
Influence of Aliphatic Chain Length on Structural, Thermal and Electrochemical Properties of n-alkylene Benzyl Alcohols: A Study of the Odd–Even Effect

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. X-ray Crystallography
2.2. Interaction Energy Calculation and Hirshfeld Surface Analysis
2.3. Synthesis
3. Results
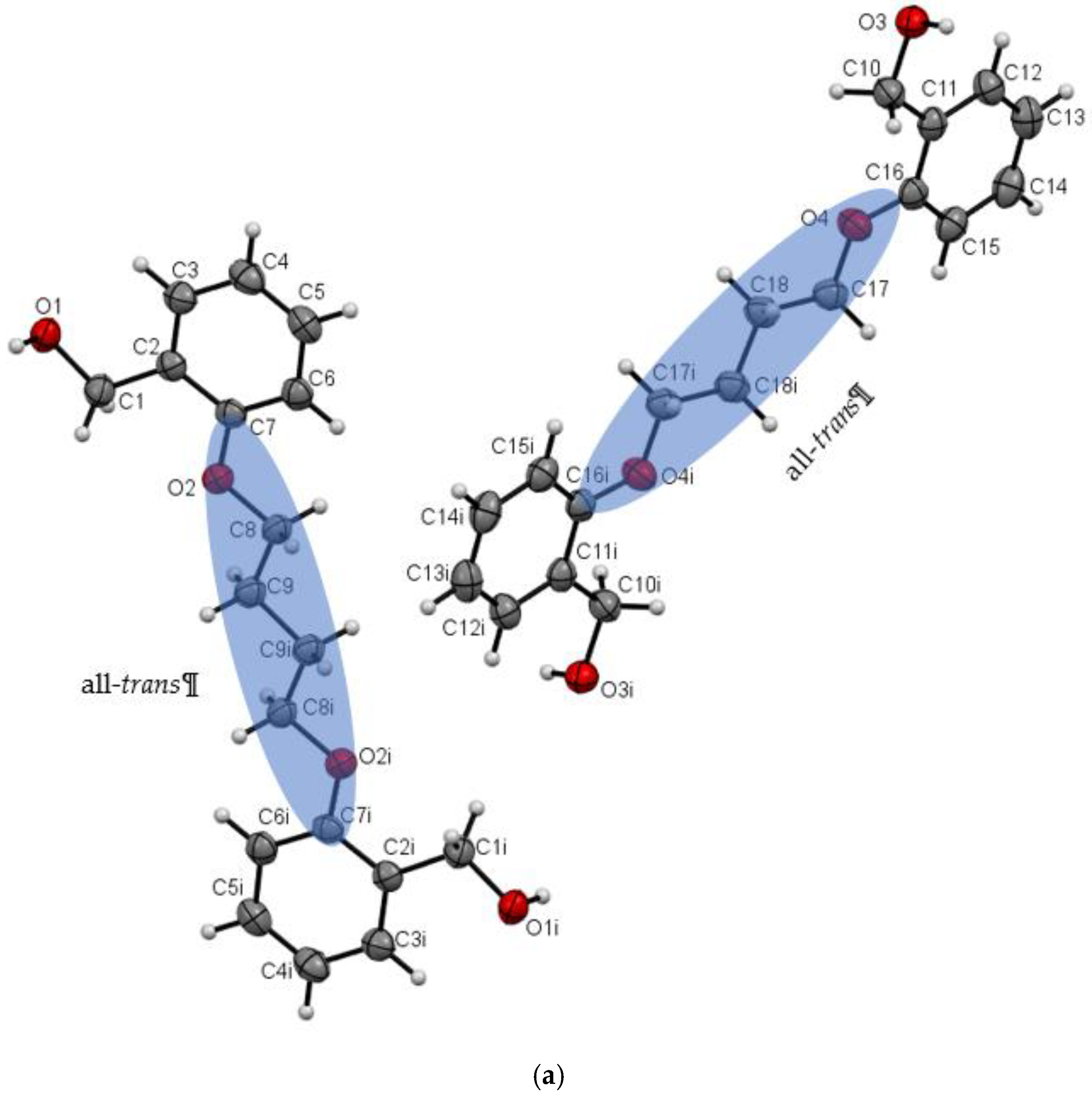
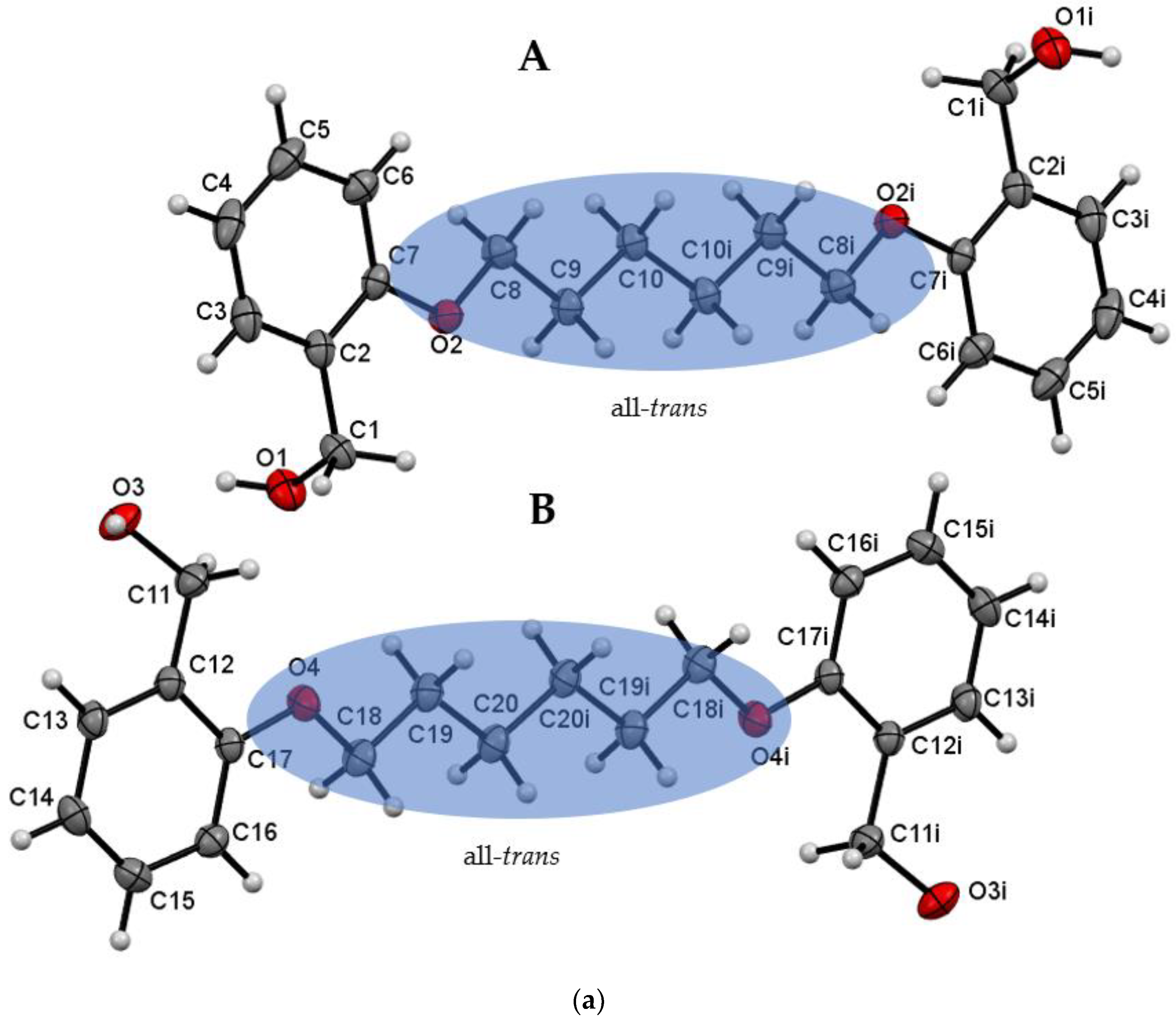
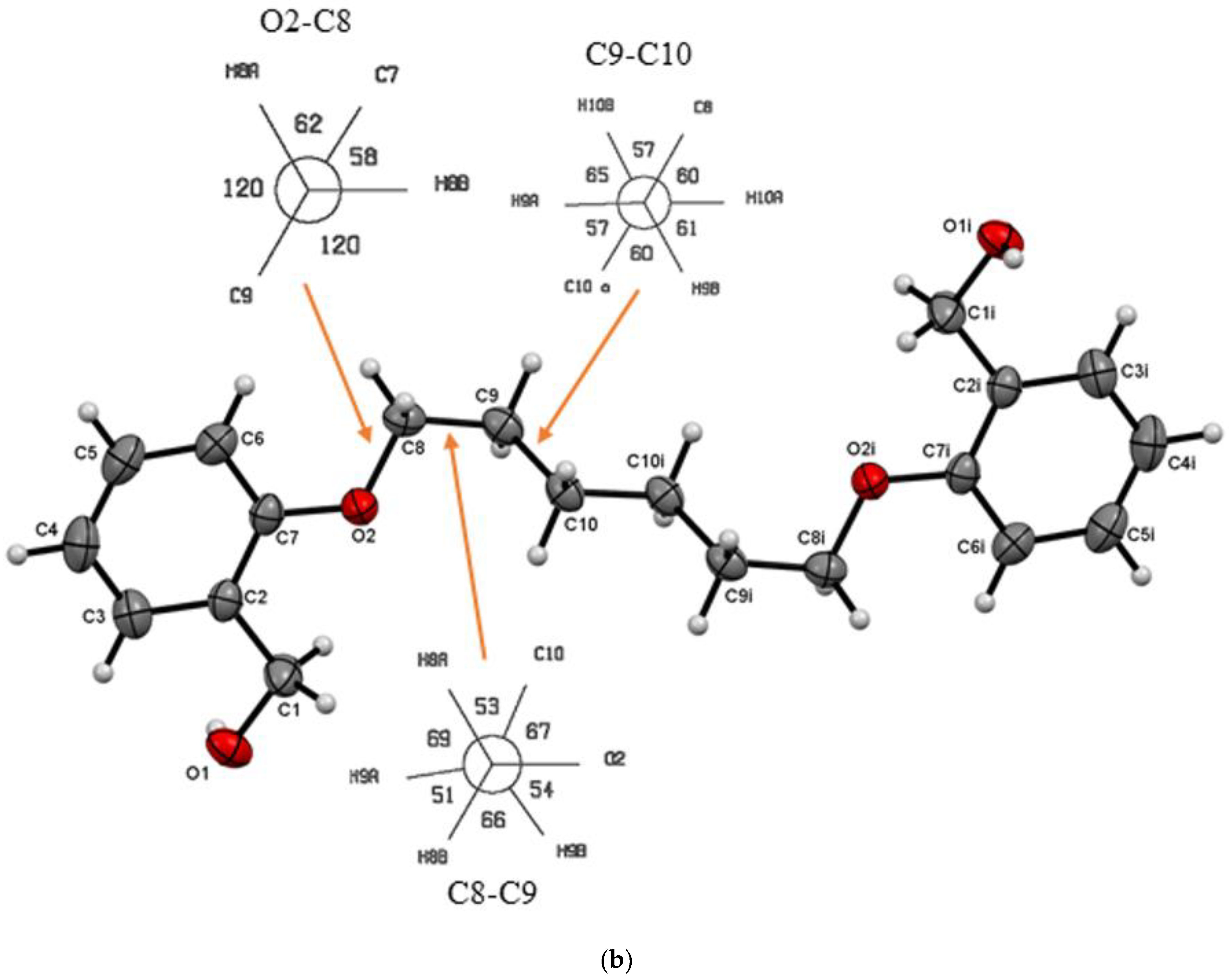
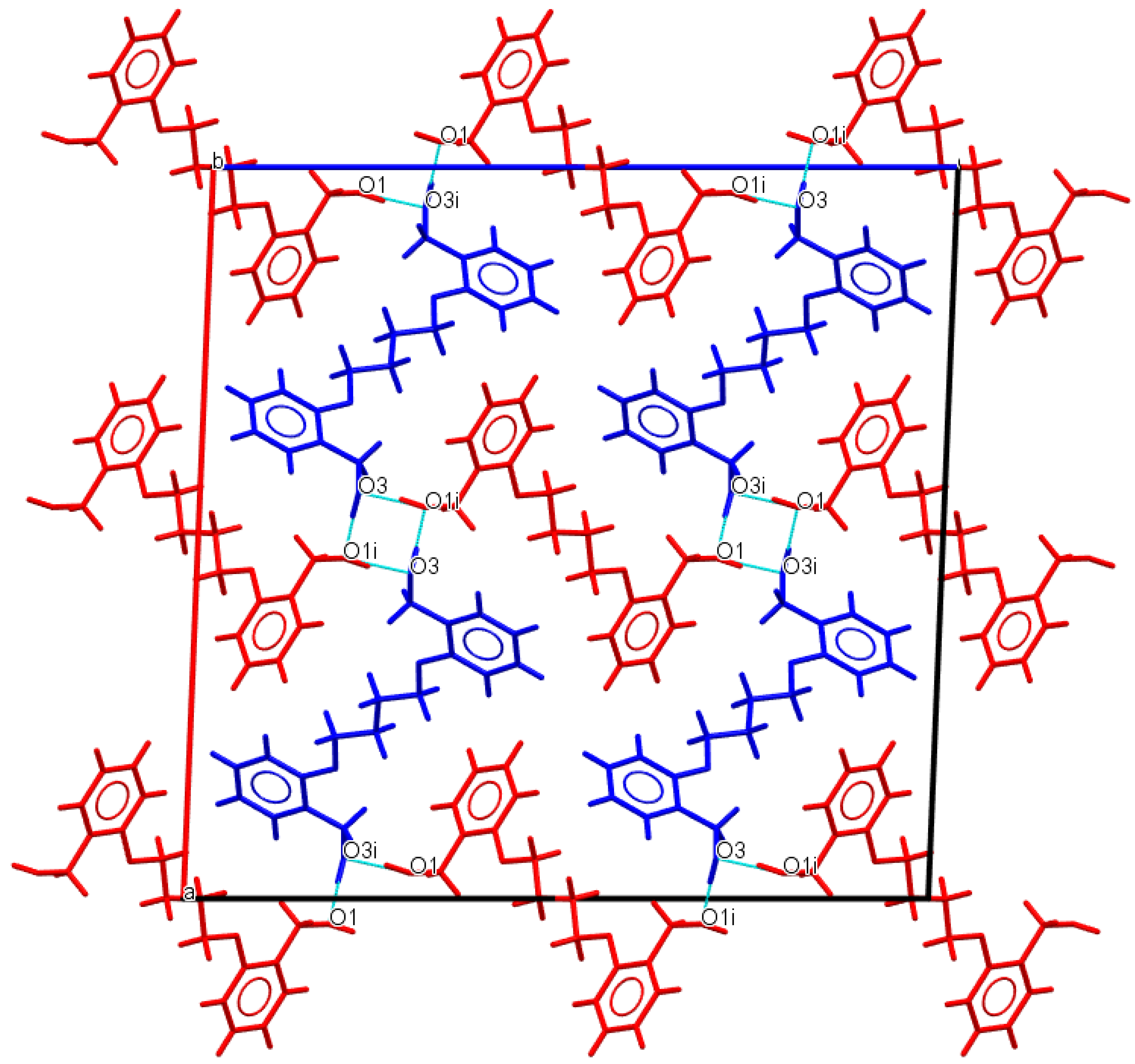
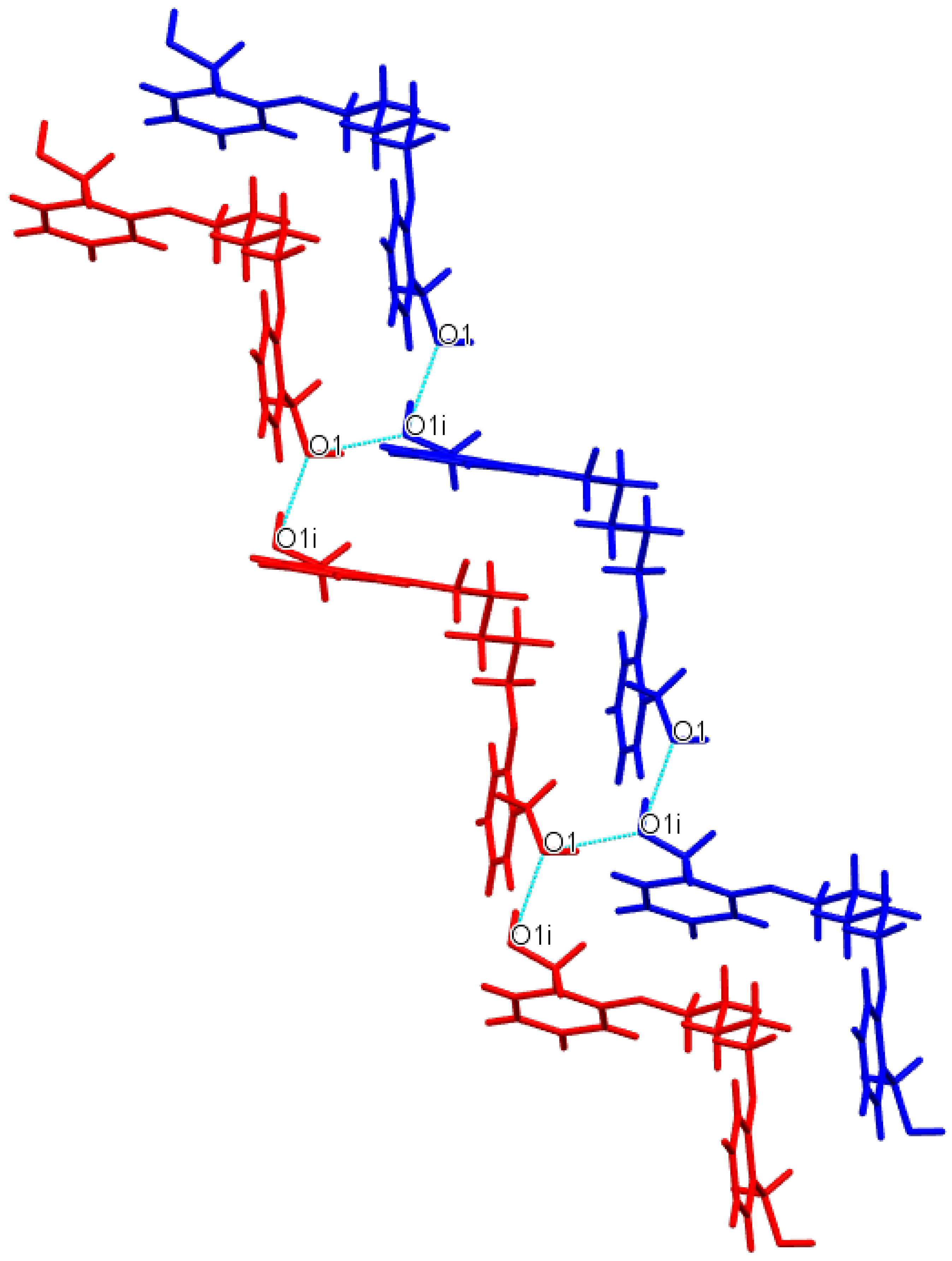
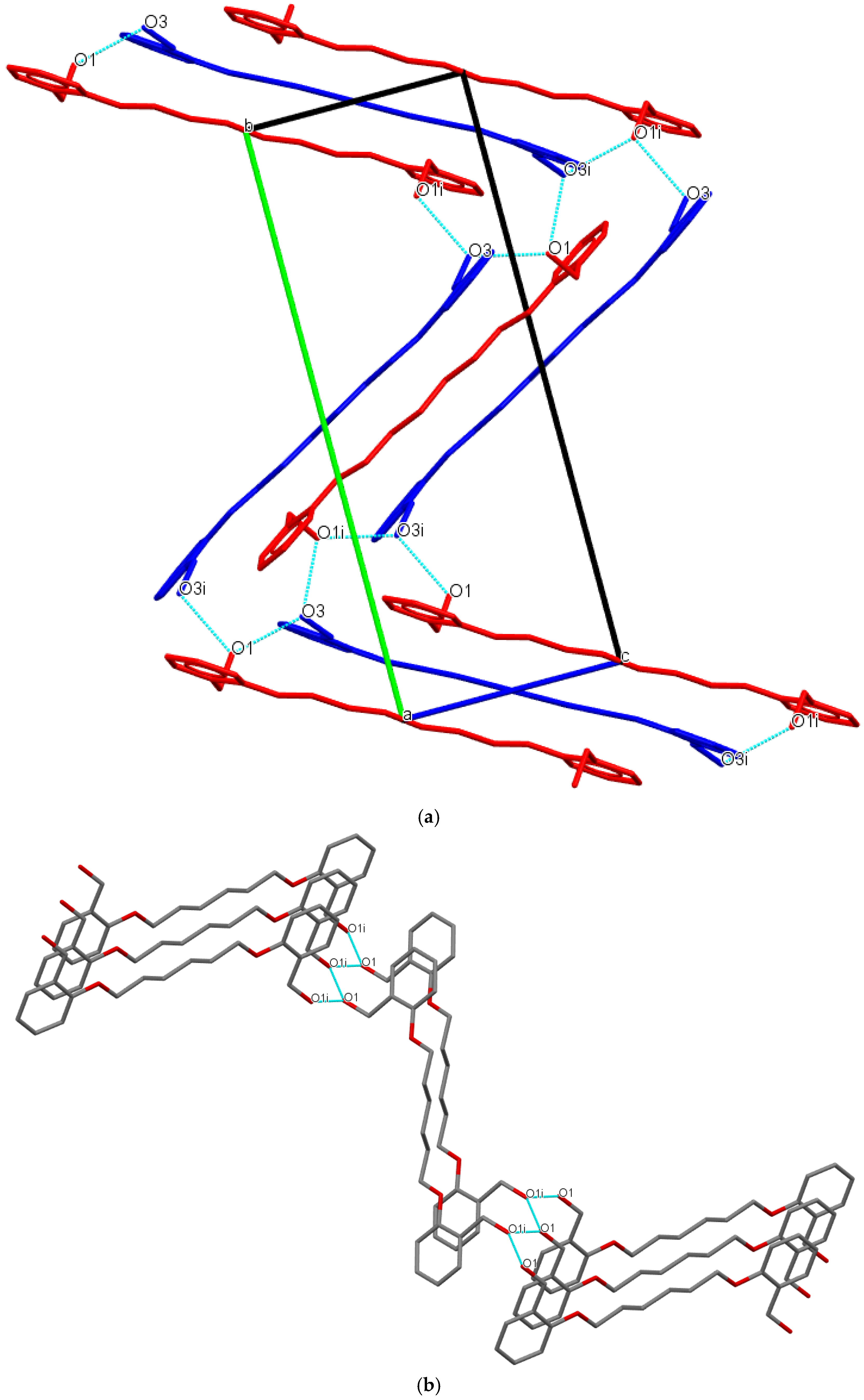
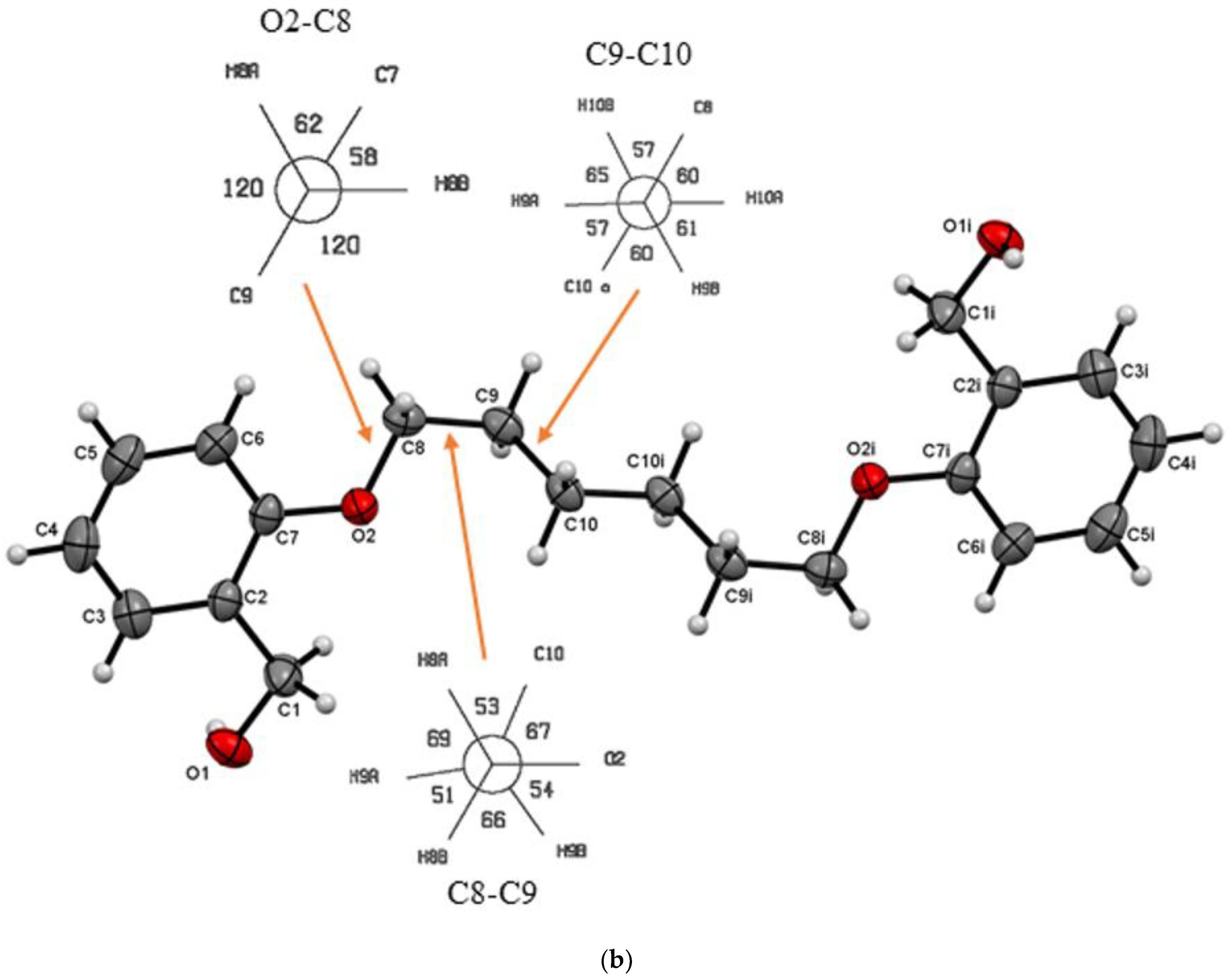
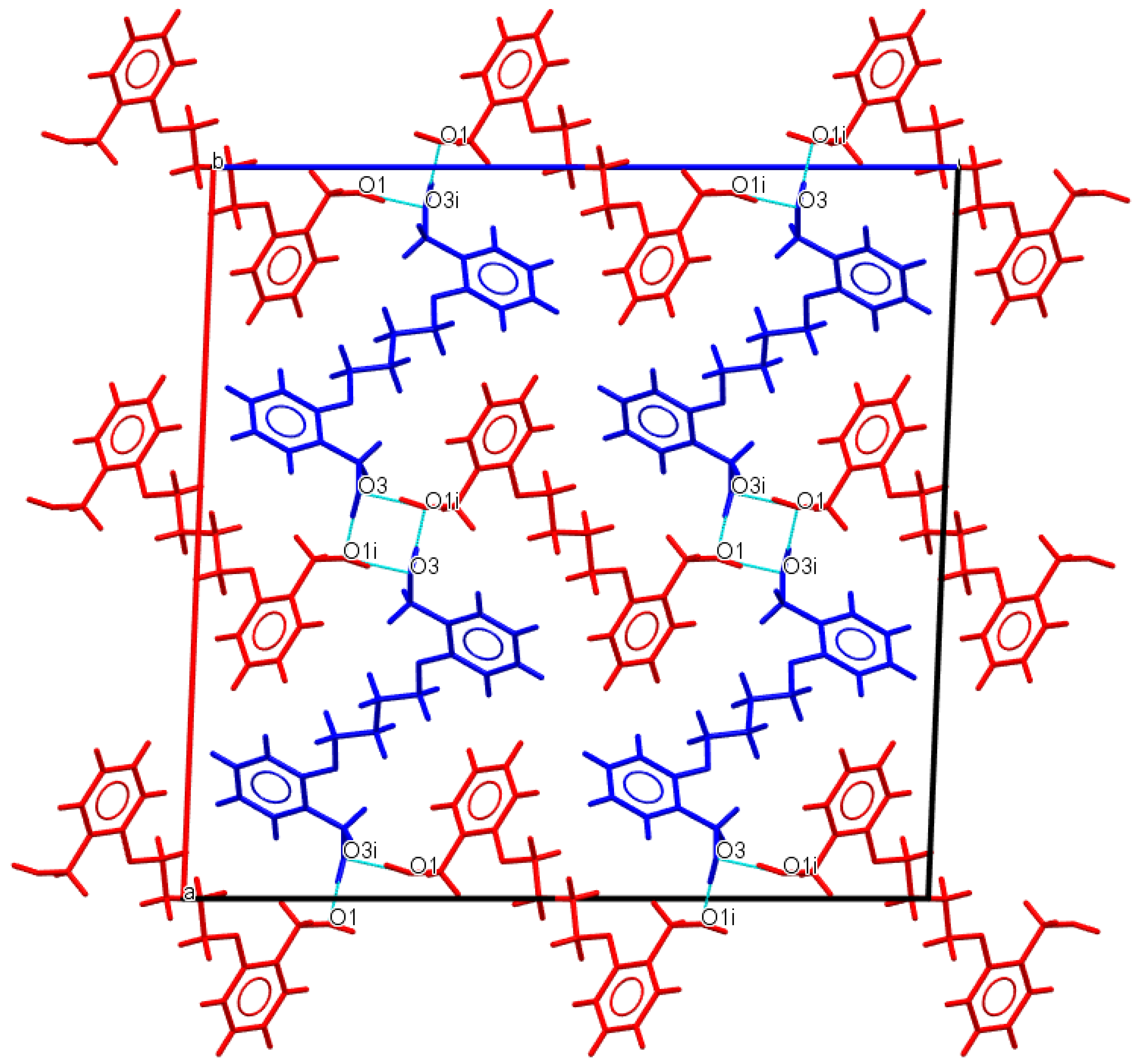
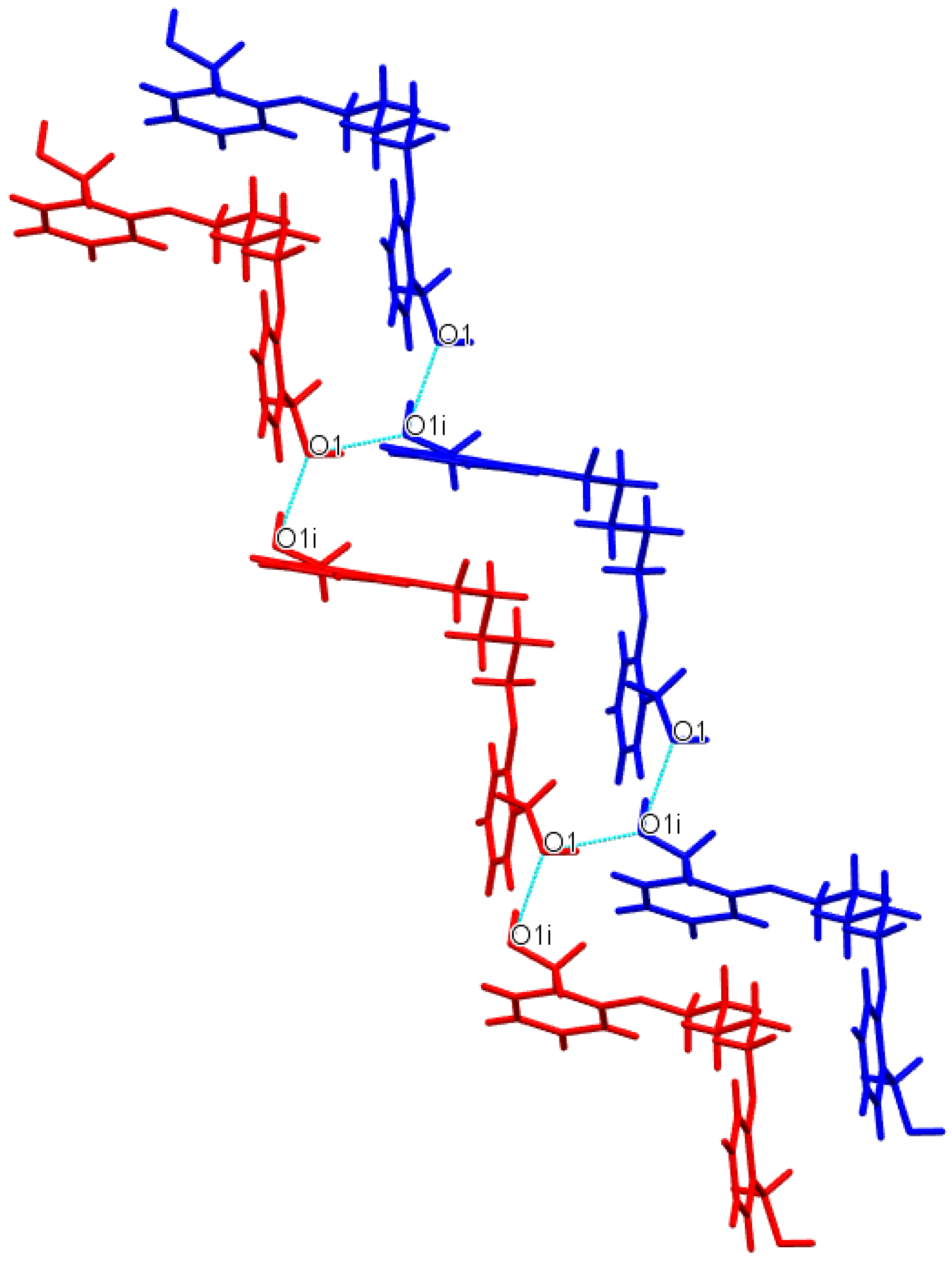
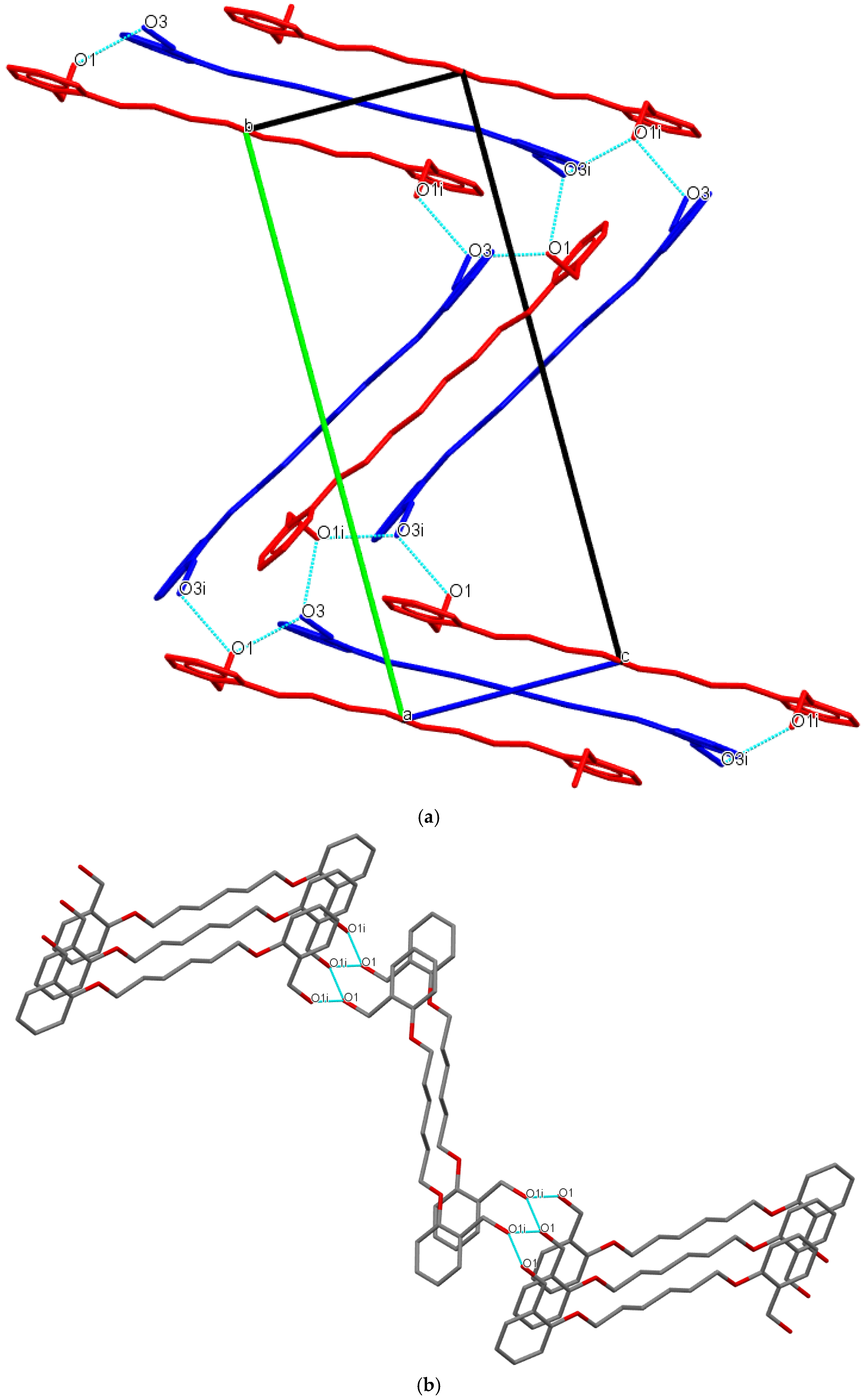
3.1. Description of Crystal Structures
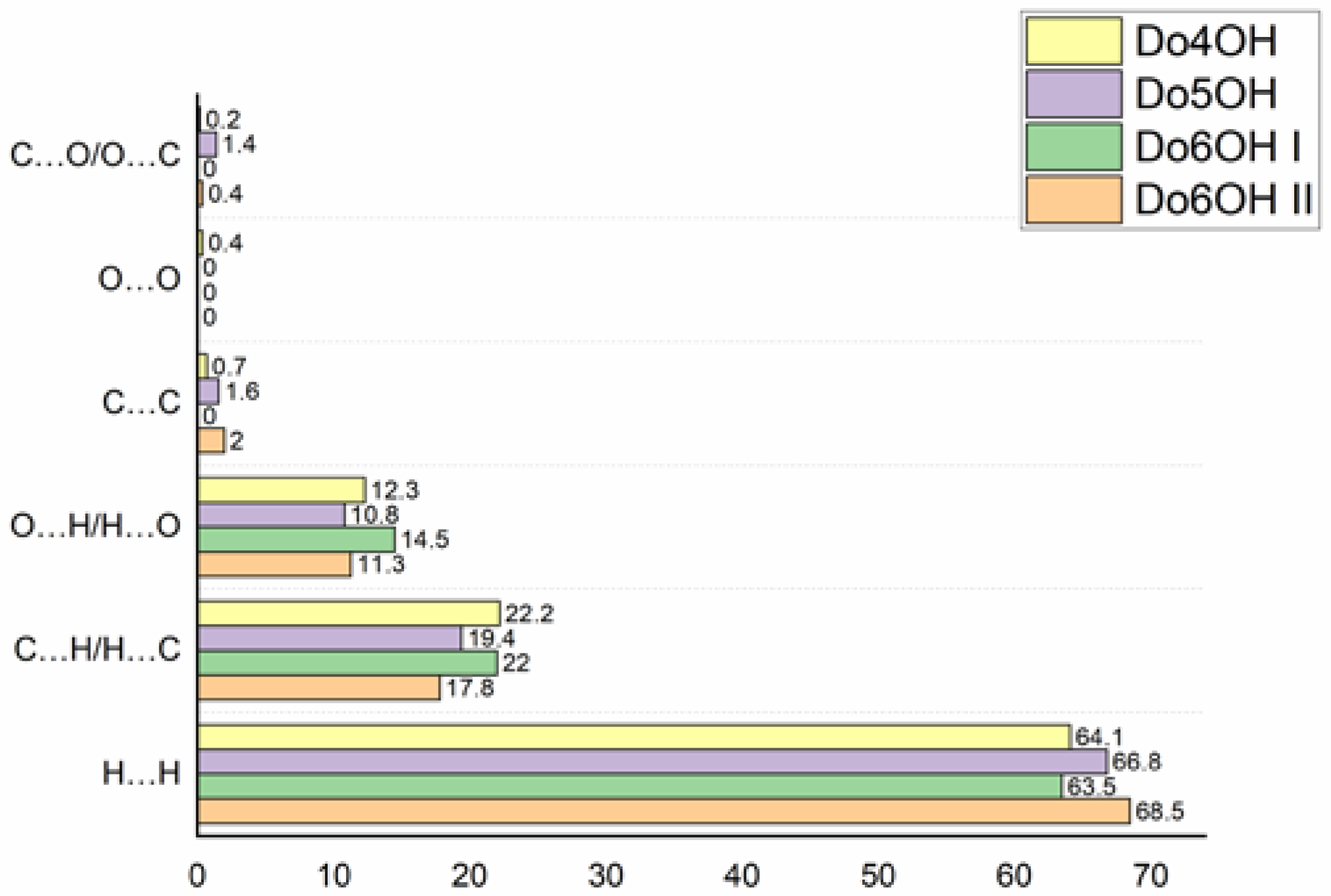
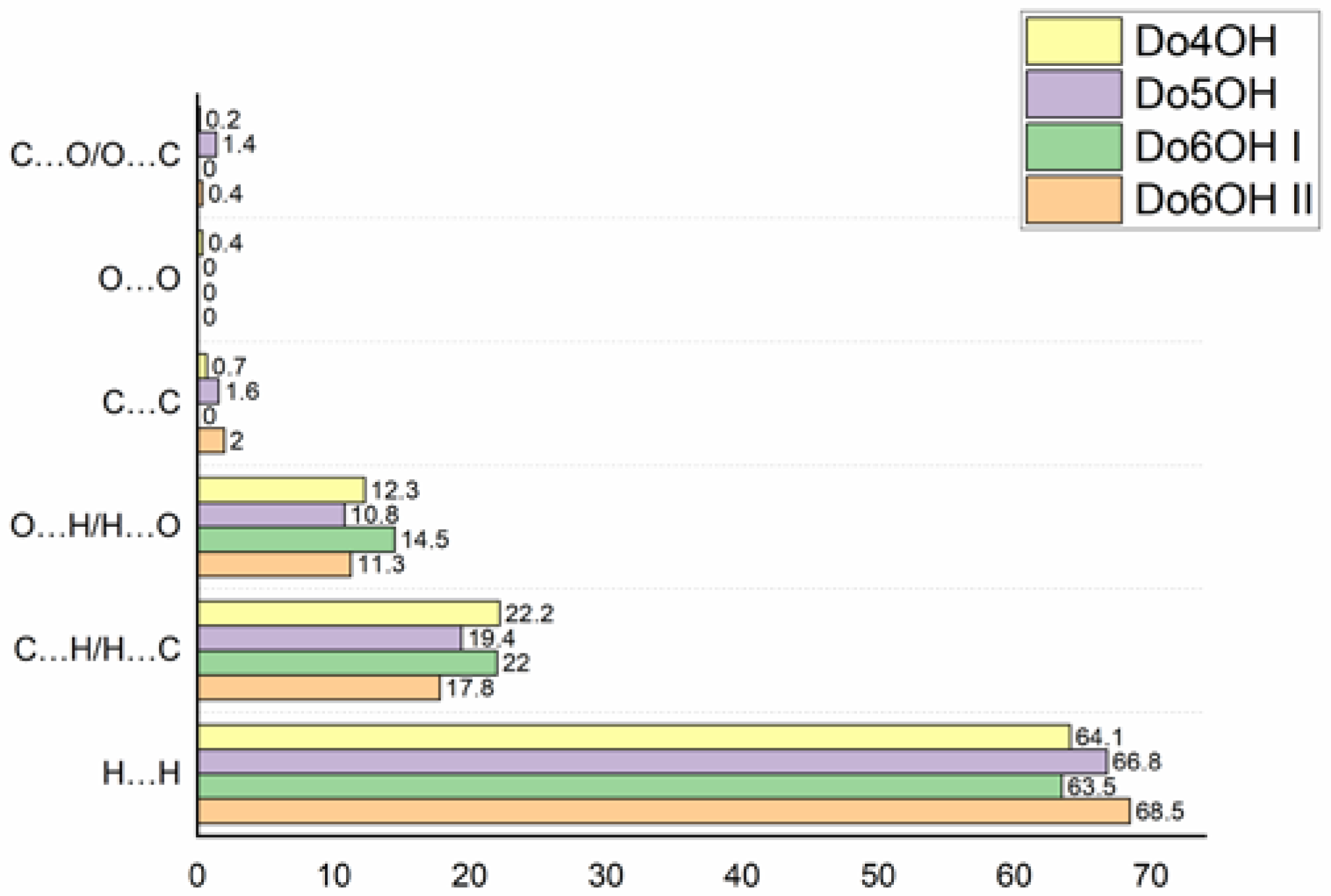
3.2. Hirshfeld Surface (HS) Analysis and Interaction Energy Calculation
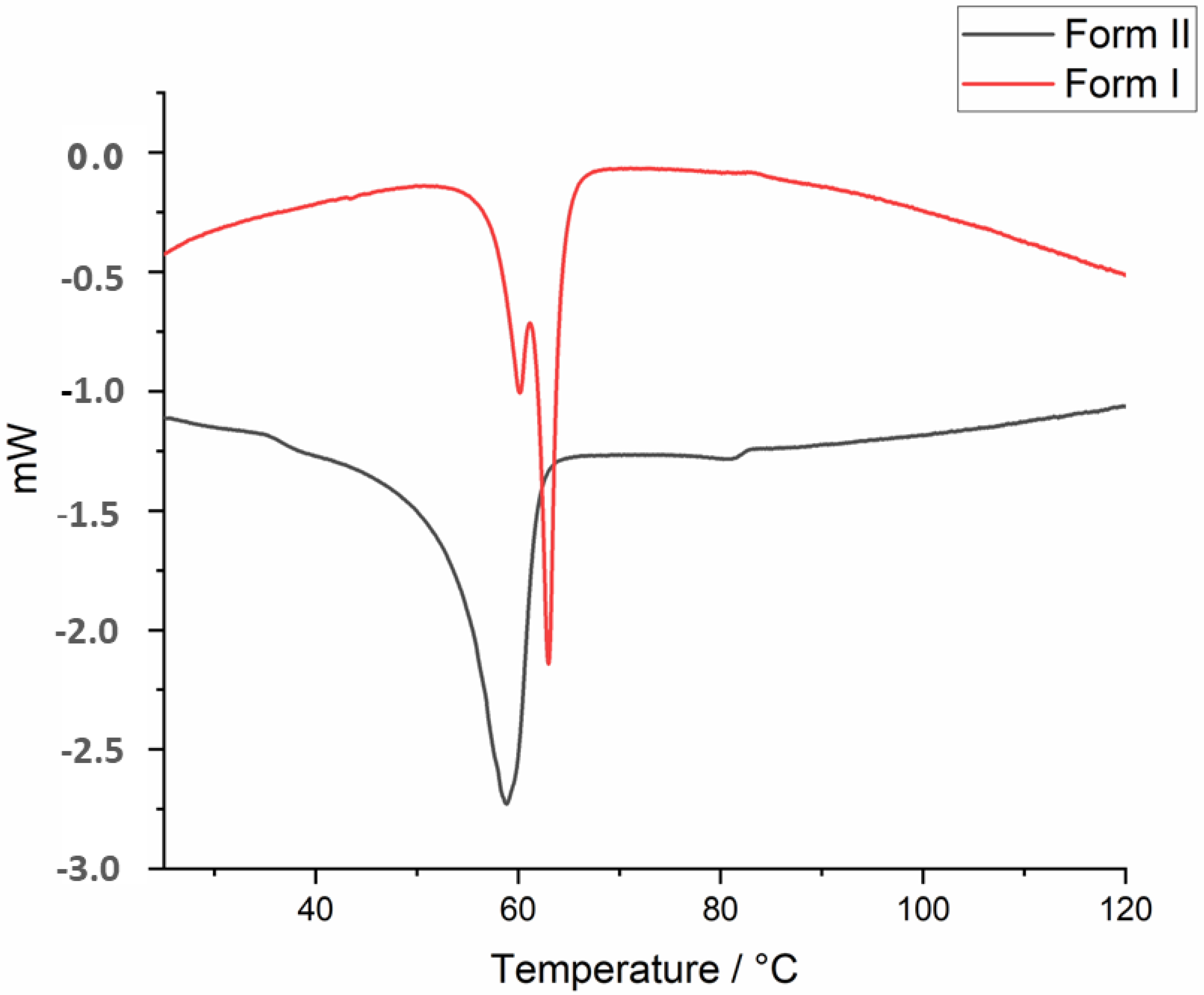
3.3. Thermal Analysis (TG/DSC)
3.4. Spectroscopy
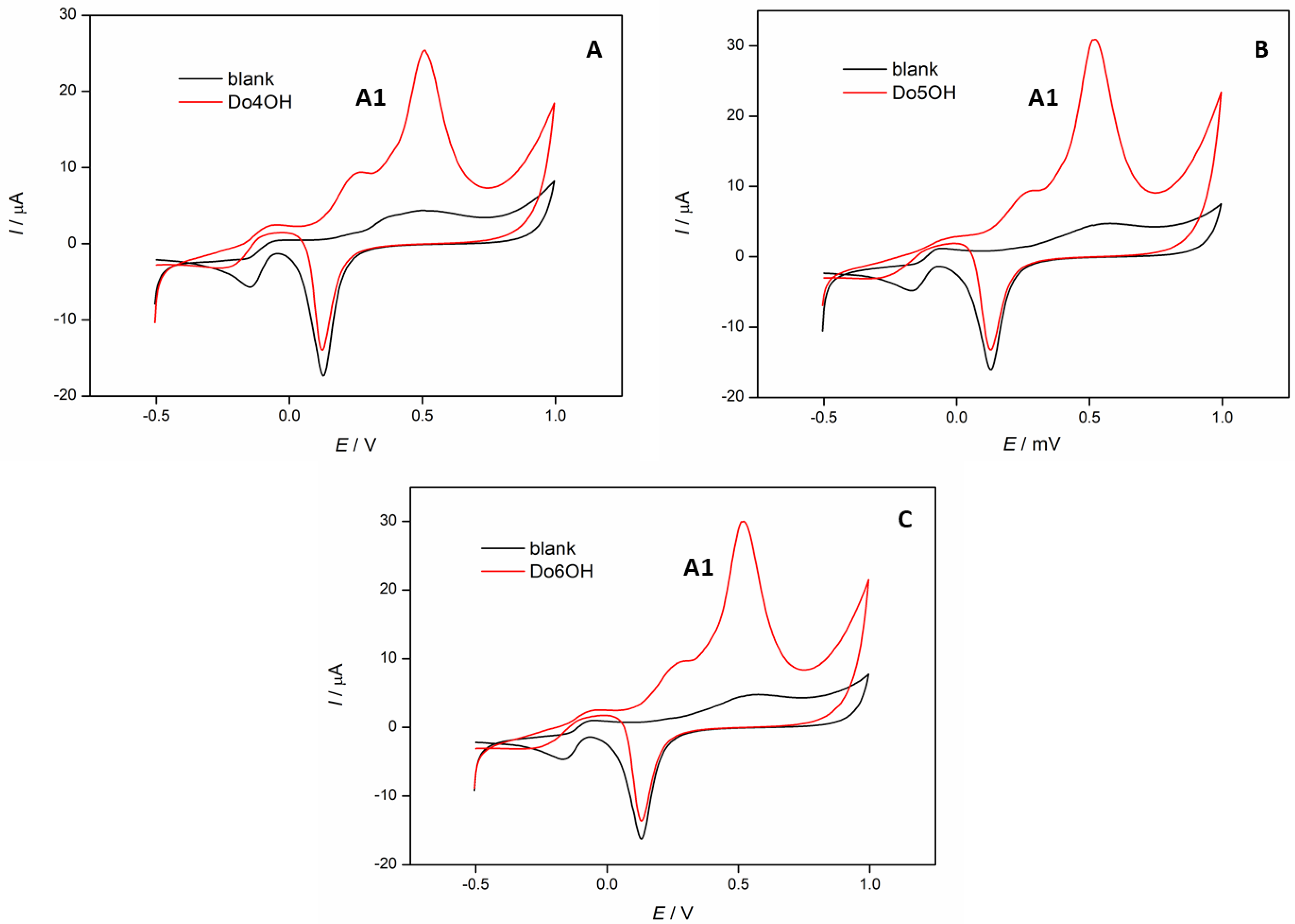
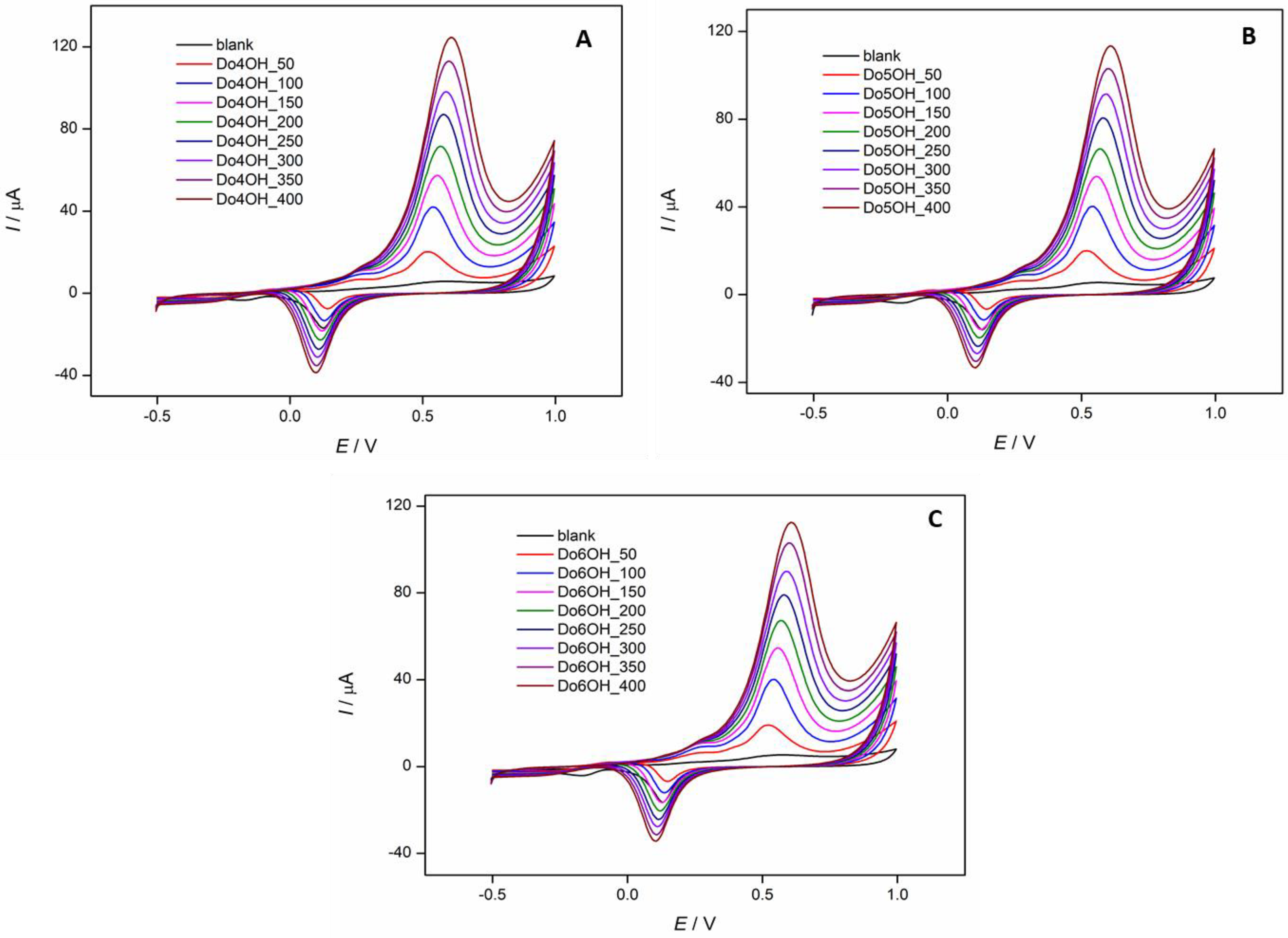
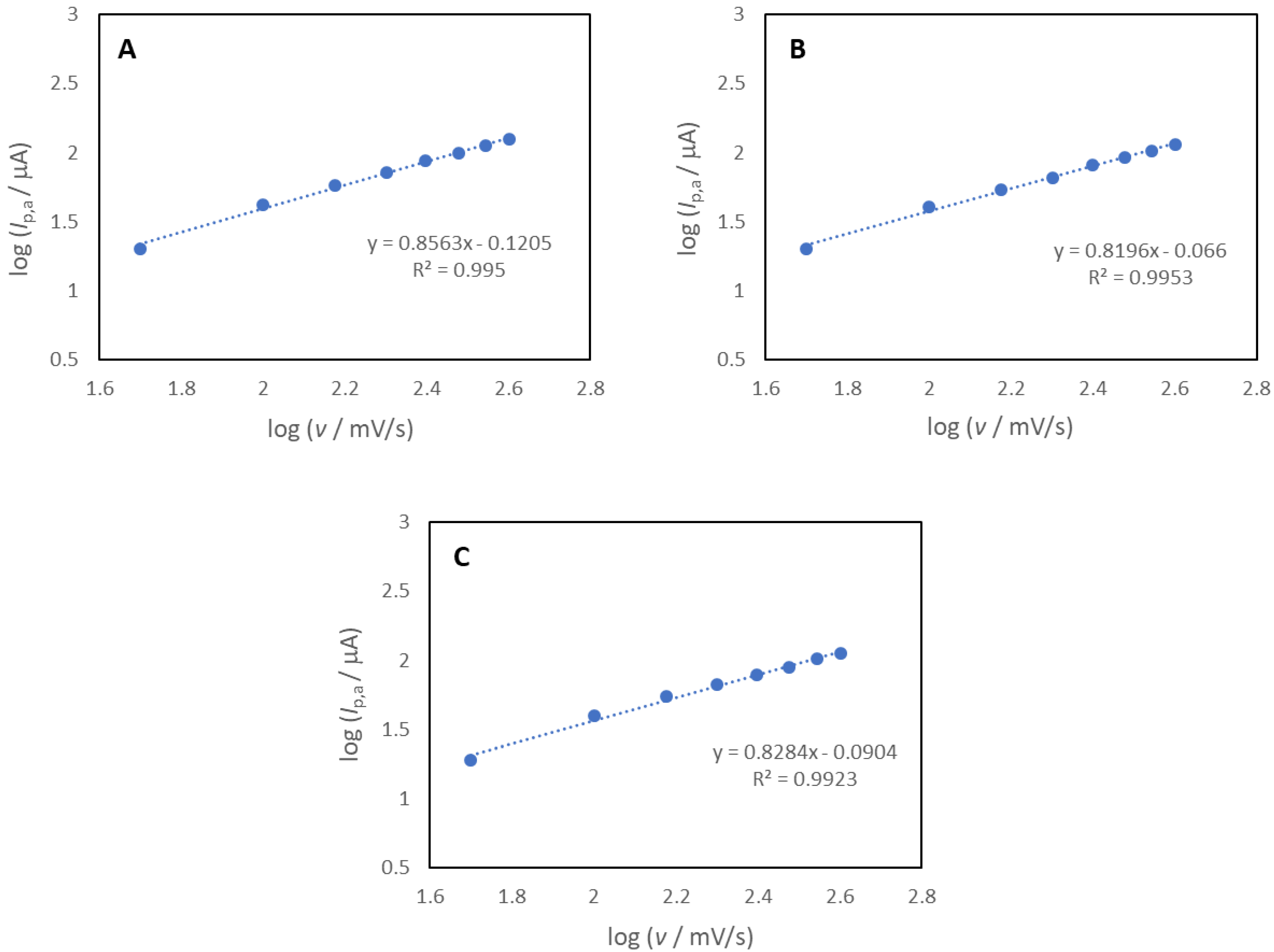
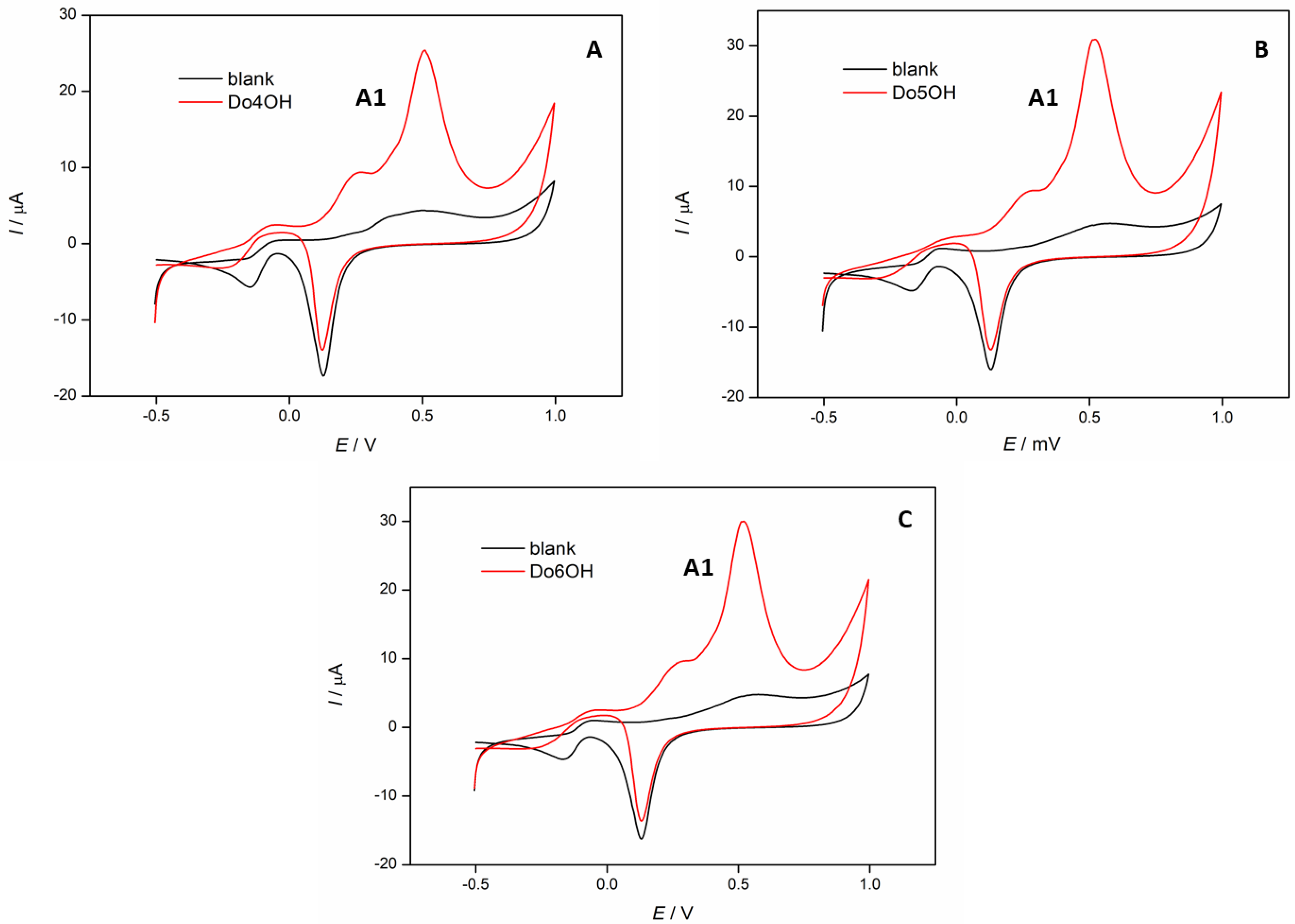
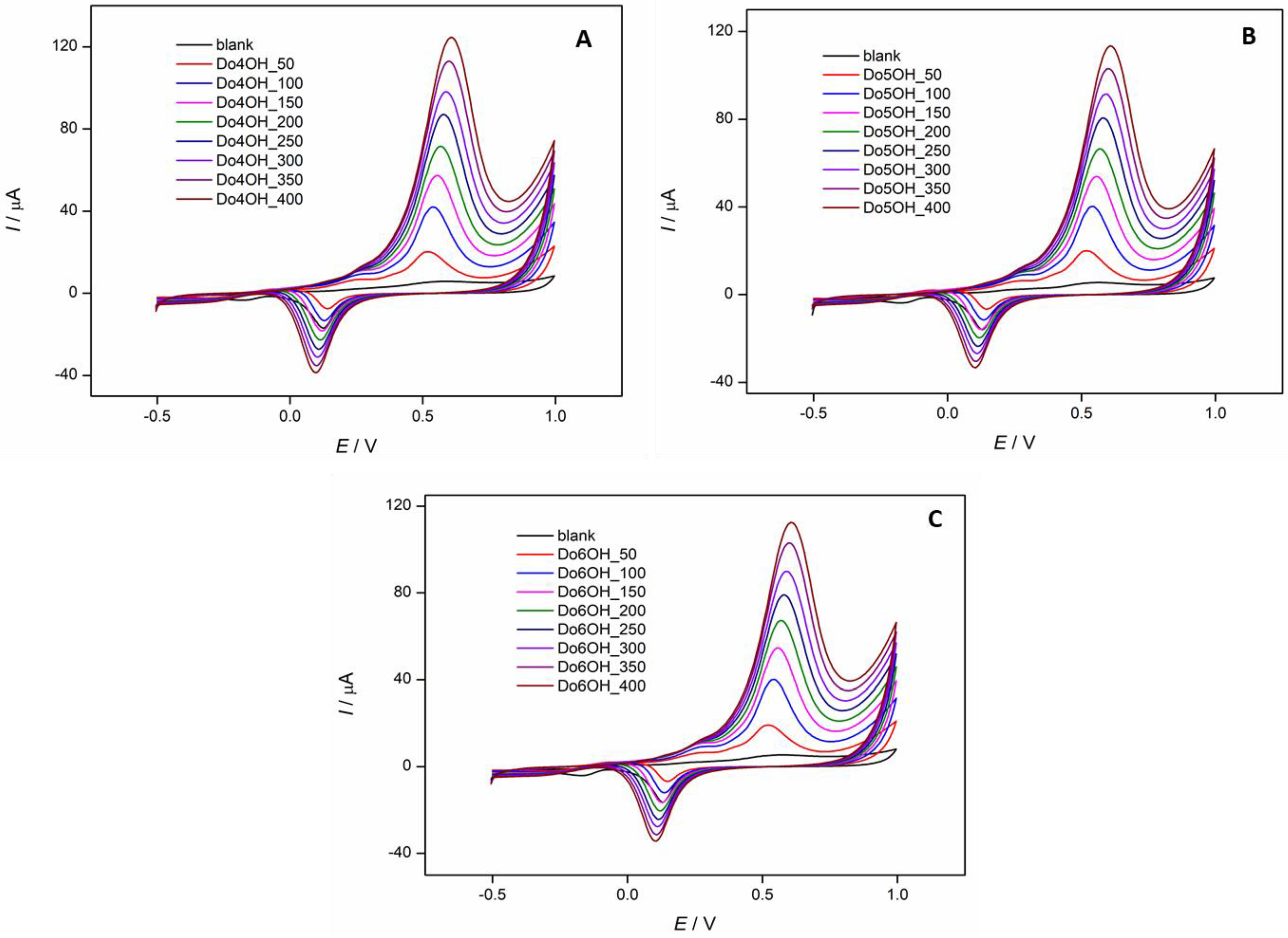
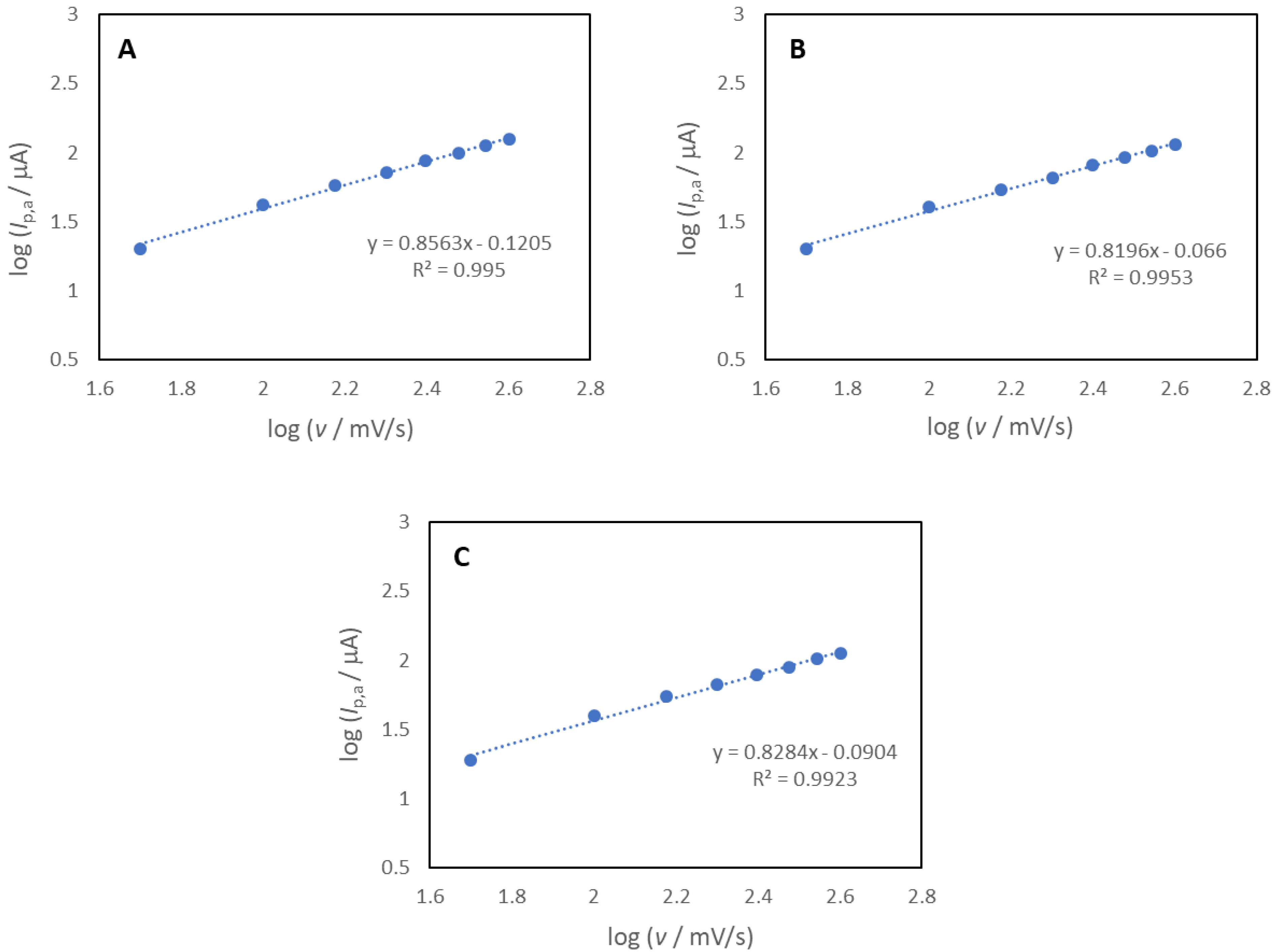
3.5. Cyclic Voltammetry
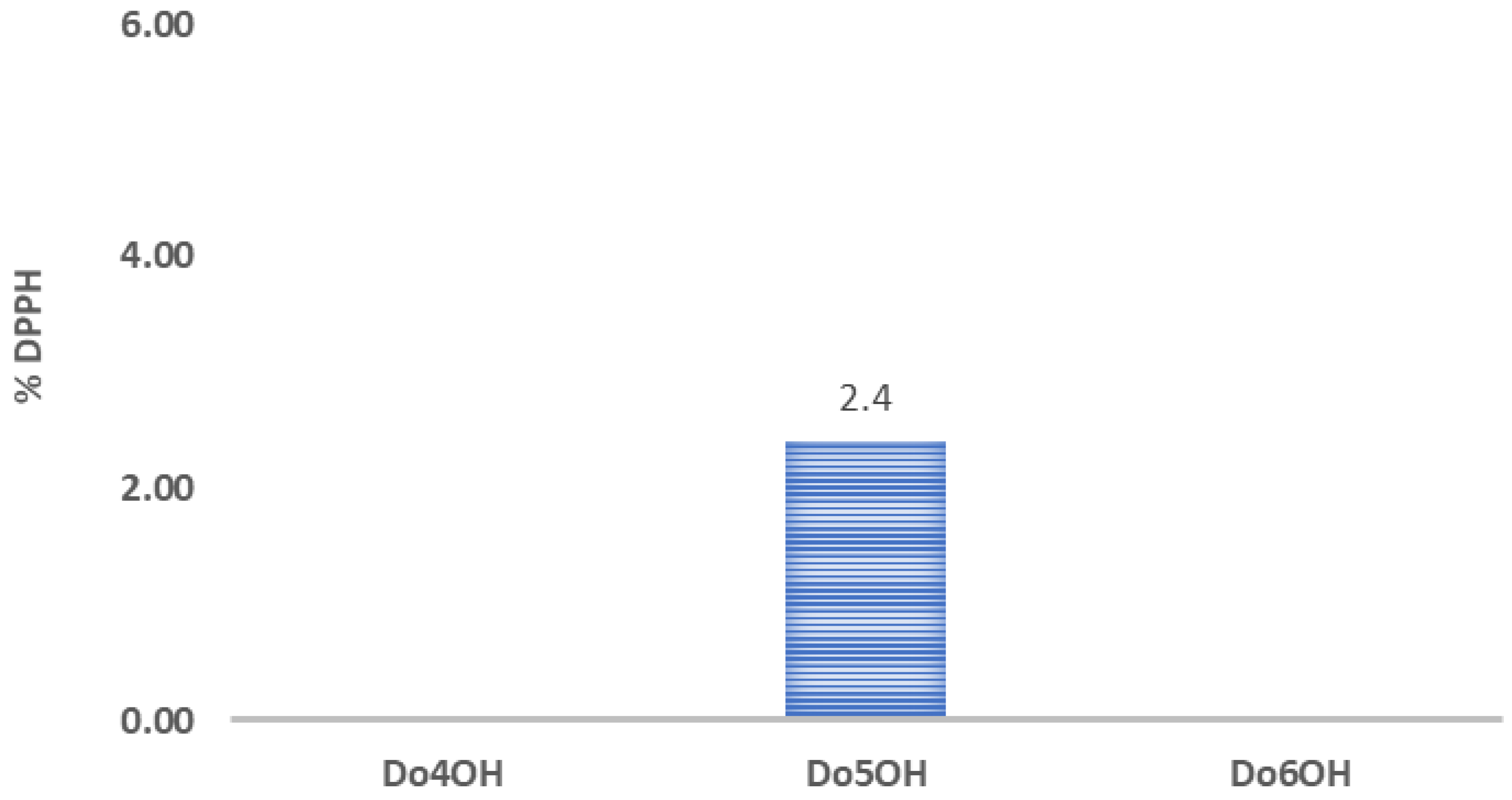
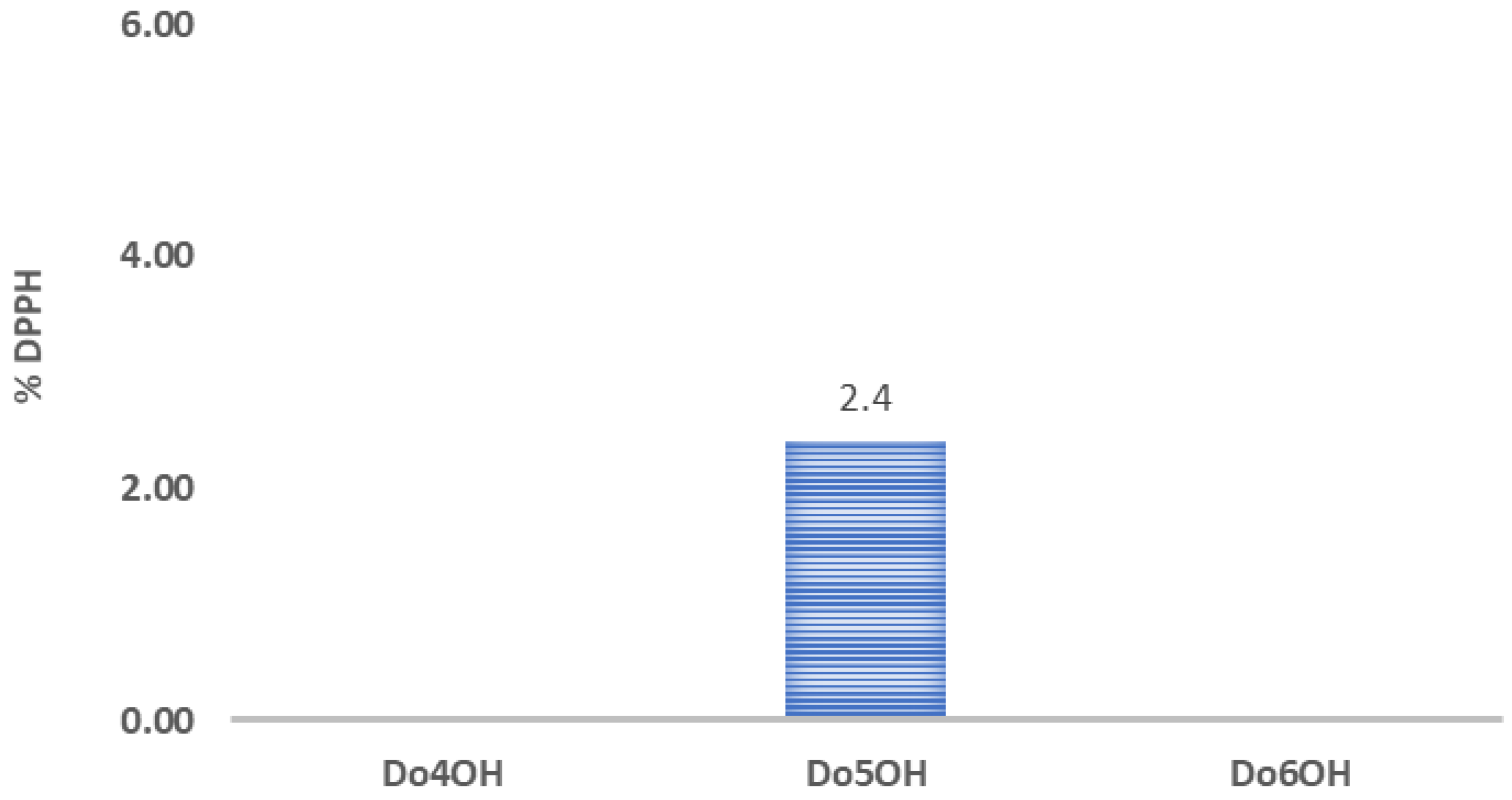
3.6. DPPH Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Baldwin, D.S.; Bowden, B.F.; Duckworth, P.A.; Lindoy, L.F.; McCool, B.J.; Meehan, G.V.; Vasilescu, I.M.; Wild, S.B. New Macrocyclic Ligands. XIV. Synthesis and X-Ray Structures of Potentially Pentadentate Ligands Incorporating Non-Symmetrically Arranged N4S-, N3OS-, N2O2S- and N2S2O-Heteroatoms. Aust. J. Chem. 2002, 55, 597–603. [Google Scholar] [CrossRef]
- Paurević, M.; Dandić, A.; Šrajer Gajdošik, M.; Vidović, B.; Perdih, F.; Balić, T. Efficient Synthesis of New 17-, 18-, 19- and 20-Membered N2O2-Donor Macrocycles by NaBH4 Reduction and Metal Picrate Extraction Studies. J. Incl. Phenom. Macro. 2020, 97, 87–98. [Google Scholar] [CrossRef]
- Balić, T.; Matasović, B.; Marković, B.; Šter, A.; Štivojević, M.; Matković-Čalogović, D. Synthesis, Structural Characterization and Extraction Studies of 17-, 18-, 19- and 20-Membered N2O2-Donor Macrocyclic Schiff Bases. J. Incl. Phenom. Macro. 2016, 85, 217–226. [Google Scholar] [CrossRef]
- Balić, T.; Perdih, F.; Mršo, T.; Balić, I. Ligand Influence on the Formation of Exo-Coordinated Silver(I) Complexes with N2O2 Schiff Base Macrocycles and the Role of Anion in Supramolecular Aggregation. Polyhedron 2020, 190, 114774. [Google Scholar] [CrossRef]
- Baeyer, A. Ueber Regelmässigkeiten Im Schmelzpunkt Homologer Verbindungen. Ber. Dtsch. Chem. Ges. 1877, 10, 1286–1288. [Google Scholar] [CrossRef]
- Pradeilles, J.A.; Zhong, S.; Baglyas, M.; Tarczay, G.; Butts, C.P.; Myers, E.L.; Aggarwal, V.K. Odd–Even Alternations in Helical Propensity of a Homologous Series of Hydrocarbons. Nat. Chem. 2020, 12, 475–480. [Google Scholar] [CrossRef]
- Bond, A.D. On the Crystal Structures and Melting Point Alternation of the N-Alkyl Carboxylic Acids. New J. Chem. 2004, 28, 104–114. [Google Scholar] [CrossRef]
- Thalladi, V.R.; Boese, R.; Weiss, H.-C. The Melting Point Alternation in α,ω-Alkanediols and α,ω-Alkanediamines: Interplay between Hydrogen Bonding and Hydrophobic Interactions. Angew. Chem. Int. Edit. 2000, 39, 918–922. [Google Scholar] [CrossRef]
- Thalladi, V.R.; Boese, R.; Weiss, H.-C. The Melting Point Alternation in α,ω-Alkanedithiols. J. Am. Chem. Soc. 2000, 122, 1186–1190. [Google Scholar] [CrossRef]
- Jiang, L.; Sangeeth, C.S.S.; Nijhuis, C.A. The Origin of the Odd–Even Effect in the Tunneling Rates across EGaIn Junctions with Self-Assembled Monolayers (SAMs) of n-Alkanethiolates. J. Am. Chem. Soc. 2015, 137, 10659–10667. [Google Scholar] [CrossRef]
- STARe, Software 10.0; Mettler-Toledo GmbH; Mettler-Toledo Inc.: Greifensee, Switzerland, 2009.
- Rigaku Oxford Diffraction. CrystAlis PRO; Rigaku Corporation: Tokyo, Japan, 2018. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON: An Integrated Tool for the Analysis of the Results of a Single Crystal Structure Determination. Acta Crystallogr. A 1990, 46, 34. [Google Scholar]
- Spek, A.L. PLATON: A Multipupose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 1998. [Google Scholar]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Crawley, WA, Australia, 2017. [Google Scholar]
- Gavezzotti, A. Calculation of Lattice Energies of Organic Crystals: The PIXEL Integration Method in Comparison with More Traditional Methods. Z. Kristallogr. 2005, 220, 499–510. [Google Scholar] [CrossRef]
- Neese, F. WIREs. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- McArdle, P. Pixel Calculations Using Orca or GAUSSIAN for Electron Density Automated within the Oscail Package. J. Appl. Crystallogr. 2021, 54, 1535–1541. [Google Scholar] [CrossRef]
- Liu, Q.-X.; Zhao, Z.-X.; Zhao, X.-J.; Wei, Q.; Chen, A.-H.; Li, H.-L.; Wang, X.-G. Structures of NHC Hg(II) and Ag(I) Complexes and Selective Recognition of Nitrate Anion. CrystEngComm 2015, 17, 1358–1373. [Google Scholar] [CrossRef]
- Balić, T.; Marković, B. Crystal Structure of 2,2′-[Pentane-1,5-Diylbis(Oxy)]Dibenzaldehyde, C19H20O4. Z. Krist.—New Cryst. St. 2016, 231, 619–621. [Google Scholar] [CrossRef]
- Balić, T.; Marković, B.; Jaźwiński, J.; Matković-Čalogović, D. Synthesis and Structural Characterization of New N2O2-Donor Schiff Base Macrocycles and Their Silver(I) Coordination Polymers. Inorg. Chim. Acta 2015, 435, 283–291. [Google Scholar] [CrossRef]
- Darabi, H.R.; Sobhani, L.; Rastgar, S.; Aghapoor, K.; Amini, S.K.; Zadmard, R.; Jadidi, K.; Notash, B. Synthesis, Characterization and Selective Cu2+ Recognition of Novel E– and Z–Stilbenophanes. Supramol. Chem. 2019, 31, 45–51. [Google Scholar] [CrossRef]
- Seo, J.; Jin, Y.; Lee, J.-E.; Park, K.-M.; Lee, B.Y.; Lee, S.S. Crystal Structures of 1,3-Bis(2-Benzyl Alcohol)-1,3-Dioxapropane and 1,5-Bis(2-Benzyl Alcohol)-1,5-Dioxapentane. Anal. Sci. 2004, 20, x15–x16. [Google Scholar] [CrossRef]
- Bailey, N.A.; Fenton, D.E.; Williams, M.G.; Winter, D.J. The Structure of 2,2’-[1,2-Ethanediylbis(Oxy)]Bis(Benzenemethanol). Acta Crystallogr. C 1989, 45, 1778–1780. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Burger, A.; Ramberger, R. On the Polymorphism of Pharmaceuticals and Other Molecular Crystals. II. Microchim. Acta 1979, 72, 273–316. [Google Scholar] [CrossRef]
- Marti, E. Applied Chemical Thermodynamics and Kinetics on Pharmaceutical Compounds. J. Therm. Anal. 1988, 33, 37–45. [Google Scholar] [CrossRef]
- Balić, T.; Perdih, F.; Počkaj, M.; Molnar, M.; Komar, M.; Balić, I. Polymorphism of Coumarin Thione-Triazole—4-Methyl-7-[(4-Phenyl-5-Thioxo-4,5-Dihydro-1H-1,2,4-Triazol-3-Yl)Methoxy]-2H-Chromen-2-One. J. Mol. Struct. 2021, 1231, 129957. [Google Scholar] [CrossRef]
- Othman, M.I.A.; Said, S.; Marin, M. A novel model of plane waves of two-temperature fiber-reinforced thermoelastic medium under the effect of gravity with three-phase-lag model. Int. J. Numer. Method. H. 2019, 29, 4788–4806. [Google Scholar] [CrossRef]
- Dhiman, S.B.; Kamat, J.P.; Naik, D.B. Antioxidant activity and free radical scavenging reactions of hydroxybenzyl alcohols. Chem.-Biol. Interact. 2009, 182, 119–127. [Google Scholar] [CrossRef]
- Wang, B.; Tao, L.; Cheng, Y.; Yang, F.; Jin, Y.; Zhou, C.; Yu, H.; Yang, Y. Electrocatalytic Oxidation of Small Molecule Alcohols over Pt, Pd, and Au Catalysts: The Effect of Alcohol’s Hydrogen Bond Donation Ability and Molecular Structure Properties. Catalysts 2019, 9, 387. [Google Scholar] [CrossRef]
- Wu, J. Electrochemical Behavior and Direct Quantitative Determination of Tanshinone IIA in Micro-Emulsion. Int. J. Electrochem. Sc. 2016, 11, 5165–5179. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
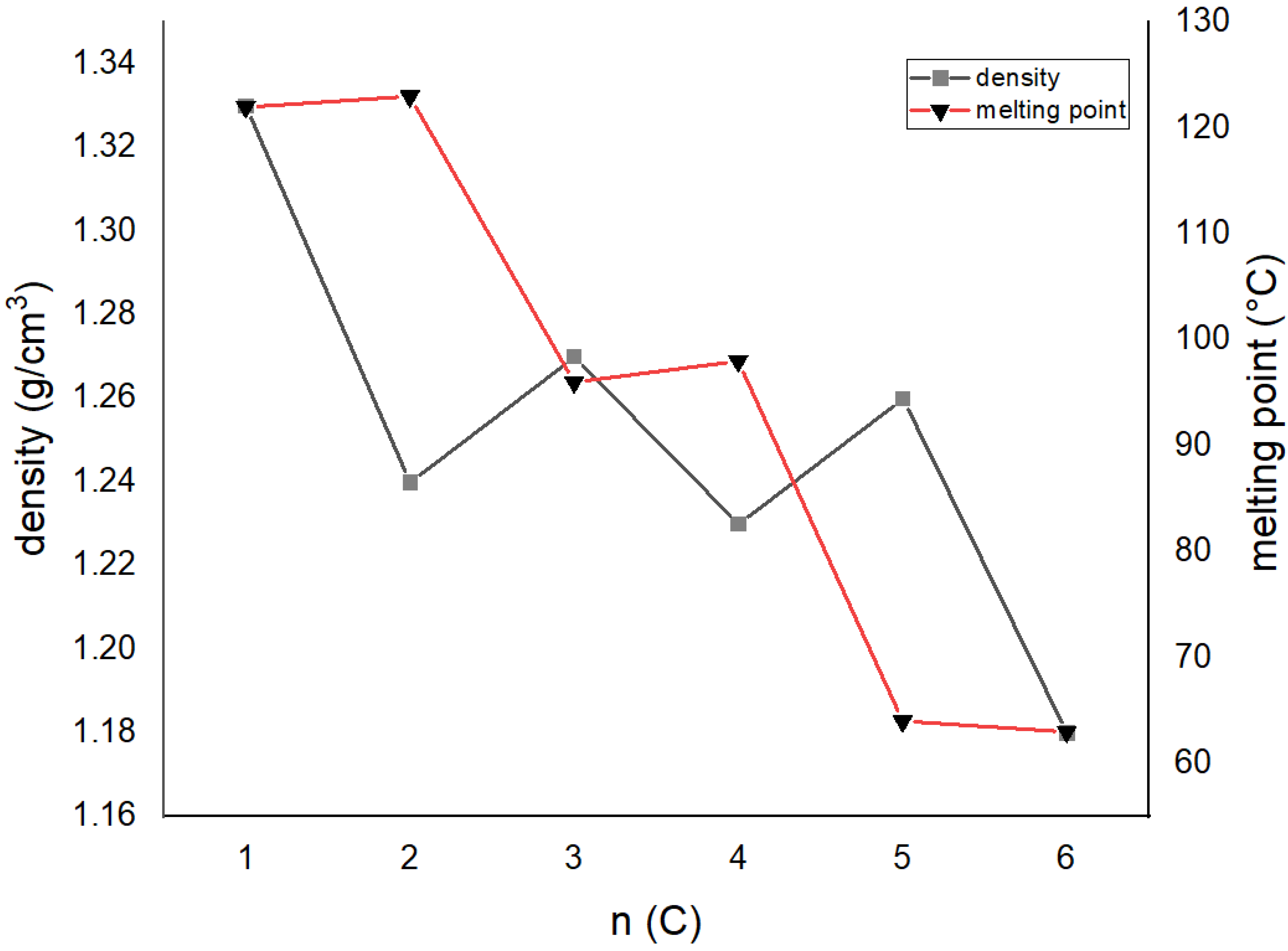
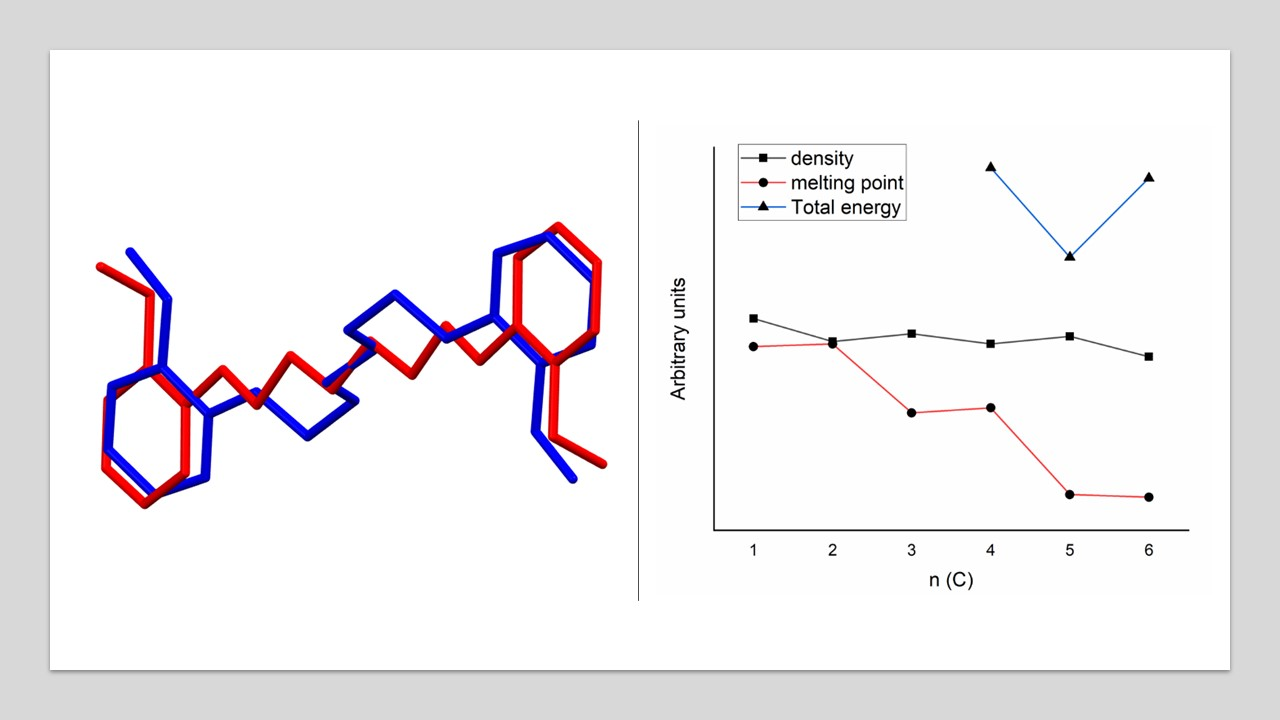
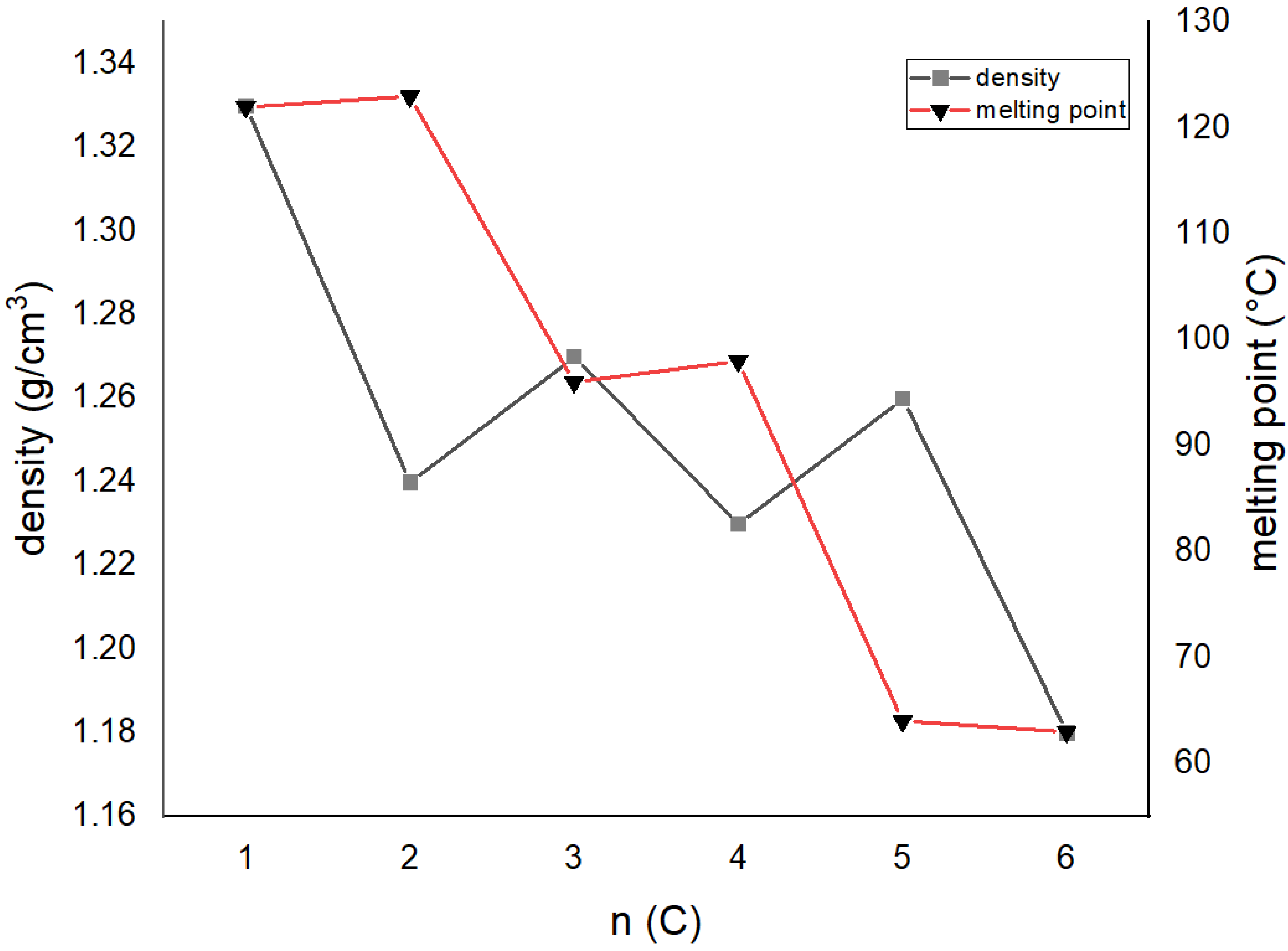
| Compound | The Dihedral Angle between Benzene Rings/° | Aliphatic Chain Conformation | OH‧‧‧OH Distance /Å | Density/g cm−3 | Melting Point/°C | CSD Code |
|---|---|---|---|---|---|---|
| 1,2-bis[2-(hydroxyformyl)phenoxy]methylene | 59.43 | gauche | 9.815(2) | 1.332 | 121–123 ref. [26] | FASCUY [27] |
| 1,2-bis[2-(hydroxyformyl)phenoxy]ethylene | 60.89 | trans–gauche–trans | 6.446(4) | 1.242 | 122–124 ref. [23] | SAZCIF [28] |
| 1,2-bis[2-(hydroxyformyl)phenoxy]propylene | 80.73 84.59 | trans–gauche-gauche–trans | 9.659(2) 9.725(2) | 1.274 | 96 ref. [23] | FASDAF [27] |
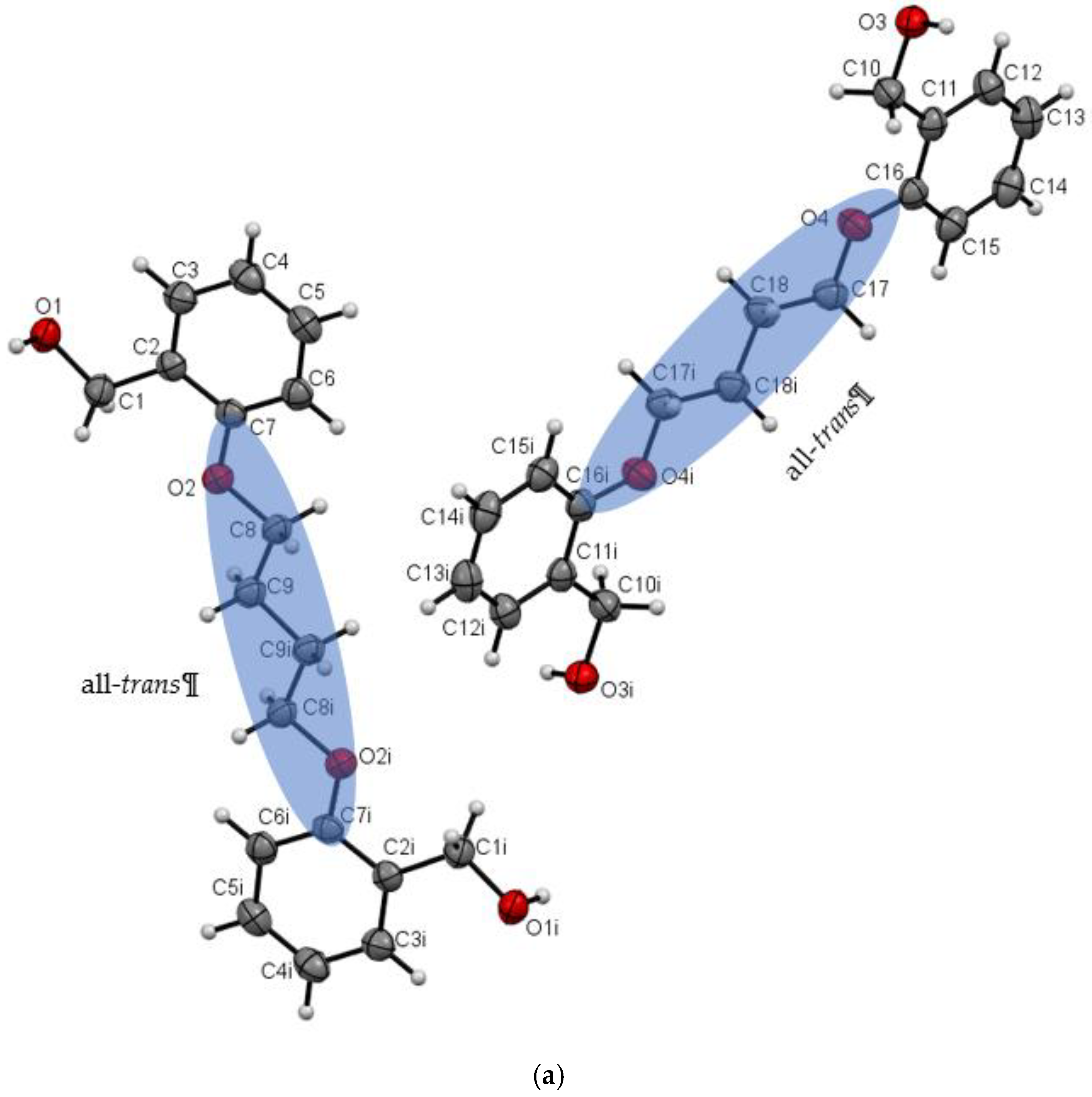
| Do4OH | 0 | all-trans | 13.754(2) | 1.236 | 98 | This work |
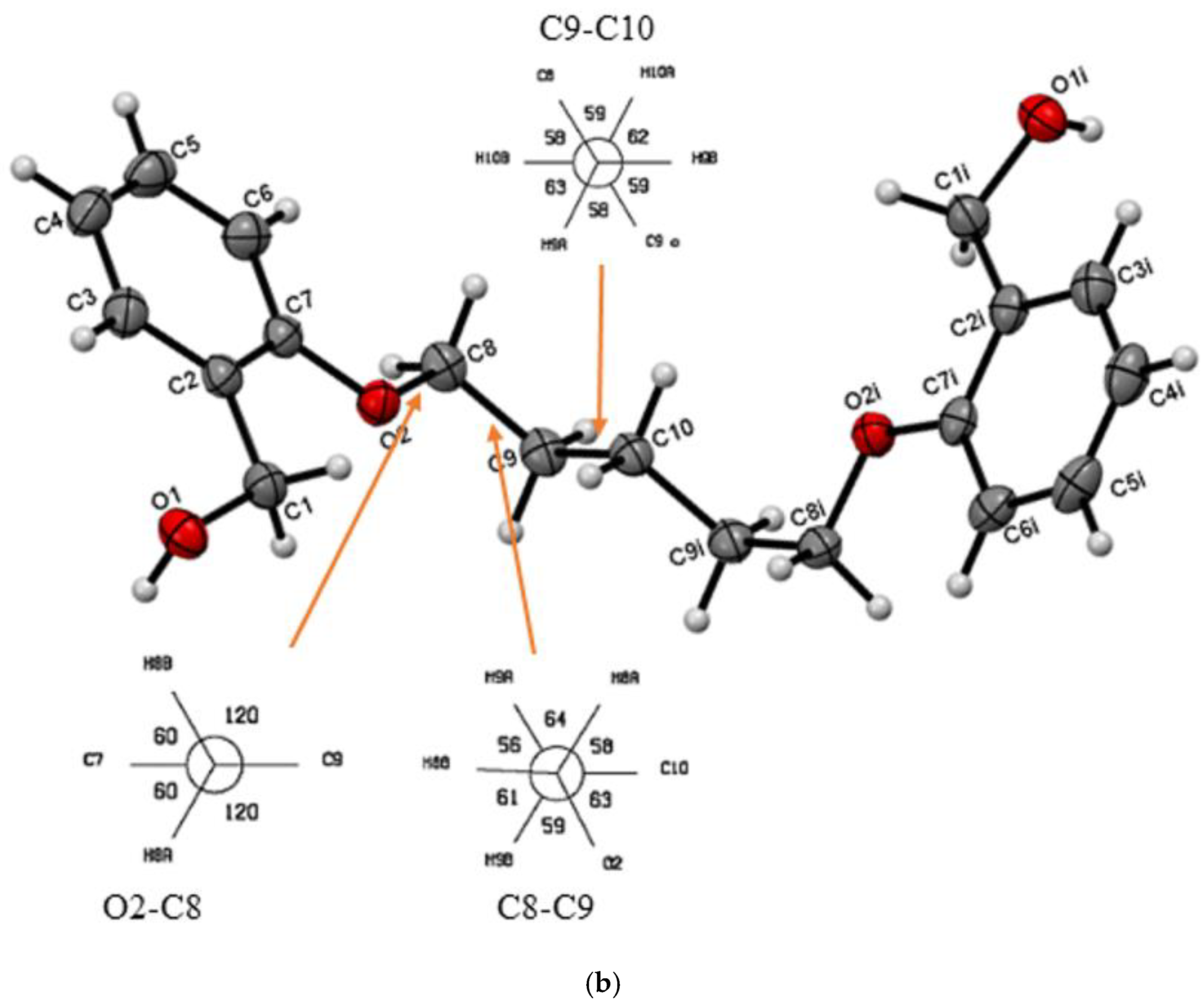
| Do5OH | 84.2(1) | trans–gauche–trans–gauche–trans | 11.133(2) | 1.268 | 64 | This work |
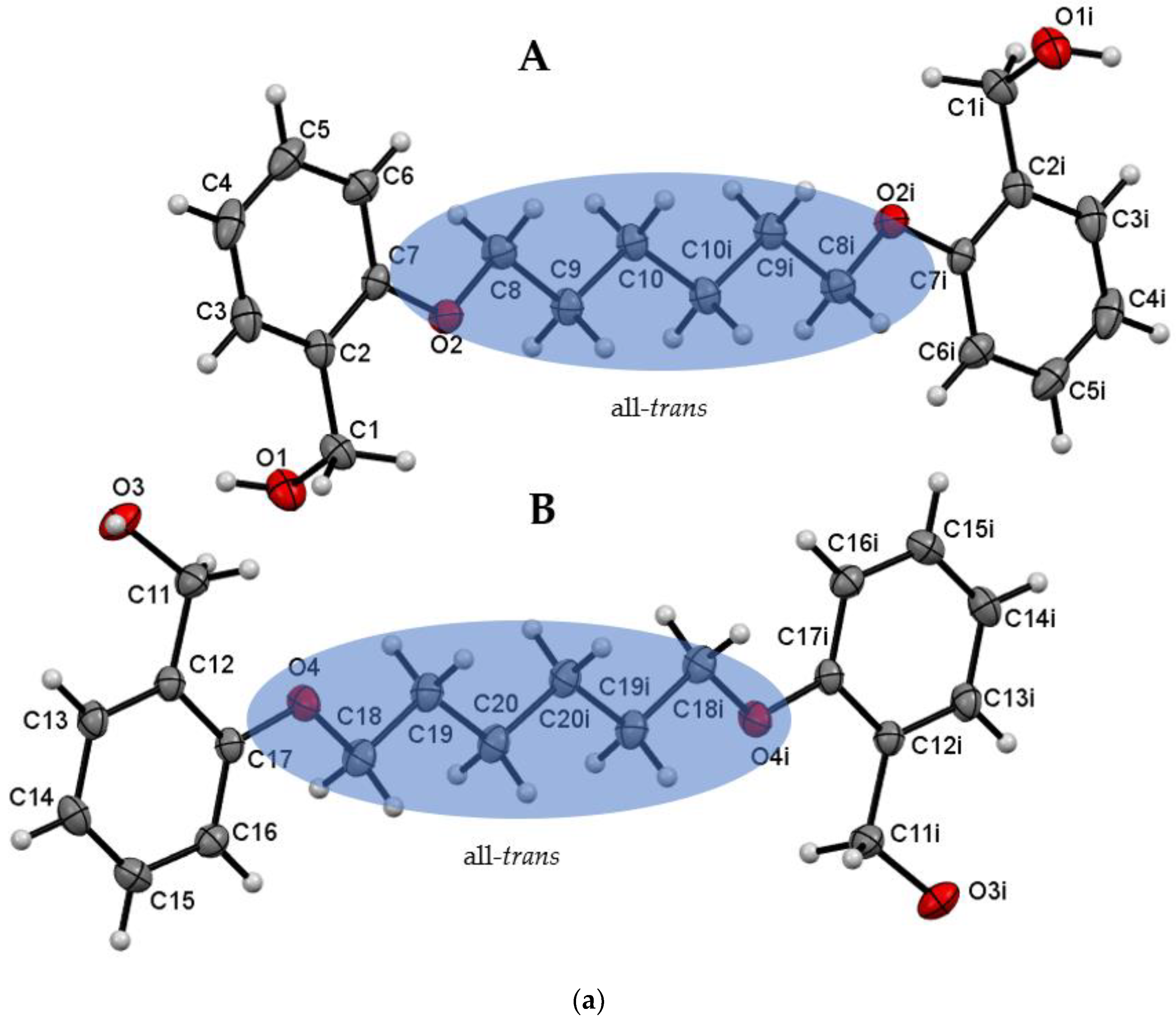
| Do6OH I | 0 | all-trans | 13.954(2)–conformer A 15.799(2)–conformer B | 1.188 | 63 | This work |
| Do6OH II | 0 | trans–gauche–trans–trans–trans–gauche-trans | 13.461(2) | 1.242 | 60 | This work |
| D−H∙∙∙A | d(D—H) | d(H⋅⋅⋅A) | d(D⋅⋅⋅A) | ∠(D—H⋅⋅⋅A) | Symmetry code |
| Do4OH | |||||
| O1—H1···O3 | 0.976(3) | 1.678(3) | 2.648(3) | 172(2) | -x+1,+y-1/2,-z+1/2 |
| O3—H3A···O1 | 0.946(3) | 1.708(3) | 2.653(2) | 176(3) | x,+y+1,+z |
| Do5OH | |||||
| O1—H1···O1 | 0.883(2) | 1.850(2) | 2.710(1) | 164(2) | -x+1/2,+y-1/2,+z |
| Do6OH I | |||||
| O3—H3A···O1 | 0.875(2) | 1.802(2) | 2.669(1) | 170(2) | x,+y,+z-1 |
| O1—H1···O3 | 0.884(2) | 1.813(2) | 2.687(1) | 169(2) | x,-y+1/2,+z+1/2 |
| Do6OH II | |||||
| O3—H1···O1 | 0.880(2) | 1.790(2) | 2.658(1) | 168(2) | x-1/2,+y,-z+1/2+1 |
| Compound | Eele | Epol | Edis | Erep | Etot | Elatt | PixelC |
|---|---|---|---|---|---|---|---|
| Do4OH | −151.99 (44.5%) | −24.79 (7.3%) | −164.97 (48.2%) | 148.81 | −192.94 | −192 | −188.7 |
| Do5OH | −80.01 (31.5%) | −13.39 (5.3%) | −161.39 (63.2%) | 97.39 | −157.40 | −193 | −215.2 |
| Do6OH I | −150.09 (44.9%) | −23.45 (7.2%) | −160.96 (47.9%) | 146.22 | −188.29 | −208 | −205.2 |
| Do6OH II | −77.48 (35.3%) | −12.13 (5.2%) | −129.08 (59.5%) | 90.16 | −128.53 | −194 | −200.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balić, T.; Paurević, M.; Počkaj, M.; Medvidović-Kosanović, M.; Goman, D.; Széchenyi, A.; Preisz, Z.; Kunsági-Máté, S. Influence of Aliphatic Chain Length on Structural, Thermal and Electrochemical Properties of n-alkylene Benzyl Alcohols: A Study of the Odd–Even Effect. Molecules 2022, 27, 3781. https://doi.org/10.3390/molecules27123781
Balić T, Paurević M, Počkaj M, Medvidović-Kosanović M, Goman D, Széchenyi A, Preisz Z, Kunsági-Máté S. Influence of Aliphatic Chain Length on Structural, Thermal and Electrochemical Properties of n-alkylene Benzyl Alcohols: A Study of the Odd–Even Effect. Molecules. 2022; 27(12):3781. https://doi.org/10.3390/molecules27123781
Chicago/Turabian StyleBalić, Tomislav, Marija Paurević, Marta Počkaj, Martina Medvidović-Kosanović, Dominik Goman, Aleksandar Széchenyi, Zsolt Preisz, and Sándor Kunsági-Máté. 2022. "Influence of Aliphatic Chain Length on Structural, Thermal and Electrochemical Properties of n-alkylene Benzyl Alcohols: A Study of the Odd–Even Effect" Molecules 27, no. 12: 3781. https://doi.org/10.3390/molecules27123781
APA StyleBalić, T., Paurević, M., Počkaj, M., Medvidović-Kosanović, M., Goman, D., Széchenyi, A., Preisz, Z., & Kunsági-Máté, S. (2022). Influence of Aliphatic Chain Length on Structural, Thermal and Electrochemical Properties of n-alkylene Benzyl Alcohols: A Study of the Odd–Even Effect. Molecules, 27(12), 3781. https://doi.org/10.3390/molecules27123781