
TLNPMD: Prediction of miRNA-Disease Associations Based on miRNA-Drug-Disease Three-Layer Heterogeneous Network


 ,
,
Abstract
:1. Introduction
2. Results
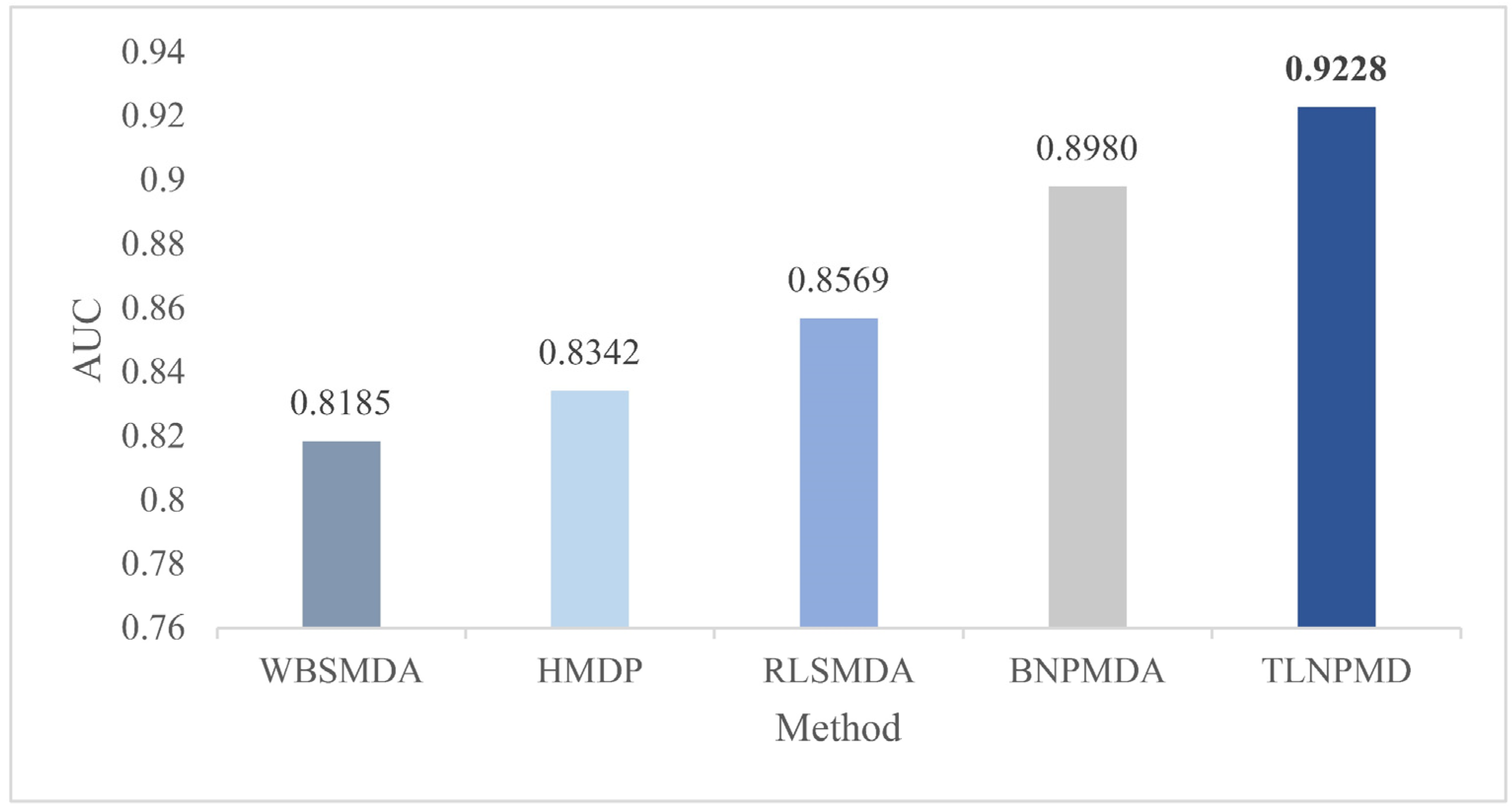
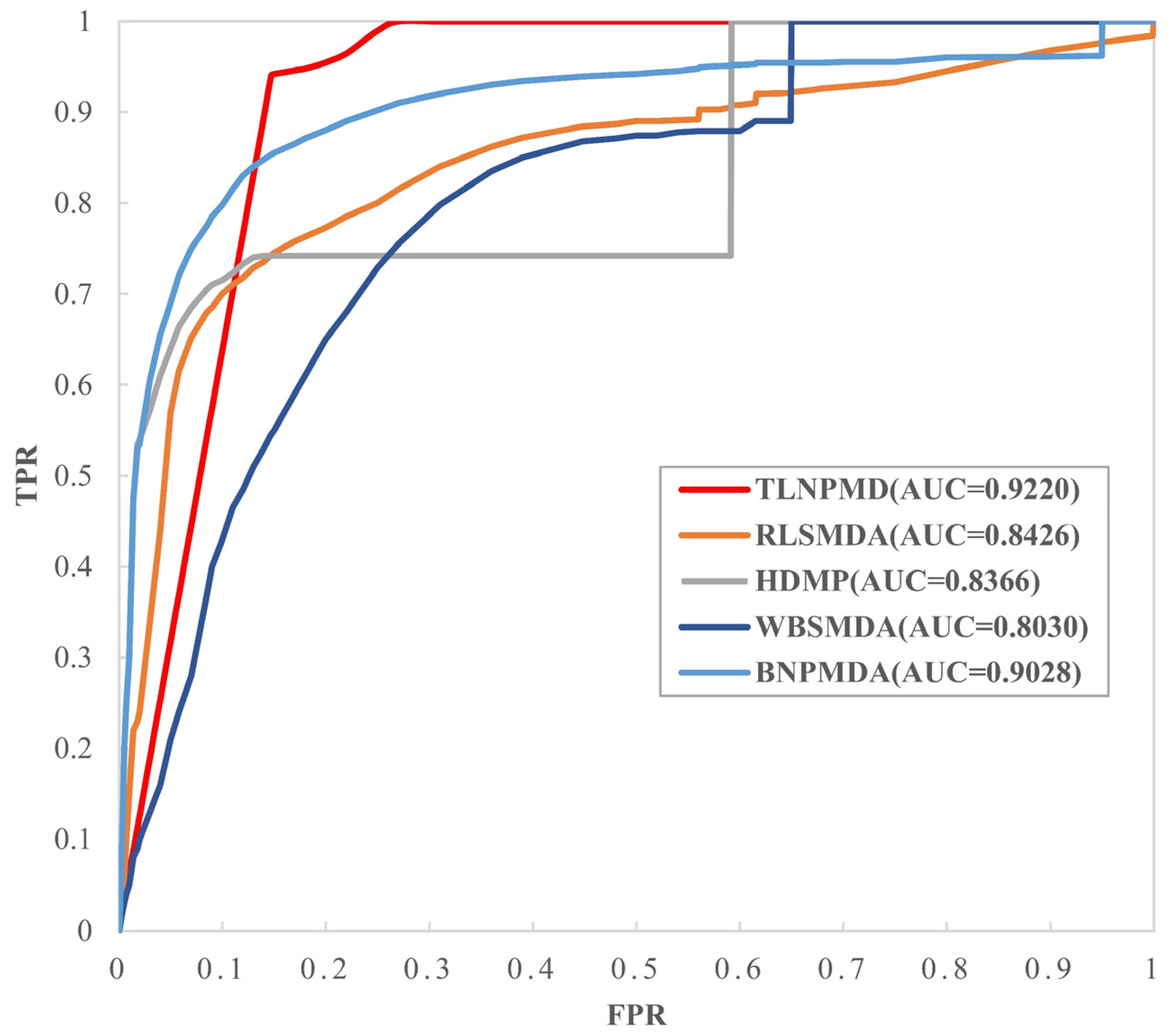
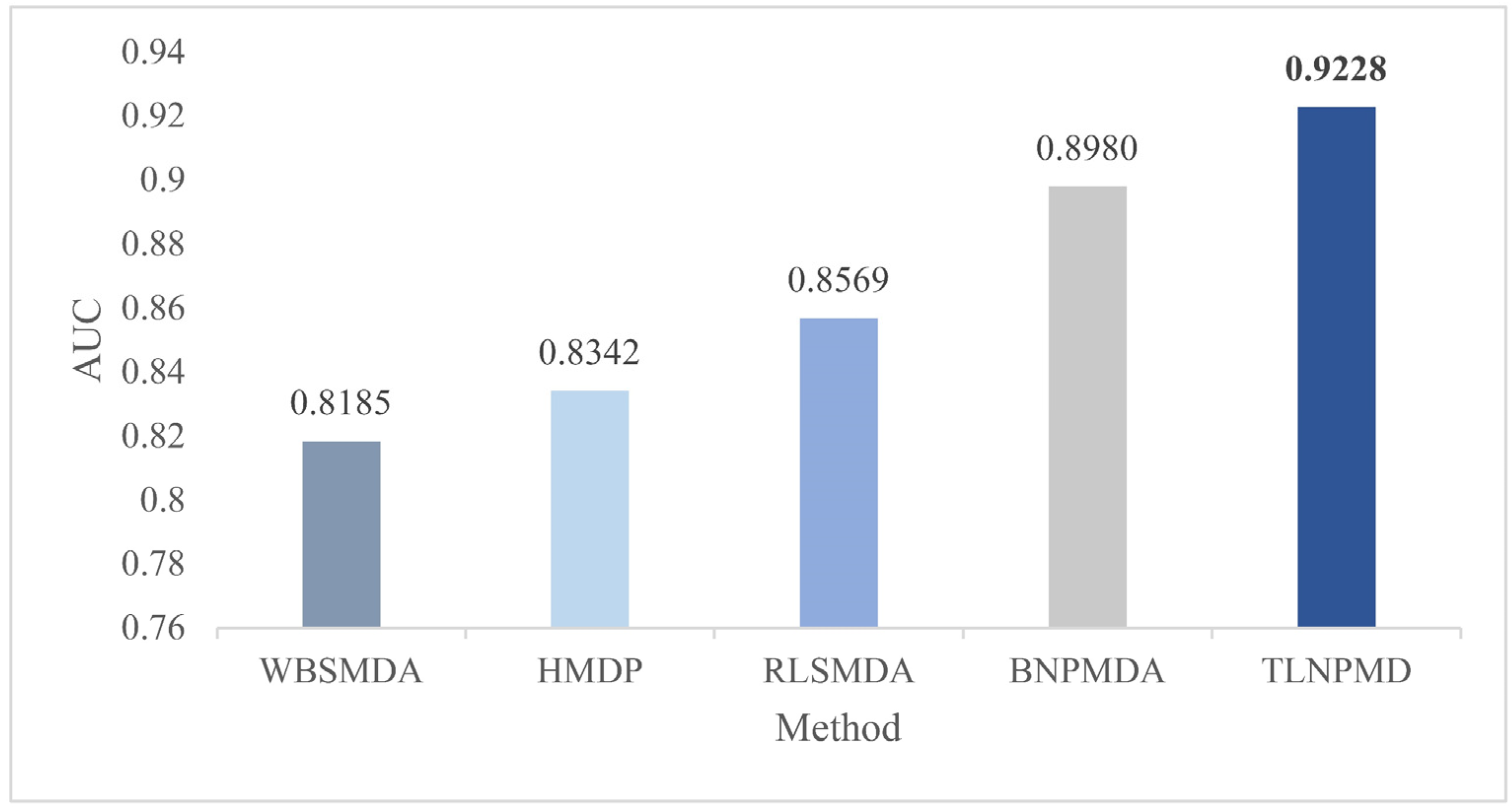
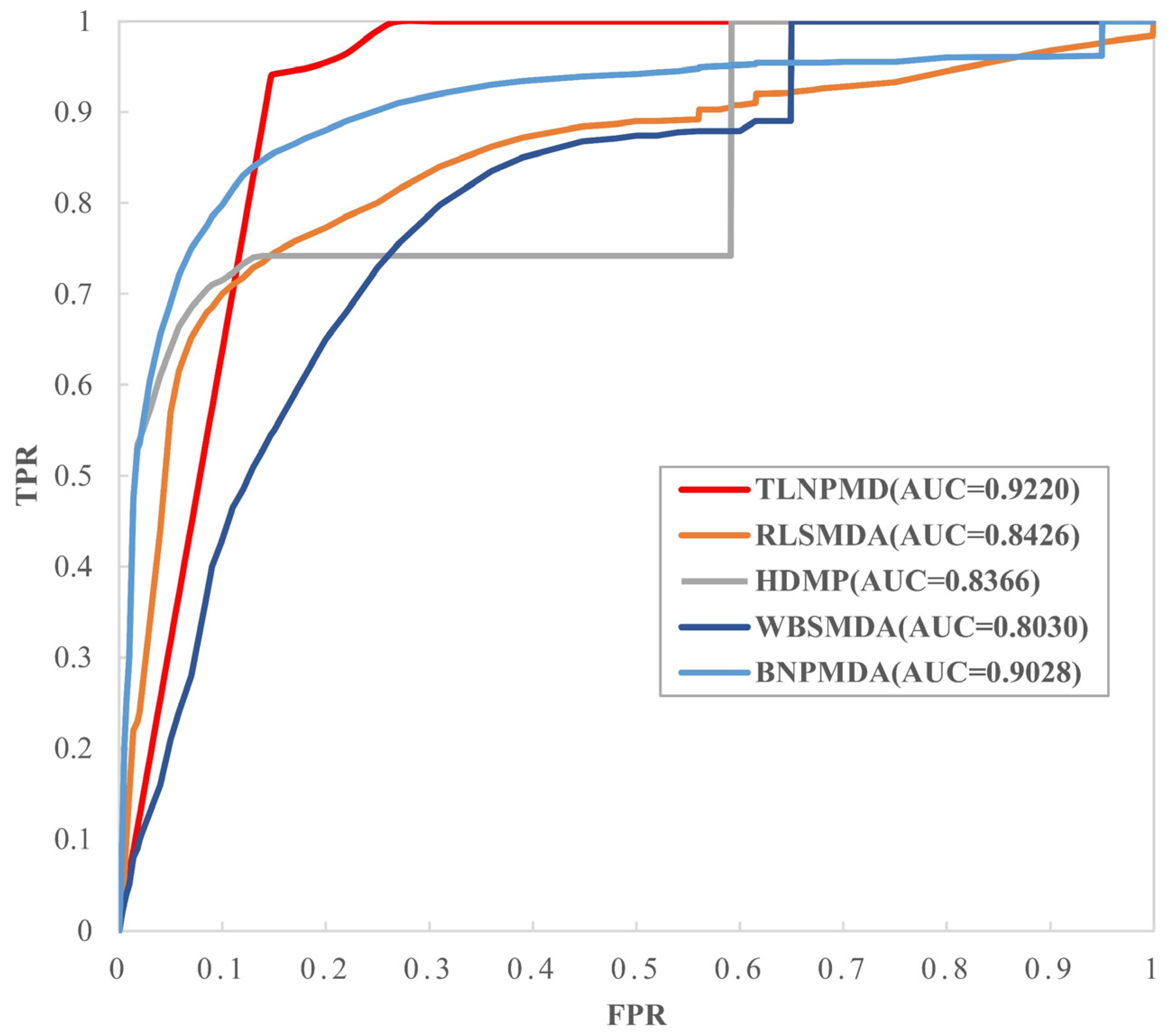
2.1. Evaluation Metrics and Performance Comparison
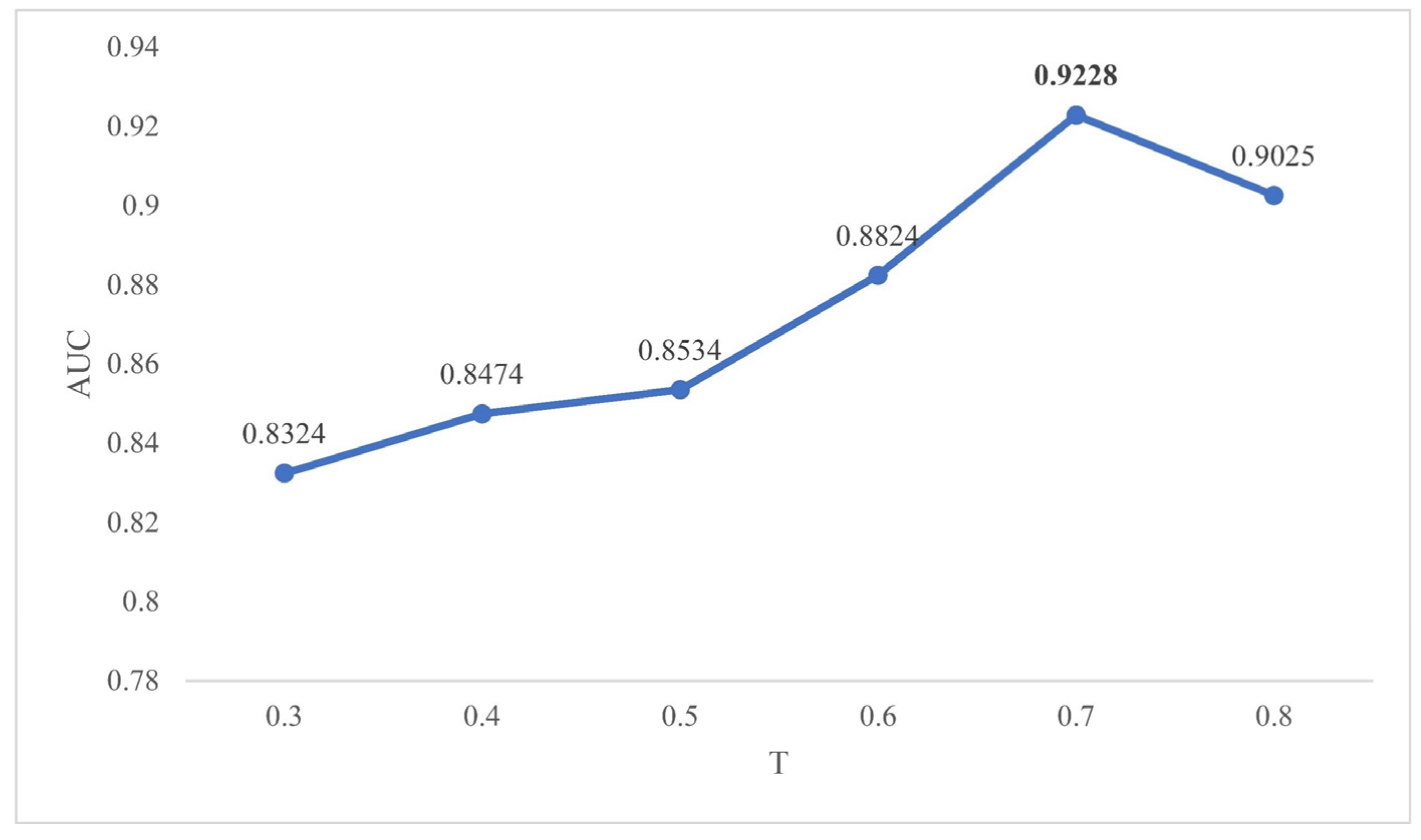
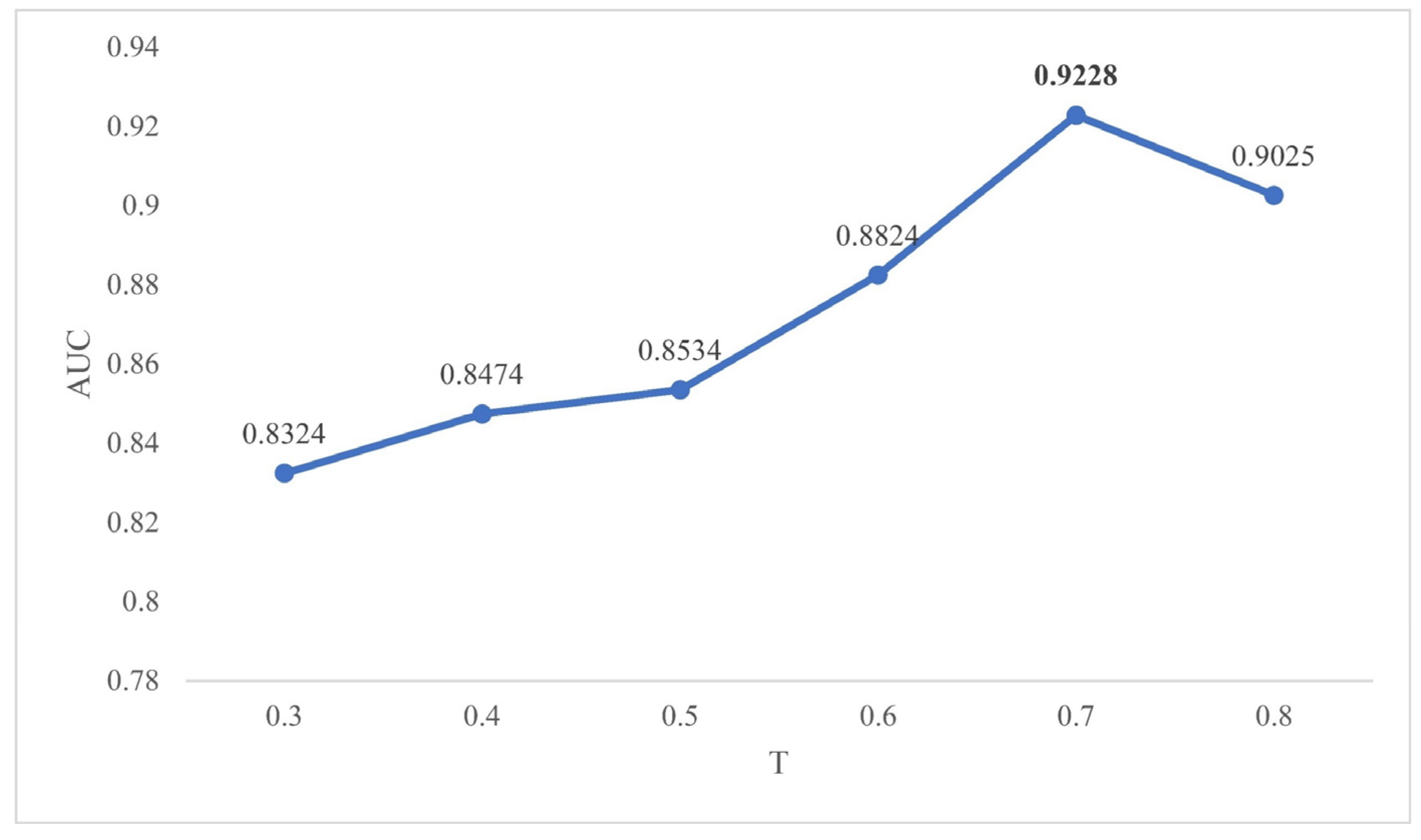
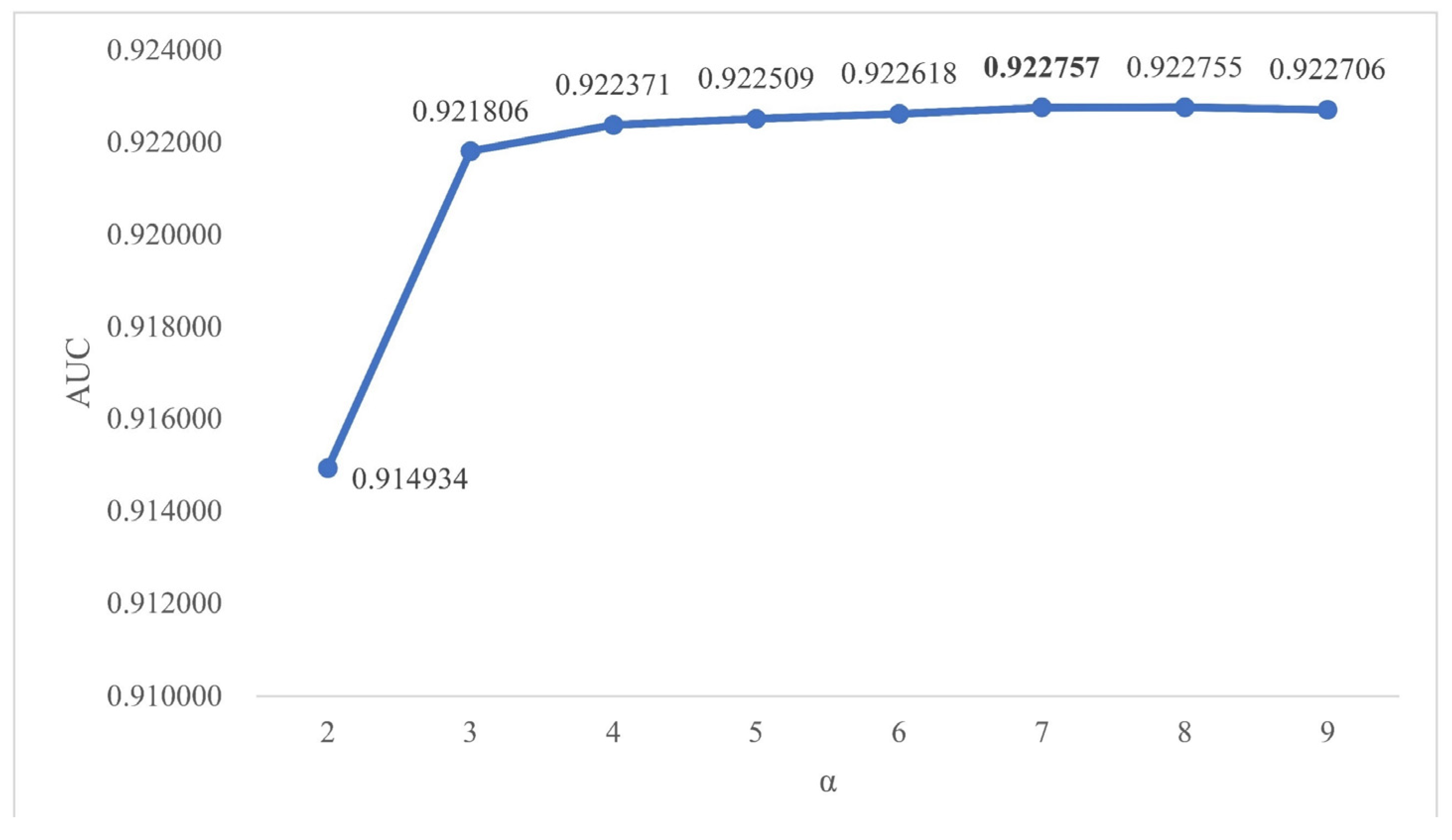
2.2. Effects of Parameters
2.3. Case Study
3. Discussion
4. Materials and Methods
4.1. The Space of MiRNA
4.2. The Space of Drug
4.3. The Space of Disease
4.3.1. Model 1
4.3.2. Model 2
4.4. The Space of Interaction
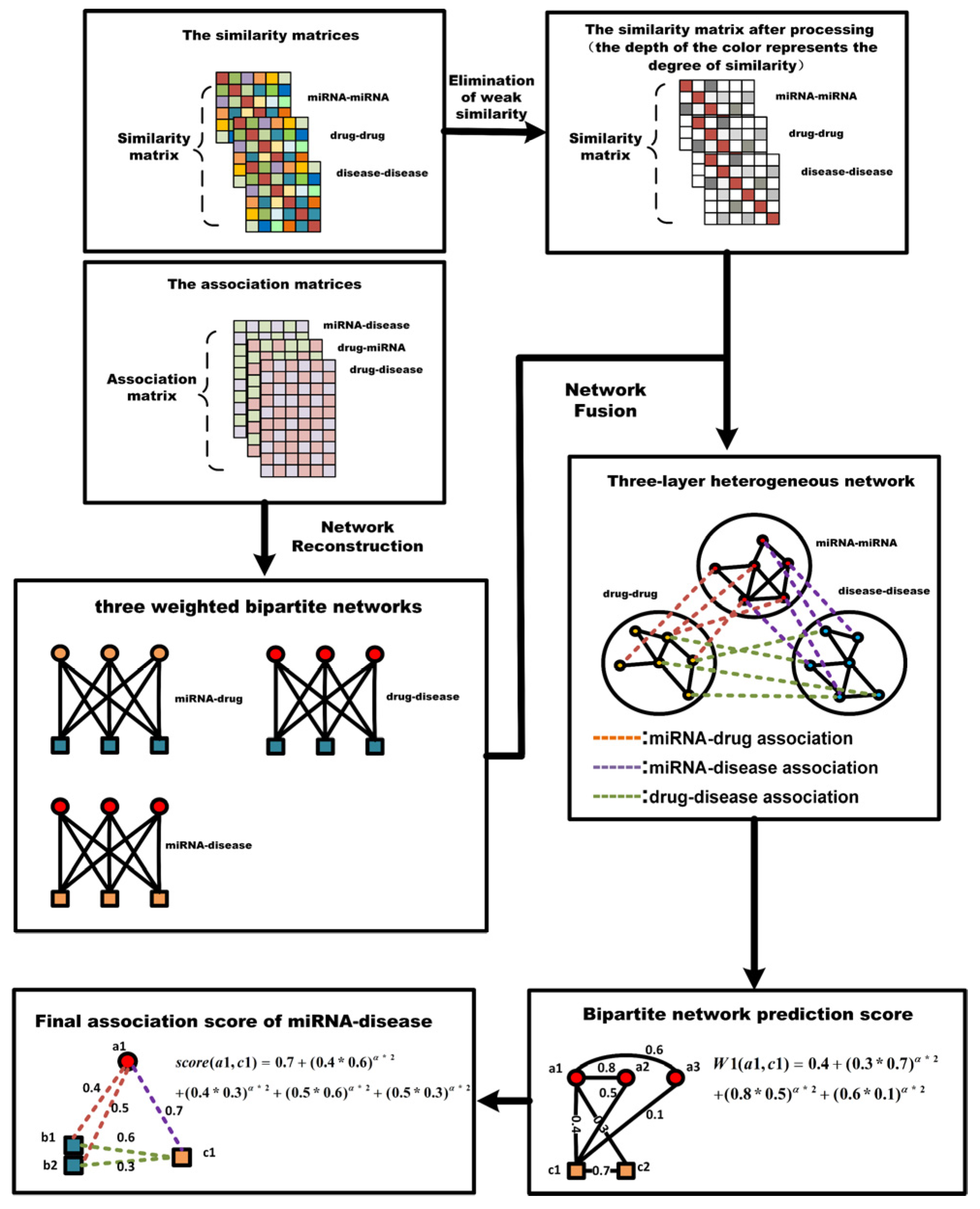
4.5. Network Reconstruction
4.6. Three-Layer Heterogeneous Network Construction
4.7. Prediction of M-DAs
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pelosi, A.; Careccia, S.; Lulli, V.; Romania, P.; Marziali, G.; Testa, U.; Lavorgna, S.; Lo-Coco, F.; Petti, M.C.; Calabretta, B.; et al. miRNA let-7cpromotes granulocytic differentiation in acute myeloid leukemia. Oncogene 2012, 32, 3648–3654. (In English) [Google Scholar] [CrossRef] [PubMed]
- Hergenreider, E.; Heydt, S.; Tréguer, K.; Boettger, T.; Horrevoets, A.J.D.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012, 14, 249. (In English) [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Huang, X.; Zhang, X.; Roensch, K.; Cao, Q.; Nakayama, K.; Blazar, B.R.; Zeng, Y.; Zhou, X. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood J. Am. Soc. Hematol. 2011, 117, 4293–4303. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartig, S.M.; Hamilton, M.P.; Bader, D.A.; Mcguire, S.E. The miRNA Interactome in Metabolic Homeostasis. Trends Endocrinol. Metab. 2016, 26, 733–745. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2008, 19, 92–105. (In English) [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, 68–73. (In English) [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, Y.; Geng, G.; Yu, J. Delay-Optimized Multicast Tree Packing in Software-Defined Networks. IEEE Trans. Serv. Comput. 2021, 1–14. [Google Scholar] [CrossRef]
- Xuan, P.; Li, L.; Zhang, T.; Zhang, Y.; Song, Y. Prediction of Disease-related microRNAs through Integrating Attributes of mi-croRNA Nodes and Multiple Kinds of Connecting Edges. Molecules 2019, 24, 3099. (In English) [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Xu, J.; Zhang, G.; Xu, L.; Xia, L. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst. Biol. 2013, 7, 101. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z.; et al. Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS ONE 2013, 8, e70204. (In English) [Google Scholar] [CrossRef]
- Chen, X.; Yan, G.Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 2014, 4, 5501. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.H.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci. Rep. 2016, 6, 21106. (In English) [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.H.; Huang, Y.A.; Yan, G.Y. HGIMDA: Heterogeneous graph inference for miR-NA-disease association prediction. Oncotarget 2016, 7, 65257–65269. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Kuang, L.; Feng, X.; Zou, Q.; Wang, L. A Novel Approach Based on a Weighted Interactive Network to Predict Associations of MiRNAs and Diseases. Int. J. Mol. Sci. 2018, 20, 110. (In English) [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.H.; Liu, H. BNPMDA: Bipartite network projection for MiRNA–Disease association prediction. Bioinformatics 2018, 34, 3178–3186. (In English) [Google Scholar] [CrossRef] [Green Version]
- Laiyi, F.; Qinke, Q. A deep ensemble model to predict miRNA-disease association. Sci. Rep. 2017, 7, 14482. (In English) [Google Scholar]
- Chen, X.; Gong, Y.; Zhang, D.H.; You, Z.H.; Li, Z.W. DRMDA: Deep representations-based miRNA–disease association prediction. J. Cell. Mol. Med. 2017, 22, 472–485. (In English) [Google Scholar] [CrossRef]
- Chen, X.; Li, T.H.; Zhao, Y.; Wang, C.C.; Zhu, C.C. Deep-belief network for predicting potential miRNA-disease associations. Brief. Bioinform. 2020, 22, bbaa186. (In English) [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Yin, J. Adaptive boosting-based computational model for predicting potential miRNA-disease associations. Bioinformatics 2019, 35, 4730–4738. (In English) [Google Scholar] [CrossRef]
- Nguyen, P.; Ma, S.; Phua, C.; Kaya, N.; Chew, V. Intratumoural immune heterogeneity as a hallmark of tumour evolution and progression in hepatocellular carcinoma. Nat. Commun. 2021, 12, 227. (In English) [Google Scholar] [CrossRef]
- Peter, R.G.; Alejandro, F. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. (In English) [Google Scholar]
- Shen, D.; Zhao, H.Y.; Gu, A.D.; Wu, Y.H.; Weng, Y.H.; Li, S.J.; Song, J.Y.; Gu, X.F.; Qiu, J.; Zhao, W. miRNA-10a-5p inhibits cell metastasis in hepatocellular carcinoma via targeting SKA1. Kaohsiung. J. Med. Sci. 2021, 37, 784–794. (In English) [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Liu, S.; Zhang, W.; Jia, B.; Tan, L.; Jin, Z.; Liu, Y. miR-494 promotes cell proliferation, migration and invasion, and increased sorafenib resistance in hepatocellular carcinoma by targeting PTEN. Oncol. Rep. 2015, 34, 1003–1010. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122-A key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Laversanne, M.; Brewster, D.H.; Mbalawa, C.G.; Kohler, B.; PiEros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Antoni, S. Cancer Incidence in Five Continents: Inclusion criteria, highlights from Volume X and the global status of cancer registration. Int. J. Cancer 2015, 137, 2060–2071. (In English) [Google Scholar] [CrossRef]
- Fang, Q.; Qiong, W.; Zhang, X. Analysis of the Prevalence of the Breast Cancer. Chin. J. Soc. Med. 2013, 295, 333–335. (In English) [Google Scholar]
- Mansoori, B.; Duijf, P.H.; Mohammadi, A.; Safarzadeh, E.; Ditzel, H.J.; Gjerstorff, M.F.; Cho, W.C.-S.; Baradaran, B. MiR-142-3p targets HMGA2 and suppresses breast cancer malignancy. Life Sci. 2021, 276, 119431. (In English) [Google Scholar] [CrossRef]
- Silvia, G.; Federica, V.; Stefano, C.; Federica, B.; Valeria, B. Targeting the Vav1/miR29b axis as a potential approach for treating selected molecular subtypes of triplenegative breast cancer. Oncol. Rep. 2021, 45, 7. (In English) [Google Scholar]
- Cui, Q. Inferring the human microRNA functional similarity and functional network based on mi-croRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. (In English) [Google Scholar]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for Chemo and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. (In English) [Google Scholar] [CrossRef] [Green Version]
- Lipscomb, C. Medical Subject Headings (MeSH). Bull. Med. Libr. Assoc. 2000, 88, 265–266. (In English) [Google Scholar] [PubMed]
- Chen, X.; Yan, C.; Luo, C.; Ji, W.; Zhang, Y.; Dai, Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci. Rep. 2015, 5, 11338. (In English) [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Sharan, R. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2012, 7, 496. (In English) [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. McKusick’s Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res. 2009, 37, D793–D796. (In English) [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Z. Prediction of Drug-Disease Associations for Drug Repositioning Through Drug-miRNA-Disease Heterogeneous Network. IEEE Access 2018, 6, 45281–45287. (In English) [Google Scholar] [CrossRef]
- You, Z.H.; Huang, Z.A.; Zhu, Z.; Yan, G.Y.; Li, Z.W.; Wen, Z.; Chen, X. PBMDA: A novel and effective path-based compu-tational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005455. (In English) [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Evidence |
|---|---|
| ‘mir-515’ | HMDD v3.2 |
| ‘mir-520a’ | HMDD v3.2; miRcancer |
| ‘mir-520h’ | HMDD v3.2 |
| ‘mir-526a’ | HMDD v3.2 |
| ‘mir-330′ | HMDD v3.2 |
| ‘mir-512′ | HMDD v3.2 |
| ‘mir-520e’ | HMDD v3.2 |
| ‘mir-526b’ | HMDD v3.2; miRcancer |
| ‘mir-297’ | HMDD v3.2 |
| ‘mir-325’ | HMDD v3.2; miRcancer |
| ‘mir-520f’ | miRcancer |
| ‘mir-520g’ | HMDD v3.2 |
| ‘mir-136’ | HMDD v3.2 |
| ‘mir-300’ | HMDD v3.2 |
| ‘mir-507’ | unconfirm |
| ‘mir-523’ | unconfirm |
| ‘mir-525′ | HMDD v3.2 |
| ‘mir-331’ | HMDD v3.2; miRcancer |
| ‘mir-658’ | unconfirm |
| ‘mir-134’ | HMDD v3.2; miRcancer |
| miRNA | Evidence |
|---|---|
| ‘mir-519b’ | miRcancer |
| ‘mir-922’ | HMDD v3.2 |
| ‘mir-92’ | HMDD v3.2; miRcancer |
| ‘mir-1254’ | HMDD v3.2 |
| ‘mir-630’ | HMDD v3.2; miRcancer |
| ‘mir-624’ | unconfirm |
| ‘mir-369’ | unconfirm |
| ‘mir-661’ | HMDD v3.2 |
| ‘mir-329’ | miRcancer |
| ‘mir-134’ | HMDD v3.2; miRcancer |
| ‘mir-574’ | HMDD v3.2; miRcancer |
| ‘mir-124a’ | HMDD v3.2; miRcancer |
| ‘mir-516a’ | HMDD v3.2 |
| ‘mir-516b’ | HMDD v3.2 |
| ‘mir-197’ | HMDD v3.2 |
| ‘mir-324’ | HMDD v3.2 |
| ‘mir-629’ | HMDD v3.2 |
| ‘mir-337’ | unconfirm |
| ‘mir-662’ | unconfirm |
| ‘mir-486’ | HMDD v3.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Shang, J.; Sun, Y.; Li, F.; Zhang, Y.; Kong, X.-Z.; Li, S.; Liu, J.-X. TLNPMD: Prediction of miRNA-Disease Associations Based on miRNA-Drug-Disease Three-Layer Heterogeneous Network. Molecules 2022, 27, 4371. https://doi.org/10.3390/molecules27144371
Yang Y, Shang J, Sun Y, Li F, Zhang Y, Kong X-Z, Li S, Liu J-X. TLNPMD: Prediction of miRNA-Disease Associations Based on miRNA-Drug-Disease Three-Layer Heterogeneous Network. Molecules. 2022; 27(14):4371. https://doi.org/10.3390/molecules27144371
Chicago/Turabian StyleYang, Yi, Junliang Shang, Yan Sun, Feng Li, Yuanyuan Zhang, Xiang-Zhen Kong, Shengjun Li, and Jin-Xing Liu. 2022. "TLNPMD: Prediction of miRNA-Disease Associations Based on miRNA-Drug-Disease Three-Layer Heterogeneous Network" Molecules 27, no. 14: 4371. https://doi.org/10.3390/molecules27144371
APA StyleYang, Y., Shang, J., Sun, Y., Li, F., Zhang, Y., Kong, X.-Z., Li, S., & Liu, J.-X. (2022). TLNPMD: Prediction of miRNA-Disease Associations Based on miRNA-Drug-Disease Three-Layer Heterogeneous Network. Molecules, 27(14), 4371. https://doi.org/10.3390/molecules27144371