
Potential Efficacy of β-Amyrin Targeting Mycobacterial Universal Stress Protein by In Vitro and In Silico Approach

 ,
,  ,
, 
 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Identification and Procurement of the Plant Materials
2.2. Preparation of Plant Extracts
2.3. Phytochemical Screening of Plant Extracts
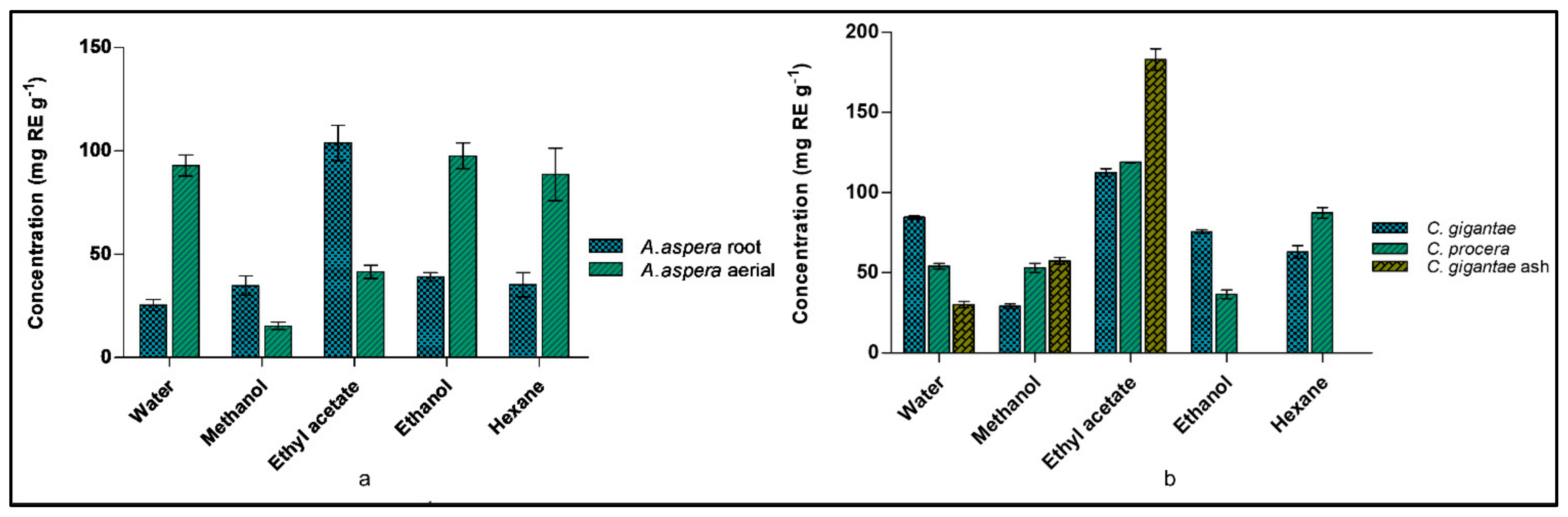
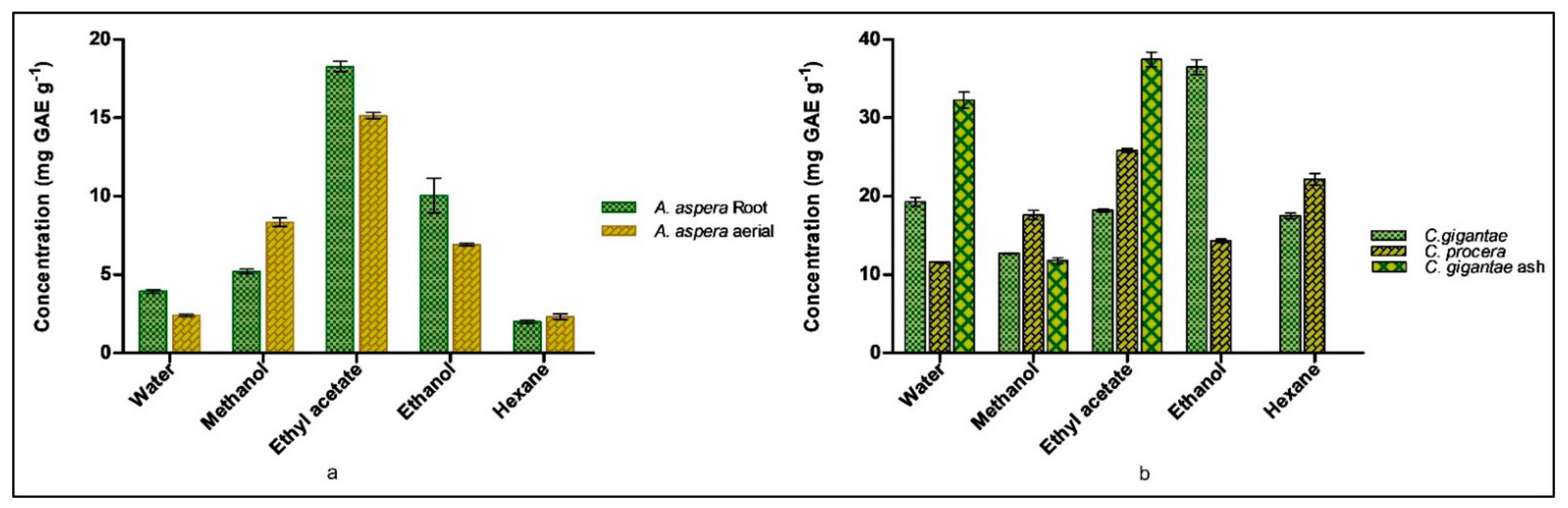
2.4. Detection of Total Flavonoid Content (TFC) of Plants
2.5. Detection of Total Polyphenolic Content (TPC) of Plants
2.6. Minimum Inhibitory Concentrations (MICs)
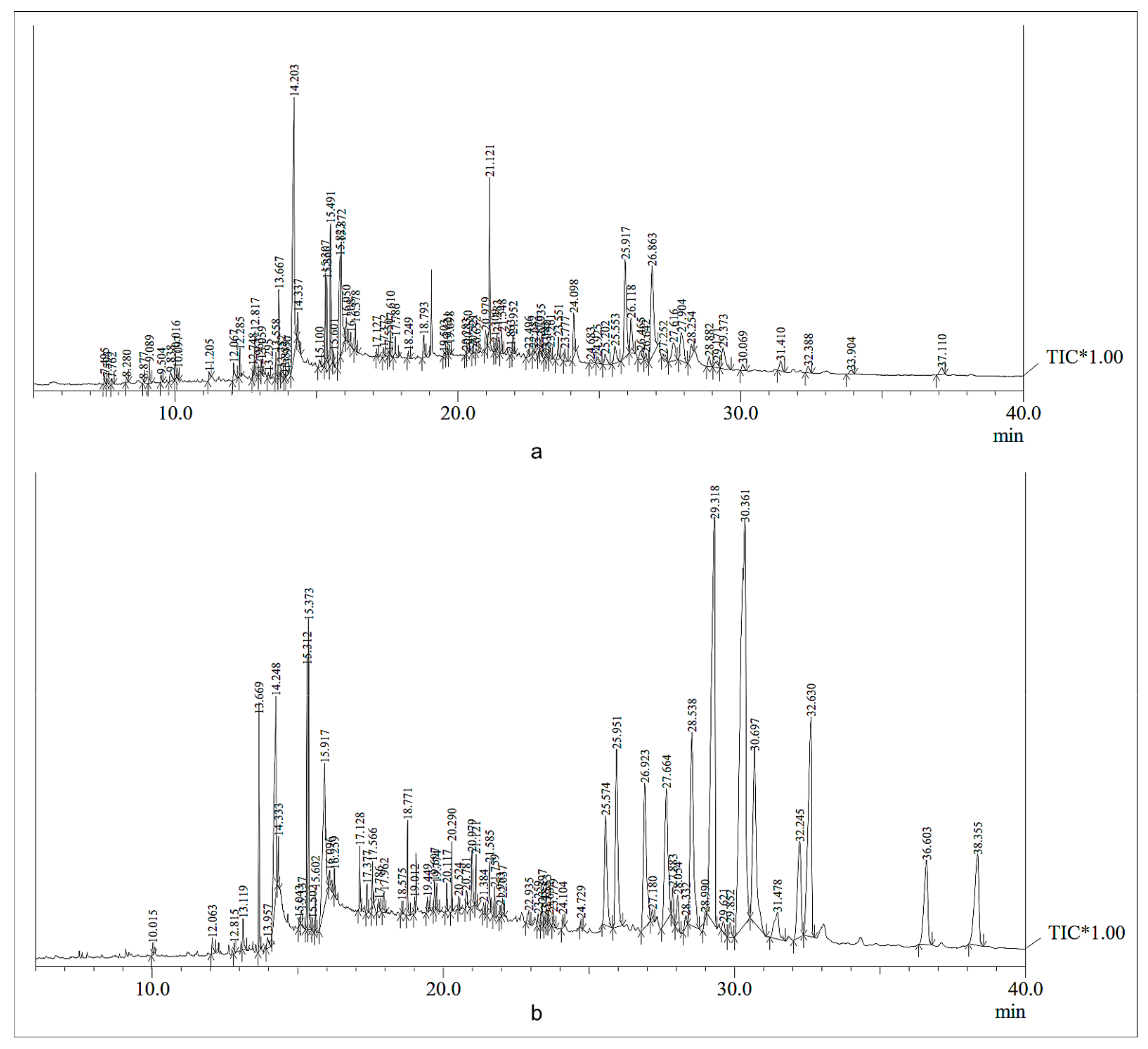
2.7. Gas Chromatography-Mass Spectrometry (GC-MS) Studies
2.8. Assessment of Multitarget Signature Mtb Proteins: An In-Silico Approach
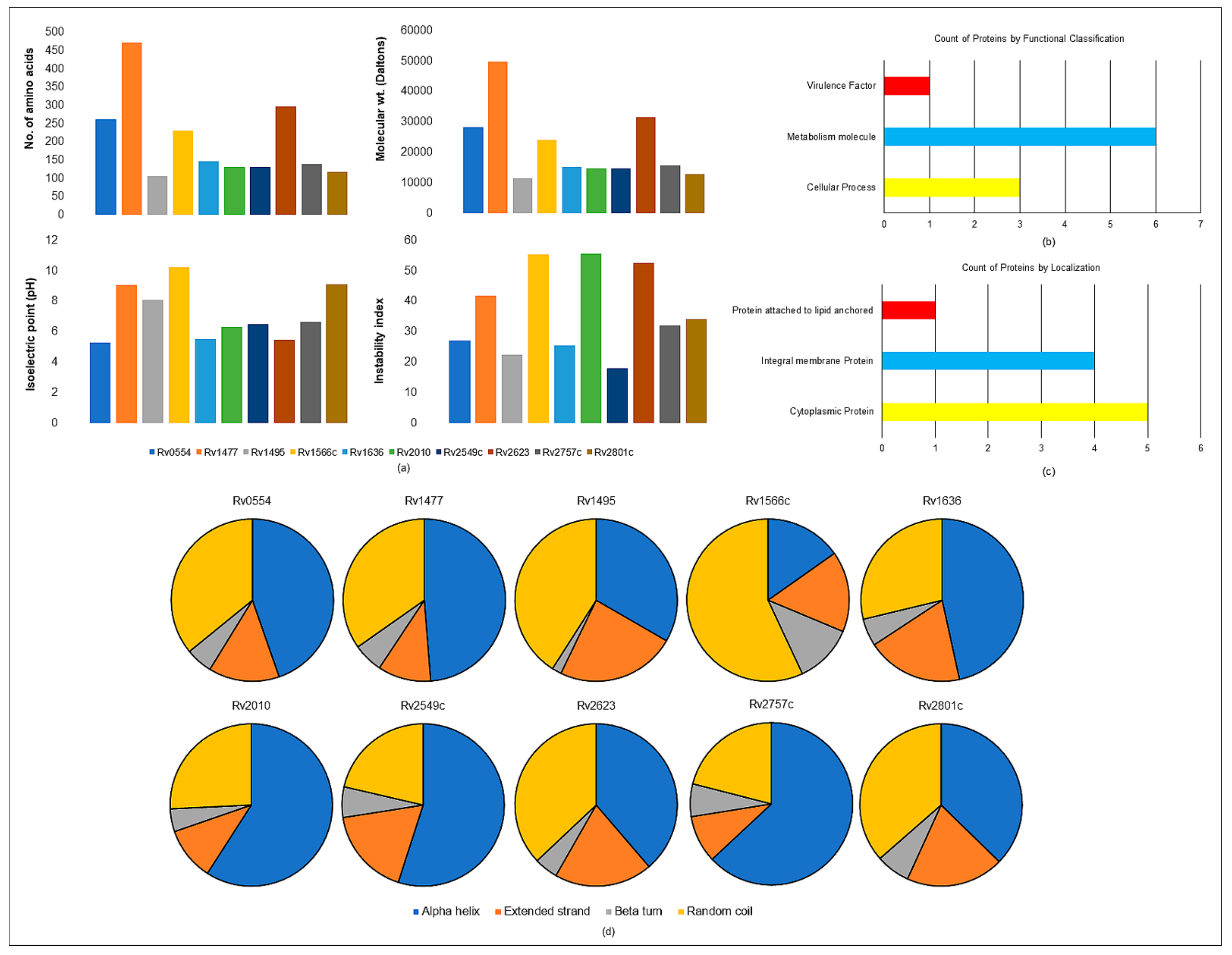
2.8.1. Physiochemical Parameters
2.8.2. Functional Classification
2.8.3. Subcellular Localization
2.8.4. Secondary Structure Prediction
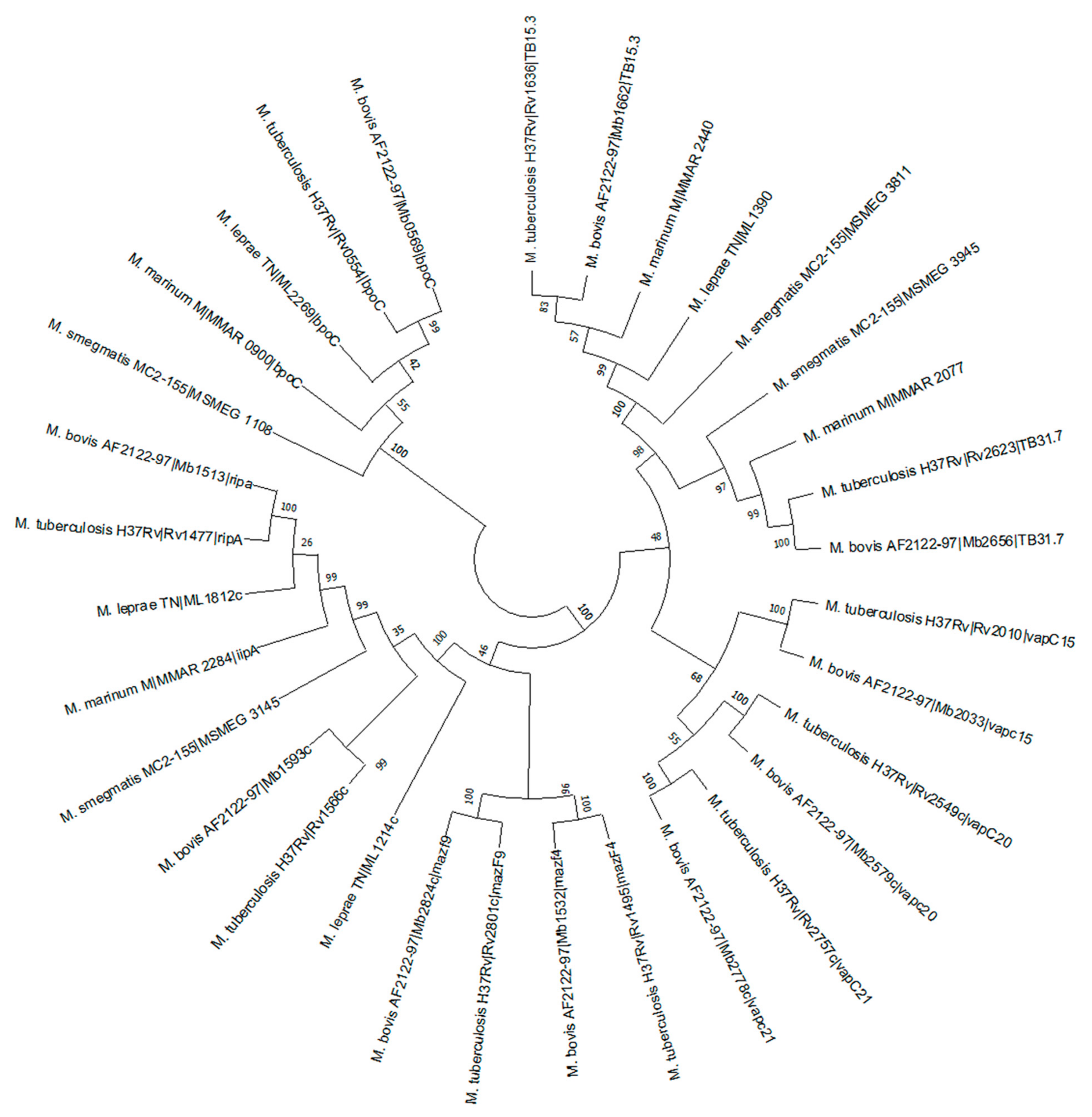
2.8.5. Phylogenetic Analysis
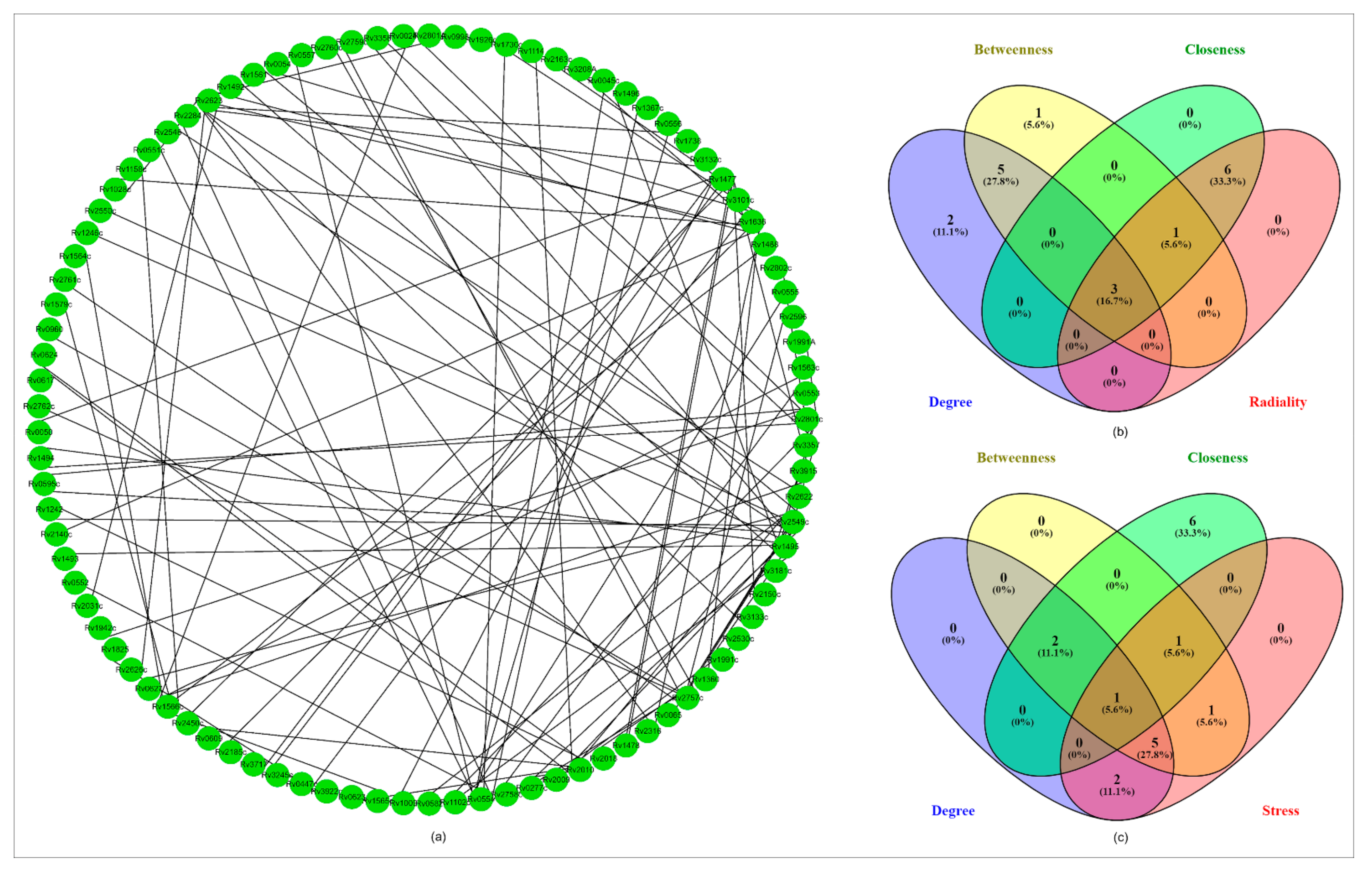
2.8.6. PPI Network Analysis
2.8.7. Structural Classification of the Selected VDA Proteins
2.8.8. Validation of the Selected Proteins’ Structures
2.8.9. Molecular Docking
2.8.10. Determination of the Selected Phytoconstituents’ Drug Abilities
2.9. MD Simulation
2.9.1. Average Potential Energy of System
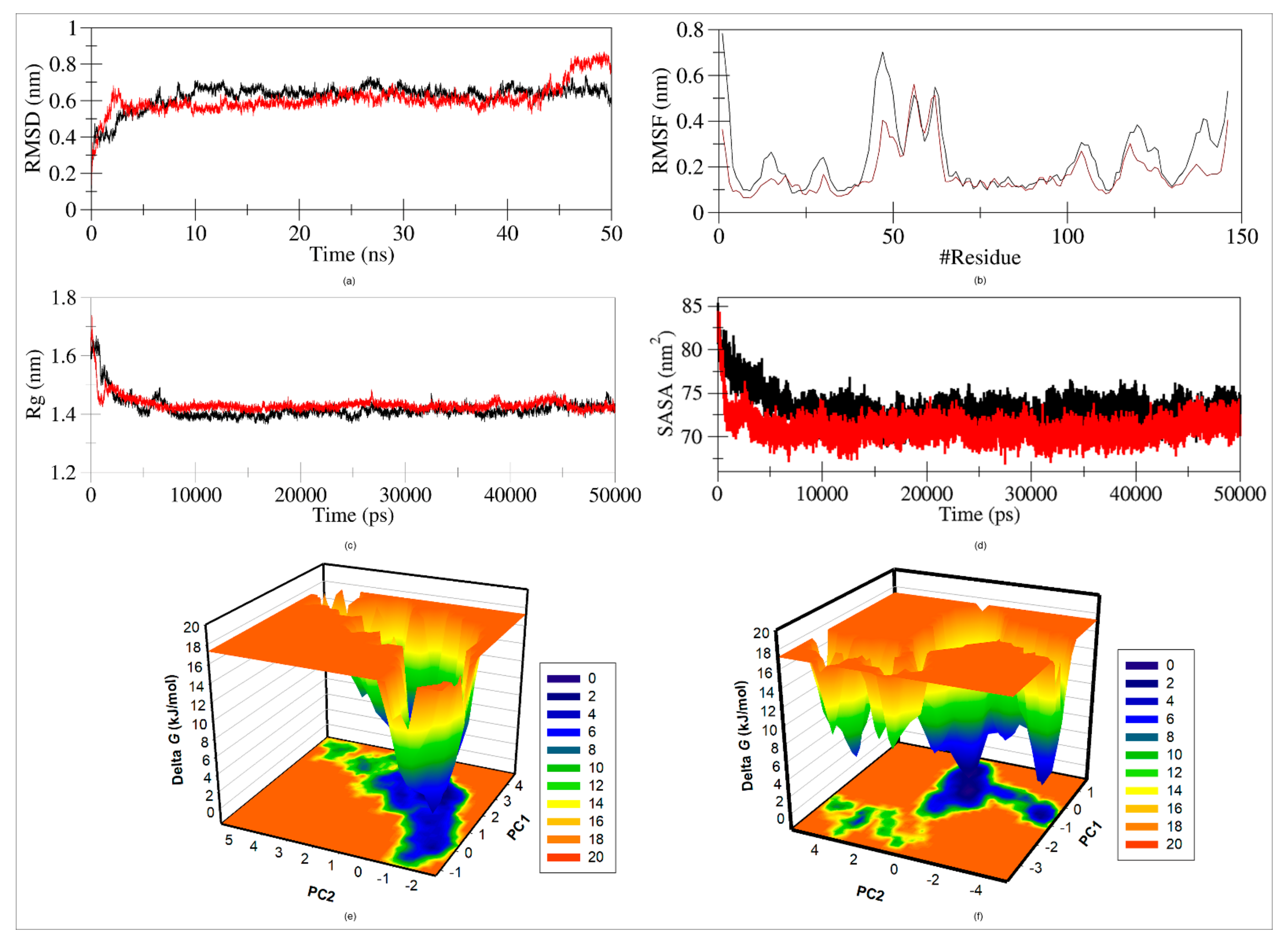
2.9.2. Root Mean Square Distance (RMSD)
2.9.3. Root Mean Square Fluctuation (RMSF)
2.9.4. Radius of Gyration
2.9.5. Solvent Accessible Surface Area (SASA)
2.9.6. Free Energy Landscape (FEL)
3. Discussion
4. Materials and Methods
4.1. Plant Collection and Identification
4.2. Plant Extraction
4.3. Secondary Metabolite Identification
4.3.1. Alkaloids Presence: Mayer’s Reagent Test
4.3.2. Tannins Presence: Ferric Chloride Test
4.3.3. Saponins Presence: Frothing Test
4.3.4. Terpenoids Presence: Salkowski Test
4.4. Total Flavonoid Content (TFC)
4.5. Total Polyphenolic Content (TPC)
4.6. Minimal Inhibitory Concentration (MIC) Assay
4.7. GC-MS Analysis of Plant Extracts
4.8. Determination of the Target Proteins: Using In Silico Approaches
4.8.1. Retrieval of the Protein Sequence
4.8.2. Physiochemical Parameters
4.8.3. Functional Classification
4.8.4. Subcellular Localization
4.8.5. Secondary (2D) Structure Prediction
4.8.6. Phylogenetic Analysis
4.8.7. Virulent Genes Regulating Network Analysis
4.8.8. Retrieval of the 3D Protein’s Structure
4.8.9. Validation of the Selected Protein’s Structure
4.8.10. Molecular Docking
4.8.11. Determination of the Selected Phytoconstituent Drug-Ability
4.9. MD Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Paik, S.; Kim, J.K.; Chung, C.; Jo, E.-K. Autophagy: A new strategy for host-directed therapy of tuberculosis. Virulence 2019, 10, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Sieniawska, E.; Sawicki, R.; Swatko-Ossor, M.; Napiorkowska, A.; Przekora, A.; Ginalska, G.; Augustynowicz-Kopec, E. The effect of combining natural terpenes and antituberculous agents against reference and clinical Mycobacterium tuberculosis strains. Molecules 2018, 23, 176. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.A.; Draper, P.; Hart, P.A. Mycobacteria and lysosomes: A paradox. Nature 1969, 221, 658–660. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Report of Tuberculosis 2021. Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 10 March 2022).
- WHO. WHO Report of Tuberculosis 2020. Available online: https://www.who.int/publications/i/item/9789240013131 (accessed on 10 March 2022).
- Dou, H.-Y.; Tseng, F.-C.; Lin, C.-W.; Chang, J.-R.; Sun, J.-R.; Tsai, W.-S.; Lee, S.-Y.; Su, I.-J.; Lu, J.-J. Molecular epidemiology and evolutionary genetics of Mycobacterium tuberculosis in Taipei. BMC Infect. Dis. 2008, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Brosch, R.; Gordon, S.V.; Marmiesse, M.; Brodin, P.; Buchrieser, C.; Eiglmeier, K.; Garnier, T.; Gutierrez, C.; Hewinson, G.; Kremer, K.; et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. USA 2002, 99, 3684–3689. [Google Scholar] [CrossRef]
- Gutierrez, M.C.; Brisse, S.; Brosch, R.; Fabre, M.; Omaïs, B.; Marmiesse, M.; Supply, P.; Vincent, V. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 2005, 1, e5. [Google Scholar] [CrossRef]
- Ge, F.; Zeng, F.; Liu, S.; Guo, N.; Ye, H.; Song, Y.; Fan, J.; Wu, X.; Wang, X.; Yu, L.; et al. In vitro synergistic interactions of oleanolic acid in combination with isoniazid, rifampicin or ethambutol against Mycobacterium tuberculosis. J. Med. Microbiol. 2010, 59, 567–572. [Google Scholar] [CrossRef]
- Knezevic, P.; Aleksic, V.; Simin, N.; Svircev, E.; Petrovic, A.; Mimica-Dukic, N. Antimicrobial activity of Eucalyptus camaldulensis essential oils and their interactions with conventional antimicrobial agents against multi-drug resistant Acinetobacter baumannii. J. Ethnopharmacol. 2016, 178, 125–136. [Google Scholar] [CrossRef]
- Hasan, S. Pharmacological and medicinal uses of Achyranthes aspera. Int. J. Sci. Environ. Technol. 2014, 3, 123–129. [Google Scholar]
- Sharma, V.; Chaudhary, U. An overview on indigenous knowledge of Achyranthes aspera. J. Crit. Rev. 2015, 2, 7–19. [Google Scholar]
- Maiden, J.H. The Useful Native Plants of Australia: (Including Tasmania); Turner and Henderson: Sydney, Australia, 1889. [Google Scholar]
- Sharma, J.; Gairola, S.; Gaur, R.D.; Painuli, R.M.; Siddiqi, T.O. Ethnomedicinal plants used for treating epilepsy by indigenous communities of sub-Himalayan region of Uttarakhand, India. J. Ethnopharmacol. 2013, 150, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Gawande, D.Y.; Druzhilovsky, D.; Gupta, R.C.; Poroikov, V.; Goel, R.K. Anticonvulsant activity and acute neurotoxic profile of Achyranthes aspera Linn. J. Ethnopharmacol. 2017, 202, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Barua, C.C.; Talukdar, A.; Begum, S.A.; Borah, P.; Lahkar, M. Anxiolytic activity of methanol leaf extract of Achyranthes aspera Linn in mice using experimental models of anxiety. Indian J. Pharmacol. 2012, 44, 63. [Google Scholar] [CrossRef] [PubMed]
- Barua, C.C.; Talukdar, A.; Begum, S.A.; Lahon, L.C.; Sarma, D.K.; Pathak, D.C.; Borah, P. Antinociceptive activity of methanolic extract of leaves of Achyranthes aspera Linn. (Amaranthaceae) in animal models of nociception. Indian J. Exp. Biol. 2010, 48, 817–821. [Google Scholar]
- Bhosale, U.; Yegnanarayan, R.; Prachi, P.; Zambare, M.; Somani, R.S. Study of CNS depressant and behavioral activity of an ethanol extract of Achyranthes aspera (Chirchita) in mouse model. Ann. Neurosci. 2011, 18, 44. [Google Scholar] [CrossRef]
- Khan, N.; Akhtar, M.S.; Khan, B.A.; de Andrade Braga, V.; Reich, A. Antiobesity, hypolipidemic, antioxidant and hepatoprotective effects of Achyranthes aspera seed saponins in high cholesterol fed albino rats. Arch. Med. Sci. AMS 2015, 11, 1261–1271. [Google Scholar] [CrossRef]
- Mainasara, M.M.; Aliero, B.L.; Aliero, A.A.; Yakubu, M. Phytochemical and antibacterial properties of root and leaf extracts of Calotropis procera. Niger. J. Basic Appl. Sci. 2012, 20, 1–6. [Google Scholar]
- Rathore, M.; Meena, R.K. Potential of utilizing Calotropis procera flower biomass as a renewable source of energy. J. Phytol. 2010, 2, 78–83. [Google Scholar]
- Dwivedi, B.; Singh, A.; Mishra, S.; Singh, R.; Pant, P.; Thakur, L.K.; Padhi, M.M. Evaluation of phytochemical constituents by gas chromatography-mass spectroscopy & HPTLC of Calotropis procera. World J. Pharm. Res. 2014, 3, 708–715. [Google Scholar]
- Kumar, V.L.; Shivkar, Y.M. In vivo and in vitro effect of latex of Calotropis procera on gastrointestinal smooth muscles. J. Ethnopharmacol. 2004, 93, 377–379. [Google Scholar] [CrossRef]
- Jalalpure, S.S.; Salahuddin, M.; Imtiyaz Shaikh, M.; Manvi, F.V. Anticonvulsant effects of Calotropis procera root in rats. Pharm. Biol. 2009, 47, 162–167. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Sahoo, A.K.; Narayanan, N.; Kumar, N.S.; Ponnusankar, S. Lead finding from medicinal plants with hepatoprotective potentials. Expert Opin. Drug Discov. 2009, 4, 545–576. [Google Scholar] [CrossRef] [PubMed]
- Patra, A.; Jha, S.; Murthy, P.N.; Vaibhav, A.D.; Chattopadhyay, P.; Panigrahi, G.; Roy, D. Anti-inflammatory and antipyretic activities of Hygrophila spinosa T. Anders leaves (Acanthaceae). Trop. J. Pharm. Res. 2009, 8, 133–137. [Google Scholar] [CrossRef]
- Mahmoud, M.E.; Shiina, T.; Hirayama, H.; Iwami, M.; Miyazawa, S.; Nikami, H.; Takewaki, T.; Shimizu, Y. Extract from Calotropis procera latex activates murine macrophages. J. Nat. Med. 2009, 63, 297–303. [Google Scholar]
- Barrett, B.; Kieffer, D. Medicinal plants, science, and health care. J. Herbs Spices Med. Plants 2001, 8, 1–36. [Google Scholar] [CrossRef]
- Oluwaniyi, O.O.; Ibiyemi, S.A. Extractability of Thevetia peruviana glycosides with alcohol mixture. Afr. J. Biotechnol. 2007, 6, 2166–2170. [Google Scholar]
- Larhsini, M.; Oumoulid, L.; Lazrek, H.B.; Wataleb, S.; Bousaid, M.; Bekkouche, K.; Jana, M. Antibacterial activity of some Moroccan medicinal plants. Phytother. Res. 2001, 15, 250–252. [Google Scholar] [CrossRef]
- Ibrar, M. Ethobotanic study of the weeds of five crops in district Abbottabad, N-WPakistan. Pak. J. Weed Sci. Res. 2003, 9, 229–240. [Google Scholar]
- Ramos, M.V.; Freitas, C.D.; Stanisçuaski, F.; Macedo, L.L.; Sales, M.P.; Sousa, D.P.; Carlini, C.R. Performance of distinct crop pests reared on diets enriched with latex proteins from Calotropis procera: Role of laticifer proteins in plant defense. Plant Sci. 2007, 173, 349–357. [Google Scholar] [CrossRef]
- Priyamvada, P.M.; Sha, A.; Mohapatra, A.K. Evaluation of antidiabetic and antioxidant activities of Achyranthes aspera leaf extracts: An in vitro study. J. Pharmacogn. Phytochem. 2021, 10, 103–110. [Google Scholar]
- Singh, N.; Jain, N.K.; Kannojia, P.; Garud, N.; Pathak, A.K.; Mehta, S.C. In vitro antioxidant activity of Calotropis gigantea hydroalcohlic leaves extract. Der Pharm. Lett. 2010, 2, 95–100. [Google Scholar]
- Pattnaik, P.K.; Kar, D.; Chhatoi, H.; Shahbazi, S.; Ghosh, G.; Kuanar, A. Chemometric profile & antimicrobial activities of leaf extract of Calotropis procera and Calotropis gigantea. Nat. Prod. Res. 2017, 31, 1954–1957. [Google Scholar] [PubMed]
- Moustafa, A.M.Y.; Ahmed, S.H.; Nabil, Z.I.; Hussein, A.A.; Omran, M.A. Extraction and phytochemical investigation of Calotropis procera: Effect of plant extracts on the activity of diverse muscles. Pharm. Biol. 2010, 48, 1080–1190. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.N.; Rana, S.A.; Mahmood-Ul-Hassan, M.; Rana, N.; Iqbal, M. Phytochemical constituents of weeds: Baseline study in mixed crop zone agroecosystem. Pak. J. Weed Sci. Res. 2013, 19, 231–238. [Google Scholar]
- Dhale, D.A.; Bhoi, S. Pharmacognostic Characterization and Phytochemical Screening of Achyranthes aspera Linn. Curr. Agric. Res. J. 2013, 1, 51. [Google Scholar] [CrossRef]
- Siddhuraju, P.; Becker, K. Antioxidant properties of various solvent extracts of total phenolic constituents from three different agroclimatic origins of drumstick tree (Moringa oleifera Lam.) leaves. J. Agric. Food Chem. 2003, 51, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, D.P.A.; Sivaganesh, S.; Tissera, M.H.A.; Handunnetti, S.M.; Arawwawala, L.D.A.M. Comparison of antioxidant properties of Cyathula prostrata Linn and Achyranthes aspera Linn grown in Sri Lanka. Res. Rev. Insights 2018, 2, 1–3. [Google Scholar] [CrossRef]
- Chopra, S.; Matsuyama, K.; Hutson, C.; Madrid, P. Identification of antimicrobial activity among FDA-approved drugs for combating Mycobacterium abscessus and Mycobacterium chelonae. J. Antimicrob. Chemother. 2011, 66, 1533–1536. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Rani, B.; Gaikwad, N.B.; Ahmad, M.N.; Kaul, G.; Shukla, M.; Nanduri, S.; Dasgupta, A.; Chopra, S.; Yaddanapudi, V.M. Synthesis and structure-activity relationship of new chalcone linked 5-phenyl-3-isoxazolecarboxylic acid methyl esters potentially active against drug resistant Mycobacterium tuberculosis. Eur. J. Med. Chem. 2021, 222, 113580. [Google Scholar] [CrossRef]
- Mészáros, B.; Tóth, J.; Vértessy, B.G.; Dosztányi, Z.; Simon, I. Proteins with complex architecture as potential targets for drug design: A case study of Mycobacterium tuberculosis. PLoS Comput. Biol. 2011, 7, e1002118. [Google Scholar] [CrossRef]
- Beg, M.A.; Hejazi, I.I.; Thakur, S.C.; Athar, F. Domain-wise differentiation of Mycobacterium tuberculosis H37 Rv hypothetical proteins: A roadmap to discover bacterial survival potentials. Biotechnol. Appl. Biochem. 2022, 69, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.M.; Jiang, M.; Guo, Z.; Baker, E.N. Structural and functional analysis of Rv0554 from Mycobacterium tuberculosis: Testing a putative role in menaquinone biosynthesis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 Pt 8, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, A.; Marasco, D.; Squeglia, F.; Soldini, S.; Pedone, E.; Pedone, C.; Berisio, R. Structure and functional regulation of RipA, a mycobacterial enzyme essential for daughter cell separation. Structure 2010, 18, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.-H.; Lee, K.-Y.; Lee, S.J.; Park, S.J.; Yoon, H.-J.; Kim, S.-J.; Lee, B.-J. Structural analyses of the MazEF4 toxin-antitoxin pair in Mycobacterium tuberculosis provide evidence for a unique extracellular death factor. J. Biol. Chem. 2017, 292, 18832–18847. [Google Scholar] [CrossRef] [PubMed]
- Böth, D.; Steiner, E.M.; Izumi, A.; Schneider, G.; Schnell, R. RipD (Rv1566c) from Mycobacterium tuberculosis: Adaptation of an NlpC/p60 domain to a non-catalytic peptidoglycan-binding function. Biochem. J. 2014, 457, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Adolph, R.S.; Gopalakrishnapai, J.; Kleinboelting, S.; Emmerich, C.; Steegborn, C.; Visweswariah, S.S. A universal stress protein (USP) in mycobacteria binds cAMP. J. Biol. Chem. 2015, 290, 12731–12743. [Google Scholar] [CrossRef]
- Das, U.; Pogenberg, V.; Subhramanyam, U.K.; Wilmanns, M.; Gourinath, S.; Srinivasan, A. Crystal structure of the VapBC-15 complex from Mycobacterium tuberculosis reveals a two-metal ion dependent PIN-domain ribonuclease and a variable mode of toxin-antitoxin assembly. J. Struct. Biol. 2014, 188, 249–258. [Google Scholar] [CrossRef]
- Deep, A.; Kaundal, S.; Agarwal, S.; Singh, R.; Thakur, K.G. Crystal structure of Mycobacterium tuberculosis VapC20 toxin and its interactions with cognate antitoxin, VapB20, suggest a model for toxin-antitoxin assembly. FEBS J. 2017, 284, 4066–4082. [Google Scholar] [CrossRef]
- Glass, L.N.; Swapna, G.; Chavadi, S.S.; Tufariello, J.M.; Mi, K.; Drumm, J.E.; Lam, T.T.; Zhu, G.; Zhan, C.; Vilchéze, C.; et al. Mycobacterium tuberculosis universal stress protein Rv2623 interacts with the putative ATP binding cassette (ABC) transporter Rv1747 to regulate mycobacterial growth. PLoS Pathog. 2017, 13, e1006515. [Google Scholar] [CrossRef]
- Jardim, P.; Santos, I.C.; Barbosa, J.A.; de Freitas, S.M.; Valadares, N.F. Crystal structure of VapC21 from Mycobacterium tuberculosis at 1.31 Å resolution. Biochem. Biophys. Res. Commun. 2016, 478, 1370–1375. [Google Scholar] [CrossRef]
- Chen, R.; Zhou, J.; Sun, R.; Du, C.; Xie, W. Conserved Conformational Changes in the Regulation of Mycobacterium tuberculosis MazEF-mt1. ACS Infect. Dis. 2020, 6, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Kapopoulou, A.; Lew, J.M.; Cole, S.T. The MycoBrowser portal: A comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 2011, 91, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.K.; Avashthi, H.; Tiwari, A.; Jain, P.A.; Ramkete, P.W.; Kayastha, A.M.; Singh, V.K. MFPPI—Multi FASTA ProtParam Interface. Bioinformation 2016, 12, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P. VICMpred: An SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genom. Proteom. Bioinform. 2006, 4, 42–47. [Google Scholar] [CrossRef]
- Rashid, M.; Saha, S.; Raghava, G.P. Support Vector Machine-based method for predicting subcellular localization of mycobacterial proteins using evolutionary information and motifs. BMC Bioinform. 2007, 8, 337. [Google Scholar] [CrossRef] [PubMed]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. CABIOS 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Sharma, K.; Singh, P.; Beg, M.A.; Dohare, R.; Athar, F.; Syed, M.A. Revealing new therapeutic opportunities in hypertension through network-driven integrative genetic analysis and drug target prediction approach. Gene 2021, 801, 145856. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Wlodawer, A. Stereochemistry and Validation of Macromolecular Structures. Methods Mol. Biol. 2017, 1607, 595–610. [Google Scholar] [PubMed]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wan, H.; Shi, Y.; Ouyang, P. Personal experience with four kinds of chemical structure drawing software: Review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J. Chem. Inf. Comput. Sci. 2004, 44, 1886–1890. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, T.; Mathur, Y.; Hassan, M.I. InstaDock: A single-click graphical user interface for molecular docking-based virtual high-throughput screening. Brief. Bioinform. 2021, 22, bbaa279. [Google Scholar] [CrossRef]
- Athar, F.; Ansari, S.; Beg, M.A. Molecular docking studies of Calotropis gigantea phytoconstituents against Staphylococcus aureus tyrosyl-tRNA synthetase protein. J. Bacteriol. Mycol. Open Access. 2020, 8, 78–91. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Baswar, D.; Sharma, A.; Mishra, A. In silico Screening of Pyridoxine Carbamates for Anti-Alzheimer’s Activities. Cent. Nerv. Syst. Agents Med. Chem. 2021, 21, 39–52. [Google Scholar] [CrossRef]
- Hejazi, I.I.; Beg, M.A.; Imam, M.A.; Athar, F.; Islam, A. Glossary of phytoconstituents: Can these be repurposed against SARS CoV-2? A quick in silico screening of various phytoconstituents from plant Glycyrrhiza glabra with SARS CoV-2 main protease. Food Chem. Toxicol. 2021, 150, 112057. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 Pt 8, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mohammad, T.; Shamsi, A.; Hussain, A.; Alajmi, M.F.; Husain, S.A.; Iqbal, M.A.; Hassan, M.I. Identification of plant-based hexokinase 2 inhibitors: Combined molecular docking and dynamics simulation studies. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A. Relative solvent accessible surface area predicts protein conformational changes upon binding. Structure 2011, 19, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Ramadwa, T.E.; Awouafack, M.D.; Sonopo, M.S.; Eloff, J.N. Antibacterial and Antimycobacterial Activity of Crude Extracts, Fractions, and Isolated Compounds from Leaves of Sneezewood, Ptaeroxylon obliquum (Rutaceae). Nat. Prod. Commun. 2019, 14, 1–7. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Extracted Fractions of A. aspera Aerial Part (1.5 kg) | EC (in g) | Extracted Fractions A. aspera of Roots (300 g) | EC (in g) |
|---|---|---|---|---|
| 1 | Methanol | 58.85 | Methanol | 39.93 |
| 2 | Aqueous | 54.60 | Aqueous | 35.86 |
| 3 | Hexane | 7.14 | Hexane | 1.82 |
| 4 | Ethyl acetate | 3.18 | Ethyl acetate | 0.93 |
| 5 | Ethanol | 1.73 | Ethanol | 0.46 |
| Extracted fractions of C. procera Flower (200 g) | EC (in g) | Extracted Fractions C. gigantea Flower (200 g) | EC (in g) | |
| 1 | Methanol | 13.26 | Methanol | 11.33 |
| 2 | Aqueous | 10.83 | Aqueous | 9.66 |
| 3 | Hexane | 1.02 | Hexane | 3.82 |
| 4 | Ethyl acetate | 3.56 | Ethyl acetate | 3.33 |
| 5 | Ethanol | 1.20 | Ethanol | 1.26 |
| Extracted fractions C. gigantea Flower (200 g) | EC (in g) | |||
| 1 | Methanol | 12.56 | ||
| 2 | Aqueous | 9.89 | ||
| 3 | Ethyl acetate | 3.69 |
| S. No. | Phytochemicals | Methanol | Aqueous | Ethanol | EtOAc | Hexane |
|---|---|---|---|---|---|---|
| Aerial Part Extracts of Achyranthes aspera | ||||||
| 1 | Alkaloids | + | − | + | − | − |
| 2 | Tannins | + | − | − | + | − |
| 3 | Saponins | − | − | + | + | − |
| 4 | Terpenoids | + | − | + | − | − |
| Roots Extracts of Achyranthes aspera | ||||||
| 1 | Alkaloids | + | + | − | + | − |
| 2 | Tannins | + | − | + | + | − |
| 3 | Saponins | + | + | + | − | − |
| 4 | Terpenoids | − | + | − | + | − |
| Flower Extracts of Calotropis procera | ||||||
| 1 | Alkaloids | + | + | + | − | − |
| 2 | Tannins | + | + | + | + | + |
| 3 | Saponins | + | − | − | − | + |
| 4 | Terpenoids | − | + | − | + | + |
| Flower Extracts of Calotropis gigantea | ||||||
| 1 | Alkaloids | − | + | + | + | − |
| 2 | Tannins | + | − | − | − | − |
| 3 | Saponins | + | + | − | − | + |
| 4 | Terpenoids | + | − | − | + | − |
| S. No. | Rv No. | Name | PDB ID | Information | Ref. |
|---|---|---|---|---|---|
| 1 | 0554 | BpoC | 7LD8 | Possible peroxidase BpoC (Non-essential gene for in vitro growth of H37Rv) | [45] |
| 2 | 1477 | RipA | 4Q4N | Peptidoglycan hydrolase; (essential gene for in vitro growth of H37Rv) | [46] |
| 3 | 1495 | MazF4 | 5XE2 | Possible toxin MazF4 (non-essential gene for in vitro growth of H37Rv) | [47] |
| 4 | 1566c | RipD | 4LJ1 | Possible Inv protein (non-essential gene for in vitro growth of H37Rv) | [48] |
| 5 | 1636 | TB15.3 | 1TQ8 | Iron-regulated universal stress protein family protein TB15.3 (non-essential gene for in vitro growth of H37Rv) | [49] |
| 6 | 2010 | VapC15 | 4CHG | Toxin VapC15 (Non-essential gene for in vitro growth of H37Rv) | [50] |
| 7 | 2549c | VapC20 | 5WZ4 | Possible toxin VapC20 (non-essential gene for in vitro growth of H37Rv) | [51] |
| 8 | 2623 | TB31.7 | 2JAX | Universal stress protein family protein TB31.7 (non-essential gene for in vitro growth of H37Rv) | [52] |
| 9 | 2757c | VapC21 | 5SV2 | Possible toxin VapC21 (non-essential gene for in vitro growth of H37Rv) | [53] |
| 10 | 2801c | MazF9 | 6L2A | Toxin MazF9 (non-essential gene for in vitro growth of H37Rv) | [54] |
| S. No. | Protein | Most Favoured Region | Additional Allowed Region | Generously Allowed Region | Disallowed Region |
|---|---|---|---|---|---|
| 1 | Rv0554 | 92.1% | 7.5% | 0.0% | 0.4% |
| 2 | Rv1477 | 92.5% | 6.9% | 0.0% | 0.6% |
| 3 | Rv1495 | 95.3% | 4.7% | 0.0% | 0.0% |
| 4 | Rv1566c | 98.0% | 2.0% | 0.0% | 0.0% |
| 5 | Rv1636 | 91.0% | 9.0% | 0.0% | 0.0% |
| 6 | Rv2010 | 94.2% | 5.0% | 0.0% | 0.7% |
| 7 | Rv2549c | 95.8% | 4.2% | 0.0% | 0.0% |
| 8 | Rv2623 | 74.5% | 22.7% | 1.8% | 0.9% |
| 9 | Rv2757c | 96.7% | 3.3% | 0.0% | 0.0% |
| 10 | Rv2801c | 94.8% | 5.2% | 0.0% | 0.0% |
| PubChem ID | Name | #M.W. | #Rot. Bond | #HBA | #HBD | LogP |
|---|---|---|---|---|---|---|
| 225689 | Beta-Amyrin | 426 | 0 | 1 | 1 | 4.74 |
| 500213 | Handianol | 426.72 | 4 | 1 | 1 | 5.17 |
| 584269 | - | 472.79 | 5 | 0 | 0 | 5.88 |
| 6436660 | Dehydroergosterol | 394.63 | 4 | 1 | 1 | 4.68 |
| 124061 | Olean-12-ene-3, 22-diol | 442.72 | 0 | 2 | 2 | 4.65 |
| 605144 | - | 468.75 | 3 | 2 | 0 | 4.95 |
| 92158 | Lupenone | 424.70 | 1 | 1 | 0 | 4.54 |
| 345510 | Beta-Amyrenyl acetate | 468.75 | 2 | 2 | 0 | 5.19 |
| 91537342 | 24-Norursa-3, 12-diene | 394.68 | 0 | 0 | 0 | 4.76 |
| 91692798 | Stigmasta-4,7,22-triene-3. alpha.-ol | 410.67 | 5 | 1 | 1 | 4.70 |
| PubChem ID | Absorption | Distribution (BBB/CNS Permeation) | Metabolism (CYP2D6 Inhibitor) | Excretion OCT2 Substrate | Toxicity A/H/S | |
|---|---|---|---|---|---|---|
| GI abs. | W.S. | |||||
| 225689 | 93.733 | −6.531 | No | No | No | No |
| 500213 | 95.248 | −5.762 | No | No | No | No |
| 584269 | 97.43 | −4.664 | No | No | No | No |
| 6436660 | 94.999 | −7.112 | No | No | No | No |
| 124061 | 92.522 | −6.351 | No | No | No | No |
| 605144 | 98.182 | −5.878 | No | No | No | No |
| 92158 | 98.467 | −5.828 | No | No | No | No |
| 345510 | 97.342 | −6.649 | No | No | No | No |
| 91537342 | 95.778 | −6.925 | No | No | No | No |
| 91692798 | 95.604 | −6.696 | No | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beg, M.A.; Shivangi; Afzal, O.; Akhtar, M.S.; Altamimi, A.S.A.; Hussain, A.; Imam, M.A.; Ahmad, M.N.; Chopra, S.; Athar, F. Potential Efficacy of β-Amyrin Targeting Mycobacterial Universal Stress Protein by In Vitro and In Silico Approach. Molecules 2022, 27, 4581. https://doi.org/10.3390/molecules27144581
Beg MA, Shivangi, Afzal O, Akhtar MS, Altamimi ASA, Hussain A, Imam MA, Ahmad MN, Chopra S, Athar F. Potential Efficacy of β-Amyrin Targeting Mycobacterial Universal Stress Protein by In Vitro and In Silico Approach. Molecules. 2022; 27(14):4581. https://doi.org/10.3390/molecules27144581
Chicago/Turabian StyleBeg, Md Amjad, Shivangi, Obaid Afzal, Md Sayeed Akhtar, Abdulmalik S. A. Altamimi, Afzal Hussain, Md Ali Imam, Mohammad Naiyaz Ahmad, Sidharth Chopra, and Fareeda Athar. 2022. "Potential Efficacy of β-Amyrin Targeting Mycobacterial Universal Stress Protein by In Vitro and In Silico Approach" Molecules 27, no. 14: 4581. https://doi.org/10.3390/molecules27144581
APA StyleBeg, M. A., Shivangi, Afzal, O., Akhtar, M. S., Altamimi, A. S. A., Hussain, A., Imam, M. A., Ahmad, M. N., Chopra, S., & Athar, F. (2022). Potential Efficacy of β-Amyrin Targeting Mycobacterial Universal Stress Protein by In Vitro and In Silico Approach. Molecules, 27(14), 4581. https://doi.org/10.3390/molecules27144581