
On the Dynamics of the Carbon–Bromine Bond Dissociation in the 1-Bromo-2-Methylnaphthalene Radical Anion

 ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
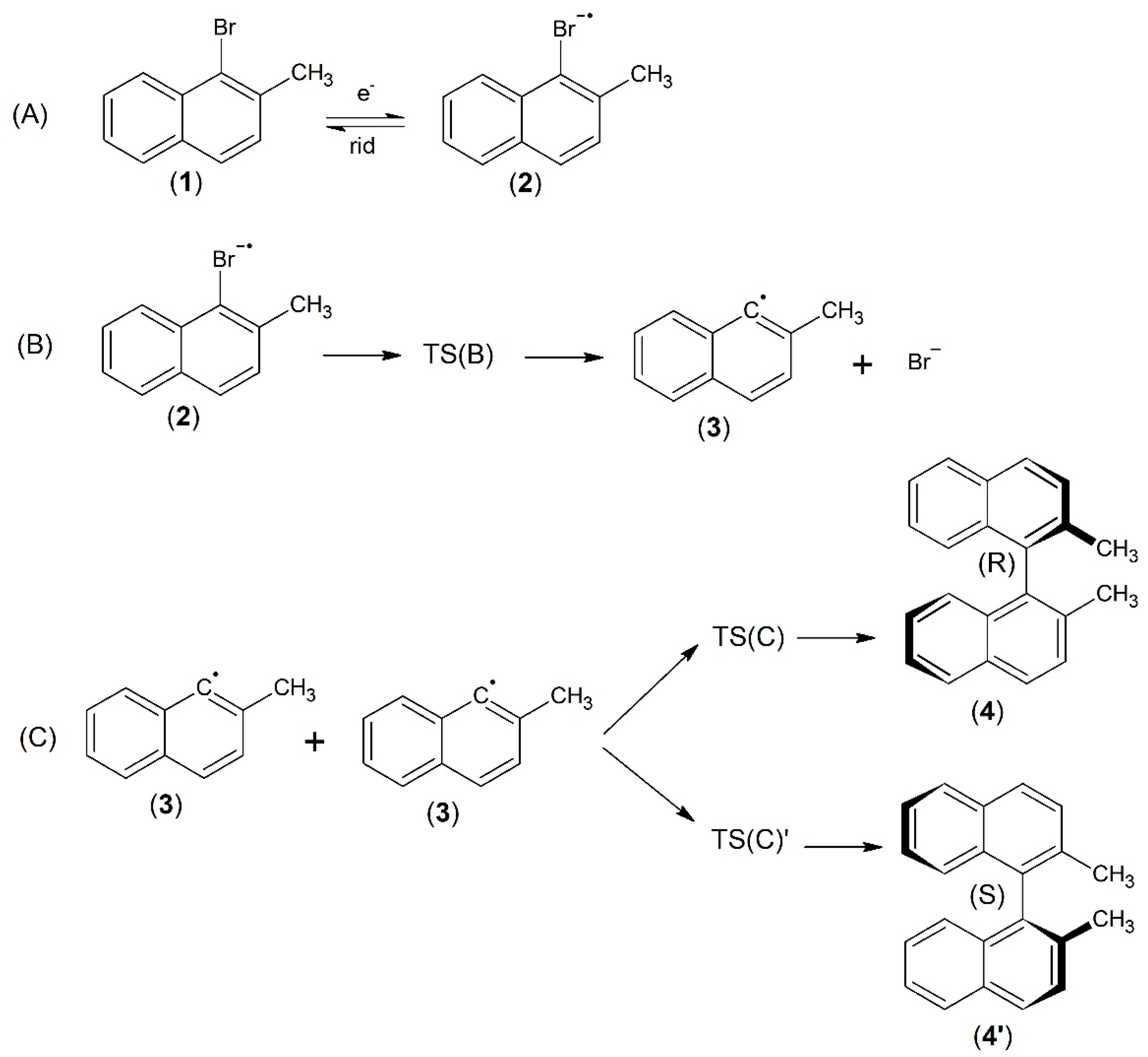
2.1. The Mechanism
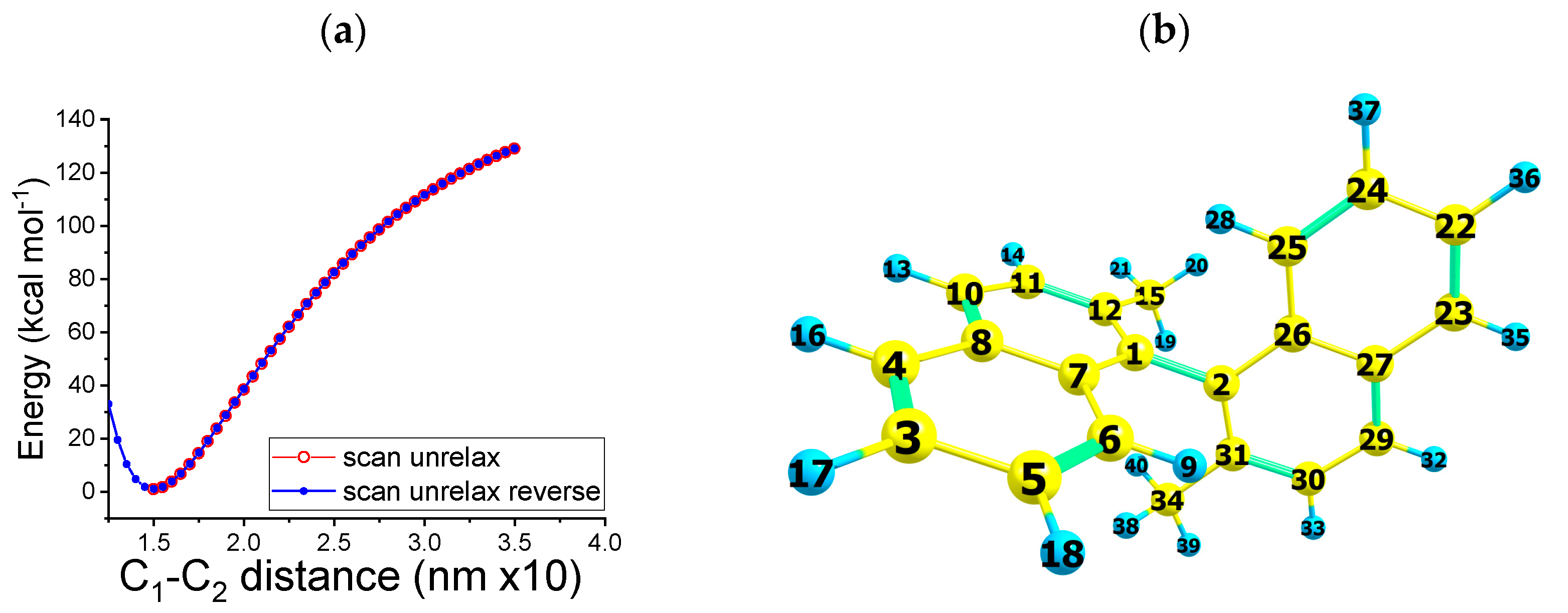
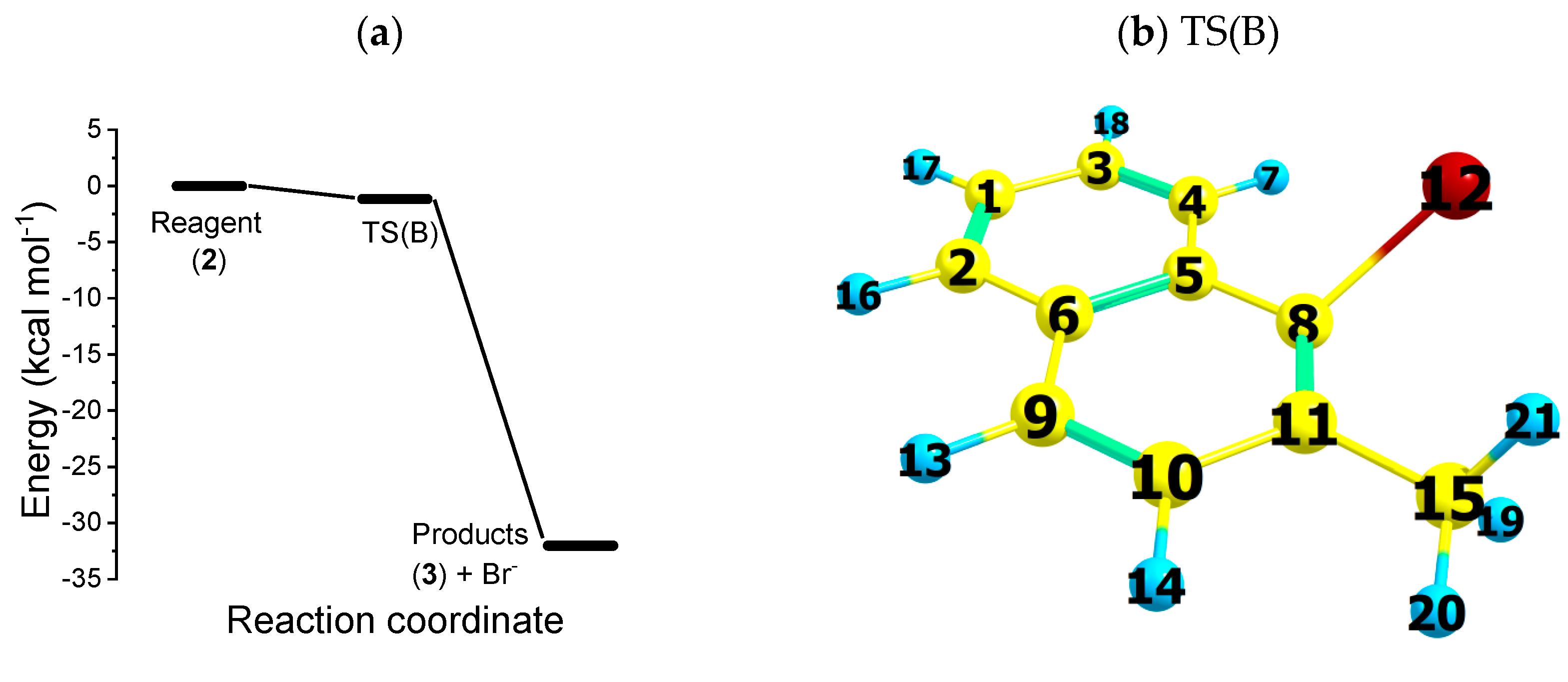
2.2. Reaction Path B
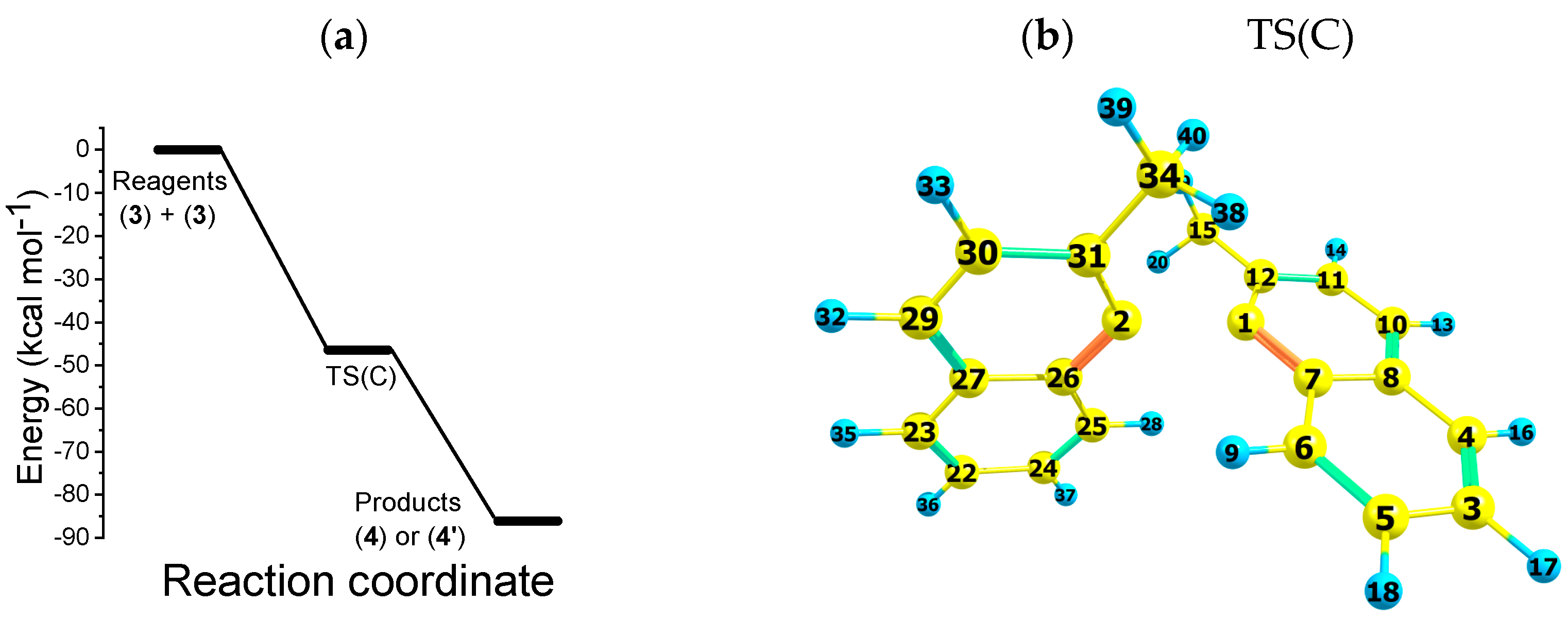
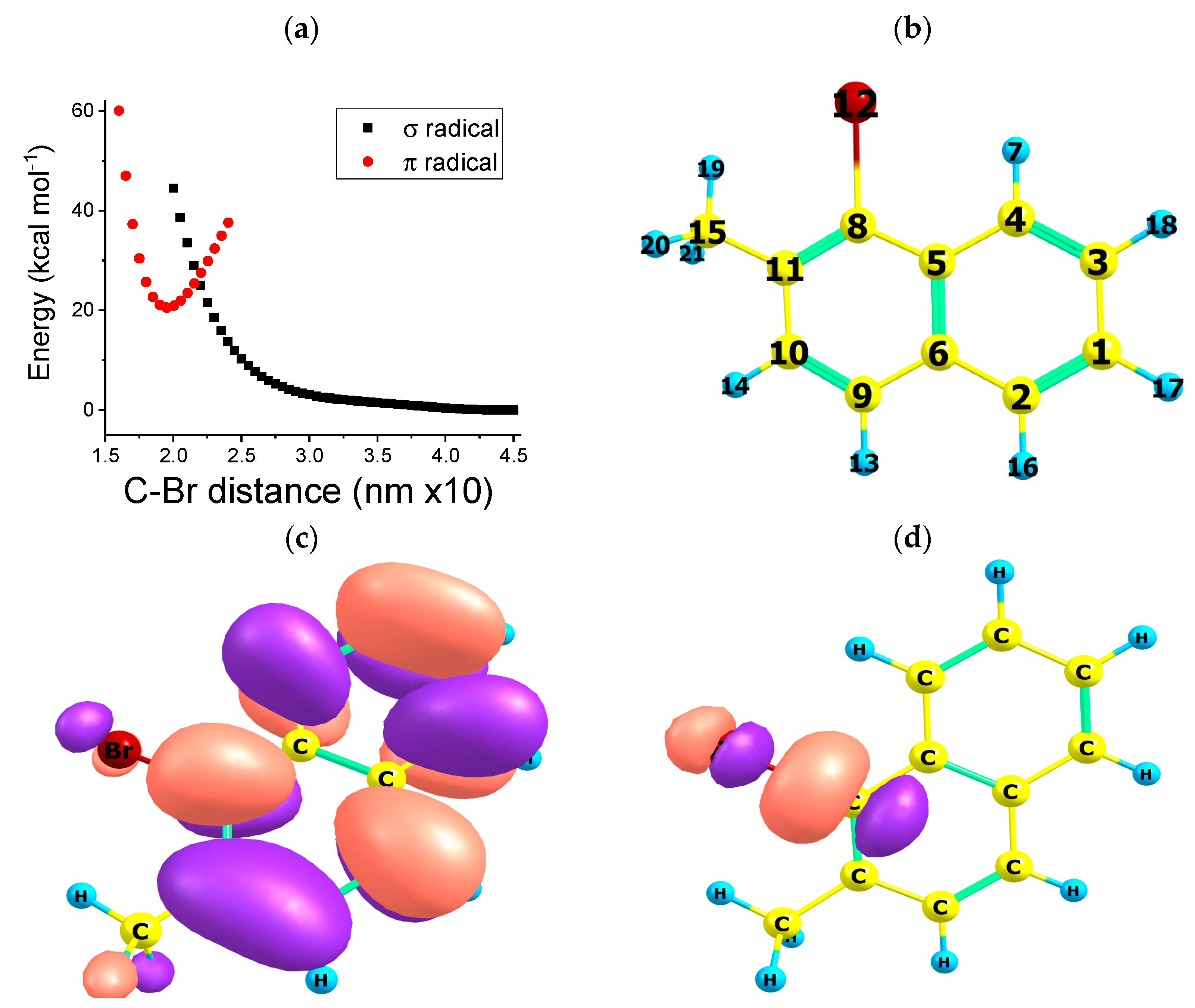
2.3. Path C
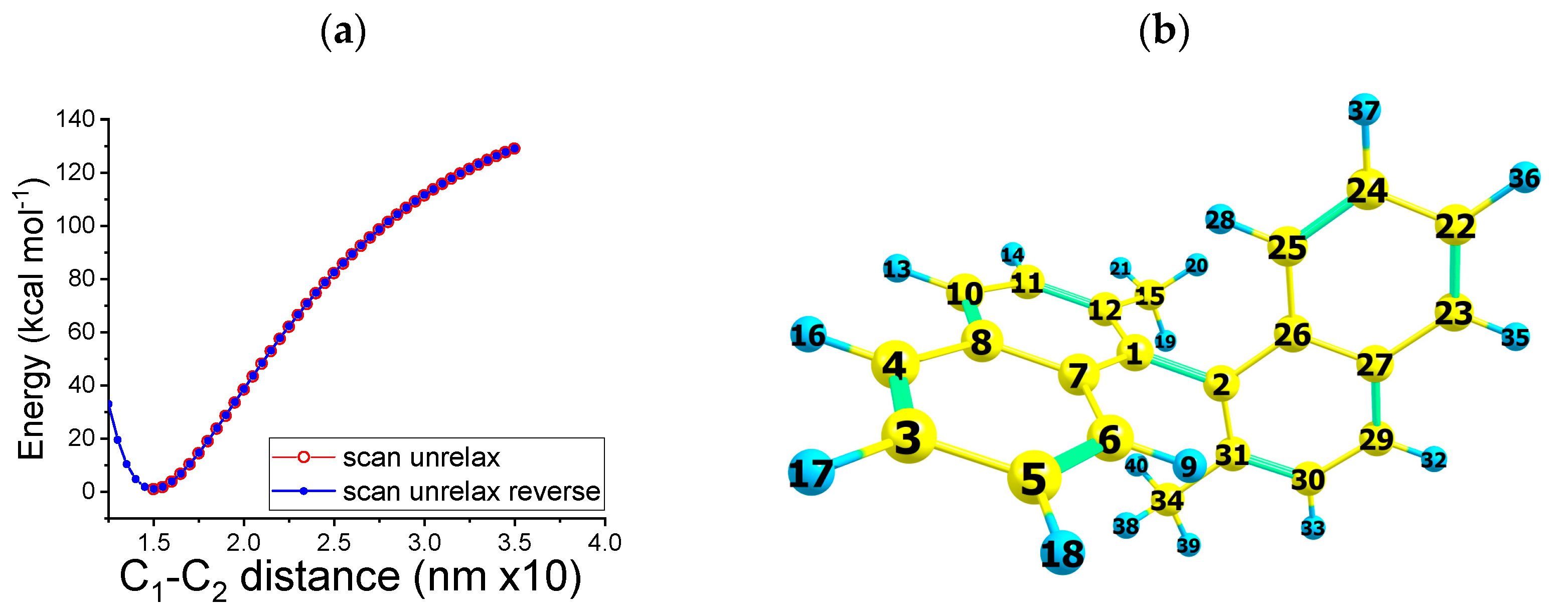
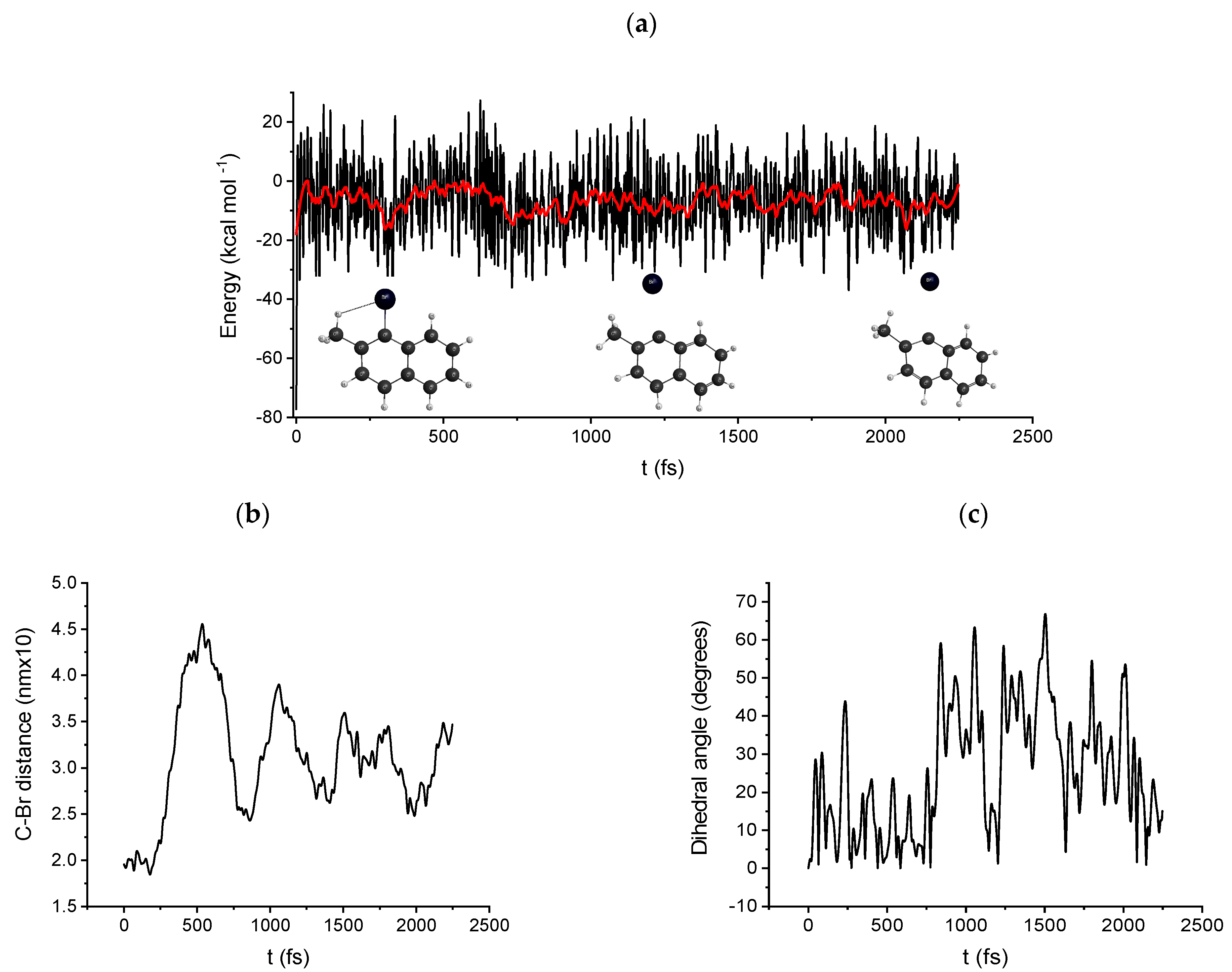
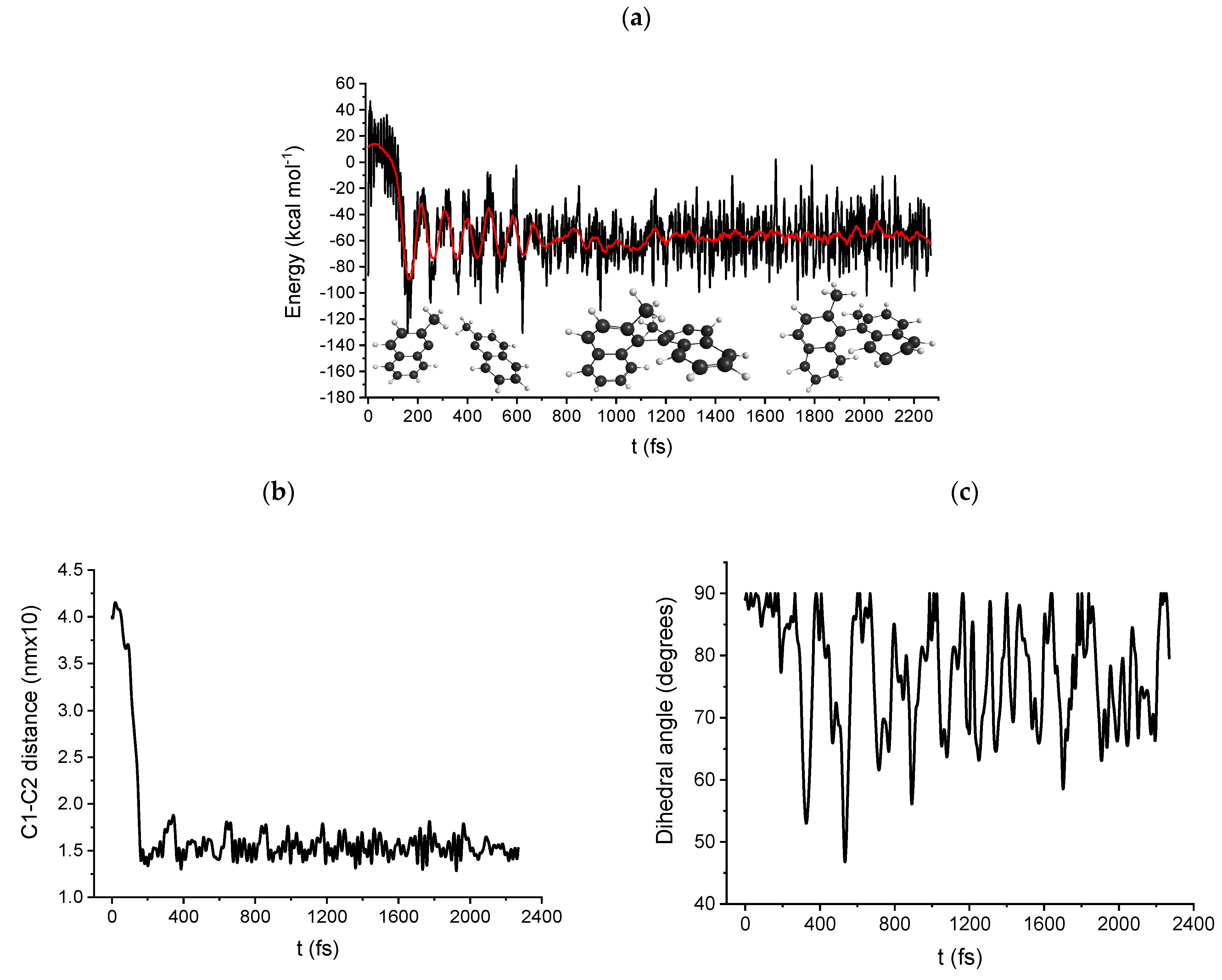
2.4. Molecular Dynamics: Path B and C DRC
3. Methods
4. Conclusions
- (1)
- The one-electron reduction in 1-bromo-2-methylnaphthalene yields a transient radical anion. The latter subsequently undergoes a cleavage of the bond with the formation of the bromide anion and an organic radical.
- (2)
- Indeed, the neutral radicals produced following the dissociation of the radical anion parent species react to form a 1,1′-binaphthalene, 2,2′-dimethyl compound, an intrinsically chiral dimer.
- (3)
- UV-VIS, IR and CD spectra calculated at the UB3LYP/cc-pVTZ level of theory suggest possible experimental methods for monitoring the time evolution of the electrochemical reaction, i.e., by conducting a spectro-electrochemical in situ experiment.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rondinini, S.; Vertova, A. Electroreduction of Halogenated Organic Compounds. In Electrochemistry for the Environment; Comninellis, C., Chen, G., Eds.; Springer: New York, NY, USA, 2010; pp. 279–306. ISBN 978-0-387-36922-8. [Google Scholar]
- Amatore, C. Basic Concepts. In Organic Electrochemistry: Revised and Expanded; Ole Hammerich and Bernd Speiser; CRC Press Taylor & Francis Group: London, UK, 2016. [Google Scholar]
- Martin, E.T.; McGuire, C.M.; Mubarak, M.S.; Peters, D.G. Electroreductive Remediation of Halogenated Environmental Pollutants. Chem. Rev. 2016, 116, 15198–15234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shi, Q.; Song, X.; Wang, H.; Bian, Z. Recent Electrochemical Methods in Electrochemical Degradation of Halogenated Organics: A Review. Environ. Sci. Pollut. Res. 2019, 26, 10457–10486. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, M.A.; Oturan, N.; Oturan, M.A. Electrochemically Assisted Remediation of Pesticides in Soils and Water: A Review. Chem. Rev. 2014, 114, 8720–8745. [Google Scholar] [CrossRef] [PubMed]
- McGrath, T.J.; Ball, A.S.; Clarke, B.O. Critical Review of Soil Contamination by Polybrominated Diphenyl Ethers (PBDEs) and Novel Brominated Flame Retardants (NBFRs); Concentrations, Sources and Congener Profiles. Environ. Pollut. 2017, 230, 741–757. [Google Scholar] [CrossRef]
- Huang, L.; Qian, H.; Deng, S.; Guo, J.; Li, Y.; Zhao, W.; Yue, Y. Urban Residential Indoor Volatile Organic Compounds in Summer, Beijing: Profile, Concentration and Source Characterization. Atmos. Environ. 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Pollok, D.; Waldvogel, S.R. Electro-Organic Synthesis—A 21st Century Technique. Chem. Sci. 2020, 11, 12386–12400. [Google Scholar] [CrossRef]
- Beil, S.B.; Pollok, D.; Waldvogel, S.R. Reproducibility in Electroorganic Synthesis—Myths and Misunderstandings. Angew. Chem. Int. Ed. 2021, 60, 14750–14759. [Google Scholar] [CrossRef]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic Electrosynthesis: A Promising Green Methodology in Organic Chemistry. Green Chem. 2010, 12, 2099. [Google Scholar] [CrossRef]
- Bonechi, M.; Giurlani, W.; Vizza, M.; Savastano, M.; Stefani, A.; Bianchi, A.; Fontanesi, C.; Innocenti, M. On the Oxygen Reduction Reaction Mechanism Catalyzed by Pd Complexes on 2D Carbon. A Theoretical Study. Catalysts 2021, 11, 764. [Google Scholar] [CrossRef]
- Gazzotti, M.; Stefani, A.; Bonechi, M.; Giurlani, W.; Innocenti, M.; Fontanesi, C. Influence of Chiral Compounds on the Oxygen Evolution Reaction (OER) in the Water Splitting Process. Molecules 2020, 25, 3988. [Google Scholar] [CrossRef]
- Lei, C.; Liang, F.; Li, J.; Chen, W.; Huang, B. Electrochemical Reductive Dechlorination of Chlorinated Volatile Organic Compounds (Cl-VOCs): Effects of Molecular Structure on the Dehalogenation Reactivity and Mechanisms. Chem. Eng. J. 2019, 358, 1054–1064. [Google Scholar] [CrossRef]
- Rowlands, G.J. Radicals in Organic Synthesis: Part 2. Tetrahedron 2010, 66, 1593–1636. [Google Scholar] [CrossRef]
- Metzger, T.S.; Mishra, S.; Bloom, B.P.; Goren, N.; Neubauer, A.; Shmul, G.; Wei, J.; Yochelis, S.; Tassinari, F.; Fontanesi, C.; et al. The Electron Spin as a Chiral Reagent. Angew. Chem. Int. Ed. 2020, 59, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Kudaş, Z.; Gür, E.; Ekinci, D. Synthesis of Graphene-like Films by Electrochemical Reduction of Polyhalogenated Aromatic Compounds and Their Electrochemical Capacitor Applications. Langmuir 2018, 34, 7958–7970. [Google Scholar] [CrossRef]
- Fontanesi, C. Theoretical Study of the Dissociative Process of the 4-Chlorotoluene Radical Anion. J. Mol. Struct. 1997, 392, 87–94. [Google Scholar] [CrossRef]
- Fontanesi, C.; Baraldi, P.; Marcaccio, M. On the Dissociation Dynamics of the Benzyl Chloride Radical Anion. An Ab Initio Dynamic Reaction Coordinate Analysis Study. J. Mol. Struct. 2001, 548, 13–20. [Google Scholar] [CrossRef]
- Neukermans, S.; Vorobjov, F.; Kenis, T.; De Wolf, R.; Hereijgers, J.; Breugelmans, T. Electrochemical Reduction of Halogenated Aromatic Compounds at Metal Cathodes in Acetonitrile. Electrochim. Acta 2020, 332, 135484. [Google Scholar] [CrossRef]
- Savéant, J.-M. Single Electron Transfer and Nucleophilic Substitution. In Advances in Physical Organic Chemistry; Bethell, D., Ed.; Academic Press: Cambridge, MA, USA, 1990; Volume 26, pp. 1–130. [Google Scholar]
- Cardinale, A.; Isse, A.A.; Gennaro, A.; Robert, M.; Savéant, J.-M. Dissociative Electron Transfer to Haloacetonitriles. An Example of the Dependency of In-Cage Ion-Radical Interactions upon the Leaving Group. J. Am. Chem. Soc. 2002, 124, 13533–13539. [Google Scholar] [CrossRef]
- Andrieux, C.P.; Savéant, J.-M.; Tallec, A.; Tardivel, R.; Tardy, C. Concerted and Stepwise Dissociative Electron Transfers. Oxidability of the Leaving Group and Strength of the Breaking Bond as Mechanism and Reactivity Governing Factors Illustrated by the Electrochemical Reduction of α-Substituted Acetophenones. J. Am. Chem. Soc. 1997, 119, 2420–2429. [Google Scholar] [CrossRef]
- Zhuikov, V.V. Step Wise and Dissociative Mechanisms of the Electron Transfer in Electrochemical Reactions Involving Organosilicon Compounds: Molecular-Thermodynamic Approach1. Russ. J. Electrochem. 2000, 36, 117–127. [Google Scholar] [CrossRef]
- Bonechi, M.; Innocenti, M.; Vanossi, D.; Fontanesi, C. The Fundamental and Underrated Role of the Base Electrolyte in the Polymerization Mechanism. The Resorcinol Case Study. J. Phys. Chem. A 2021, 125, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Stefani, A.; Giurlani, W.; Bonechi, M.; Marchetti, A.; Preda, G.; Pasini, D.; Innocenti, M.; Fontanesi, C. On the Savéant’s Concerted/Stepwise Model. The Electroreduction of Halogenated Naphthalene Derivatives as a Case Study. ChemElectroChem 2021, 8, 4337–4344. [Google Scholar] [CrossRef]
- Jaworski, J.S. Estimation of Bond Energy in Radical Anions of Arylmethyl Halides as the Controlling Factor of Stepwise versus Concerted Reductive Cleavage. J. Electroanal. Chem. 1996, 407, 237–241. [Google Scholar] [CrossRef]
- Andrieux, C.P.; Differding, E.; Robert, M.; Saveant, J.M. Controlling Factors of Stepwise versus Concerted Reductive Cleavages. Illustrative Examples in the Electrochemical Reductive Breaking of Nitrogen-Halogen Bonds in Aromatic N-Halosultams. J. Am. Chem. Soc. 1993, 115, 6592–6599. [Google Scholar] [CrossRef]
- Bruno, C.; Benassi, R.; Passalacqua, A.; Paolucci, F.; Fontanesi, C.; Marcaccio, M.; Jackson, E.A.; Scott, L.T. Electrochemical and Theoretical Investigation of Corannulene Reduction Processes. J. Phys. Chem. B 2009, 113, 1954–1962. [Google Scholar] [CrossRef]
- Aquilanti, V.; Coutinho, N.D.; Carvalho-Silva, V.H. Kinetics of Low-Temperature Transitions and a Reaction Rate Theory from Non-Equilibrium Distributions. Phil. Trans. R. Soc. A 2017, 375, 20160201. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P.; Davis, L.P.; Burggraf, L.W. Semi-Empirical Calculations of Molecular Trajectories: Method and Applications to Some Simple Molecular Systems. J. Comput. Chem. 1987, 8, 1117–1123. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. J Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2017. [Google Scholar]
- Granovsky, A. Firefly; 2016. Available online: http://classic.chem.msu.su/ (accessed on 11 July 2022).
- Andrienko, G.A. Chemcraft-Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com/index.html (accessed on 11 July 2022).
- Bode, B.M.; Gordon, M.S. Macmolplt: A Graphical User Interface for GAMESS. J. Mol. Graph. Model. 1998, 16, 133–138. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. The Atoms Aluminum through Argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.K.; van Mourik, T.; Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. VI. Sextuple Zeta Correlation Consistent Basis Sets for Boron through Neon. J. Mol. Struct. 1996, 388, 339–349. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Binning, R.C.; Curtiss, L.A. Compact Contracted Basis Sets for Third-Row Atoms: Ga-Kr. J. Comput. Chem. 1990, 11, 1206–1216. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Model Chemistry. I. The Total Energies of Closed-shell Atoms and Hydrides of the First-row Elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A Complete Basis Set Model Chemistry. II. Open-shell Systems and the Total Energies of the First-row Atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods II. Applications. J. Comput. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonechi, M.; Giurlani, W.; Innocenti, M.; Pasini, D.; Mishra, S.; Giovanardi, R.; Fontanesi, C. On the Dynamics of the Carbon–Bromine Bond Dissociation in the 1-Bromo-2-Methylnaphthalene Radical Anion. Molecules 2022, 27, 4539. https://doi.org/10.3390/molecules27144539
Bonechi M, Giurlani W, Innocenti M, Pasini D, Mishra S, Giovanardi R, Fontanesi C. On the Dynamics of the Carbon–Bromine Bond Dissociation in the 1-Bromo-2-Methylnaphthalene Radical Anion. Molecules. 2022; 27(14):4539. https://doi.org/10.3390/molecules27144539
Chicago/Turabian StyleBonechi, Marco, Walter Giurlani, Massimo Innocenti, Dario Pasini, Suryakant Mishra, Roberto Giovanardi, and Claudio Fontanesi. 2022. "On the Dynamics of the Carbon–Bromine Bond Dissociation in the 1-Bromo-2-Methylnaphthalene Radical Anion" Molecules 27, no. 14: 4539. https://doi.org/10.3390/molecules27144539
APA StyleBonechi, M., Giurlani, W., Innocenti, M., Pasini, D., Mishra, S., Giovanardi, R., & Fontanesi, C. (2022). On the Dynamics of the Carbon–Bromine Bond Dissociation in the 1-Bromo-2-Methylnaphthalene Radical Anion. Molecules, 27(14), 4539. https://doi.org/10.3390/molecules27144539