
Identification of Potential Parkinson’s Disease Drugs Based on Multi-Source Data Fusion and Convolutional Neural Network



Abstract
:1. Introduction
2. Results and Discussion
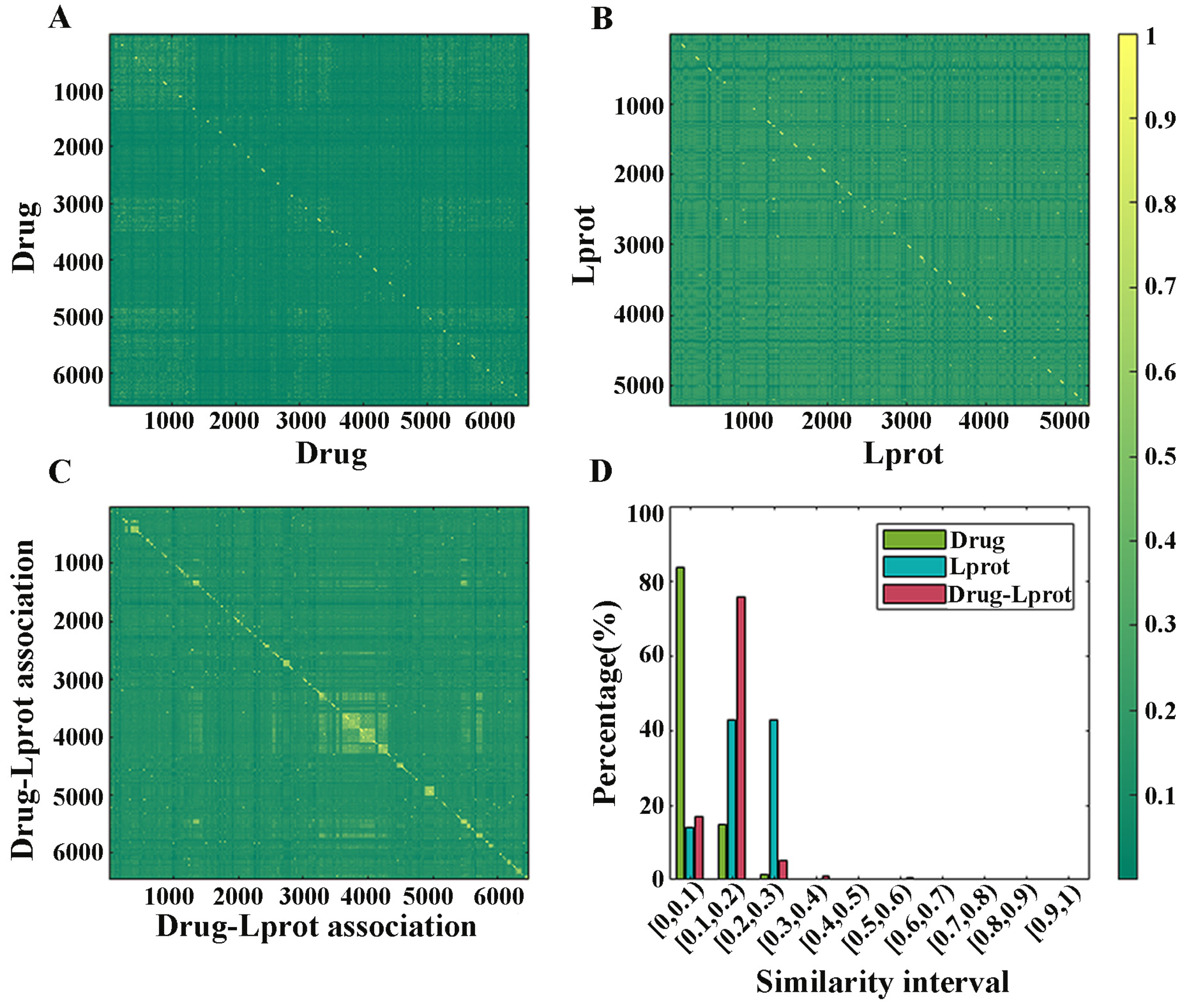
2.1. Redundancy Analysis of Dataset
2.2. Optimal Feature Dimension of Diffusion Component Analysis
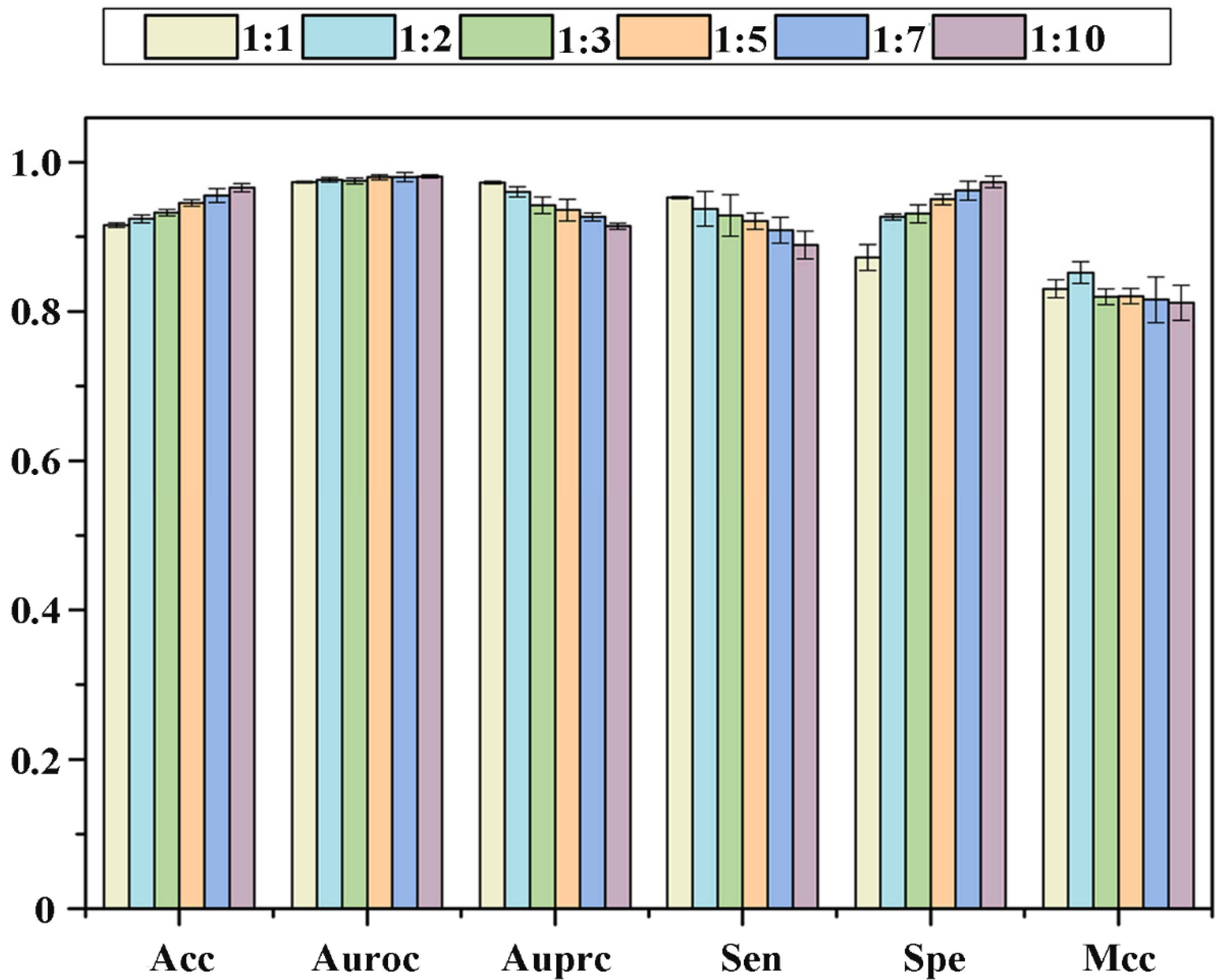
2.3. Effect of the Proportion of Positive and Negative Samples on Performance
2.4. Identification Ability of New Drugs
2.5. Recognition Ability of New Targets
2.6. Discriminatory Performance of Potential Drug-LProt Associations
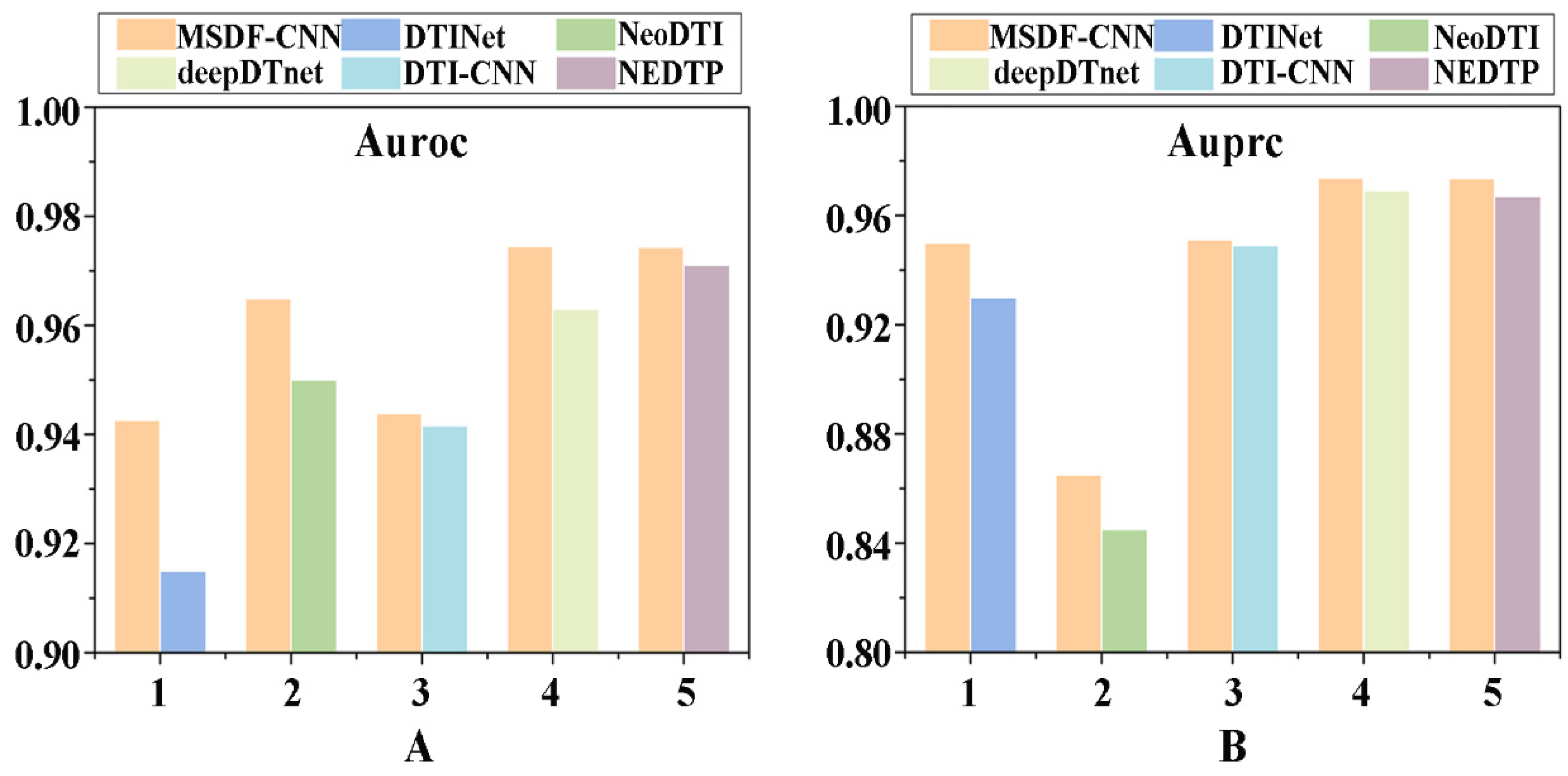
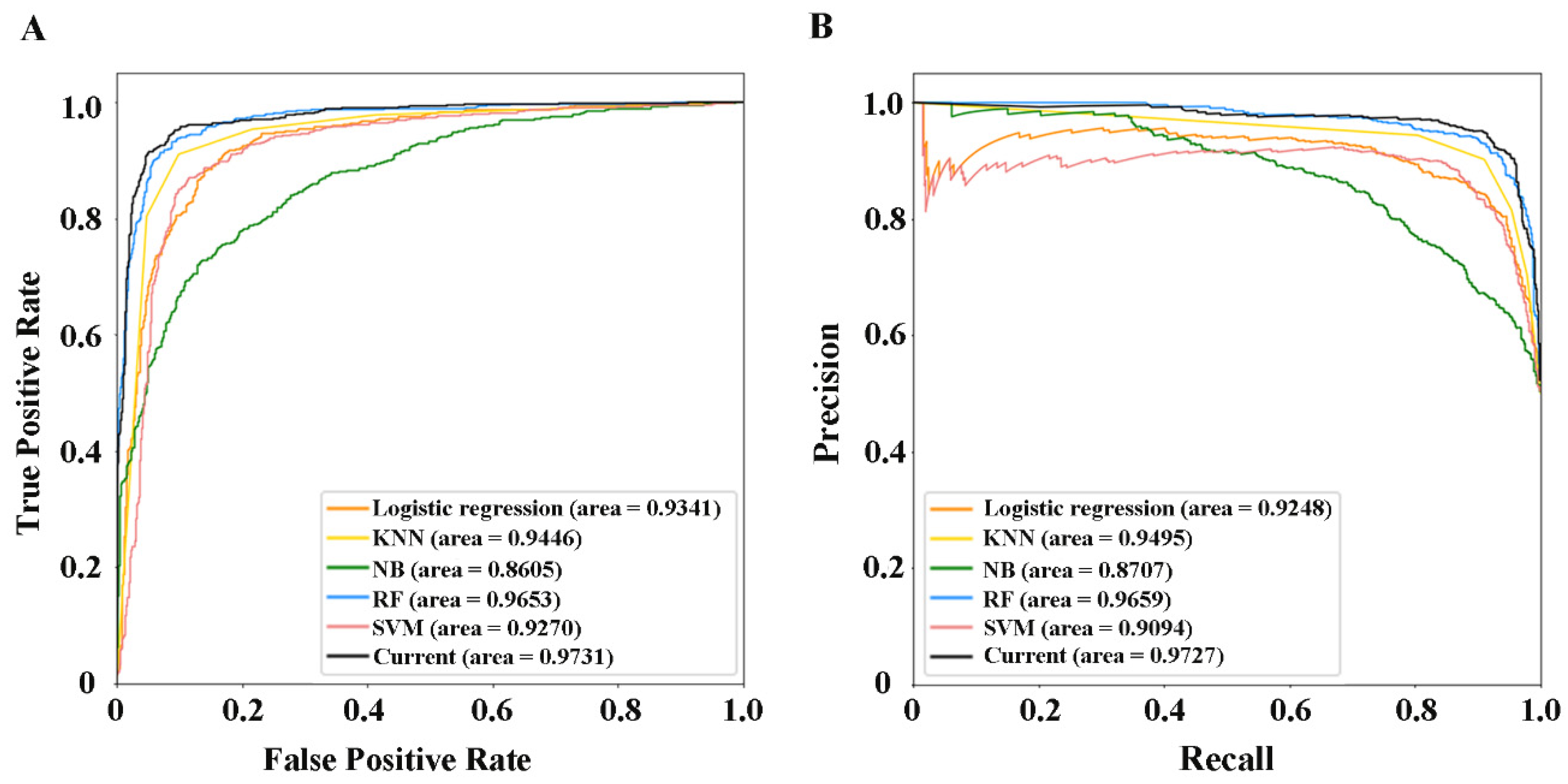
2.7. Performance Evaluation of Current Methods
2.8. Comparison with Existing Methods
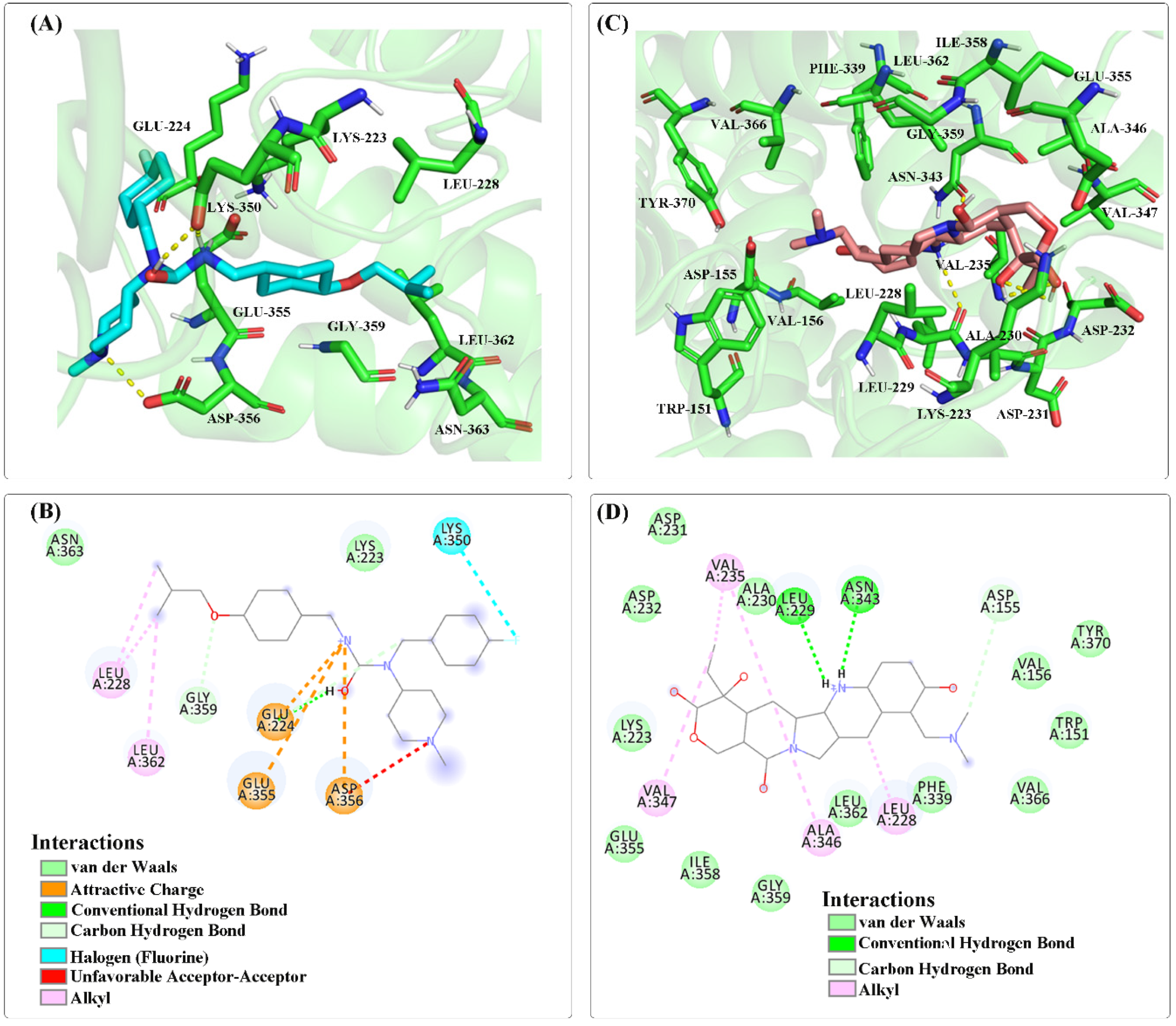
2.9. Molecular Docking
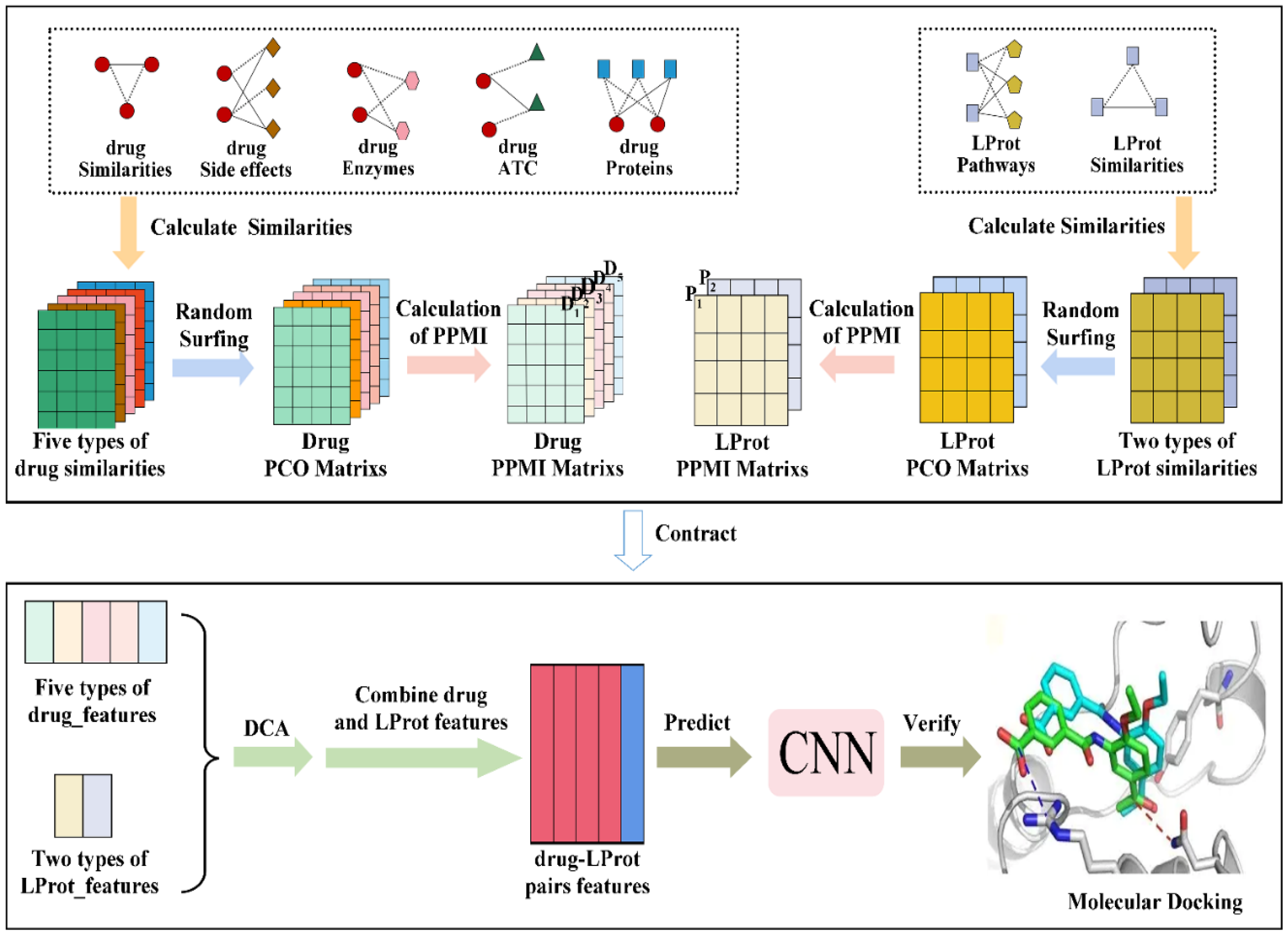
3. Materials and Methods
3.1. Collection and Processing of Data
3.2. Characterization of Drugs and LProt
3.3. Extraction and Selection of Feature
3.4. Construction and Evaluation of Models
- (1)
- Set a threshold based on the PPI network and known PD targets to screen out LProts (PD-associated proteins) with high correlation.
- (2)
- Construct multiple drug and LProt networks according to multi-source data and characterized by similarity networks.
- (3)
- Obtain high-dimensional features of drugs and LProt by capturing global and local topological information in the network.
- (4)
- Employ diffusion component analysis to reduce dimensionality and obtain low-dimensional and rich features for drug and LProt.
- (5)
- Construct a convolutional neural network model to predict potential association pairs.
- (6)
- Evaluate and verify the prediction and application performance of the developed method by using the 10-fold cross-validation test and molecular docking research, respectively.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statements
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Silva, R.C.; Domingues, H.S.; Salgado, A.J.; Teixeira, F.G. From regenerative strategies to pharmacological approaches: Can we fine-tune treatment for parkinson’s disease? Neural Regen. Res. 2022, 17, 933–936. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Exarchos, T.; Vlamos, P. Bioinformatics approaches for parkinson’s disease in clinical practice: Data-driven biomarkers and pharmacological treatment. Adv. Exp. Med. Biol. 2021, 1338, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.B.; Storstein, A. Epidemiology of parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Etiology and pathogenesis of parkinson disease. Neurol. Clin. 2009, 27, 583–603. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, J.; Xue, T.; Yang, Y.; Wu, D.; Chen, Z.; You, W.; Wang, Z. Different catechol-o-methyl transferase inhibitors in parkinson’s disease: A bayesian network meta-analysis. Front. Neurol. 2021, 12, 707723. [Google Scholar] [CrossRef]
- Grall-Bronnec, M.; Victorri-Vigneau, C.; Donnio, Y.; Leboucher, J.; Rousselet, M.; Thiabaud, E.; Zreika, N.; Derkinderen, P.; Challet-Bouju, G. Dopamine agonists and impulse control disorders: A complex association. Drug Saf. 2018, 41, 19–75. [Google Scholar] [CrossRef] [Green Version]
- Cerri, S.; Blandini, F. An update on the use of non-ergot dopamine agonists for the treatment of parkinson’s disease. Expert Opin. Pharmacother. 2020, 21, 2279–2291. [Google Scholar] [CrossRef]
- Özdemir, Z.; Alagöz, M.A.; Bahçecioğlu, Ö.F.; Gök, S. Monoamine oxidase-B (MAO-B) inhibitors in the treatment of alzheimer’s and parkinson’s disease. Curr. Med. Chem. 2021, 28, 6045–6065. [Google Scholar] [CrossRef]
- Koeglsperger, T.; Palleis, C.; Hell, F.; Mehrkens, J.H.; Bötzel, K. Deep brain stimulation programming for movement disorders: Current concepts and evidence-based strategies. Front. Neurol. 2019, 10, 410. [Google Scholar] [CrossRef] [Green Version]
- Swann, N.C.; de Hemptinne, C.; Miocinovic, S.; Qasim, S.; Ostrem, J.L.; Galifianakis, N.B.; Luciano, M.S.; Wang, S.S.; Ziman, N.; Taylor, R.; et al. Chronic multisite brain recordings from a totally implantable bidirectional neural interface: Experience in 5 patients with Parkinson’s disease. J. Neurosurg. 2018, 128, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okun, M.S. Deep-brain stimulation—Entering the era of human neural-network modulation. N. Engl. J. Med. 2014, 371, 1369–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, S.K.; Sankar, T.; Lozano, A.M. Deep brain stimulation for parkinson’s disease and other movement disorders. Curr. Opin. Neurol. 2013, 26, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Götz, M.; Parmar, M. New approaches for brain repair-from rescue to reprogramming. Nature 2018, 557, 329–334. [Google Scholar] [CrossRef]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human trials of stem cell-derived dopamine neurons for parkinson’s disease: Dawn of a new era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Pogarell, O.; Hagell, P.; Piccini, P.; Widner, H.; Schrag, A.; Kupsch, A.; Crabb, L.; Odin, P.; Gustavii, B.; et al. Bilateral caudate and putamen grafts of embryonic mesencephalic tissue treated with lazaroids in parkinson’s disease. Brain 2000, 123, 1380–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting alpha-synuclein as a therapy for parkinson’s disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [Green Version]
- Sapru, M.K.; Yates, J.W.; Hogan, S.; Jiang, L.; Halter, J.; Bohn, M.C. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol. 2006, 198, 382–390. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Saarma, M. Can growth factors cure parkinson’s disease? Trends Pharmacol. Sci. 2020, 41, 909–922. [Google Scholar] [CrossRef]
- Mullard, A. 2020 FDA drug approvals. Nat. Rev. Drug Discov. 2021, 20, 85–90. [Google Scholar] [CrossRef]
- Mullard, A. 2021 FDA approvals. Nat. Rev. Drug Discov. 2022, 21, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Sakate, R.; Kimura, T. Drug repositioning trends in rare and intractable diseases. Drug Discov. Today 2022, 27, 1789–1795. [Google Scholar] [CrossRef]
- Shah, S.; Dooms, M.M.; Amaral-Garcia, S.; Igoillo-Esteve, M. Current drug repurposing strategies for rare neurodegenerative disorders. Front. Pharmacol. 2021, 12, 768023. [Google Scholar] [CrossRef] [PubMed]
- Khanfar, M.A.; Al-Qtaishat, S.; Habash, M.; Taha, M.O. Discovery of potent adenosine A2a antagonists as potential anti-parkinson disease agents. non-linear QSAR analyses integrated with pharmacophore modeling. Chem. Biol. Interact. 2016, 254, 93–101. [Google Scholar] [CrossRef]
- Sebastián-Pérez, V.; Martínez, M.J.; Gil, C.; Campillo, N.E.; Martínez, A.; Ponzoni, I. QSAR modelling to identify LRRK2 inhibitors for parkinson’s disease. J. Integr. Bioinform. 2019, 16, 20180063. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.R.; Picanço, L.C.S.; Brito, M.F.B.; Almeida, M.R.S.; Marino, B.L.B.; Sousa, K.P.A.; Ferreira, J.V.; Dos Santos, C.B.R.; Silva, G.M.; Silva, C.H.T.P.; et al. Theoretical study of monoamine oxidase B inhibitors as drug candidates for treatment of parkinson’s disease. Cent. Nerv. Syst. Agents Med. Chem. 2020, 20, 128–143. [Google Scholar] [CrossRef]
- Meco, G.; Bernardi, S. Antidepressant use in treatment of psychosis with comorbid depression in parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Chiba, Y.; Katsuse, O.; Hirayasu, Y. A case of parkinson disease with both visual hallucination and pain improved by gabapentin. Clin. Neuropharmacol. 2016, 39, 55–56. [Google Scholar] [CrossRef]
- Palencia, G.; Garcia, E.; Osorio-Rico, L.; Trejo-Solís, C.; Escamilla-Ramírez, A.; Sotelo, J. Neuroprotective effect of thalidomide on MPTP-induced toxicity. Neurotoxicology 2015, 47, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Mante, M.; Anderson, S.; Rockenstein, E.; Masliah, E. Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of parkinson’s disease. J. Neuroinflammation 2015, 12, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalive, P.H.; Neuhaus, O.; Benkhoucha, M.; Burger, D.; Hohlfeld, R.; Zamvil, S.S.; Weber, M.S. Glatiramer acetate in the treatment of multiple sclerosis: Emerging concepts regarding its mechanism of action. CNS Drugs 2011, 25, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uc, E.Y. Salbutamol potentiates effect of levodopa in parkinson’s disease. Inpharma Wkly. 2003, 1406, 27. [Google Scholar] [CrossRef]
- Zhang, W.; Yue, X.; Lin, W.; Wu, W.; Liu, R.; Huang, F.; Liu, F. Predicting drug-disease associations by using similarity constrained matrix factorization. BMC Bioinform. 2018, 19, 233. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Huang, F.; Yue, X.; Lu, X.; Yang, W.; Li, Z.; Liu, F. Prediction of drug-disease associations and their effects by signed network-based nonnegative matrix factorization. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine, Madrid, Spain, 3–6 December 2018; pp. 798–802. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, P.; Yan, L.; Fu, Y.; Peng, F.; Qu, L.; Shao, M.; Chen, Y.; Chen, Z. LRSSL: Predict and interpret drug–disease associations based on data integration using sparse subspace learning. Bioinformatics 2017, 33, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.N.; You, Z.H.; Li, L.P.; Chen, Z.H.; Xie, X.J. WGMFDDA: A novel weighted-based graph regularized matrix factorization for predicting drug-disease associations. In International Conference on Intelligent Computing; Springer: Cham, Switzerland, 2020; pp. 542–551. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Ni, P.; Zhao, K.; Wu, F.X.; Pan, Y. Computational drug repositioning with random walk on a heterogeneous network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 1890–1900. [Google Scholar] [CrossRef]
- Li, Z.; Huang, Q.; Chen, X.; Wang, Y.; Li, J.; Xie, Y.; Dai, Z.; Zou, X. Identification of drug-disease associations using information of molecular structures and clinical symptoms via deep convolutional neural network. Front. Chem. 2020, 7, 924. [Google Scholar] [CrossRef]
- Peng, J.; Li, J.; Shang, X. A learning-based method for drug-target interaction prediction based on feature representation learning and deep neural network. BMC Bioinform. 2020, 21, 394. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinform. 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, F.; Hong, L.; Xiao, A.; Jiang, T.; Zeng, J. NeoDTI: Neural integration of neighbor information from a heterogeneous network for discovering new drug-target interactions. Bioinformatics 2019, 35, 104–111. [Google Scholar] [CrossRef]
- An, Q.; Yu, L. A heterogeneous network embedding framework for predicting similarity-based drug-target interactions. Brief. Bioinform. 2021, 22, bbab275. [Google Scholar] [CrossRef]
- Kim, K.; Che, T.; Panova, O.; DiBerto, J.F.; Lyu, J.; Krumm, B.E.; Wacker, D.; Robertson, M.J.; Seven, A.B.; Nichols, D.E.; et al. Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 2020, 182, 1574–1588.e19. [Google Scholar] [CrossRef] [PubMed]
- Lihua, P.; Chen, X.Y.; Wu, T.X. Topotecan for ovarian cancer. Cochrane Database Syst. Rev. 2008, 2008, CD005589. [Google Scholar] [CrossRef]
- Hart, L.L.; Ferrarotto, R.; Andric, Z.G.; Beck, J.T.; Subramanian, J.; Radosavljevic, D.Z.; Zaric, B.; Hanna, W.T.; Aljumaily, R.; Owonikoko, T.K.; et al. Myelopreservation with trilaciclib in patients receiving topotecan for small cell lung cancer: Results from a randomized, double-blind, placebo-controlled phase II study. Adv. Ther. 2021, 38, 350–365. [Google Scholar] [CrossRef]
- Ackermann, S.; Beckmann, M.W.; Thiel, F.; Bogenrieder, T. Topotecan in cervical cancer. Int. J. Gynecol. Cancer 2007, 17, 1215–1223. [Google Scholar] [CrossRef]
- Pastrana, T.; Meißner, W. Behandlung der diarrhö mit loperamid in der palliativmedizin. Eine systematische literaturübersicht [Treatment of diarrhea with loperamide in palliative medicine. A systematic review]. Schmerz 2013, 27, 182–189. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, C.J.; Chia, W.N.; Loh, C.C.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.X.; Lim, T.K.; Liu, M.; et al. Haem-activated promiscuous targeting of artemisinin in plasmodium falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. 2016 FDA drug approvals. Nat. Rev. Drug Discov. 2017, 16, 73–76. [Google Scholar] [CrossRef]
- Hu, K.; Zhang, M.; Wu, D.; Xie, Y.; Ren, J. A novel synthesis of pimavanserin: A selective serotonin 5-HT2A receptor inverse agonist. Org. Prep. Proced. Int. 2020, 52, 69–76. [Google Scholar] [CrossRef]
- Kitten, A.K.; Hallowell, S.A.; Saklad, S.R.; Evoy, K.E. Pimavanserin: A novel drug approved to treat Parkinson’s Disease psychosis. Innov. Clin. Neurosci. 2018, 15, 16–22. [Google Scholar] [PubMed]
- Kianirad, Y.; Simuni, T. Pimavanserin, a novel antipsychotic for management of parkinson’s disease psychosis. Expert Rev. Clin. Pharmacol. 2017, 10, 1161–1168. [Google Scholar] [CrossRef]
- Cruz, M.P. Pimavanserin (Nuplazid): A treatment for hallucinations and delusions associated with Parkinson’s Disease. Pharm. Ther. 2017, 42, 368–371. [Google Scholar]
- Markham, A. Pimavanserin: First global approval. Drugs 2016, 76, 1053–1057. [Google Scholar] [CrossRef]
- Stahl, S.M. Mechanism of action of pimavanserin in Parkinson’s disease psychosis: Targeting serotonin 5HT2A and 5HT2C receptors. CNS Spectr. 2016, 21, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Tampi, R.R.; Tampi, D.J.; Young, J.J.; Balachandran, S.; Hoq, R.A.; Manikkara, G. Evidence for using pimavanserin for the treatment of parkinson’s disease psychosis. World J. Psychiatry 2019, 9, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.M. Parkinson’s disease psychosis as a serotonin-dopamine imbalance syndrome. CNS Spectr. 2016, 21, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Yunusa, I.; El Helou, M.L.; Alsahali, S. Pimavanserin: A novel antipsychotic with potentials to address an unmet need of older adults with dementia-related psychosis. Front. Pharmacol. 2020, 11, 87. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jeon, B.S.; Kim, H.J.; Park, S.S. Genetic variant of HTR2A associates with risk of impulse control and repetitive behaviors in Parkinson’s disease. Parkinsonism. Relat. Disord. 2012, 18, 76–78. [Google Scholar] [CrossRef]
- Shukla, R.; Pahal, S.; Gupta, A.; Choudhary, P.; Misra, K.; Singh, S. Modulation of GPCR receptors common to gut inflammatory diseases and neuronal disorders, Alzheimer’s and Parkinson’s diseases as druggable targets through Withania somnifera bioactives: An in silico study. J. Biomol. Struct. Dyn. 2022, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Samanthula, B.K.; Jiang, W. Secure multiset intersection cardinality and its application to jaccard coefficient. IEEE Trans. Dependable Secur. Comput. 2015, 13, 591–604. [Google Scholar] [CrossRef]
- Cao, S.; Lu, W.; Xu, Q. Deep neural networks for learning graph representations. In Proceedings of the AAAI Conference on Artificial Intelligence, Phoenix, AZ, USA, 12–17 February 2016; Volume 30. [Google Scholar] [CrossRef]
- Bullinaria, J.A.; Levy, J.P. Extracting semantic representations from word co-occurrence statistics: Stop-lists, stemming, and SVD. Behav. Res. Methods 2012, 44, 890–907. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Berger, B.; Peng, J. Compact integration of multi-network topology for functional analysis of genes. Cell Syst. 2016, 3, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Fan, C.; Li, Y.; Wu, Q.; Ming, Y. Improving deep neural network with multiple parametric exponential linear units. Neurocomputing 2018, 301, 11–24. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimension | Acc (%) | Spe (%) | Sen (%) | Mcc | Auroc | Auprc | |
|---|---|---|---|---|---|---|---|
| Drug | LProt | ||||||
| 100 | 200 | 90.76 | 94.54 | 86.98 | 0.8187 | 0.9709 | 0.9709 |
| 100 | 300 | 91.51 | 94.88 | 88.13 | 0.8328 | 0.9730 | 0.9729 |
| 100 | 400 | 91.57 | 95.26 | 87.24 | 0.8303 | 0.9731 | 0.9726 |
| 100 | 600 | 91.25 | 95.43 | 87.08 | 0.8287 | 0.9721 | 0.9708 |
| 200 | 300 | 91.08 | 94.51 | 89.65 | 0.8252 | 0.9711 | 0.9710 |
| 300 | 500 | 91.06 | 95.49 | 86.63 | 0.8251 | 0.9715 | 0.9702 |
| 500 | 500 | 91.50 | 94.77 | 88.15 | 0.8320 | 0.9706 | 0.9702 |
| Threshold | Acc (%) | Sen (%) | Spe (%) | Mcc | Auroc | Auprc |
|---|---|---|---|---|---|---|
| 0.9 | 91.57 | 95.27 | 87.24 | 0.8304 | 0.9732 | 0.9727 |
| 0.8 | 91.81 | 95.21 | 88.41 | 0.8387 | 0.9735 | 0.9726 |
| 0.7 | 91.34 | 94.92 | 87.76 | 0.8294 | 0.9708 | 0.9691 |
| 0.6 | 90.74 | 95.41 | 86.07 | 0.8193 | 0.9699 | 0.9691 |
| 0.5 | 90.11 | 94.61 | 85.61 | 0.8065 | 0.9658 | 0.9654 |
| 0.4 | 88.49 | 94.73 | 82.24 | 0.7769 | 0.9598 | 0.9587 |
| Threshold | Acc (%) | Sen (%) | Spe (%) | Mcc | Auroc | Auprc |
|---|---|---|---|---|---|---|
| 0.9 | 91.92 | 94.85 | 89.05 | 0.8408 | 0.9726 | 0.9728 |
| 0.8 | 90.23 | 94.32 | 86.13 | 0.8087 | 0.9666 | 0.9661 |
| 0.7 | 90.24 | 94.37 | 86.11 | 0.8090 | 0.9667 | 0.9654 |
| 0.6 | 90.73 | 93.62 | 87.85 | 0.8171 | 0.9652 | 0.9658 |
| 0.5 | 90.59 | 94.20 | 86.98 | 0.8151 | 0.9660 | 0.9654 |
| 0.4 | 91.90 | 90.84 | 92.95 | 0.8395 | 0.9645 | 0.9692 |
| Threshold | Acc (%) | Sen (%) | Spe (%) | Mcc | Auroc | Auprc |
|---|---|---|---|---|---|---|
| 0.9 | 91.87 | 95.58 | 88.56 | 0.8405 | 0.9746 | 0.9764 |
| 0.8 | 91.84 | 95.58 | 88.25 | 0.8397 | 0.9757 | 0.9760 |
| 0.7 | 91.75 | 94.67 | 88.11 | 0.8366 | 0.9720 | 0.9741 |
| Acc (%) | Sen (%) | Spe (%) | Mcc | Auroc | Auprc | |
|---|---|---|---|---|---|---|
| Logistic regression | 86.51 | 86.84 | 86.19 | 0.7303 | 0.9341 | 0.9248 |
| KNN | 87.29 | 94.40 | 80.16 | 0.7534 | 0.9446 | 0.9495 |
| NB | 77.62 | 71.46 | 83.82 | 0.5569 | 0.8605 | 0.8707 |
| RF | 90.57 | 91.12 | 90.02 | 0.8114 | 0.9653 | 0.9659 |
| SVM | 86.92 | 88.12 | 85.72 | 0.7386 | 0.9270 | 0.9094 |
| Current | 91.57 | 95.27 | 87.24 | 0.8304 | 0.9731 | 0.9727 |
| Number | Drug | Indication |
|---|---|---|
| 1 | Topotecan | Treat ovarian cancer, small cell lung cancer or cervical cancer. |
| 2 | Loperamide | Control nonspecific and chronic diarrhea caused by inflammatory bowel disease or gastroenteritis. |
| 3 | Artenimol | Treatment of artemisinin derivatives and the antimalarial agent Plasmodium falciparum infection. |
| 4 | Mitotane | Treatment of inoperable adrenal cortical tumors; Cushing’s syndrome. |
| 5 | Estramustine | The palliative treatment of patients with metastatic and/or progressive carcinoma of the prostate. |
| 6 | Quercetin | A flavonol widely distributed in plants. It is an antioxidant, like many other phenolic heterocyclic compounds. |
| 7 | Nortriptyline | A tricyclic antidepressant used to treat major depressive disorder and also to treat chronic pain and other conditions. |
| 8 | Bacitracin | Topical preparations for acute and chronic topical skin infections. |
| 9 | Minocycline | Treatment of inflammatory lesions of acne vulgaris. |
| 10 | Doxepin | A psychotropic agent with antidepressant and anxiolytic properties. |
| Ligand | Target Protein | Binding Energy (kcal/mol) | Inhibition Constant (μM) |
|---|---|---|---|
| Pimavanserin | HTR2A | −6.4 | 20.49 |
| Loperamide | −7.76 | 2.05 | |
| Topotecan | −7.96 | 1.47 | |
| Artenimol | −7.65 | 2.46 |
| Information | Number | Sources |
|---|---|---|
| drug–chemical structure | 6587 | DrugBank Database |
| drug–ATC | 4636 | |
| drug–enzyme | 4828 | |
| drug–target | 15,504 | |
| drug–side effect | 755,165 | SIDES Database |
| PPI | 353,550 | HIPPIES Database |
| PD targets | 157 | TTD Database CTD Database Uniprot Database DrugBank Database |
| PD drugs | 30 | |
| PD associated targets (LProt) | 5295 | PPI PD targets |
| LProt–pathway | 13,947 | CTD Database |
| LProt–sequence | 5295 | Uniprot Database |
| drugs | 6587 | DrugBank Database |
| Layer | Size |
|---|---|
| Input | 500*1 |
| Convolutional | 4 filters with 5*1, stride 1*1 |
| ReLU | - |
| Convolutional | 8 filters with 10*1, stride 1*1 |
| ReLU | - |
| Max-Pooling | 2*1, stride 2*1 |
| ReLU | - |
| Fully connected | 256, dropout = 0.5 |
| Sigmoid | - |
| Classification | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Peng, D.; Li, J.; Dai, Z.; Zou, X.; Li, Z. Identification of Potential Parkinson’s Disease Drugs Based on Multi-Source Data Fusion and Convolutional Neural Network. Molecules 2022, 27, 4780. https://doi.org/10.3390/molecules27154780
Liu J, Peng D, Li J, Dai Z, Zou X, Li Z. Identification of Potential Parkinson’s Disease Drugs Based on Multi-Source Data Fusion and Convolutional Neural Network. Molecules. 2022; 27(15):4780. https://doi.org/10.3390/molecules27154780
Chicago/Turabian StyleLiu, Jie, Dongdong Peng, Jinlong Li, Zong Dai, Xiaoyong Zou, and Zhanchao Li. 2022. "Identification of Potential Parkinson’s Disease Drugs Based on Multi-Source Data Fusion and Convolutional Neural Network" Molecules 27, no. 15: 4780. https://doi.org/10.3390/molecules27154780
APA StyleLiu, J., Peng, D., Li, J., Dai, Z., Zou, X., & Li, Z. (2022). Identification of Potential Parkinson’s Disease Drugs Based on Multi-Source Data Fusion and Convolutional Neural Network. Molecules, 27(15), 4780. https://doi.org/10.3390/molecules27154780