
Homology Modeling and Molecular Docking Approaches for the Proposal of Novel Insecticides against the African Malaria Mosquito (Anopheles gambiae)




Abstract
:1. Introduction
2. Materials and Methods
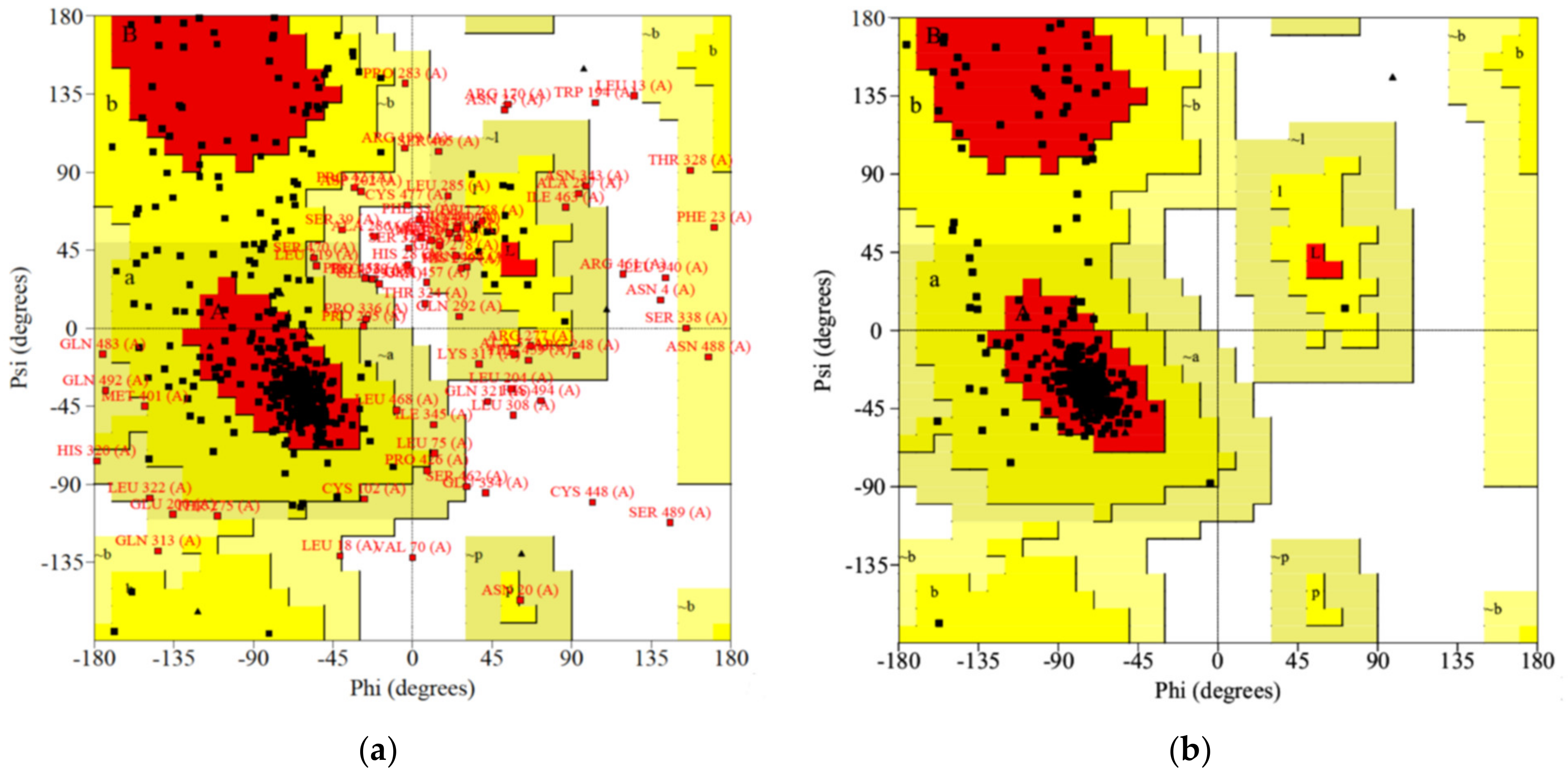
2.1. Homology Modeling
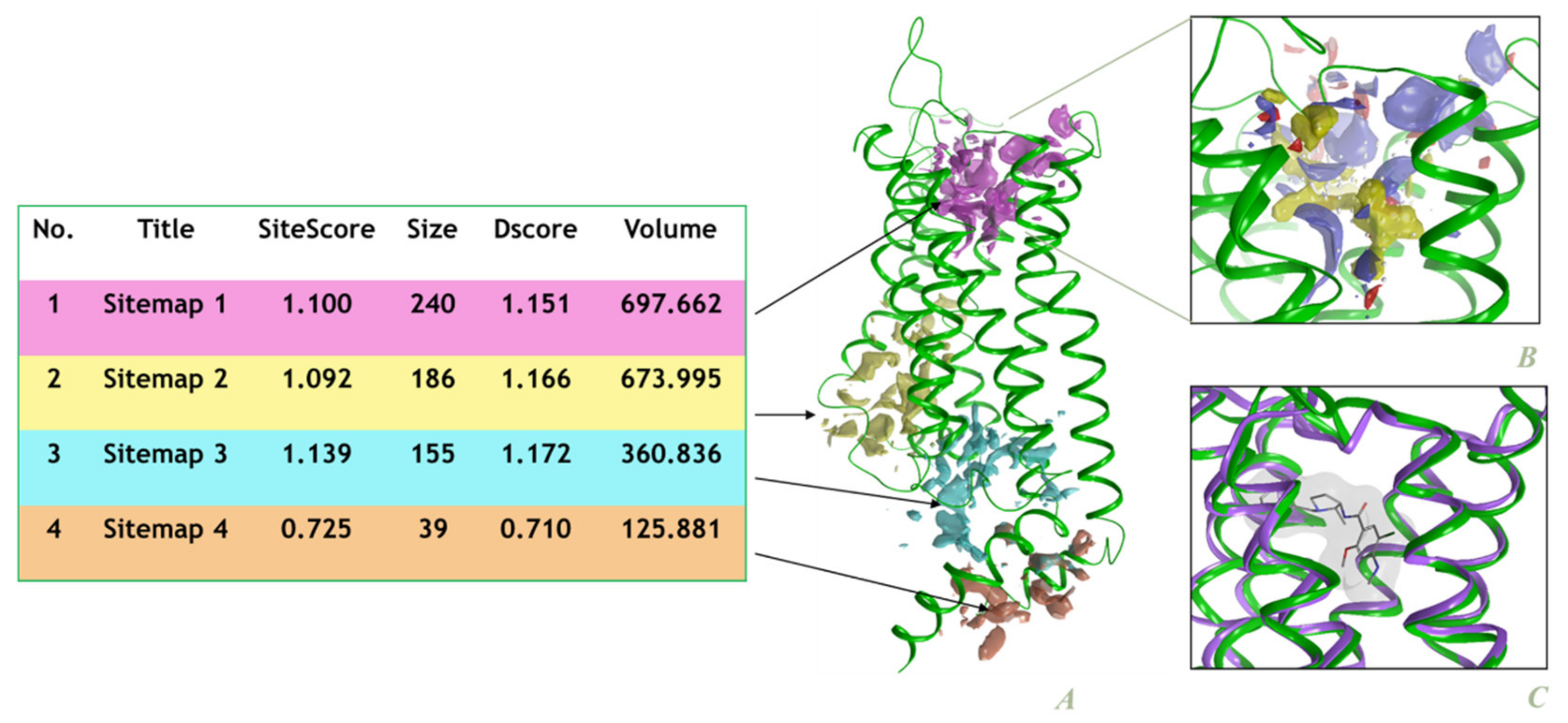
2.2. Site Identification
2.3. Docking Protocol
2.4. Insecticide-Likeness Prediction
2.5. Toxicity and Environmental Hazard Predictions
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Factsheet. Vector Borne Diseases. 2017. Available online: https://www.who.int/en/news-room/fact-sheets/detail/vector-borne-diseases (accessed on 19 April 2022).
- Sinka, M.E.; Bangs, M.J.; Manguin, S.; Coetzee, M.; Mbogo, C.M.; Hemingway, J.; Patil, A.P.; Temperley, W.H.; Gething, P.W.; Kabaria, C.W.; et al. The dominant Anopheles vectors of human malaria in Africa, Europe and the Middle East: Occurrence data, distribution maps and bionomic précis. Parasites Vectors 2010, 3, 117–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. World Malaria Report 2019; WHO, Ed.; WHO: Geneva, Switzerland, 2019; ISBN 978-92-4-156572-1. Available online: https://www.who.int/publications/i/item/9789241565721 (accessed on 12 March 2020).
- Opondo, K.O.; Jawara, M.; Cham, S.; Jatta, E.; Jarju, L.; Camara, M.; Sanneh, F.; Gaye, P.M.; Jadama, L.; Ceesay, S.; et al. Status of insecticide resistance in Anopheles gambiae (s.l.) of The Gambia. Parasites Vectors 2019, 12, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemingway, J. The role of vector control in stopping the transmission of malaria: Threats and opportunities. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharan, S.; Hill, C.A. Potential of GPCR-Targeting Insecticides for Control of Arthropod Vectors. In Advances in Agrochemicals: Ion Channels and G Protein-Coupled Receptors (GPCRs) as Targets for Pest Control, Volume 2: GPCRs and Ion Channels; Gross, A.D., Ozoe, Y., Coats, J.R., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2017; Volume 1265, pp. 55–84. [Google Scholar]
- Mustard, J.A.; Beggs, K.T.; Mercer, A.R. Molecular biology of the invertebrate dopamine receptors. Arch. Insect Biochem. Physiol. 2005, 59, 103–117. [Google Scholar] [CrossRef]
- Meyer, J.M.; Ejendal, K.F.K.; Avramova, L.V.; Garland-Kuntz, E.E.; Giraldo-Calderon, G.I.; Brust, T.F.; Watts, V.J.; Hill, C.A. A “genome-to-lead” approach for insecticide discovery: Pharmacological characterization and screening of Aedes aegypti D1-like dopamine receptors. PLoS Negl. Trop. Dis. 2012, 6, e1478. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, S.; Rende, E.; Crisanti, A.; Nolan, T. Disruption of aminergic signaling reveals novel compounds with distinct inhibitory effects on mosquito reproduction, locomotor function and survival. Sci. Rep.-UK 2014, 4, 5526. [Google Scholar] [CrossRef] [Green Version]
- Bai, H.; Zhu, F.; Shah, K.; Palli, S.R. Large-scale RNAi screen of G protein-coupled receptors involved in larval growth, molting and metamorphosis in the red flour beetle. BMC Genom. 2011, 12, 388. [Google Scholar] [CrossRef] [Green Version]
- Nuss, A.B.; Ejendal, K.F.K.; Doyle, T.B.; Meyer, J.M.; Lang, E.G.; Watts, V.J.; Hill, C.A. Dopamine receptor antagonists as new mode-of-action insecticide leads for control of Aedes and Culex mosquito vectors. PLoS Negl. Trop. Dis. 2015, 9, e0003515. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Espinoza, S.; Gainetdinov, R.R. Dopamine receptors-IUPHAR Review 13. Br. J. Pharmacol. 2015, 172, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.M.; Mai, T.L.; Chen, C.M. Visualizing the GPCR network: Classification and evolution. Sci. Rep. 2017, 7, 15495. [Google Scholar] [CrossRef]
- Hill, C.A.; Doyle, T.; Nuss, A.B.; Ejendal, K.F.K.; Meyer, J.M.; Watts, V.J. Comparative pharmacological characterization of D1-like dopamine receptors from Anopheles gambiae, Aedes aegypti and Culex quinquefasciatus suggests pleiotropic signaling in mosquito vector lineages. Parasites Vectors 2016, 9, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.A.; Meyer, J.M.; Ejendal, K.F.K.; Echeverry, D.F.; Lang, E.G.; Avramova, L.V.; Conley, J.M.; Watts, V.J. Re-invigorating the insecticide discovery pipeline for vector control: GPCRs as targets for the identification of next gen insecticides. Pestic. Biochem. Phys. 2013, 106, 141–148. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem. 2004, 25, 865–871. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-1: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, 2016.
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-1; Prime, Schrödinger, LLC: New York, NY, USA, 2019.
- Akhter, M.; Tasleem, M.; Mumtaz Alam, M.; Ali, S. In silico approach for bioremediation of arsenic by structure prediction and docking studies of arsenite oxidase from Pseudomonas stutzeri TS44. Int. Biodeterior. Biodegrad. 2017, 122, 82–91. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- FRED 3.5.0.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2020. Available online: www.eyesopen.com (accessed on 26 March 2022).
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible shape-guided docking for pose prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Crisan, L.; Borota, A.; Bora, A.; Pacureanu, L. Diarylthiazole and diarylimidazole selective COX-1 inhibitor analysis through pharmacophore modeling, virtual screening, and DFT-based approaches. Struct. Chem. 2019, 30, 2311–2326. [Google Scholar] [CrossRef]
- Funar-Timofei, S.; Borota, A.; Crisan, L. Combined molecular docking and QSAR study of fused heterocyclic herbicide inhibitors of D1 protein in photosystem II of plants. Mol. Divers. 2017, 21, 437–454. [Google Scholar] [CrossRef]
- OMEGA 4.0.0.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2020. Available online: http://www.eyesopen.com (accessed on 26 March 2022).
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and the Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- MakeReceptor 3.2.0.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2019. Available online: http://www.eyesopen.com (accessed on 26 March 2022).
- Schrödinger Release 2022-1; LigPrep, Schrödinger, LLC: New York, NY, USA, 2021.
- BIOVIA Discovery Studio Visualizer, version 20.1.0.19295; Dassault Systèmes: San Diego, CA, USA, 2019; Available online: http://www.3dsbiovia.com (accessed on 26 March 2022).
- Avram, S.; Funar-Timofei, S.; Borota, A.; Chennamaneni, S.R.; Manchala, A.K.; Muresan, S. Quantitative estimation of pesticide-likeness for agrochemical discovery. J. Cheminformatics 2014, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Instant JChem, Version 21.20.0; ChemAxon: Budapest, Hungary, 2021. Available online: http://www.chemaxon.com (accessed on 26 March 2022).
- Banerjee, P.; Eckert, O.A.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://tox-new.charite.de/protox_II/index.php?site=compound_input (accessed on 26 April 2021).
- Available online: http://chemyang.ccnu.edu.cn/ccb/server/beetox/index.php/prediction/index (accessed on 16 March 2022).
- Wang, F.; Yang, J.F.; Wang, M.Y.; Jia, C.Y.; Shi, X.X.; Hao, G.F.; Yang, G.F. Graph attention convolutional neural network model for chemical poisoning of honey bees’ prediction. Sci. Bull. 2020, 65, 1184–1191. [Google Scholar] [CrossRef]
- US EPA. Estimation Programs Interface Suite™ for Microsoft Windows; version 4.11; United States Environmental Protection Agency: Washington, DC, USA, 2012.
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Tasleem, M.; Alrehaily, A.; Almeleebia, T.M.; Alshahrani, M.Y.; Ahmad, I.; Asiri, M.; Alabdallah, N.M.; Saeed, M. Investigation of Antidepressant Properties of Yohimbine by Employing Structure-Based Computational Assessments. Curr. Issues Mol. Biol. 2021, 43, 127. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wacker, D.; Levit, A.; Che, T.; Betz, R.M.; McCorvy, J.D.; Venkatakrishnan, A.J.; Huang, X.P.; Dror, R.O.; Shoichet, B.K.; et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 2017, 358, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. A Protocol for Computer-Based Protein Structure and Function Prediction. J. Vis. Exp. 2011, 57, e3259. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Villoutreix, B.O.; Ecker, G.F. Rigorous sampling of docking poses unveils binding hypothesis for the halogenated ligands of L-type Amino Acid Transporter 1 (LAT1). Sci. Rep.-UK 2019, 9, 15061. [Google Scholar] [CrossRef] [Green Version]
- Borota, A.; Halip, L.; Curpan, R.; Bora, A.; Avram, S.; Mracec, M.; Mracec, M. Structure- and ligand-based studies to gain insight into the pharmacological implications of histamine H3 receptor. Struct. Chem. 2021, 32, 1141–1149. [Google Scholar] [CrossRef]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Tice, C.M. Selecting the right compounds for screening: Does Lipinski’s Rule of 5 for pharmaceuticals apply to agrochemicals? Pest Manag. Sci. 2001, 57, 3–16. [Google Scholar] [CrossRef]
- Wang, W.-X. Bioaccumulation and Biomonitoring. In Marine Ecotoxicology: Current Knowledge and Future Issues, 1st ed.; Blasco, J., Chapman, P., Campana, O., Hampel, M., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 99–119. [Google Scholar]
- Yamashita, N.; Etoh, H.; Sakata, K.; Ina, H.; Ina, K. New Acylated Rhaponticin Isolated from Eucalyptus rubida as a Repellent against the Blue Mussel Mytilus edulis. Agric. Biol. Chem. Tokyo 1989, 53, 2827–2829. [Google Scholar]
- Che, Z.-P.; Yang, J.-M.; Zhang, S.; Sun, D.; Tian, Y.-E.; Liu, S.-M.; Lin, X.-M.; Jiang, J.; Chen, G.-Q. Synthesis of novel 9R/S-acyloxy derivatives of cinchonidine and cinchonine as insecticidal agents. J. Asian Nat. Prod. Res. 2020, 23, 163–175. [Google Scholar] [CrossRef]
- Knauer, A.; Sirichaisinthop, J.; Reinthaler, F.F.; Wiedermann, G.; Wernsdorfer, G.; Wernsdorfer, W.H. In-vitro response of Plasmodium falciparum to the main alkaloids of Cinchona in northwestern Thailand. Wien. Klin. Wochenschr. 2003, 115 (Suppl. S3), 39–44. [Google Scholar]
- Sowunmi, A.; Salako, L.A.; Laoye, O.J.; Aderounmu, A.F. Combination of quinine, quinidine and cinchonine for the treatment of acute falciparum malaria: Correlation with the susceptibility of Plasmodium falciparum to the cinchona alkaloids in vitro. Trans. R. Soc. Trop. Med. Hyg. 1990, 84, 626–629. [Google Scholar] [CrossRef]
- Talontsi, F.M.; Matasyoh, J.C.; Ngoumfo, R.M.; Chepkorir, R. Mosquito larvicidal activity of alkaloids from Zanthoxylum lemairei against the malaria vector Anopheles gambiae. Pestic. Biochem. Physiol. 2011, 99, 82–85. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
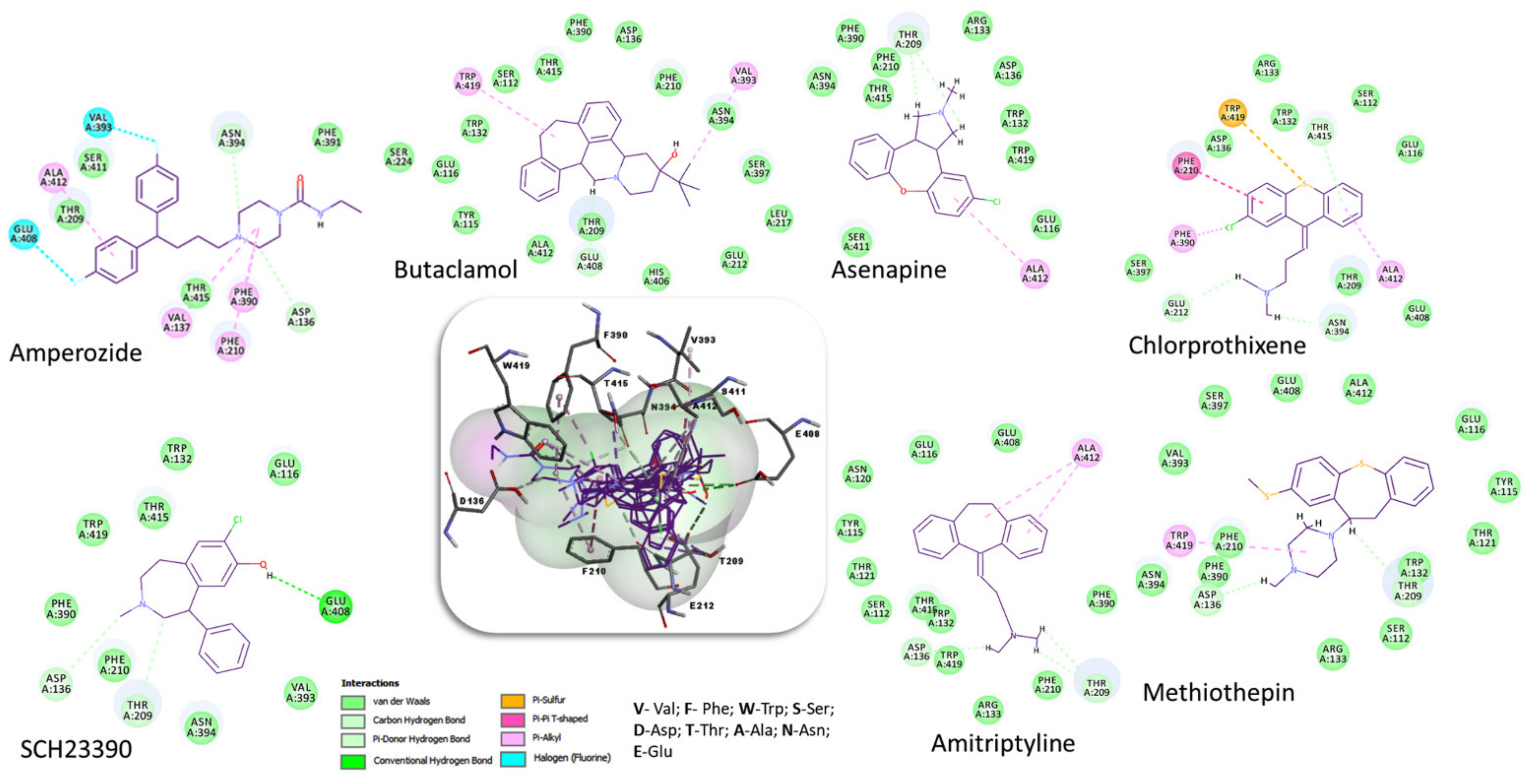
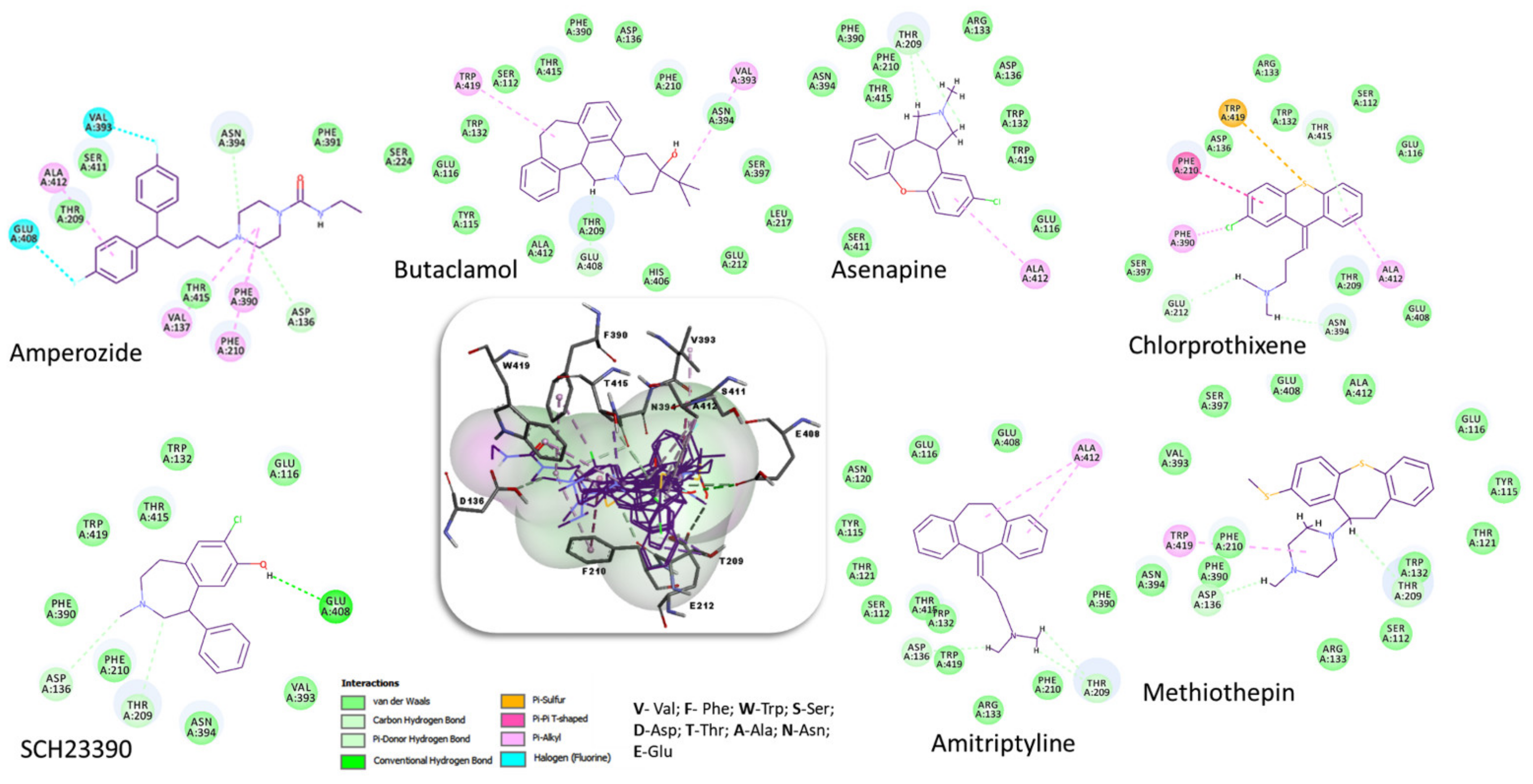
| Compound | 2D Structure | pIC50 1 | CG4 Score | H Bonds 2 | Hydrophobic Interactions 3 |
|---|---|---|---|---|---|
| Amitriptyline |  | 6.638 | −8.522 | D136, T209 | A412 |
| Amperozide | 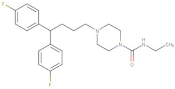 | 4.726 | −8.901 | D136, N394, | F390, F210, A412 |
| Asenapine |  | 9.155 | −8.935 | D136, T209 | A412 |
| Butaclamol | 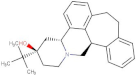 | 5.921 | −8.947 | E408 | V393, W419 |
| Chlorprothixene |  | 6.398 | −8.056 | N394, E212, T415 | F390, F210, A412 |
| Methiothepin | 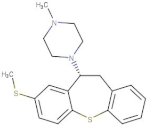 | 6.854 | −7.051 | D136, T209 | S411, W419 |
| SCH23390 | 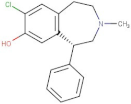 | 5.444 | −8.706 | D136, E408, T209 |
| No. | Specs ID Number | 2D Structure | Name | CG4 Score | H Bonds | Hydrophobic Interactions |
|---|---|---|---|---|---|---|
| 1 | AO-253/40760091 |  | Rhaponticin (Rhapontin) | −14.354 | N394 T209 E408 S411 L217 | L217 |
| 2 | AH-632/20791006 | 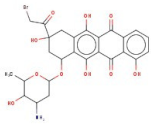 | 3-(bromoacetyl)-3,5,10,12-tetrahydroxy-6,11-dioxo-1,2,3,4,6,11-hexahydro-1-naphthacenyl 3-amino-2,3,6-trideoxyhexopyranoside | −12.776 | D136 T209 T415 L217 | F390 V393 V137 F210 |
| 3 | AE-508/21132035 |  | O-Benzoylcinchonine | −12.212 | T209 | F390 V137 F210 |
| 4 | AO-166/21204006 |  | Abysinnone | −11.947 | S140 L217 | F391 V137 W132 W419 F210 R133 V108 |
| 5 | AO-222/41148840 |  | Teuscorodonin | −11.807 | T121 N394 T415 Y115 | W132 W419 A412 |
| 6 | AM-331/20711002 |  | Indirubin | −11.676 | E408 | T415 F210 |
| 7 | AI-899/21033027 | 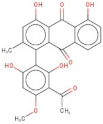 | 1-(3-acetyl-2,6-dihydroxy-4-methoxyphenyl)-4,5-dihydroxy-2-methylanthra-9,10-quinone | −11.546 | N394 S112 | F390 F210 |
| 8 | AQ-152/40869673 | 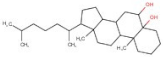 | Cholestane-3,5,6-triol | −11.445 | E408 | F391 W132 V137 W419 F210 |
| 9 | AO-774/41465391 |  | 3,7-Dihydroxycholan-24-oic acid | −11.336 | E408 T209 | F390 W132 F210 W419 L217 |
| 10 | AL-466/21162039 |  | Cinchonine | −11.277 | E408 | - |
| No. | MW | LogP | HBA | HBD | RB | arR | QEI |
|---|---|---|---|---|---|---|---|
| 1 | 420.41 | 0.98 | 9 | 6 | 6 | 2 | 0.124 |
| 2 | 593.40 | 2.45 | 10 | 6 | 4 | 2 | 0.092 |
| 3 | 399.51 | 5.17 | 2 | 1 | 6 | 3 | 0.543 |
| 4 | 392.50 | 5.95 | 4 | 2 | 5 | 2 | 0.441 |
| 5 | 400.43 | 2.53 | 3 | 0 | 4 | 1 | 0.751 |
| 6 | 261.26 | 3.32 | 3 | 1 | 1 | 3 | 0.442 |
| 7 | 433.39 | 5.22 | 8 | 3 | 2 | 3 | 0.159 |
| 8 | 404.68 | 6.67 | 2 | 2 | 5 | 0 | 0.540 |
| 9 | 404.59 | 4.07 | 3 | 1 | 5 | 0 | 0.686 |
| 10 | 295.41 | 2.67 | 2 | 2 | 3 | 2 | 0.525 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crisan, L.; Funar-Timofei, S.; Borota, A. Homology Modeling and Molecular Docking Approaches for the Proposal of Novel Insecticides against the African Malaria Mosquito (Anopheles gambiae). Molecules 2022, 27, 3846. https://doi.org/10.3390/molecules27123846
Crisan L, Funar-Timofei S, Borota A. Homology Modeling and Molecular Docking Approaches for the Proposal of Novel Insecticides against the African Malaria Mosquito (Anopheles gambiae). Molecules. 2022; 27(12):3846. https://doi.org/10.3390/molecules27123846
Chicago/Turabian StyleCrisan, Luminita, Simona Funar-Timofei, and Ana Borota. 2022. "Homology Modeling and Molecular Docking Approaches for the Proposal of Novel Insecticides against the African Malaria Mosquito (Anopheles gambiae)" Molecules 27, no. 12: 3846. https://doi.org/10.3390/molecules27123846
APA StyleCrisan, L., Funar-Timofei, S., & Borota, A. (2022). Homology Modeling and Molecular Docking Approaches for the Proposal of Novel Insecticides against the African Malaria Mosquito (Anopheles gambiae). Molecules, 27(12), 3846. https://doi.org/10.3390/molecules27123846