
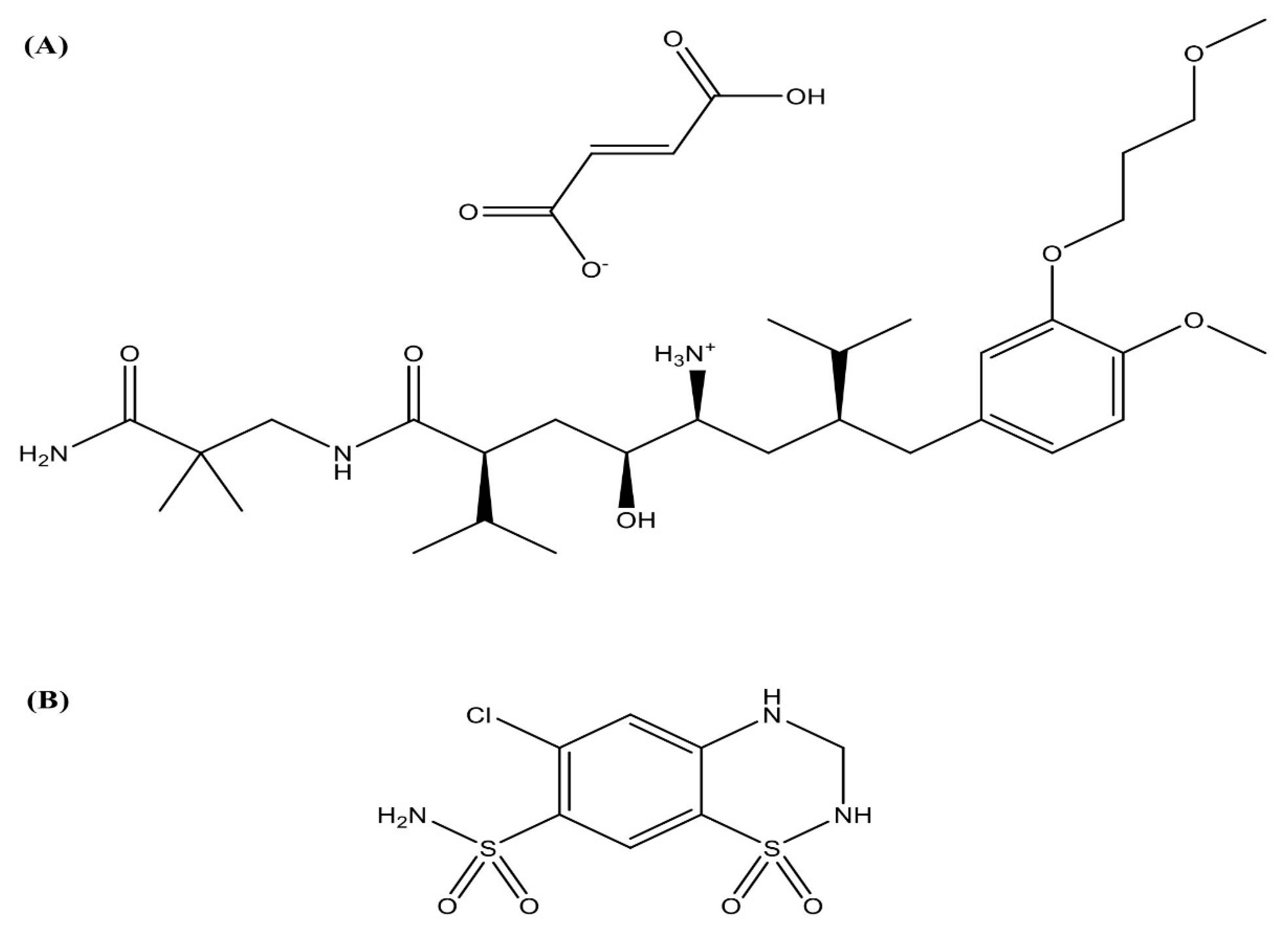
Aliskiren Hemifumarate Proliposomes for Improved Oral Drug Delivery: Formulation Development, In Vitro and In Vivo Permeability Testing

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Analytical Method Validation
2.1.1. Linearity and Range
2.1.2. Precision and Accuracy
2.1.3. Stability
2.1.4. Robustness
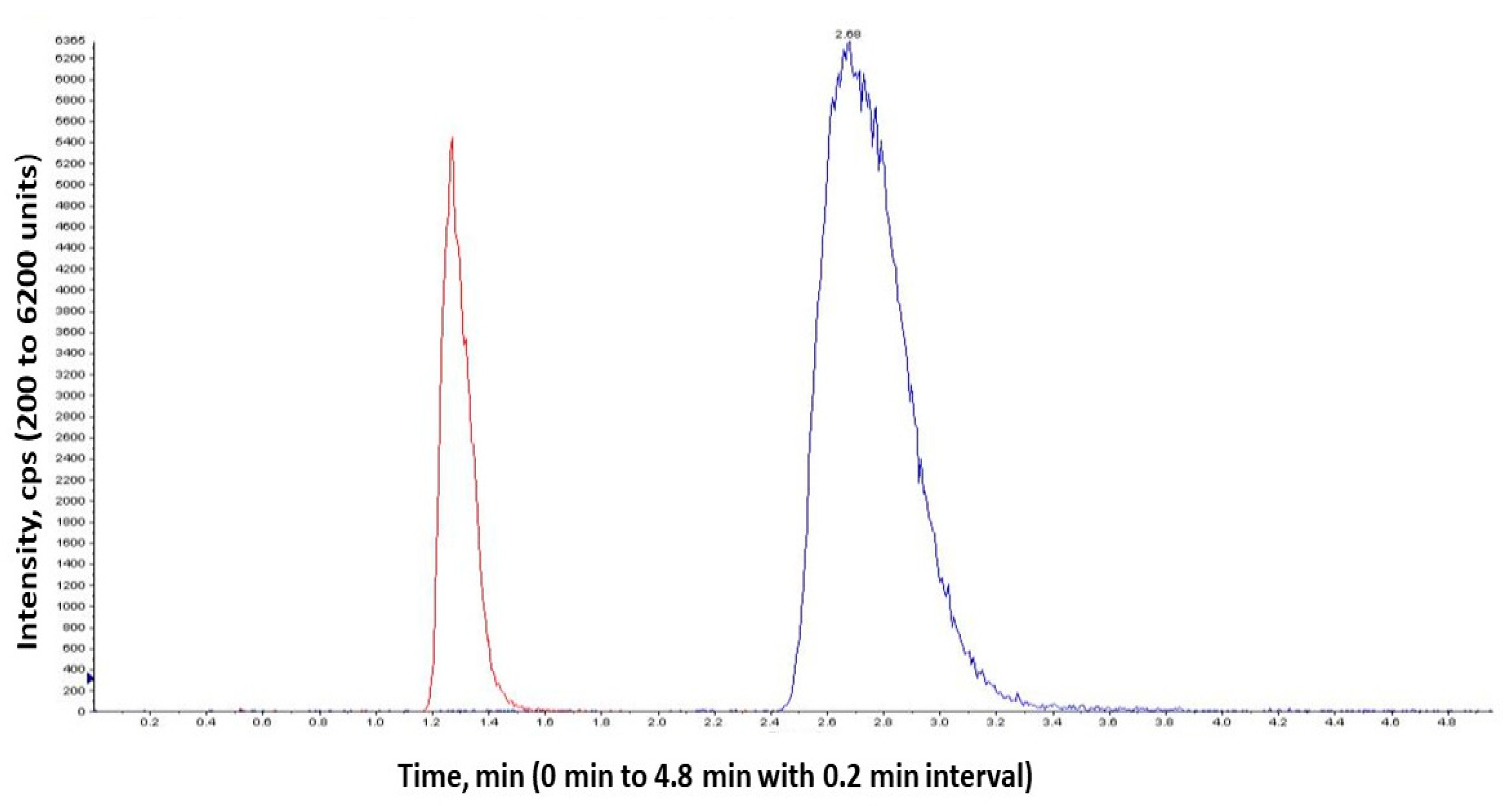
2.1.5. System Suitability
2.2. Formulation of Proliposomes
2.3. Physical Characterization of Proliposomes
2.4. Encapsulation Efficiency
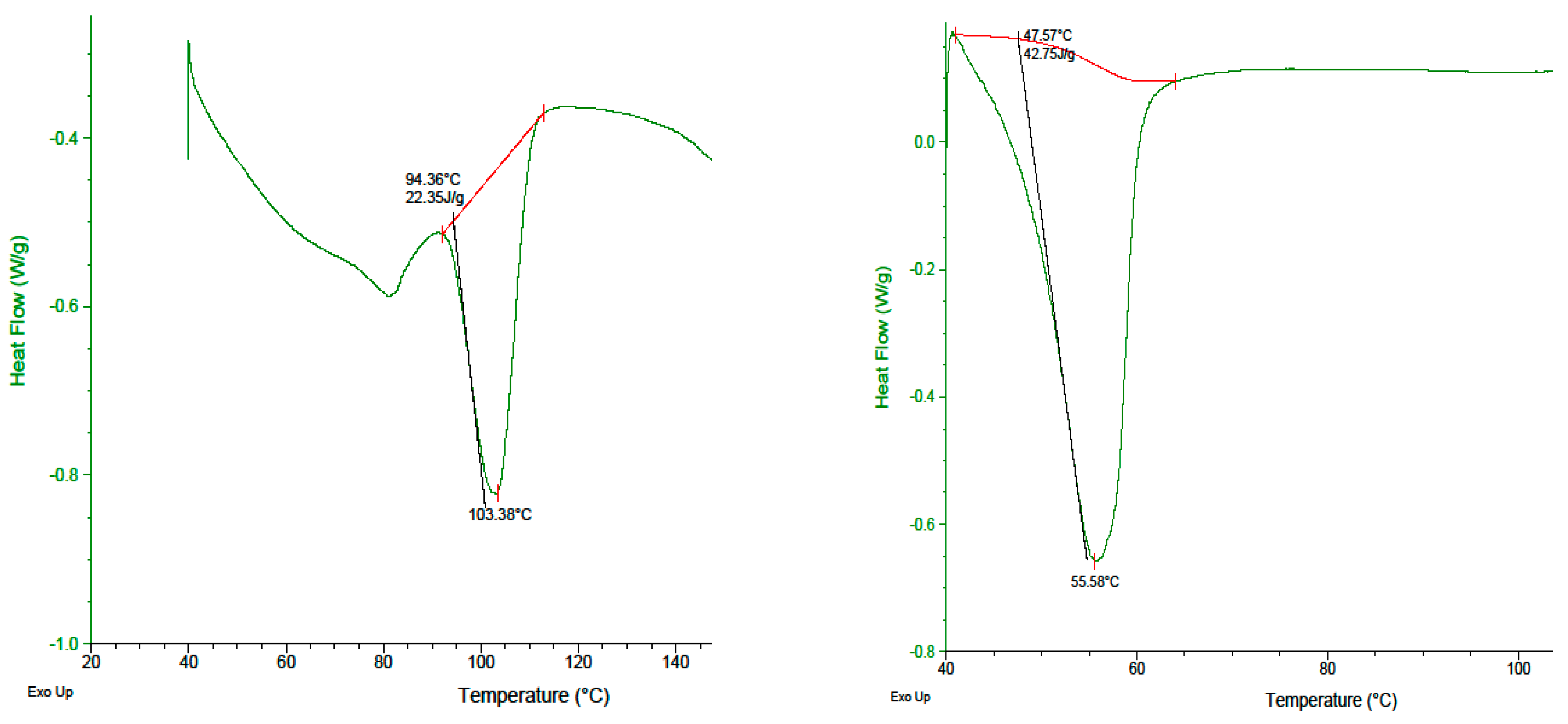
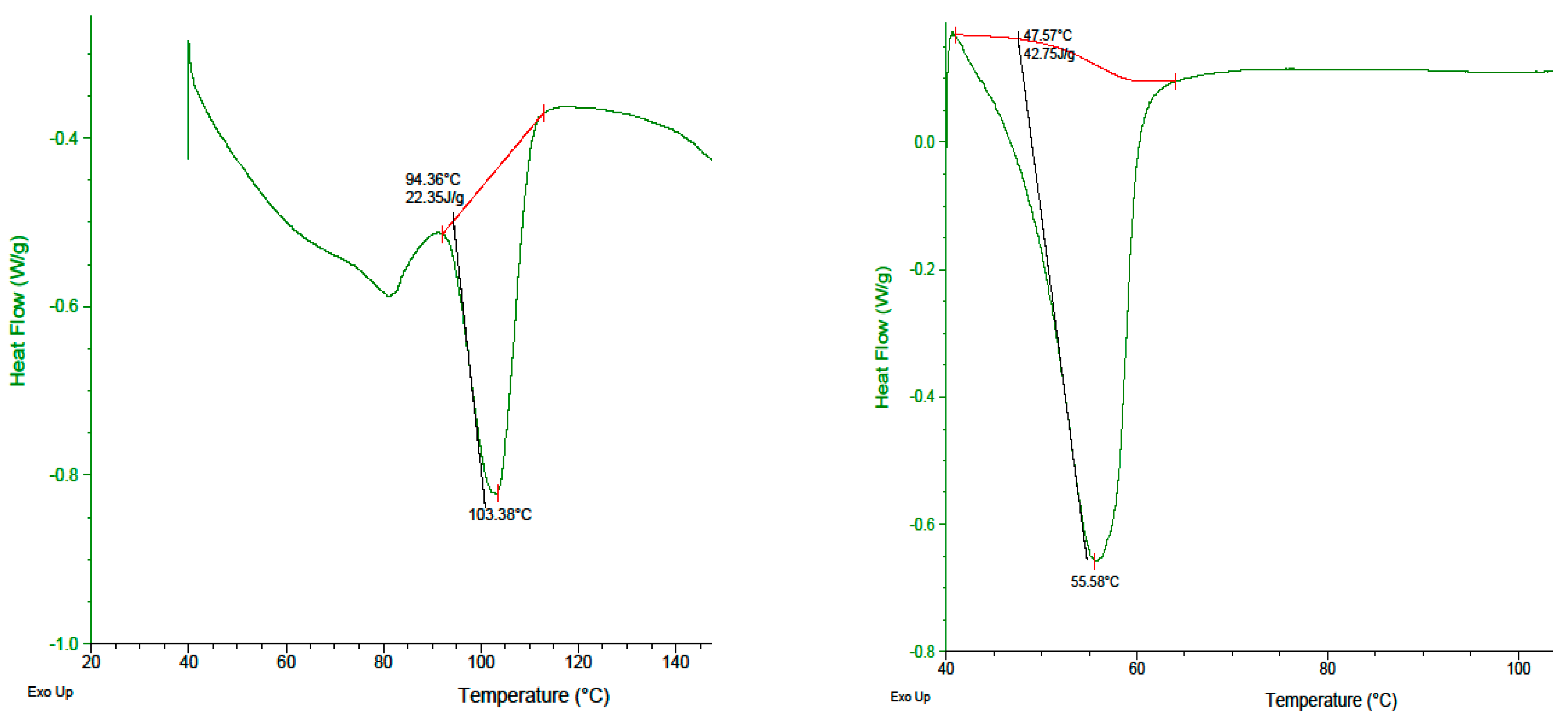
2.5. DSC Analysis
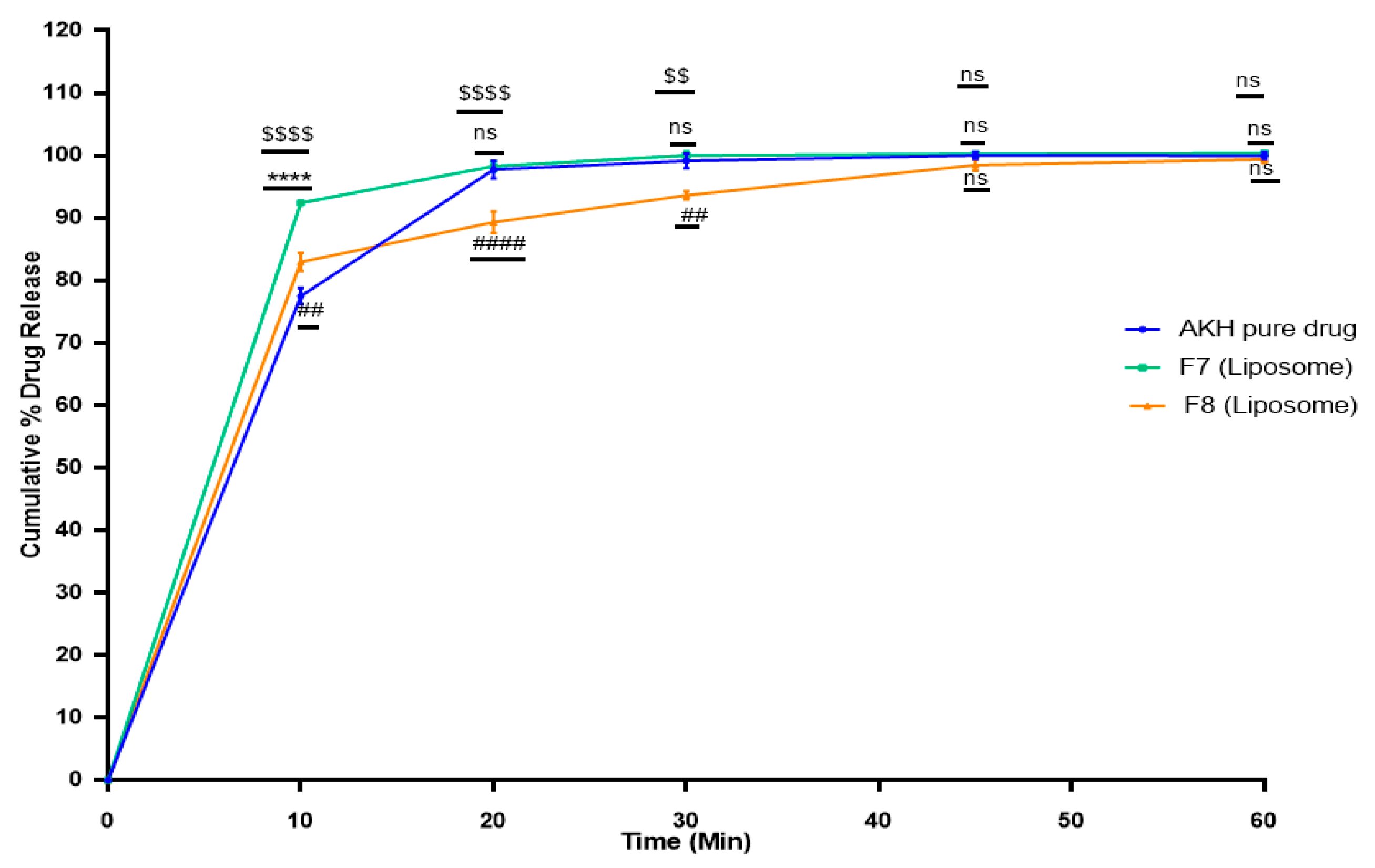
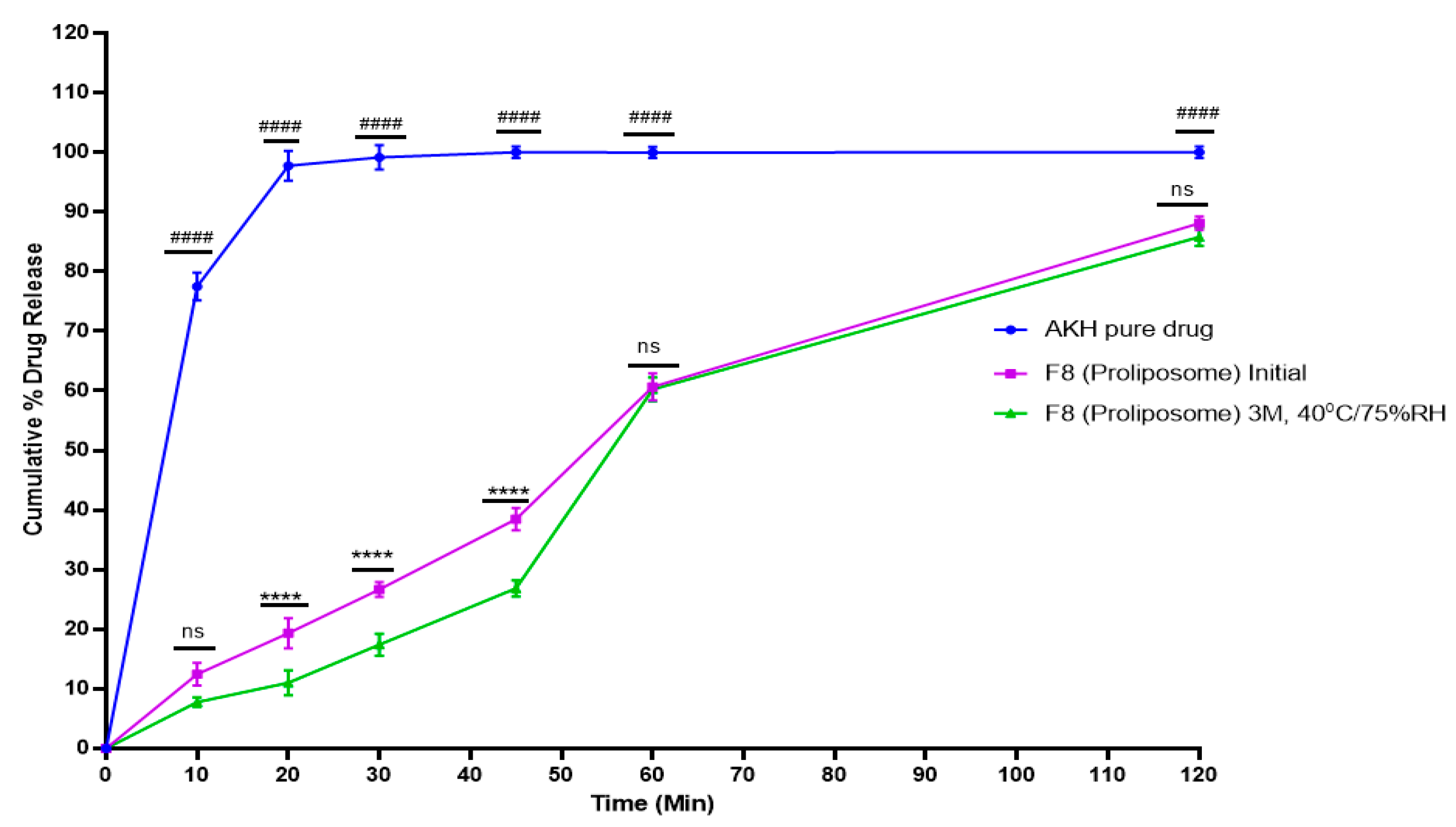
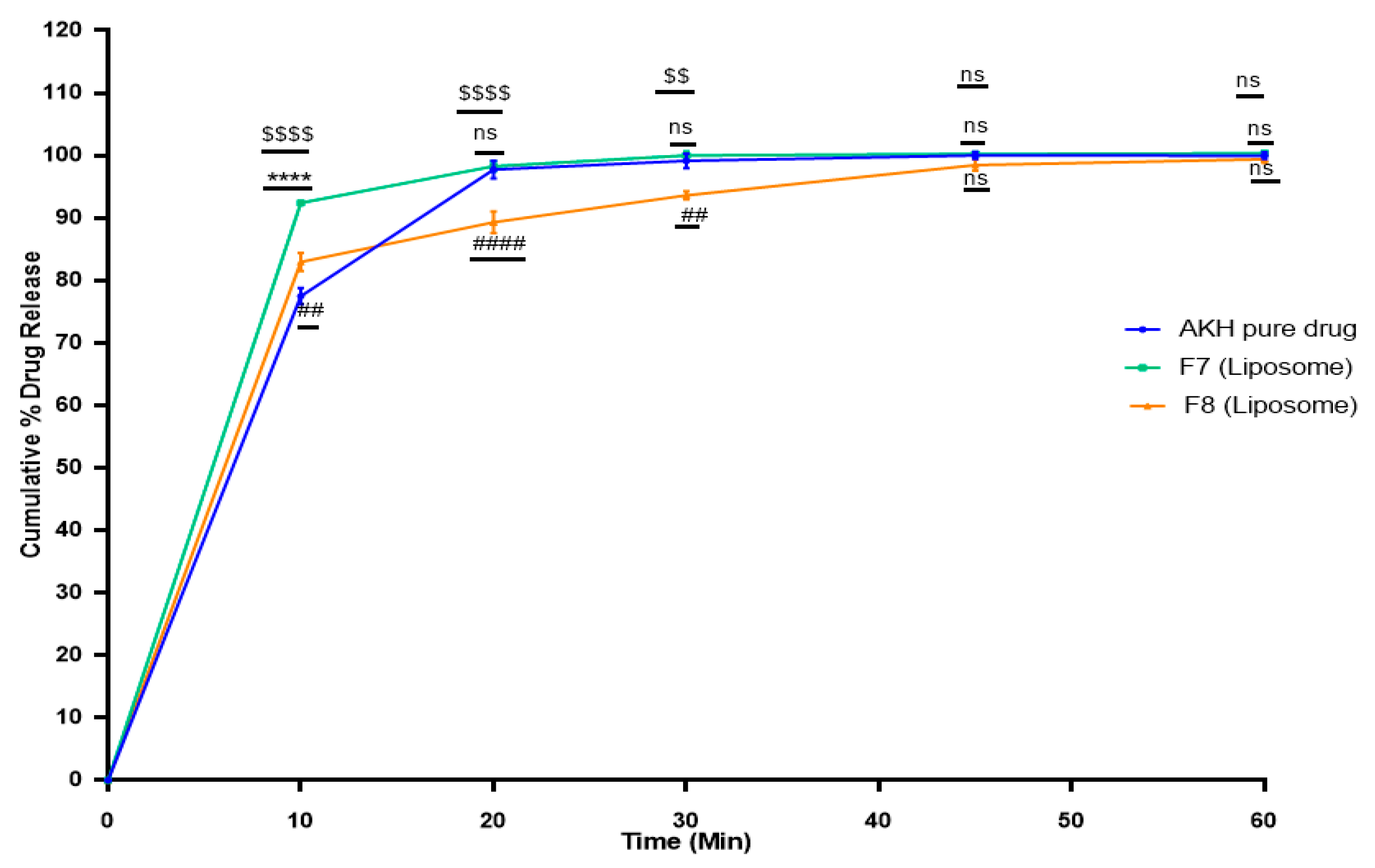
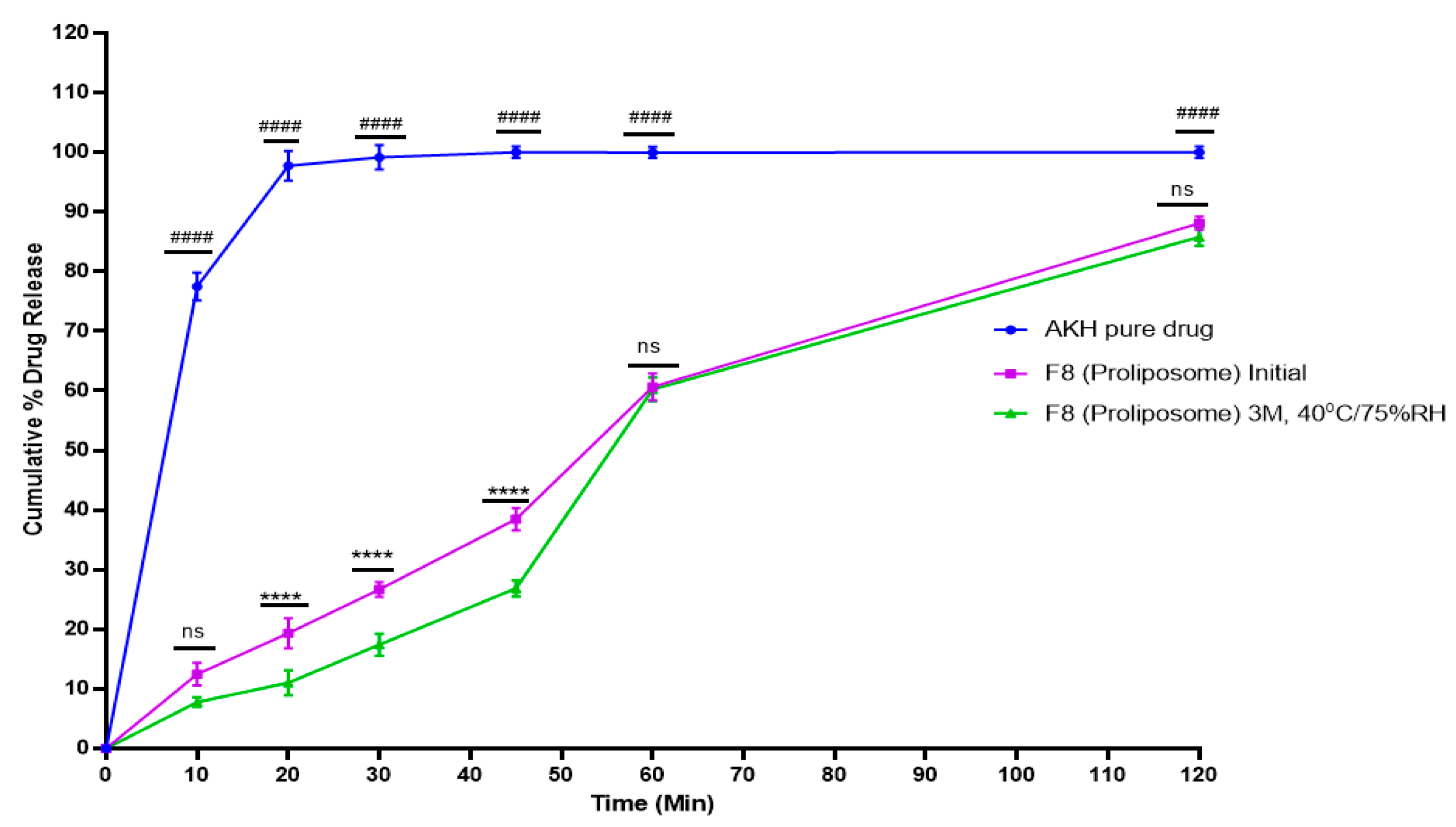
2.6. In Vitro Dissolution Studies
2.7. Parallel Artificial Membrane Permeability Assay
2.8. Caco-2 Studies
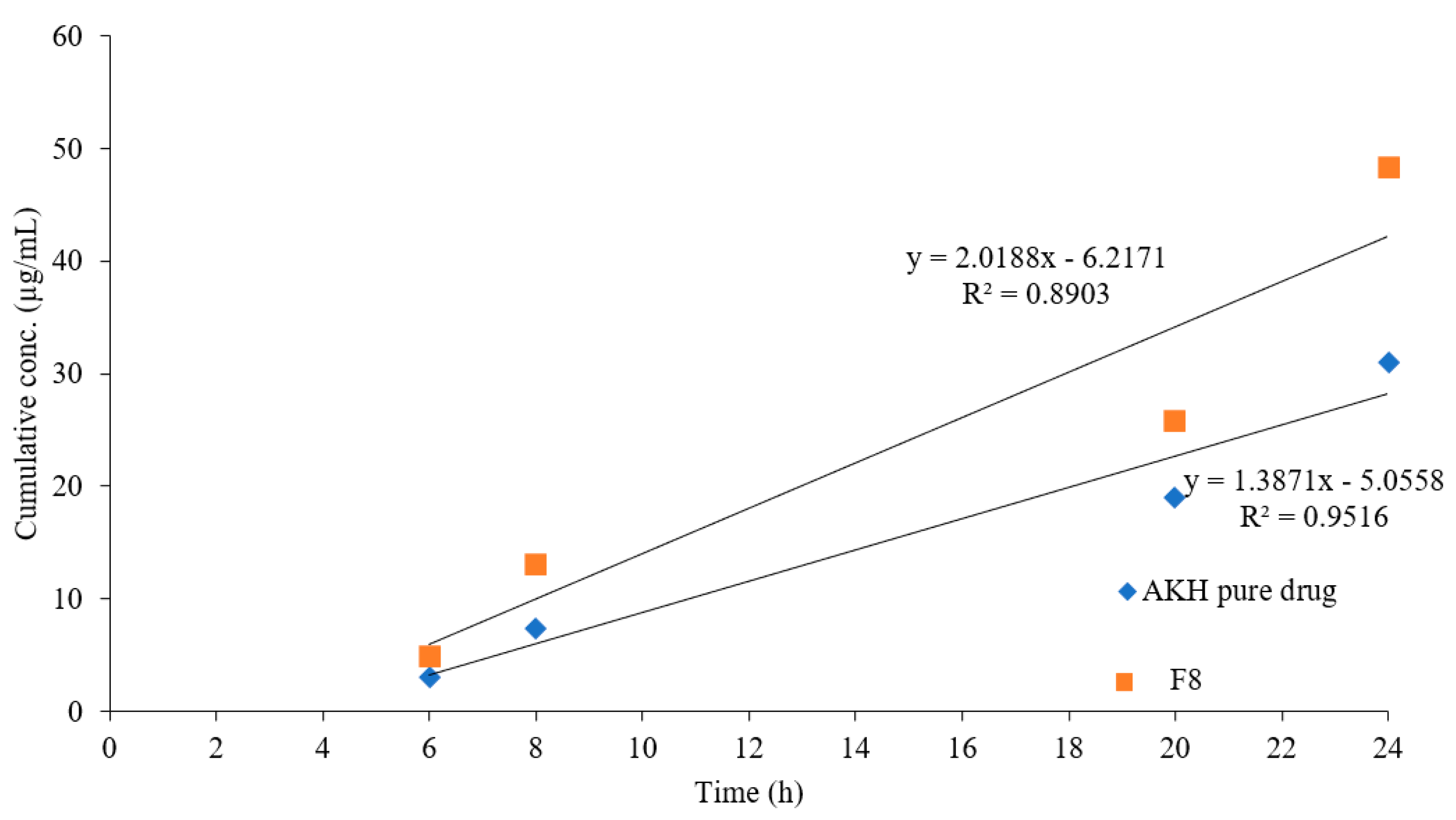
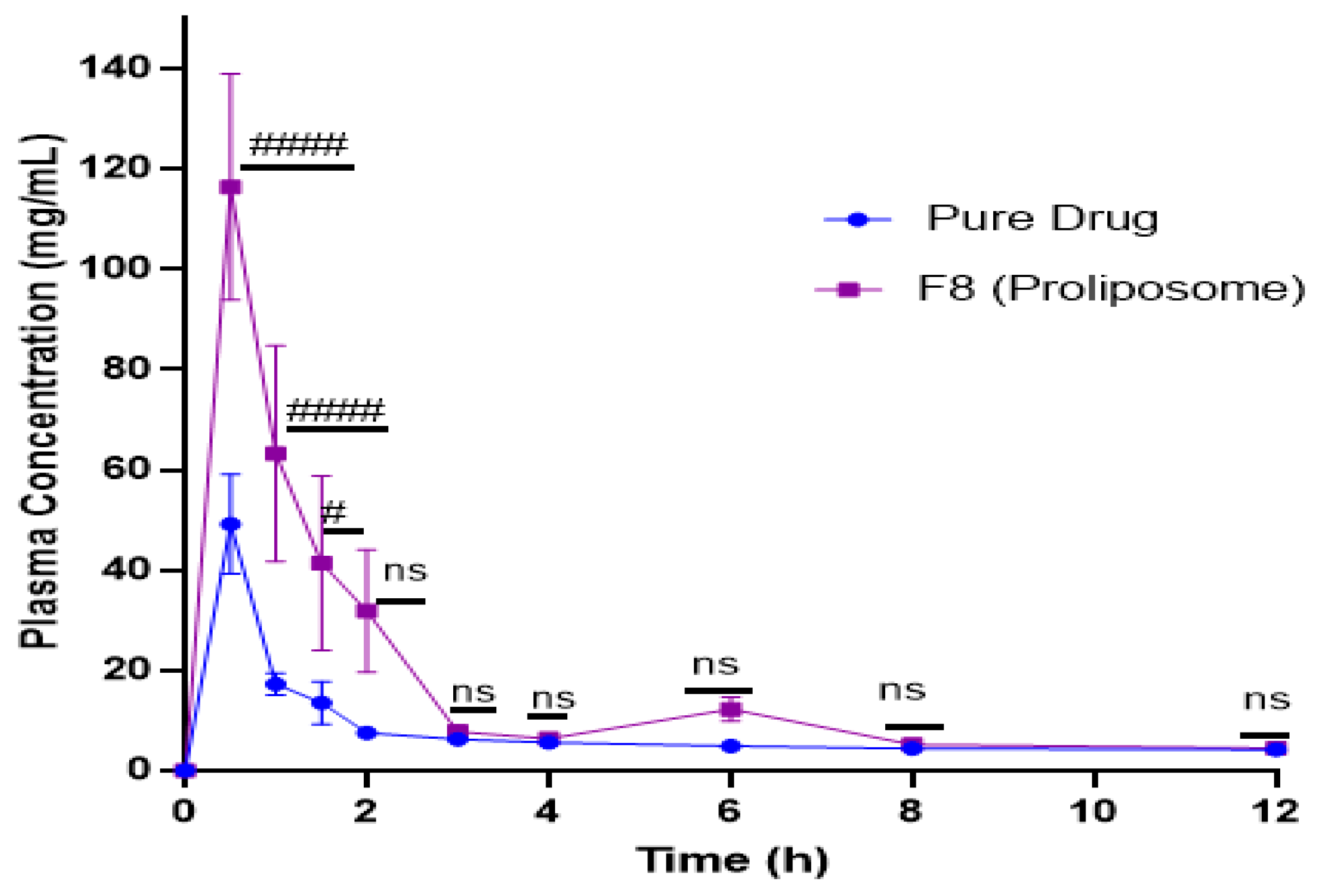
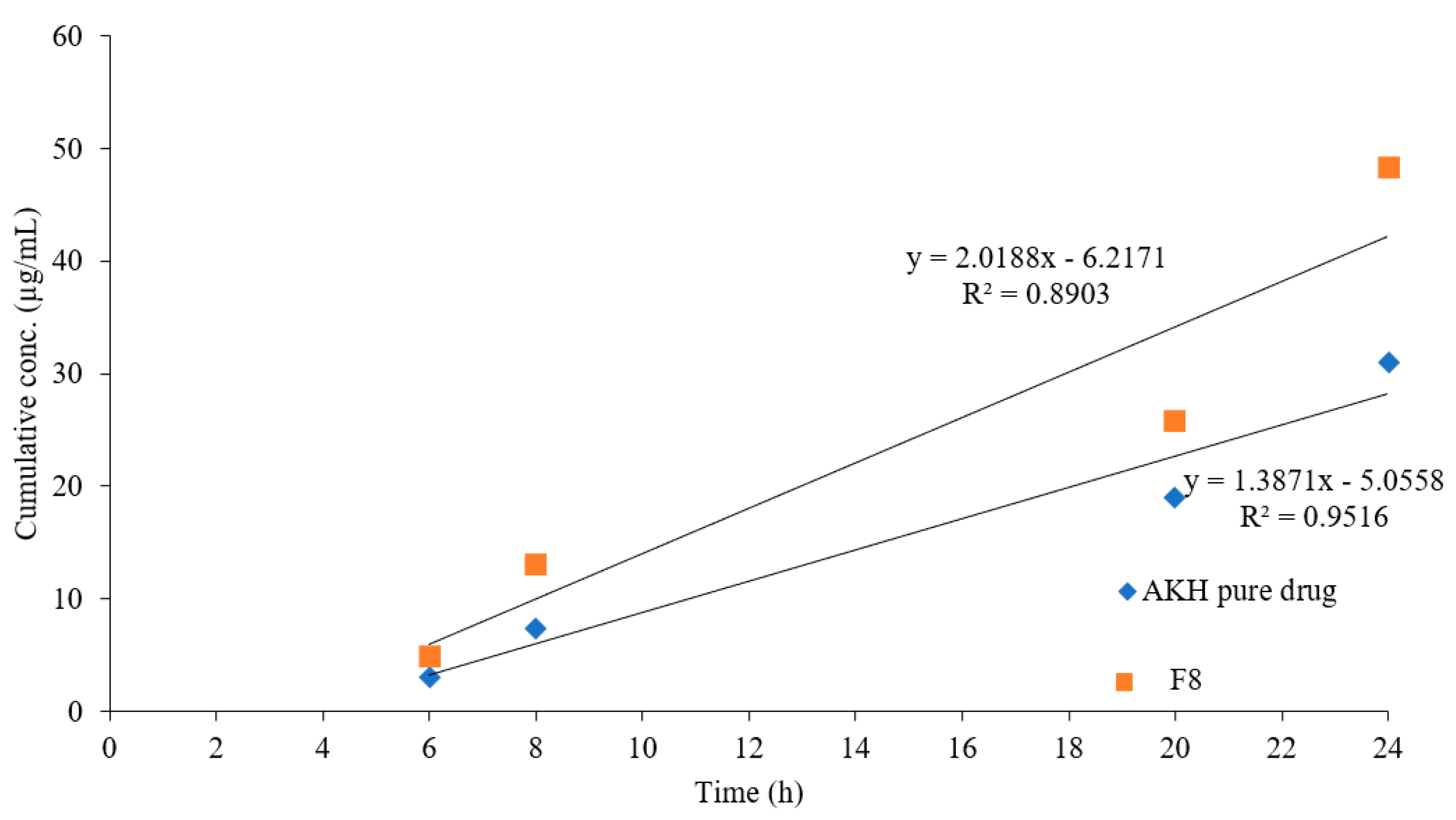
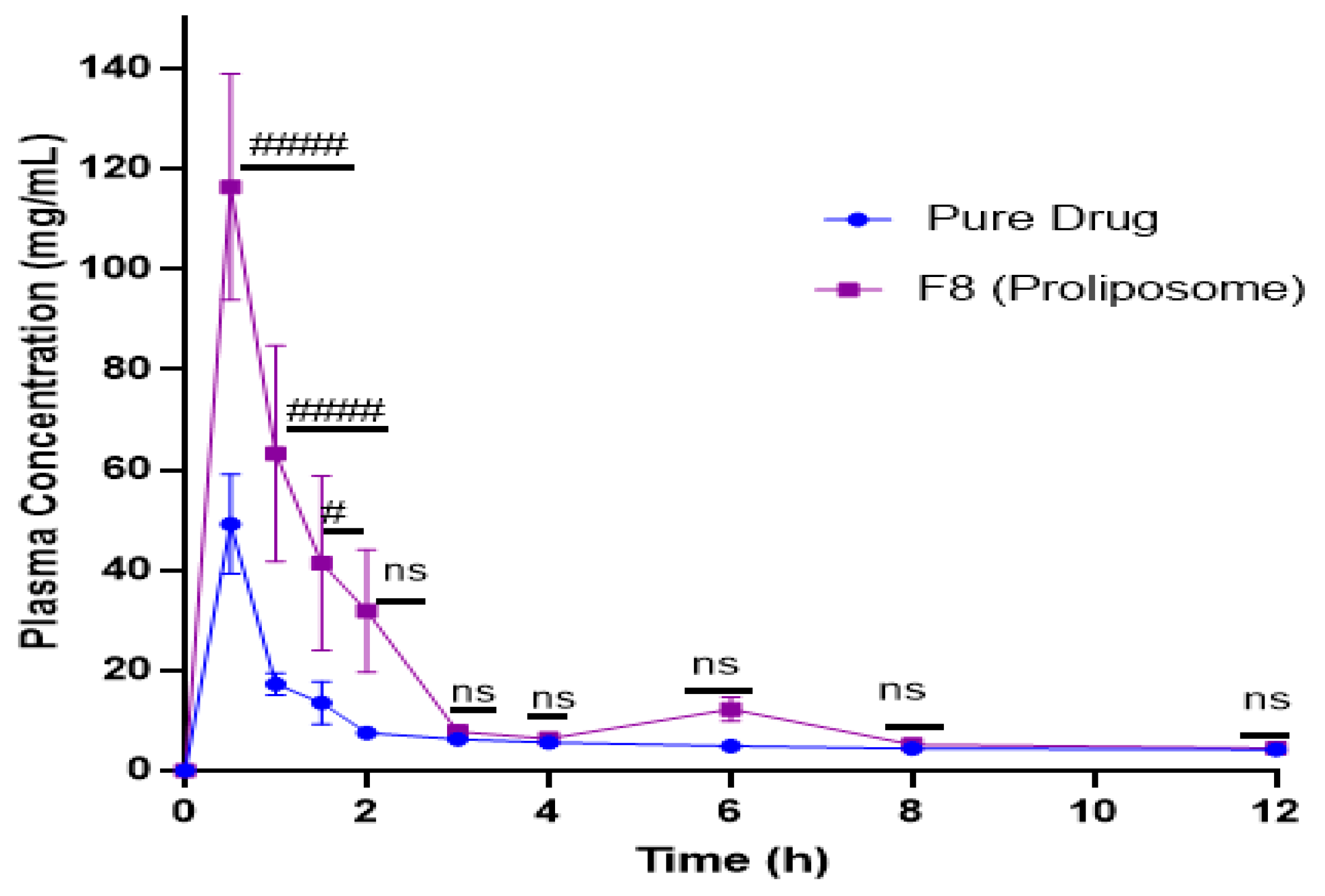
2.9. In Vivo Studies
2.10. Stability Studies
3. Discussion
4. Materials and Methods
4.1. Materials
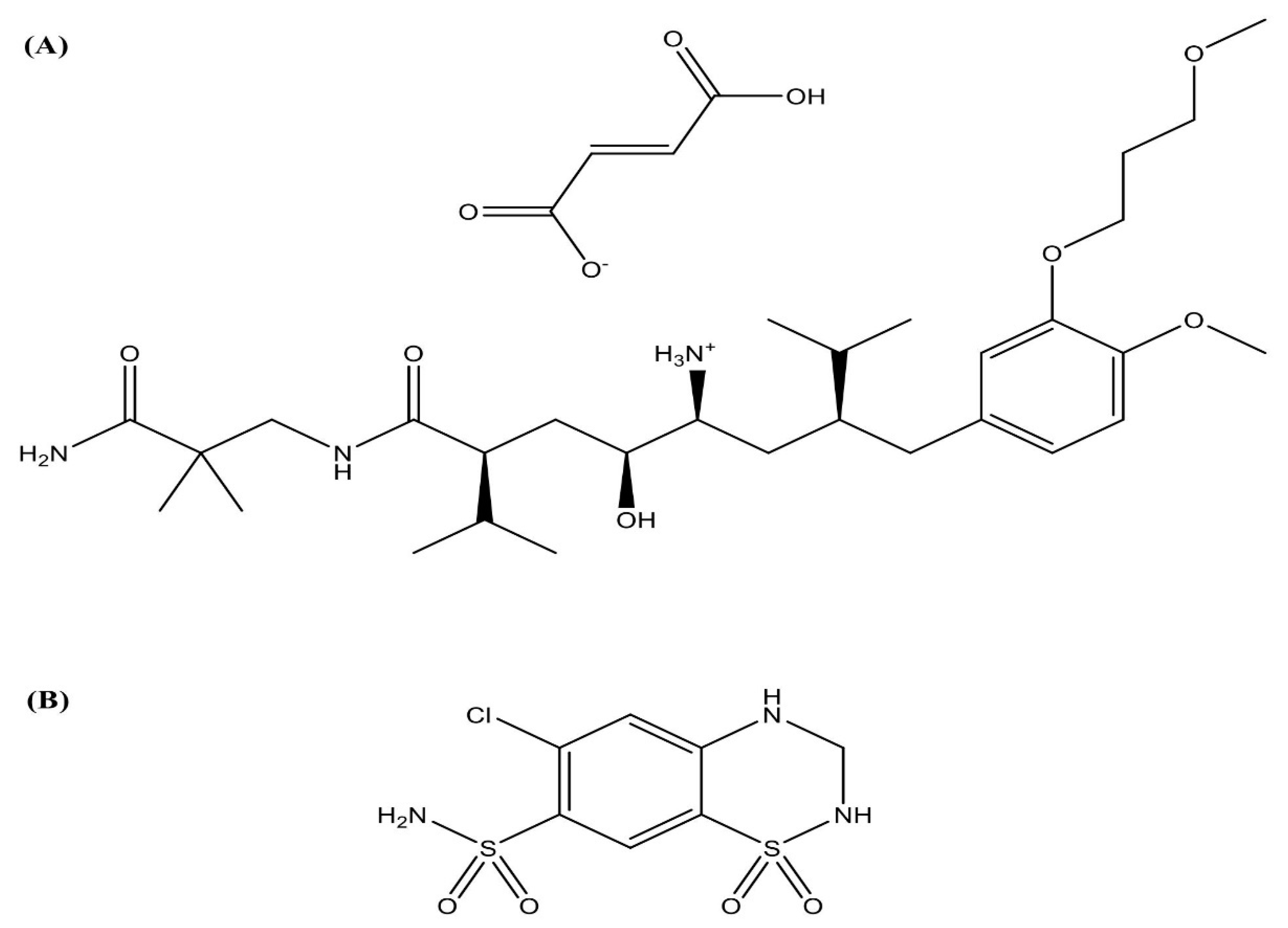
4.2. Analysis of Aliskiren Hemifumarate
Preparation of Calibration Standards and Quality Control Samples
4.3. Method Validation
4.3.1. Linearity and Range
4.3.2. Precision and Accuracy
4.3.3. Stability
4.3.4. Robustness
4.3.5. System Suitability
4.4. Preparation of Proliposomes
4.5. Characterization of Formulations
4.5.1. Particle Size
4.5.2. Morphological Evaluation
4.5.3. Encapsulation Efficiency
4.5.4. Differential Scanning Calorimetric (DSC) Analysis
4.6. Preparation of Capsule Dosage Form
4.7. In Vitro Dissolution Studies
4.8. Parallel Artificial Membrane Permeability Assay (PAMPA)
4.9. Caco-2 Permeation Studies
4.10. Stability Studies
4.11. Pharmacokinetic Study
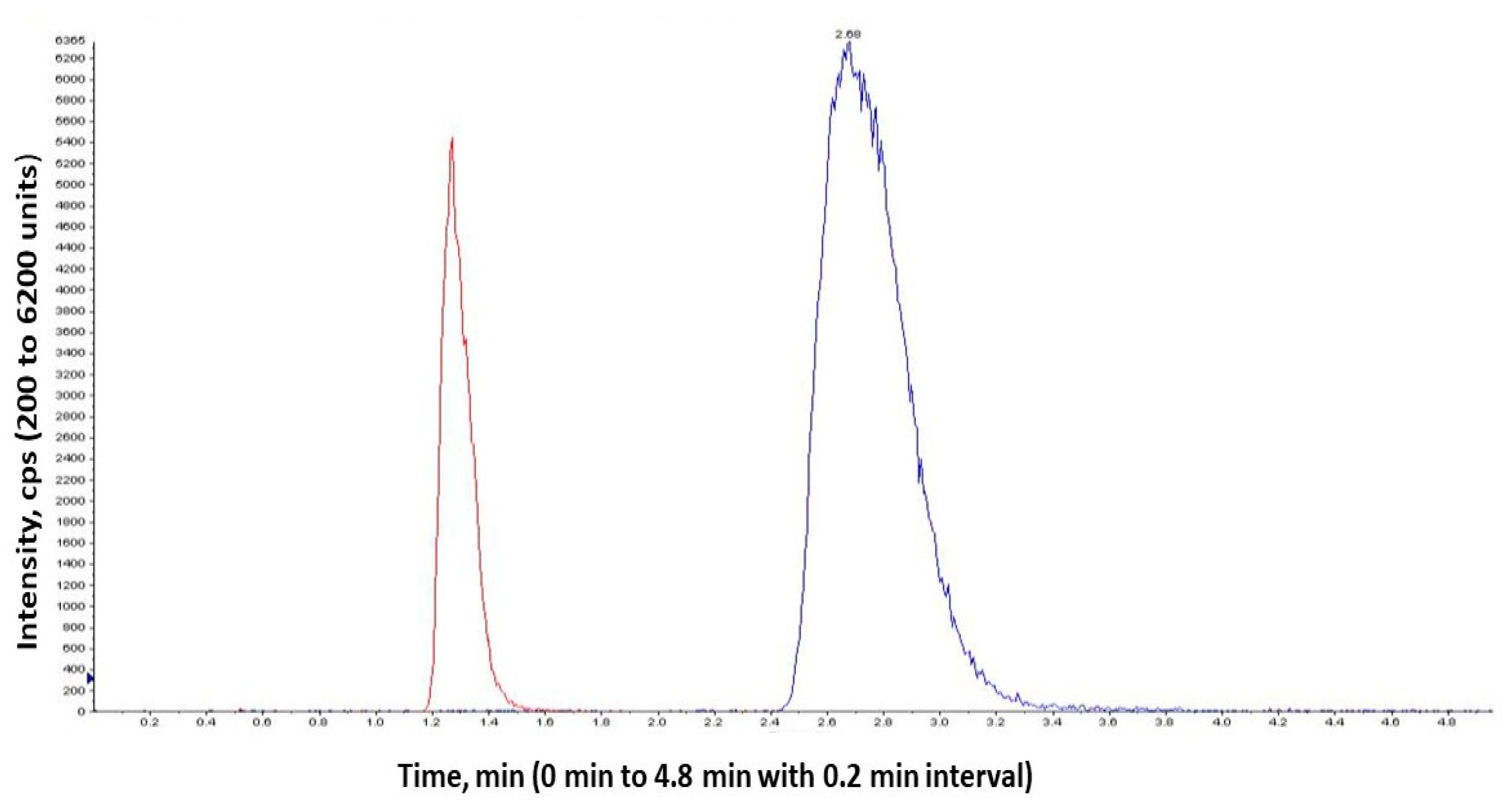
4.12. Quantification of Aliskiren in Plasma Using LC/MS/MS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| AKH | aliskiren hemifumarate |
| SPC | soy phosphatidylcholine |
| DMPC | dimyristoylphosphatidylcholine |
| DMPG Na | dimyristoylphosphatidylglycerol sodium |
| PAMPA | parallel artificial membrane permeation assay |
| RSD | relative standard deviation |
| mV | milli volts |
| DSC | differential scanning calorimetry |
| MCC | microcrystalline cellulose |
| DMSO | dimethyl sulfoxide |
| TEER | transepithelial electrical resistance |
| LCMS/MS | liquid chromatography/mass spectrometry |
| Tmax | time to reach maximum concentration |
| Cmax | maximum drug concentration |
| AUC | area under curve |
| HDPE | high density polyethylene |
| mM | milli molar |
| rpm | rotations per minute |
| FDA | Food and Drug Administration |
| PVDF | polyvinylidene difluoride |
| PTFE | polytetrafluoroethylene |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | fetal bovine serum |
| EDTA | ethylenediaminetetraacetic acid |
| HBSS | Hank’s balanced salt solution |
| IACUC | Institutional Animal Care and Use Committee |
| BCS | biopharmaceutical classification system |
References
- Understanding a Heart Attack—What Just Happened. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 11 June 2021).
- Heart Attack Related Stories—Your Road to Recovery. Available online: https://www.heart.org/en/health-topics/high-blood-pressure (accessed on 12 April 2022).
- Nguyen, Q.; Dominguez, J.; Nguyen, L.; Gullapalli, N. Hypertension management: An update. Am. Health Drug Benefits. 2010, 3, 47–56. [Google Scholar] [PubMed]
- Mende, C.W. Application of direct renin inhibition to chronic kidney disease. Cardiovasc. Drugs Ther. 2010, 24, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Aliskiren is a Direct Renin Inhibitor Used to Manage Hypertension. Available online: https://www.drugbank.ca/drugs/DB09026 (accessed on 10 May 2020).
- Aliskiren 300 mg Tablet Angiotensin Inhibiting Agents. Available online: https://www.webmd.com/drugs/2/drug-147671/aliskiren-oral/details (accessed on 10 May 2020).
- Buczko, W.; Hermanowicz, J.M. Pharmacokinetics and pharmacodynamics of aliskiren, an oral direct renin inhibitor. Pharmacol. Rep. 2008, 60, 623–631. [Google Scholar]
- Waldmeier, F.; Glaenzel, U.; Wirz, B.; Oberer, L.; Schmid, D.; Seiberling, M.; Valencia, J.; Riviere, G.J.; End, P.; Vaidyanathan, S. Absorption, distribution, metabolism, and elimination of the direct renin inhibitor aliskiren in healthy volunteers. Drug Metab. Dispos. 2007, 35, 1418–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.M. Formulation and Optimization of Aliskiren loaded Poly(Lactide-Co-Glycolide) Nanoparticles, East Tennessee State University. 2015, Undergraduate Honors Theses. Paper 275. Available online: https://dc.etsu.edu/honors/275 (accessed on 10 May 2015).
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharmaceu. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Rao, M.R.P.; Babrekar, L.S. Liposomal drug delivery for solubility and bioavailability enhancement of efavirenz. Indian J. Pharm. Sci. 2018, 80, 1115–1124. [Google Scholar]
- Bobbala, S.K.R.; Veerareddy, P.R. Formulation, evaluation, and pharmacokinetics of isradipine proliposomes for oral delivery. J. Liposome Res. 2012, 22, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Tong, S.S.; Xu, Y.; Wang, L.; Fu, M.; Ge, Y.R.; Yu, J.N.; Xu, X.M. Proliposomes for oral delivery of dehydrosilymarin: Preparation and evaluation in vitro and in vivo. Acta Pharmacol. Sin. 2011, 32, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Betageri, G.V.; Thirucote, R.; Gowda, V. Proliposomal Testosterone Formulations. International Publication Number. WO2013170012A2, 14 November 2013. [Google Scholar]
- Eloy, J.O.; De Souza, M.C.; Petrilli, R.; Barcellos, J.P.A.; Lee, R.J.; Marchetti, J.M. Liposomes as carriers of hydrophilic small molecule drugs: Strategies to enhance encapsulation and delivery. Colloids Surf. B 2014, 123, 345–363. [Google Scholar] [CrossRef]
- Hiremath, P.S.; Soppimath, K.S.; Betageri, G.V. Proliposomes of exemestane for improved oral delivery: Formulation and in vitro evaluation using PAMPA, Caco-2 and rat intestine. Int. J. Pharm. 2009, 380, 96–104. [Google Scholar] [CrossRef]
- Spinks, C.B.; Zidan, A.S.; Khan, M.A.; Habib, M.J.; Faustino, P.J. Pharmaceutical characterization of novel tenofovir liposomal formulations for enhanced oral drug delivery: In vitro pharmaceutics and Caco-2 permeability investigations. Clin. Pharmacol. Adv. Appl. 2017, 9, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Muneer, S.; Masood, Z.; Butt, S.; Anjum, S.; Zainab, H.; Anwar, N.; Ahmad, N. Proliposomes as pharmaceutical drug delivery system: A brief review. J. Nanomed. Nanotechnol. 2017, 8, 448. [Google Scholar]
- Gregoriadis, G. Engineering liposomes for drug delivery: Progress and problems. Trends Biotechnol. 1995, 13, 527–537. [Google Scholar] [CrossRef]
- Shaji, J.; Bhatia, V. Proliposomes: A brief overview of novel delivery system. Int. J. Pharma Biosci. 2013, 4, 150–160. [Google Scholar]
- Manojlovic, V.; Winkler, K.; Bunjes, V.; Neub, A.; Schubert, R.; Bugarski, B.; Leneweit, G. Membrane interactions of ternary phospholipid/cholesterol bilayers and encapsulation efficiencies of a rip ii protein. Colloids Surf B Biointerfaces 2008, 64, 284–296. [Google Scholar] [CrossRef]
- Grohmann, F.; Csempesz, F.; Szogyi, M. The influence of polymers on the physical stability and the thermal properties of dimyristoyl-phosphatidylcholine liposomes. Pharmazie 1999, 54, 52–55. [Google Scholar]
- Akbarzadeh, A.R.; Rezaei, S.S.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Jukanti, R.; Mateti, A.; Bandari, S.; Veerareddy, P.R. Transdermal delivery of acyclovir sodium via carbopol gels: Role of chemical permeation enhancers. Lett. Drug Des. Discov. 2011, 8, 381–389. [Google Scholar] [CrossRef]
- Nekkanti, V.; Kalepu, S. Recent advances in liposomal drug delivery: A review. Pharm Nanotechnol. 2015, 3, 35–55. [Google Scholar] [CrossRef]
- Janga, K.Y.; Jukanti, R.; Velpula, A.; Sunkavalli, S.; Bandari, S.; Kandadi, P.; Veerareddy, P.R. Bioavailability Enhancement of Zaleplon Via Proliposomes: Role of Surface Charge. Eur. J. Pharm. Biopharm. 2012, 80, 347–357. [Google Scholar] [CrossRef]
- Velpula, A.; Jukanti, R.; Janga, K.Y.; Sunkavalli, S.; Bandari, S.; Kandadi, P.; Veerareddy, P.R. Proliposome Powders for Enhanced Intestinal Absorption and Bioavailability of Raloxifene Hydrochloride: Effect of Surface Charge. Drug Dev. Ind. Pharm. 2013, 39, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Arregui, J.R.; Kovvasu, S.P.; Betageri, G.V. Daptomycin proliposomes for oral delivery: Formulation, characterization, and in vivo pharmacokinetics. AAPS PharmSciTech. 2018, 19, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, D.D.; Ravis, W.R.; Betageri, G.V. Improved delivery of cromolyn from oral proliposomal beads. Int. J. Pharm. 2008, 358, 128–136. [Google Scholar] [CrossRef]
- Mozafari, M.R. Liposomes: An overview of manufacturing techniques. Cell Mol. Biol. Lett. 2005, 10, 711. [Google Scholar]
- Colletier, J.P.; Chaize, B.; Winterhalter, M.; Fournier, D. Protein encapsulation in liposomes: Efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotech. 2002, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.; Sachse, A.; Bender, J.; Schubert, R.; Brandl, M. Filter extrusion of liposomes using different devices: Comparison of liposome size, encapsulation efficiency, and process characteristics. Int. J. Pharm. 2001, 223, 55–68. [Google Scholar] [CrossRef]
- Yanamandra, S.; Venkatesan, N.; Kadajji, V.G.; Wang, Z.; Issar, M.; Betageri, G.V. Proliposomes as a drug delivery system to decrease the hepatic first-pass metabolism: Case study using a model drug. Eur. J. Pharm. Sci. 2014, 64, 26–36. [Google Scholar] [CrossRef]
- Kanzer, J.; Tho, I.; Flaten, G.E.; Mägerlein, M.; Hölig, P.; Fricker, G.; Brandl, M.G.E. In-vitro permeability screening of melt extrudate formulations containing poorly water-soluble drug compounds using the phospholipid vesicle-based barrier. J. Pharm. Pharmacol. 2010, 62, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.J.; Rosenblatt, K.M.; Westedt, U.; Hölig, P.; Rosenberg, J.; Mägerlein, M.; Fricker, G.; Brandl, M. Amorphous solid dispersion enhances permeation of poorly soluble ABT-102: True supersaturation vs. apparent solubility enhancement. Int. J. Pharm. 2012, 437, 288–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubatsch, I.; Ragnarsson, E.G.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Conc. (µg/mL) | Intra-Day Precision (n = 3) (%RSD) | Inter-Day Precision (n = 3) (%RSD) | % Recovery (n = 3) |
|---|---|---|---|
| 5 | 0.38 | 0.36 | 98.84 |
| 20 | 1.65 | 1.15 | 100.39 |
| 50 | 0.32 | 0.95 | 98.88 |
| Drug Conc. (µg/mL) | % Recovery Samples Kept at RT | % Recovery Samples Kept at 2–8 °C | ||||
|---|---|---|---|---|---|---|
| (Day 1) | (Day 2) | (Day 3) | (Day 1) | (Day 2) | (Day 3) | |
| 5 | 98.08 | 98.97 | 99.24 | 99.17 | 97.56 | 100.59 |
| 20 | 100.06 | 99.95 | 99.43 | 100.41 | 101.05 | 101.74 |
| 50 | 101.35 | 99.49 | 99.33 | 99.72 | 100.61 | 98.67 |
| Flow Rate (mL/min) | Retention Time (RT) | Area | %RSD |
|---|---|---|---|
| 0.9 | 3.96 | 1256.46 | 0.25 |
| 1.0 | 4.2 | 1452.40 | 0.28 |
| 1.1 | 4.7 | 1536.53 | 0.28 |
| Mobile Phase (v/v/v) Retention Time (RT) | Area | %RSD | |
|---|---|---|---|
| 50:40:10 | 4.2 | 1452.4 | 0.28 |
| 60:30:10 | 3.9 | 1493.6 | 0.24 |
| 50:30:20 | 4.6 | 1596.2 | 0.30 |
| S.No. | Concentration of Std Taken | Area |
|---|---|---|
| 1 | 1378.4 | |
| 2 | 1384.0 | |
| 3 | 100 µg/mL | 1387.1 |
| 4 | 1379.6 | |
| 5 | 1372.9 | |
| 6 | 1375.7 | |
| Mean | 1379.61 | |
| %RSD | 0.37 | |
| Theoretical plate | 20152 | |
| Retention time | 4.2 |
| Formulations | Lipid | Drug (in Parts) | Lipid (in Parts) | Cholesterol (in Parts) | Stearylamine (in Parts) |
|---|---|---|---|---|---|
| F1 | SPC | 1 | 2 | 0.010 | - |
| F2 | SPC | 1 | 5 | 0.025 | - |
| F3 | DMPC | 1 | 2 | 0.010 | - |
| F4 | DMPC | 1 | 5 | 0.025 | - |
| F5 | DMPG Na | 1 | 2 | 0.010 | - |
| F6 | DMPG Na | 1 | 5 | 0.025 | - |
| F7 | DMPC | 1 | 5 | 0.025 | 0.025 |
| F8 | DMPC | 1 | 5 | 0.050 | 0.050 |
| Formulations | D:L:C (in Parts) | Stearylamine (in Parts) | Surface Charge (mV) | Entrapment Efficiency (%) | Particle Size (Mean ± SD, nm) |
|---|---|---|---|---|---|
| F1 | 1:2:0.010 | - | 0.034 | 17.5 | 1989 ± 87 |
| F2 | 1:5:0.025 | - | 0.236 | 23.4 | 1674 ± 58 |
| F3 | 1:2:0.010 | - | 0.019 | 22.0 | 309 ± 34 |
| F4 | 1:5:0.025 | - | 0.543 | 37.0 | 450 ± 22 |
| F5 | 1:2:0.010 | - | 0.923 | 12.1 | 647 ± 26 |
| F6 | 1:5:0.025 | - | 0.236 | 14.1 | 596 ± 120 |
| F7 | 1:5:0.025 | 0.025 | +21.3 | 40.3 | 427 ± 35 |
| F8 | 1:5:0.025 | 0.050 | +39.7 | 47.2 | 302 ± 24 |
| Formulation | Flux (J) (±SD) | No of Folds |
|---|---|---|
| Increase in Flux | ||
| Pure AKH | 0.554 (±0.003) | - |
| Proliposome (F8) | 0.829 (±0.083) | 1.497 |
| Formulation | Media | Papp (cm s−1) (±SD) |
|---|---|---|
| Pure AKH | HBSS (Normal) | 1.778 × 10−6 (±0.23) |
| Proliposome (F8) | HBSS (Normal) | 1.433 × 10−5(±0.14) |
| PK Parameters | Pure Drug | Proliposomal Formulation (F8) |
|---|---|---|
| Tmax (h) | 0.5 | 0.5 |
| Cmax (ng/mL) | 49.1 | 116.2 |
| AUC0-t (ng/mL/h) | 166.8 | 382.7 |
| Relative bioavailability (%) | - | 230 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunamaneni, P.; Kovvasu, S.; Yeung, S.; Wang, J.; Shah, S.; Betageri, G. Aliskiren Hemifumarate Proliposomes for Improved Oral Drug Delivery: Formulation Development, In Vitro and In Vivo Permeability Testing. Molecules 2022, 27, 4828. https://doi.org/10.3390/molecules27154828
Kunamaneni P, Kovvasu S, Yeung S, Wang J, Shah S, Betageri G. Aliskiren Hemifumarate Proliposomes for Improved Oral Drug Delivery: Formulation Development, In Vitro and In Vivo Permeability Testing. Molecules. 2022; 27(15):4828. https://doi.org/10.3390/molecules27154828
Chicago/Turabian StyleKunamaneni, Priyanka, Surya Kovvasu, Steven Yeung, Jeffrey Wang, Salim Shah, and Guru Betageri. 2022. "Aliskiren Hemifumarate Proliposomes for Improved Oral Drug Delivery: Formulation Development, In Vitro and In Vivo Permeability Testing" Molecules 27, no. 15: 4828. https://doi.org/10.3390/molecules27154828
APA StyleKunamaneni, P., Kovvasu, S., Yeung, S., Wang, J., Shah, S., & Betageri, G. (2022). Aliskiren Hemifumarate Proliposomes for Improved Oral Drug Delivery: Formulation Development, In Vitro and In Vivo Permeability Testing. Molecules, 27(15), 4828. https://doi.org/10.3390/molecules27154828