol. 2012, 107, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Tang, X.; Xie, L.; Meng, G.; Ji, Y. Aliskiren improves endothelium-dependent relaxation of thoracic aorta by activating PI3K/Akt/eNOS signal pathway in SHR. Clin. Exp. Pharmacol. Physiol. 2016, 43, 450–458. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Wier, W.G.; Zhang, Q.; Jiang, H.; Li, Q.; Chen, S.; Tian, Z.; Li, Y.; Yu, X.; et al. Exercise improves the dilatation function of mesenteric arteries in postmyocardial infarction rats via a PI3K/Akt/eNOS pathway-mediated mechanism. Am. J. Physiol Heart. Circ. Physiol. 2010, 299, 2097–2107. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kamata, K. Effect of chronic insulin treatment on NO production and endothelium-dependent relaxation in aortae from established STZ-induced diabetic rats. Atherosclerosis 2001, 155, 313–320. [Google Scholar] [CrossRef]
- Duncan, E.R.; Crossey, P.A.; Walker, S.; Anilkumar, N.; Poston, L.; Douglas, G.; Ezzat, V.A.; Wheatcroft, S.B.; Shah, A.M.; Kearney, M.I. Effect of endothelium-specific insulin resistance on endothelial function in vivo. Diabetes 2008, 57, 3307–3314. [Google Scholar] [CrossRef]
- Miura, H.; Wachtel, R.E.; Loberiza, F.; Saito, T.; Miura, M.; Nicolosi, A.C.; Gutterman, D.D. Diabetes Mellitus Impairs Vasodilation to Hypoxia in Human Coronary Arterioles Reduced Activity of ATP-Sensitive Potassium Channels. Circ. Res. 2003, 92, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Damrauer, S.M.; Lee, M.; Sellke, F.W.; Ferran, C.; Abid, R. Endothelium-Dependent Coronary Vasodilatation Requires NADPH Oxidase—Derived Reactive Oxygen Species. Arterioscler. Thromb. Vasc. Biol. 2009, 10, 1704–1710. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Morikawa, K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J. Mol. Cell. Cardiol. 2005, 39, 725–732. [Google Scholar] [CrossRef]
- Gao, Y.; Hirota, S.; Zhang, D.; Janssen, L.J.; Lee, R.M.K.W. Mechanisms of hydrogen-peroxide-induced biphasic response in rat mesenteric artery. Br. J. Pharmacol. 2003, 138, 1085–1092. [Google Scholar] [CrossRef]
- Fujimoto, S.; Asano, T.; Sakai, M.; Sakurai, K.; Takagi, D.; Yoshimoto, N.; Itoh, T. Mechanisms of hydrogen peroxide-induced relaxation in rabbit mesenteric small artery. Eur. J. Pharmacol. 2001, 412, 291–300. [Google Scholar] [CrossRef]
- Liu, Y.; Bubolz, A.H.; Mendoza, S.; Zhang, D.X.; Gutterman, D.D. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ. Res. 2011, 108, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Aziz, Q.; Thomas, A.M.; Gomes, J.; Ang, R.; Sones, W.R.; Li, Y.; Ng, K.E.; Gee, L.; Tinker, A. The ATP-sensitive potassium channel subunit, Kir6.1, in vascular smooth muscle plays a major role in blood pressure control. Hypertension 2014, 64, 523–529. [Google Scholar] [CrossRef]
- Hutcheson, I.R.; Griffith, T.M. Heterogeneous populations of K+ channels mediate EDRF release to flow but not agonists in rabbit aorta. Am. J. Physiol. Heart. Circ. Physiol. 1994, 266, 590–596. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.J.; Li, J.; Hosoya, K.I.; Ratan, R.; Townes, T.; Zhang, S.X. Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia 2012, 55, 2533–2545. [Google Scholar] [CrossRef]
- Li, Y.; Aziz, Q.; Anderson, N.; Ojake, L.; Tinker, A. Endothelial ATP-Sensitive Potassium Channel Protects against the Development of Hypertension and Atherosclerosis. Hypertension 2020, 76, 776–784. [Google Scholar] [CrossRef]
- Wang, X.C.; Sun, W.T.; Yu, C.M.; Pun, S.H.; Underwood, M.J.; He, G.W.; Yang, Q. ER stress mediates homocysteine-induced endothelial dysfunction: Modulation of IKCa and SKCa channels. Atherosclerosis 2015, 242, 191–198. [Google Scholar] [CrossRef]
- Kim, J.; Jang, H.; Hwang, D.H. Toll-like receptor 4-induced endoplasmic reticulum stress contributes to impairment of vasodilator action of insulin. Am. J. Physiol. Endocrinol. Metab. 2015, 309, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Spitler, K.M.; Webb, R.C. Endoplasmic reticulum stress contributes to aortic stiffening via proapoptotic and fibrotic signaling mechanisms. Hypertension 2013, 63, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Ando, M.; Watanabe, S.; Iguchi, M.; Nagata, M.; Kobayashi, S.; Taguchi, K.; Kobayashi, T. Tunicamycin-Induced Alterations in the Vasorelaxant Response in Organ-Cultured Superior Mesenteric Arteries of Rats. Biol. Pharm. Bull. 2016, 39, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Watanabe, S.; Ando, M.; Yamada, K.; Iguchi, M.; Taguchi, K.; Kobayashi, T. Diabetes and age-related differences in vascular function of renal artery: Possible involvement of endoplasmic reticulum stress. Rejuvenation Res. 2016, 19, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Vettorazzi, J.F.; Ribeiro, R.A.; Borck, P.C.; Branco, R.C.; Soriano, S.; Merino, B.; Boschero, A.C.; Nadal, A.; Quesada, I.; Carneiro, E.M. The bile acid TUDCA increases glucose-induced insulin secretion via the cAMP/PKA pathway in pancreatic beta cells. Metabolism 2016, 65, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Nair, B.J. Alteration of cellular metabolism in cancer cells and its therapeutic. J. Oral Maxillofac. Pathol. 2017, 21, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Bandeira, S.D.M.; Guedes, G.D.S.; Fonseca, L.J.S.D.; Pires, A.S.; Gelain, D.P.; Moreira, J.C.F.; Rabelo, L.A.; Vasconcelos, S.M.L.; Goulart, M.O.F. Characterization of blood oxidative stress in type 2 diabetes mellitus patients: Increase in lipid peroxidation and SOD activity. Oxid. Med. Cell. Longev. 2012, 2012, 819310. [Google Scholar] [CrossRef] [PubMed]
- Bigagli, E.; Raimondi, L.; Mannucci, E.; Colombi, C.; Bardini, G.; Rotella, C.M.; Lodovici, M. Lipid and protein oxidation products, antioxidant status and vascular complications in poorly controlled type 2 diabetes. Br. J. Diabetes Vasc. Dis. 2012, 12, 33–39. [Google Scholar] [CrossRef]
- Jiménez-Osorio, A.S.; Picazo, A.; González-Reyes, S.; Barrera-Oviedo, D.; Rodríguez-Arellano, M.E.; Pedraza-Chaverri, J. Nrf2 and redox status in prediabetic and diabetic patients. Int. J. Mol. Sci. 2014, 15, 20290–20305. [Google Scholar] [CrossRef]
- Lodovici, M.; Bigagli, E.; Luceri, C.; Mannucci, E.; Rotella, C.M.; Raimondi, L. Gender-related drug effect on several markers of oxidation stress in diabetes patients with and without complications. Eur. J. Pharmacol. 2015, 766, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.Y.; Song, J.H.; Shin, Y.K.; Han, E.S.; Lee, C.S. Protective effect of boldine on oxidative mitochondrial damage in streptozotocin-induced diabetic rats. Pharmacol. Res. 2000, 42, 361–371. [Google Scholar] [CrossRef] [PubMed]
- de Souza, D.N.; de Souza, E.M.N.; da Silva Pedrosa, M.; Nogueira, F.N.; Simões, A.; Nicolau, J. Effect of Tungstate Administration on the Lipid Peroxidation and Antioxidant Parameters in Salivary Glands of STZ-Induced Diabetic Rats. Biol. Trace Elem. Res. 2020, 199, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Adeghate, E.; D’Souza, C.M.; Saeed, Z.; Al Jaberi, S.; Tariq, S.; Kalász, H.; Tekes, K.; Adeghate, E.A. Nociceptin increases antioxidant expression in the kidney, liver and brain of diabetic rats. Biology 2021, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Que, Y.; Shen, X. Correlation of the plasma levels of soluble RAGE and endogenous secretory RAGE with oxidative stress in pre-diabetic patients. J. Diabetes Complicat. 2015, 29, 422–426. [Google Scholar] [CrossRef]
- Strom, A.; Kaul, K.; Brüggemann, J.; Ziegler, I.; Rokitta, I.; Püttgen, S.; Szendroedi, J.; Müssig, K.; Roden, M.; Ziegler, D. Lower serum extracellular superoxide dismutase levels are associated with polyneuropathy in recent-onset diabetes. Exp. Mol. Med. 2017, 49, e394. [Google Scholar] [CrossRef]
- Chou, S.T.; Tseng, S.T. Oxidative stress markers in type 2 diabetes patients with diabetic nephropathy. Clin. Exp. Nephrol. 2017, 21, 283–292. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Cheung, F.; Chow, O.; Wang, X.; Su, W.; Tong, Y. A comparative study on the hepatoprotective action of bear bile and coptidis rhizoma aqueous extract on experimental liver fibrosis in rats. Complement. Altern. Med. 2012, 12, 239. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y. Tauroursodeoxycholic Acid Alleviates H2O2-Induced Oxidative Stress and Apoptosis via Suppressing Endoplasmic Reticulum Stress in Neonatal Rat Cardiomyocytes. Dose-Response 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Alhasani, R.H.; Almarhoun, M.; Zhou, X.; Reilly, J.; Patterson, S.; Zeng, Z.; Shu, X. Tauroursodeoxycholic Acid Protects Retinal Pigment Epithelial Cells from Oxidative Injury and Endoplasmic Reticulum Stress In Vitro. Biomedicines 2020, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Luan, J.; Huang, T.; Deng, T.; Li, X.; Xiao, Z.; Zhan, J.; Luo, D.; Hou, Y.; Xu, L.; et al. Tauroursodeoxycholic acid alleviates secondary injury in spinal cord injury mice by reducing oxidative stress, apoptosis, and inflammatory response. J. NeuroInflamm. 2021, 18, 216. [Google Scholar] [CrossRef]
- Sumathi, K.; Dilliraj, G.; Chaganti, S.; Lalitha, S.; Nadu, T. Use of malondialdehyde (MDA) as a screening tool for vestibulopathy in Type 2 diabetes mellitus. Biomedicine 2021, 41, 576–579. [Google Scholar] [CrossRef]
- Azemi, A.K.; Mokhtar, S.S.; Hou, L.J.; Emilia, S.; Sharif, T.; Hanum, A.; Rasool, G. Model for type 2 diabetes exhibits changes in vascular function and structure due to vascular oxidative stress and inflammation. Biotech. Histochem. 2020, 96, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lin, Y.; Lei, Y.P.; Wang, L.; He, Z.M.; Xiong, Y. Pyrrolidine dithiocarbamate ameliorates endothelial dysfunction in thoracic aorta of diabetic rats by preserving vascular DDAH activity. PLoS ONE 2017, 12, e0179908. [Google Scholar] [CrossRef] [PubMed]
- Obafemi, T.O.; Jaiyesimi, K.F.; Olomola, A.A.; Olasehinde, O.R.; Olaoye, O.A.; Adewumi, F.D.; Afolabi, B.A.; Adewale, O.B.; Akintayo, C.O.; Ojo, O.A. Combined effect of metformin and gallic acid on inflammation, antioxidant status, endoplasmic reticulum (ER) stress and glucose metabolism in fructose-fed streptozotocin-induced diabetic rats. Toxicol. Rep. 2021, 8, 1419–1427. [Google Scholar] [CrossRef]
- Abdel-ghaffar, A.; Elhossary, G.G.; Mahmoud, A.M.; Elshazly, A.H.M.; Hassanin, O.A.; Saleh, A.; Mansour, S.M.; Metwally, F.G.; Hanafy, L.K.; Karam, S.H.; et al. Potential prophylactic effect of chemical chaperones for alleviation of endoplasmic reticulum stress in experimental diabetic cataract. Bull. Natl. Res. Cent. 2019, 43, 71. [Google Scholar] [CrossRef]
- Lu, X.; Yang, R.-R.; Hang, J.-L.; Wang, P.; Gong, Y.; Hu, W.; Wu, Y.; Gao, M.; Huang, C. Tauroursodeoxycholic acid produces antidepressant-like effects in a chronic unpredictable stress model of depression via attenuation of neuroinflammation, oxido-nitrosative stress and endoplasmic reticulum stress. Fundam. Clin. Pharmacol. 2018, 32, 363–377. [Google Scholar] [CrossRef]
- Mantopoulos, D.; Murakami, Y.; Comander, J.; Thanos, A.; Roh, M.; Miller, J.W.; Vavvas, D.G. Tauroursodeoxycholic Acid (TUDCA) Protects Photoreceptors from Cell Death after Experimental Retinal Detachment Dimosthenis. PLoS ONE 2011, 6, e24245. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Schwabe, R.F.; DeVries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6: Model of NF-κB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef]
- Samarghandian, S.; Azimi-nezhad, M.; Farkhondeh, T. Cytokine Crocin attenuate Tumor Necrosis Factor-alpha (TNF- a) and interleukin-6 (IL-6) in streptozotocin-induced diabetic rat aorta. Cytokine 2016, 88, 20–28. [Google Scholar] [CrossRef]
- Choy, K.W.; Zain, Z.; Murugan, D.D.; Giribabu, N. Effect of Hydrolyzed Bird’ s Nest on β -Cell Function and Insulin Signaling in Type 2 Diabetic Mice. Front. Pharmacol. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Lamireau, T.; Zoltowska, M.; Levy, E.; Yousef, I. Effects of bile acids on biliary epithelial cells: Proliferation, cytotoxicity, and cytokine secretion. Life Sci. 2003, 72, 1401–1411. [Google Scholar] [CrossRef]
- Ocana, G.J.; Perez, L.; Guindon, L.; Deffit, S.N.; Evans-Molina, C.; Thurmond, D.C.; Blum, J.S. Inflammatory stress of pancreatic beta cells drives release of extracellular heat shock protein 90α. J. Immunol. 2016, 38, 42–49. [Google Scholar] [CrossRef]
- Ishak, N.A.; Ismail, M.; Hamid, M.; Ahmad, Z.; Aisyah, S.; Ghafar, A. Antidiabetic and Hypolipidemic Activities of Curculigo latifolia Fruit: Root Extract in High Fat Fed Diet and Low Dose STZ Induced Diabetic Rats. Evid. Based Complement. Altern. Med. 2013, 2013, 601838. [Google Scholar] [CrossRef]
- Salmanoglu, D.S.; Gurpinar, T.; Vural, K.; Ekerbicer, N.; Dariverenli, E.; Var, A. Melatonin and L-carnitin improves endothelial disfunction and oxidative stress in Type 2 diabetic rats. Redox Biol. 2016, 8, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.R.; Shankar, R.R.; Fineberg, N.; McGill, J.; Baron, A.D. HbA1c measurement improves the detection of type 2 diabetes in high-risk individuals with nondiagnostic levels of fasting plasma glucose: The early diabetes intervention program (EDIP). Diabetes Care 2001, 24, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, K.; Suzuki, K.; Kizaki, T.; Hitomi, Y.; Ishida, H.; Katsuta, H.; Itoh, E.; Ookawara, T.; Suzuki, K.; Honke, K.; et al. Gliclazide protects pancreatic b -cells from damage by hydrogen peroxide. Biochem. Biophys. Res. Commun. 2003, 303, 112–119. [Google Scholar] [CrossRef]
- Stolk, J.; Hiltermann, T.J.N.; Dijkman, J.H.; Verhoeven, A.J. Characteristics of the Inhibition of NADPH Oxidase Activation in Neutrophils by Apocynin, a Methoxy-substituted Catechol. Am. J. Respir. Cell. Mol. Biol. 1994, 11, 95–102. [Google Scholar] [CrossRef]
- Wang, H.; Wang, A.X.; Liu, Z.; Chai, W.; Barrett, E.J. The Trafficking/Interaction of eNOS and Caveolin-1. Mol. Endocrinol 2009, 23, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, A.; Tripathi, Y.; Kumar, A. Change in Oxidative Stress of Normotensive Elderly Subjects Following Lifestyle Modifications. J. Clin. Diagn. Res. 2016, 10, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Lubkowska, A.; Chudecka, M. The Effects of Small-Volume Liposuction Surgery of Subcutaneous Adipose Tissue in the Gluteal-Femoral Region on Selected Biochemical Parameters. Int. J. Environ. Res. Public Health 2019, 16, 3298. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | CON | DM | DMT |
|---|---|---|---|
| Maximal relaxation, % | |||
| W/O inhibitor | 89.89 ± 1.63 a | 42.57 ± 1.68 | 84.91 ± 3.44 a |
| LNAME | 5.71 ± 1.91 b | 11.56 ± 2.34 b | 16.84 ± 2.17 b |
| Wortmannin | 14.76 ± 2.80 b | 20.13 ± 2.45 b | 19.13 ± 1.95 b |
| Apocynin | 13.71 ± 2.95 b | 13.24 ± 2.86 b | 20.23 ± 3.34 b |
| Gliclazide | 21.43 ± 4.86 b | 32.56 ± 4.30 | 25.71 ± 2.03 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustapha, S.; Azemi, A.K.; Wan Ahmad, W.A.N.; Rasool, A.H.G.; Mustafa, M.R.; Mokhtar, S.S. Inhibition of Endoplasmic Reticulum Stress Improves Acetylcholine-Mediated Relaxation in the Aorta of Type-2 Diabetic Rats. Molecules 2022, 27, 5107. https://doi.org/10.3390/molecules27165107
Mustapha S, Azemi AK, Wan Ahmad WAN, Rasool AHG, Mustafa MR, Mokhtar SS. Inhibition of Endoplasmic Reticulum Stress Improves Acetylcholine-Mediated Relaxation in the Aorta of Type-2 Diabetic Rats. Molecules. 2022; 27(16):5107. https://doi.org/10.3390/molecules27165107
Chicago/Turabian StyleMustapha, Sagir, Ahmad Khusairi Azemi, Wan Amir Nizam Wan Ahmad, Aida Hanum Ghulam Rasool, Mohd Rais Mustafa, and Siti Safiah Mokhtar. 2022. "Inhibition of Endoplasmic Reticulum Stress Improves Acetylcholine-Mediated Relaxation in the Aorta of Type-2 Diabetic Rats" Molecules 27, no. 16: 5107. https://doi.org/10.3390/molecules27165107
APA StyleMustapha, S., Azemi, A. K., Wan Ahmad, W. A. N., Rasool, A. H. G., Mustafa, M. R., & Mokhtar, S. S. (2022). Inhibition of Endoplasmic Reticulum Stress Improves Acetylcholine-Mediated Relaxation in the Aorta of Type-2 Diabetic Rats. Molecules, 27(16), 5107. https://doi.org/10.3390/molecules27165107