
Novel Radioiodinated and Radiofluorinated Analogues of FT-2102 for SPECT or PET Imaging of mIDH1 Mutant Tumours

 , ,
, ,  , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
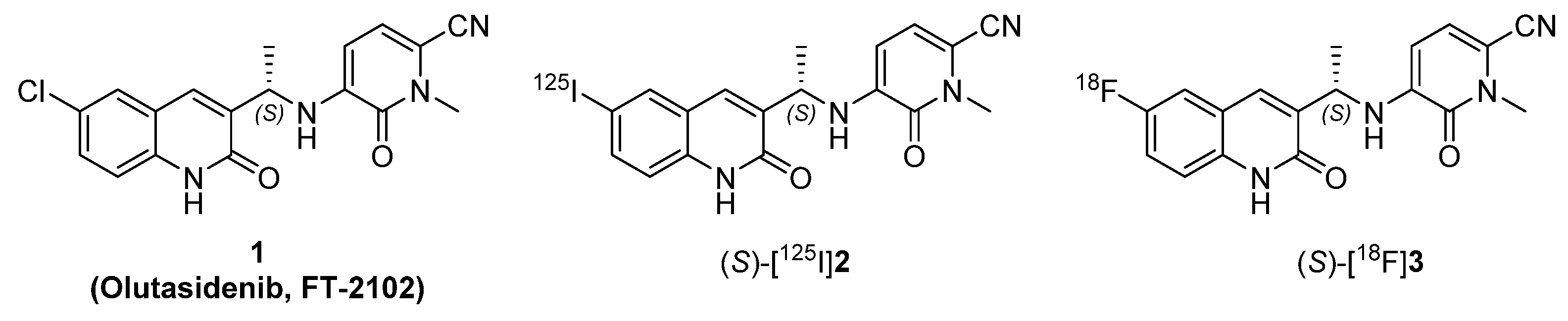
2.1. Chemistry
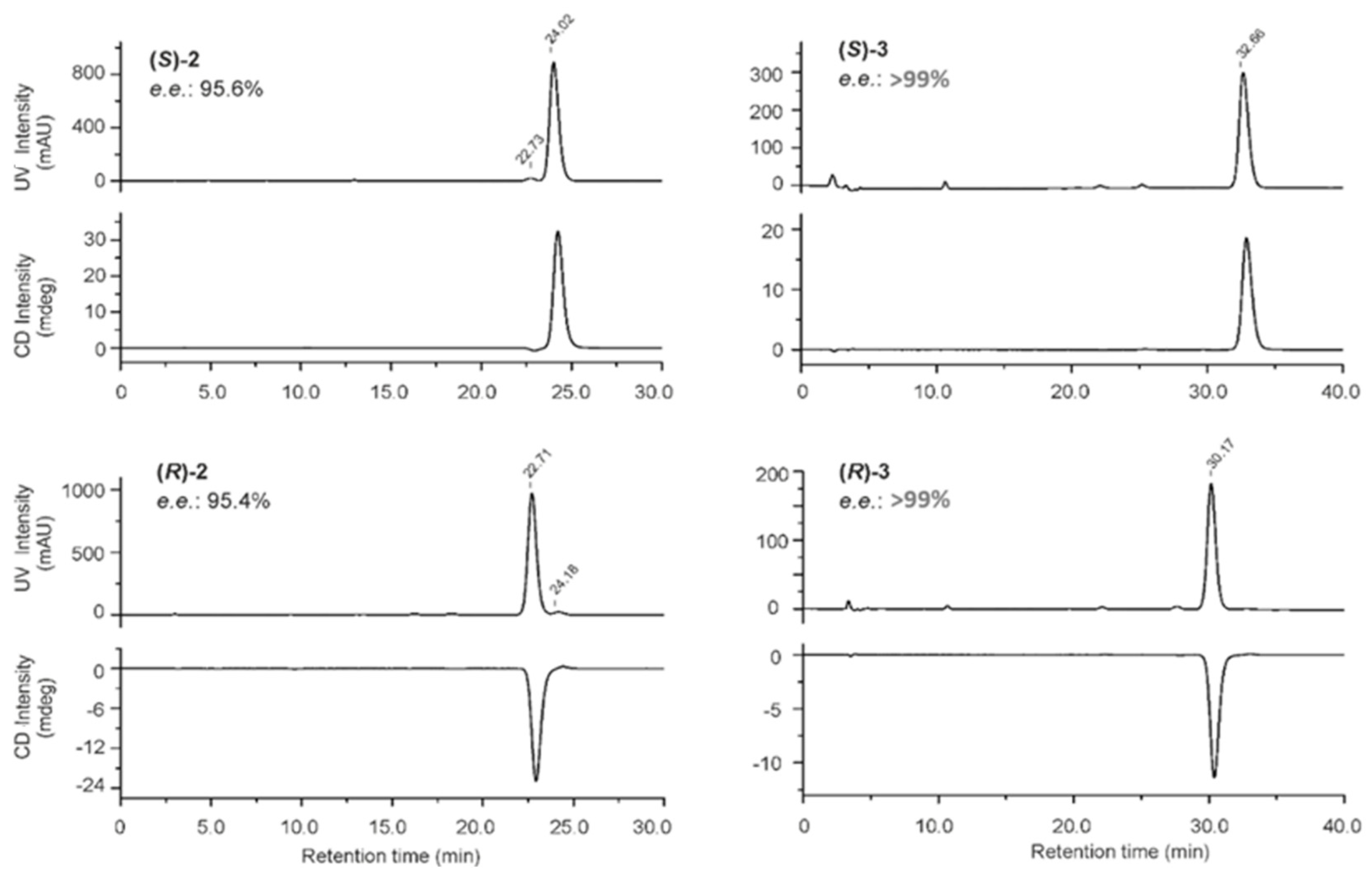
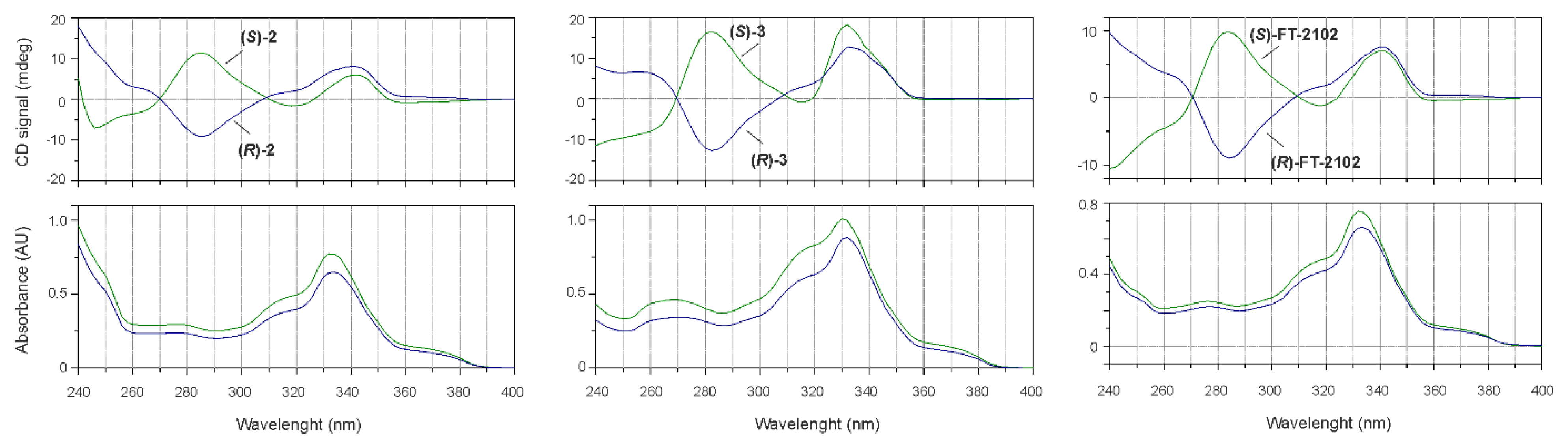
2.2. Biological Evaluation
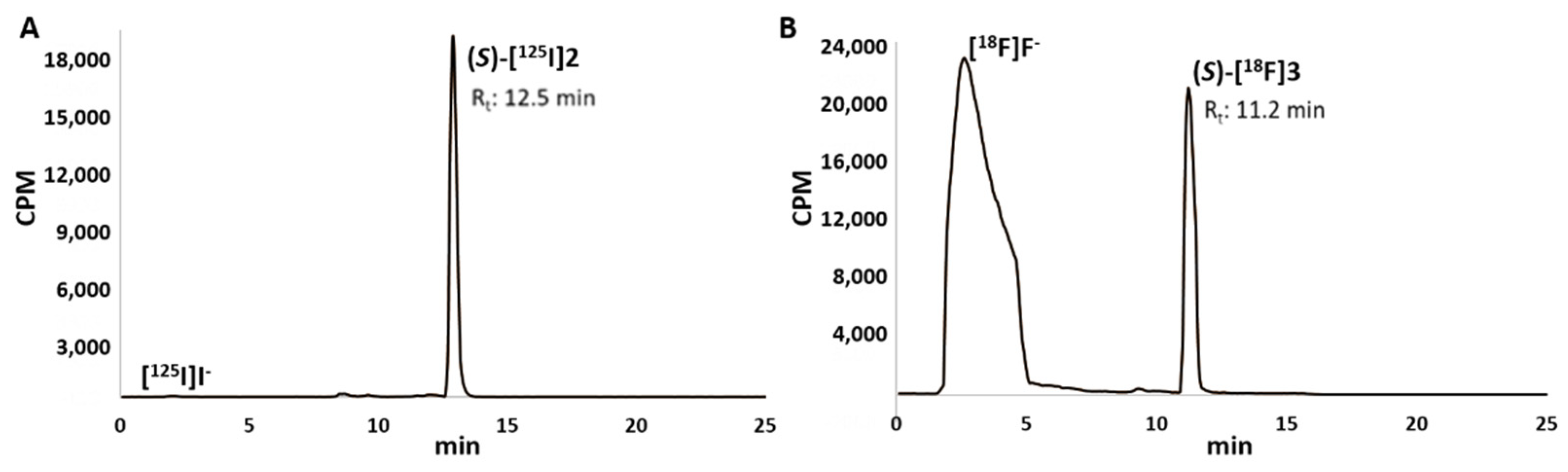
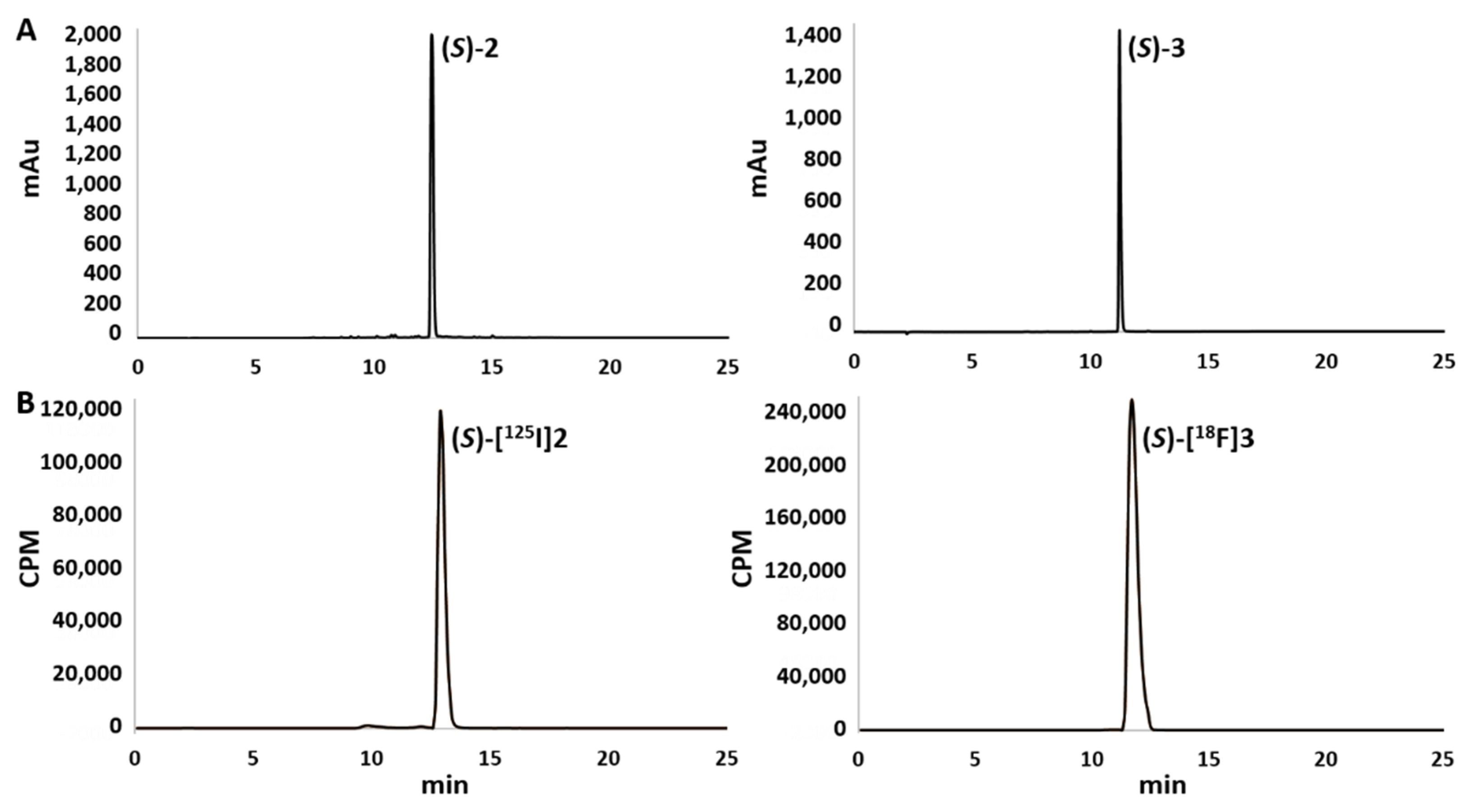
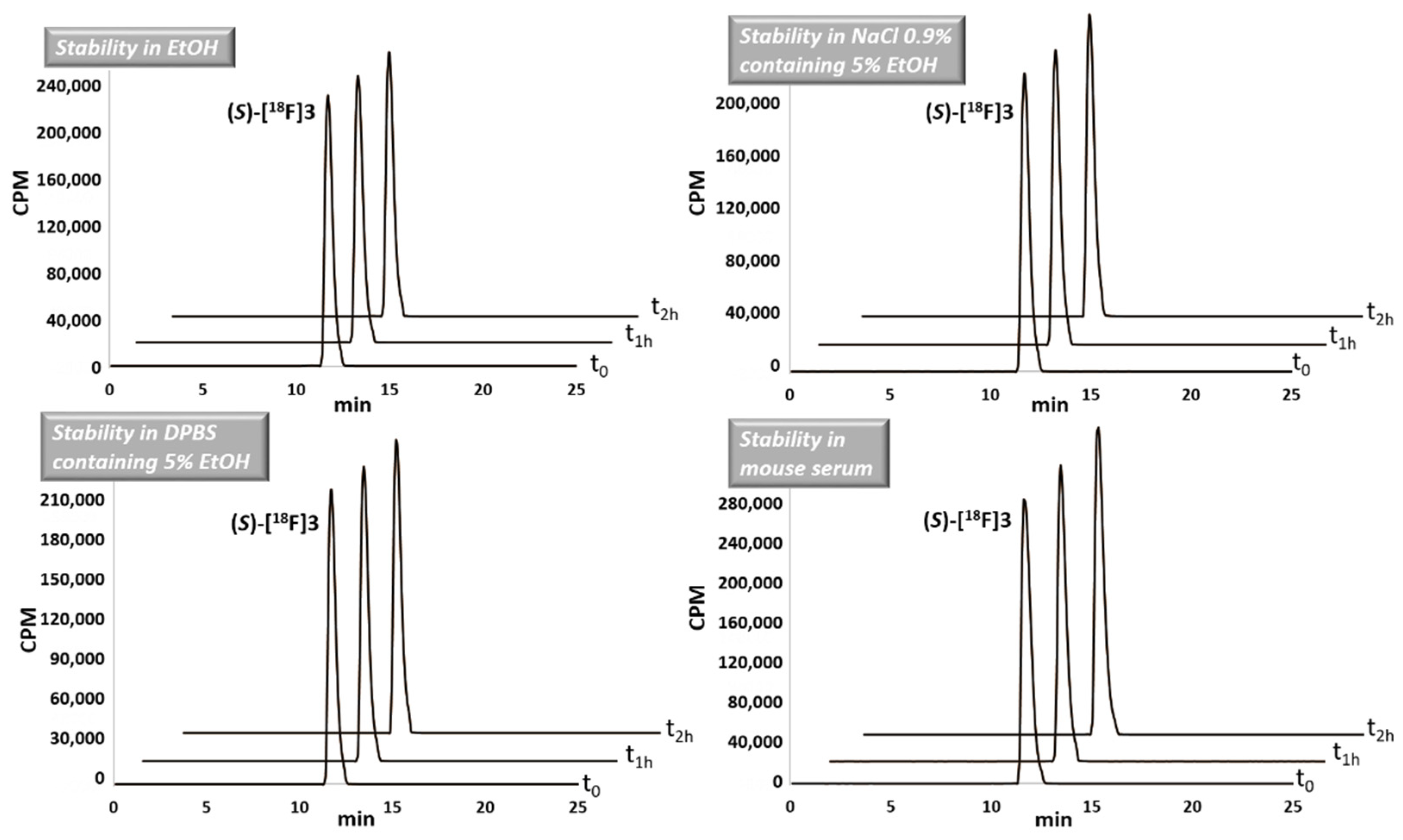
2.3. Radiochemistry
3. Materials and Methods
3.1. Chemistry
General Information
- N-(4-iodophenyl)acetamide (7). To a solution of 4-iodoaniline (1.00 g, 4.57 mmol) and DIPEA (1.6 mL, 9.19 mmol) in DCM (10 mL) was added acetic anhydride (0.5 mL, 5.49 mmol) dropwise at 0 °C. The reaction mixture was stirred at room temperature for 2.5 h. Then, the reaction was quenched with deionised water (20 mL). After decantation, the aqueous layer was extracted with DCM (6 × 10 mL). The combined organic layers were washed with deionised water (20 mL), dried on MgSO4, filtered, and concentrated under vacuum. The residue was purified by column chromatography (Al2O3, cyclohexane/EtOAc, 6/4, v/v) to provide derivative 7 (1.17 g, 4.48 mmol, 98%) as a white powder. Rf (Al2O3, cyclohexane/EtOAc, 6/4, v/v) 0.47. IR (ATR, cm−1) 3285, 3251, 3043, 1662, 1596, 1579, 1526, 1482, 1388, 814. Mp 184 ± 1 °C (Lit. mp 183 °C [38]). 1H NMR (500 MHz, CDCl3) δ 7.61 (d, 2H, 3J = 8.8 Hz, H-3, H-5), 7.28 (d, 2H, 3J = 8.8 Hz, H-2, H-6), 7.21 (br.s, 1H, NH), 2.17 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 168.39 (1C, CO), 138.06 (2C, C-3, C-5), 137.76 (1C, C-1), 121.76 (2C, C-2, C-6), 87.59 (1C, C-4), 24.82 (1C, CH3).
- N-(4-fluorophenyl)acetamide (8). To a solution of 4-fluoroaniline (1.00 g, 9.00 mmol) and DIPEA (3.14 mL, 18.0 mmol) in DCM (20 mL) was added acetic anhydride (1 mL, 10.8 mmol) dropwise at 0 °C. The reaction mixture was stirred at room temperature for 1 h. Then, the reaction was quenched with deionised water (5 mL). After decantation, the aqueous layer was extracted with DCM (3 × 10 mL). The combined organic layers were dried on MgSO4, filtered, and concentrated under vacuum. The residue was purified by column chromatography (Al2O3, cyclohexane/EtOAc, 5/5, v/v) to provide compound 8 (1.36 g, 8.88 mmol, 99%) as a white powder. Rf (Al2O3, cyclohexane/EtOAc, 5/5, v/v) 0.41. IR (ATR, cm−1) 3301, 3271, 1662, 1618, 1556, 1503, 1204, 831. Mp 153 ± 1 °C (Lit. mp 152–153 °C [39]). 1H NMR (500 MHz, CDCl3) δ 7.89 (br.s, 1H, NH), 7.44 (dd, 2H, 3JH-H = 9.0 Hz, 4JH-F = 4.8 Hz, H-2, H-6), 6.97 (t, 2H, 3JH-H = 3JH-F = 8.7 Hz, H-3, H-5), 2.14 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 168.43 (1C, CO), 159.53 (d, 1C, 1JC-F = 244 Hz, C-4), 133.97 (1C, 4JC-F = 3 Hz, C-1), 121.92 (d, 2C, 3JC-F = 8 Hz, C-2, C-6), 115.77 (d, 2C, 2JC-F = 22 Hz, C-3, C-5), 24.56 (1C, CH3). 19F NMR (470 MHz, CDCl3) δ—118.97.
- 2-chloro-3-formyl-6-iodoquinoline (9). Under anhydrous argon atmosphere, POCl3 (38 mL, 408 mmol) was added dropwise (over 20 min) to precooled (0 °C) anhydrous DMF (10 mL, 129 mmol). The orange solution was allowed to warm to room temperature (20 min) and then treated with compound 7 (10.0 g, 38.3 mmol). The round-bottom flask was sealed, and the solution was stirred at 80 °C overnight. After cooling to room temperature, the solution was then poured over ice (500 mL). The formed yellow precipitate was filtered, washed with deionised water (100 mL), and dried under vacuum. The crude product was further purified by column chromatography (SiO2, cyclohexane/EtOAc, 5/5, v/v) to provide quinoline 9 (1.53 g, 4.82 mmol, 13%) as a yellow powder. Rf (SiO2, cyclohexane/EtOAc, 9/1, v/v) 0.42. IR (ATR, cm−1) 1683, 1673, 1596, 1571, 1479, 1360, 829. Mp 169 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 10.55 (s, 1H, CHO), 8.64 (s, 1H, H-4), 8.37 (d, 1H, 4J = 2.0 Hz, H-5), 8.11 (dd, 1H, 3J = 8.9 Hz,4J = 2.0 Hz, H-7), 7.80 (d, 1H, 3J = 8.9 Hz, H-8). 13C NMR (126 MHz, CDCl3) δ 188.90 (1C, CHO), 150.73, 148.64 (2C, C-2, C-3), 142.35 (1C, C-7), 139.00 (1C, C-4), 138.31 (1C, C-5), 130.20 (1C, C-8), 128.27, 127.04 (2C, C-4a, C-8a), 93.80 (1C, C-6). HRMS (ESI) m/z 317.9171 [M+H]+ (calculated for [C11H6ClINO]+ 317.9177), 349.9432 [M+32]+ (calculated for [C11H10ClINO2]+, 349.9445 methyl hemiacetal form).
- 2-chloro-3-formyl-6-fluoroquinoline (10). Under anhydrous argon atmosphere, POCl3 (4.2 mL, 45.7 mmol) was added dropwise to precooled (0 °C) anhydrous DMF (1.25 mL, 16.3 mmol). The solution was allowed to warm to room temperature (15 min) and then treated with compound 8 (1.00 g, 6.53 mmol). The round-bottom flask was sealed, and the solution was stirred at 85 °C for 19 h. After cooling to room temperature, the solution was then poured over ice (100 mL). The yellow precipitate was filtered, washed with deionised water (20 mL), and dried under vacuum to provide compound 10 (182 mg, 0.868 mmol, 13%) as a yellow powder. Rf (SiO2, cyclohexane/EtOAc, 9/1, v/v) 0.40. IR (ATR, cm−1) 1693 (νC=O), 1581, 1497 (νC=C), 1337 (νC-N), 1220 (νC-F), 1046 (νS=O), 834 (δCH op). Mp 180 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 10.38 (s, 1H, CHO), 8.97 (s, 1H, H-4), 8.15-8.10 (m, 2H, H-5, H-7), 7.92 (td, 1H, 3JH-H = 3JH-F = 8.7 Hz,4JH-H = 3.0 Hz, H-7). 13C NMR (126 MHz, CDCl3) δ 189.08 (1C, CHO), 161.21 (d, 1C, 1JC-F = 250 Hz, C-6), 149.60 (d, 1C, 4JC-F = 3 Hz, C-8a), 146.82 (1C, C-2 or C-3), 139.65 (d, 1C, 4JC-F = 6Hz, C-4), 131.34 (d, 1C, 3JC-F = 9 Hz, C-8), 127.49 (d, 1C, 3JC-F = 10 Hz, C-4a), 127.05 (1C, C-2 or C-3), 123.98 (d, 1C, 2JC-F = 26 Hz, C-7), 112.80 (d, 1C, 2JC-F = 22 Hz, C-5). 19F NMR (470 MHz, CDCl3) δ—110.89. HRMS (ESI) m/z 210.0113 [M+H]+ (calculated for [C11H6ClFNO]+ 210,0116), 242.0375 [M+32] (calculated for [C11H10ClFNO2]+ 242.0384, methyl hemiacetal form).
- (R,E)-N-[(2-chloro-6-iodoquinolin-3-yl)methylene]-2-methylpropane-2-sulfinamide (11). To a mixture of 9 (600 mg, 1.89 mmol) and (R)-2-methylpropane-2-sulfinamide (252 mg, 2.08 mmol) in 1,2-dichloroethane (DCE, 5 mL) was added CuSO4 (455 mg, 2.85 mmol). The resulting mixture was stirred at 60 °C for 3 d. The mixture was cooled to room temperature, filtered through a pad of Celite® 545, and rinsed with EtOAc (4 × 17 mL). The filtrate was evaporated under vacuum and purified by column chromatography (SiO2, ethanol gradient in DCM from 0 to 5%) to provide 11 (530 mg, 1.26 mmol, 67%) as a yellow powder. Rf (SiO2, DCM) 0.09. IR (ATR, cm−1) 1569, 1475, 1360, 1085, 825. Mp 163 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 9.08 (s, 1H, CH=N), 8.72 (s, 1H, H-4), 8.36 (d, 1H,4J = 1.9 Hz, H-5), 8.05 (dd, 1H, 3J = 8.9 Hz, 4J = 1.9 Hz, H-7), 7.78 (d, 1H, 3J = 8.9 Hz, H-8), 1.32 (s, 9H, CH3). 13C NMR (126 MHz, CDCl3) δ 159.02 (1C, CH=N), 150.72, 147.88 (2C, C-2, C-3), 141.34 (1C, C-7), 137.64 (1C, C-5), 137.52 (1C, C-4), 130.20 (1C, C-8), 128.52, 126.8 (2C, C-4a, C-8a), 93.48 (1C, C-6), 58.74 (1C, C(CH3)3), 22.93 (3C, C(CH3)3). HRMS (ESI) m/z 420.9624 [M+H]+ (calculated for [C14H15ClIN2OS]+ 420.9632).
- (R,E)-N-[(2-chloro-6-fluoroquinolin-3-yl)methylene]-2-methylpropane-2-sulfinamide (12). To a mixture of 10 (150 mg, 0.716 mmol) and (R)-2-methylpropane-2-sulfinamide (96 mg, 0.788 mmol) in DCE (4 mL) was added CuSO4 (173 mg, 0.109 mmol). The resulting mixture was stirred at 60 °C for 17 h. The mixture was cooled to room temperature, filtered through a pad of Celite® 545, and rinsed with EtOAc (2 × 10 mL). The filtrate was evaporated under vacuum to provide 12 (194 mg, 0.620 mmol, 87%) as a white powder. Rf (SiO2, cyclohexane/EtOAc, 9/1, v/v) 0.17. IR (ATR, cm−1) 1564, 1495, 1207, 1082, 829. Mp 155 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 9.10 (s, 1H, CH=N), 8.78 (s, 1H, H-4), 8.06 (dd, 1H, 3JH-H = 9.2 Hz,4JH-F = 5.1 Hz, H-8), 7.63-7.54 (m, 2H, H-7, H-5), 1.32 (s, 9H, CH3). 13C NMR (126 MHz, CDCl3) δ 161.12 (d, 1C, 1JC-F= 251 Hz, C-6), 159.13 (1C, CH=N), 149.56 (d, 1C, 4JC-F= 3 Hz, C-8a), 146.06 (1C, C-2 or C-3), 138.13 (d, 1C, 4JC-F= 6 Hz, C-4), 131.29 (d, 1C, 3JC-F= 9 Hz, C-8), 127.66 (d, 1C, 3JC-F= 10 Hz, C-4a), 126.85 (1C, C-2 or C-3), 123.96 (d, 1C, 2JC-F= 26 Hz, C-7), 112.04 (d, 1C, 2JC-F= 22 Hz, C-5), 58.70 (1C, C(CH3)3), 22.92 (3C, C(CH3)3). 19F NMR (470 MHz, CDCl3) δ—111.60. HRMS (ESI) m/z 313.0568 [M+H]+ (calculated for [C14H15ClFN2OS]+ 313.0572).
- (R)-N-[(S)-1-(2-chloro-6-iodoquinolin-3-yl)ethyl]-2-methylpropane-2-sulfinamide ((S,R)-13) and (R)-N-[(R)-1-(2-chloro-6-iodoquinolin-3-yl)ethyl]-2-methylpropane-2-sulfinamide ((R,R)-13). To a solution of 11 (700 mg, 2.38 mmol) in anhydrous DCM (13 mL) was added dropwise, at −60 °C, MeMgBr (3 M solution in diethyl ether, 0.84 mL, 2.51 mmol) under stirring and argon atmosphere. Then, the reaction mixture was stirred at − 60 °C for 3 h and allowed to rise slowly to room temperature overnight. After cooling, a saturated aqueous solution of NH4Cl (12 mL) was slowly added. After decantation, the aqueous layer was extracted with DCM (4 × 15 mL). The combined organic layers were dried on MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, cyclohexane/EtOAc, EtOAc gradient varying from 50 to 80%) to provide compound (S,R)-13 (452 mg, 1.03 mmol, 62%) and its diastereomer (R,R)-13 (110 mg, 0.250 mmol, 15%), both as white solids. Diastereomer (S,R)-13: Rf (SiO2, cyclohexane/EtOAc, 5/5, v/v) 0.11. IR (ATR, cm−1) 3211, 1581, 1475, 1035), 822. Mp 124 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 8.20 (d, 1H,4J = 1.9 Hz, H-5), 8.12 (s, 1H, H-4), 7.96 (dd, 1H, 3J = 8.9 Hz, 4J = 1.9 Hz, H-7), 7.74 (d, 1H, 3J = 8.9 Hz, H-8), 5.10 (q, 1H, 3J = 6.7 Hz, CH), 3.47 (br.s, 1H, NH), 1.68 (d, 3H, 3J = 6.7 Hz, CH3), 1.26 (s, 9H, C(CH3)3). 13C NMR (126 MHz, CDCl3) δ 150.58, 145.91 (2C, C-2, C-3), 139.35 (1C, C-7), 136.50 (1C, C-5), 136.38 (1C, C-4), 135.07 (1C, C-8a or C-4a), 130.01 (1C, C-8), 129.02 (1C, C-8a or C-4a), 92.91 (1C, C-6), 56.26 (1C, C(CH3)3), 52.08 (1C, CHCH3), 23.58 (1C, CHCH3), 22.76 (3C, C(CH3)3). HRMS (ESI) m/z 436.9936 [M+H]+ (calculated for [C15H19ClIN2OS]+ 436.9945). Diastereomer (R,R)-13: Rf (SiO2, cyclohexane/EtOAc, 5/5, v/v) 0.18. IR (ATR, cm−1) 1581, 1474, 1043, 822. Mp 79 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 8.25 (d, 1H,4J = 1,9 Hz, H-5), 8.15 (s, 1H, H-4), 7.95 (dd, 1H, 3J = 8.9 Hz, 4J = 1.9 Hz, H-7), 7.73 (d, 1H, 3J = 8.9 Hz, H-8), 5.02 (qt, 1H, 3J = 6.3 Hz, CH), 3.77 (d, 1H,3J = 5.3 Hz, NH), 1.64 (d, 3H, 3J = 6.6 Hz, CH3), 1.25 (s, 9H, C(CH3)3). 13C NMR (126 MHz, CDCl3) δ 149.97, 145.77 (2C, C-2, C-3), 139.23 (1C, C-7), 136.63 (1C, C-5), 136.42 (1C, C-4), 134.96 (1C, C-8a or C-4a), 129.78 (1C, C-8), 128.83 (1C, C-8a or C-4a), 92.85 (1C, C-6), 56.16 (1C, C(CH3)), 51.62 (1C, CHCH3), 22.63 (1C, C(CH3)3), 22.30 (3C, CHCH3). HRMS (ESI) m/z 436.9940 [M+H]+ (calculated for [C15H19ClIN2OS]+ 436.9945).
- (R)-N-[(S)-1-(2-chloro-6-fluoroquinolin-3-yl)ethyl]-2-methylpropane-2-sulfinamide ((S,R)-14) and (R)-N-[(R)-1-(2-chloro-6-fluoroquinolin-3-yl)ethyl]-2-methylpropane-2-sulfinamide ((R,R)-14). To a solution of 12 (350 mg, 1.12 mmol) in anhydrous DCM (9 mL) at −60 °C was added dropwise MeMgBr (3 M solution in diethyl ether, 0.56 mL, 1.68 mmol) under stirring and argon atmosphere. Then, the reaction mixture was stirred at −60 °C for 3 h and allowed to rise slowly to room temperature overnight. After cooling, a saturated aqueous solution of NH4Cl (15 mL) was added slowly. The aqueous layer was extracted with DCM (3 × 20 mL). The combined organic layers were dried on MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, cyclohexane/EtOAc, 5/5, v/v) to provide derivative (S,R)-14 (242 mg, 0.735 mmol, 66%) and its diastereomer (R,R)-14 (41 mg, 0.126 mmol, 11%), both as white solids. Diastereomer (S,R)-14: Rf (SiO2, cyclohexane/EtOAc, 5/5, v/v) 0.10. IR (ATR, cm−1) 3181, 1495, 1211, 1055, 828. Mp 138 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 8.18 (s, 1H, H-4), 8.02 (dd, 1H, 3JH-H= 9.2 Hz, 4JH-F= 5.2 Hz, H-8), 7.49 (ddd, 1H, 3JH-H = 9.2 Hz, 3JH-F = 8.3 Hz, 4JH-H = 2.8 Hz, H-7), 7.43 (dd, 1H, 3JH-F = 8.6 Hz, 4JH-H = 2.8 Hz, H-5), 5.10 (qd, 1H, 3JH-H = 4.6 and 6.7 Hz, CH), 3.48 (d, 1H,3JH-H = 4.4 Hz, NH), 1.70 (d, 3H, 3JH-H = 6.7 Hz, CH3), 1.25 (s, 9H, C(CH3)3). 13C NMR (126 MHz, CDCl3) δ 160.92 (d, 1C, 1JC-F= 249 Hz, C-6), 149.31 (d, 1C, 4JC-F= 3 Hz, C-8a), 143.99, 136.46 (2C, C-2, C-3), 135.69 (d, 1C, 4JC-F= 5 Hz, C-4), 130.90 (d, 1C, 3JC-F= 9 Hz, C-8), 128.07 (d, 1C, 3JC-F= 10 Hz, C-4a), 120.77 (d, 1C, 2JC-F= 26 Hz, C-7), 110.84 (d, 1C, 2JC-F= 22 Hz, C-5), 56.24 (1C, C(CH3)3), 52.15 (1C, CHCH3), 23.53 (1C, CHCH3), 22.74 (3C, C(CH3)3). 19F NMR (470 MHz, CDCl3) δ—112.88. HRMS (ESI) m/z 329.0877 [M+H]+ (calculated for [C15H19ClFN2OS]+ 329.0885). Diastereomer (R,R)-14: Rf (SiO2, cyclohexane/EtOAc, 5/5, v/v) 0.12. IR (ATR, cm−1) 3463, 1496, 1040-1008, 822. Mp 116 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 8.20 (s, 1H, H-4), 8.01 (dd, 1H, 3JH-H= 9.2 Hz, 4JH-F= 5.2 Hz, H-8), 7.52-7.43 (m, 2H, H-5, H-7), 5.03 (qt, 1H, 3JH-H = 6.5 Hz, CH), 3.81 (d, 1H,3JH-H = 5.4 Hz, NH), 1.65 (d, 3H, 3JH-H = 6.6 Hz, CH3), 1.26 (s, 9H, C(CH3)3). 13C NMR (126 MHz, CDCl3) δ 160.95 (d, 1C, 1JC-F= 249 Hz, C-6), 148.90 (d, 1C, 4JC-F= 3 Hz, C-8a), 144.08, 136.63 (2C, C-2, C-3), 135.62 (d, 1C, 4JC-F= 5 Hz, C-4), 130.90 (d, 1C, 3JC-F= 9 Hz, C-8), 128.10 (d, 1C, 3JC-F= 10 Hz, C-4a), 120.88 (d, 1C, 2JC-F= 26 Hz, C-7), 111.00 (d, 1C, 2JC-F= 22 Hz, C-5), 56.26 (1C, C(CH3)3), 51.80 (1C, CHCH3), 22.35 (1C, CHCH3), 22.70 (3C, C(CH3)3). 19F NMR (470 MHz, CDCl3) δ—112.79. HRMS (ESI) m/z 329.0877 [M+H]+ (calculated for [C15H19ClFN2OS]+ 329.0885).
- ((S)-3-(1-aminoethyl)-6-iodoquinolin-2(1H)-one hydrochloride salt ((S)-5). A mixture of (S,R)-13 (450 mg, 1.03 mmol) in 1,4-dioxane (3.5 mL) and aqueous 1 N HCl solution (3.5 mL, 3.52 mmol) was heated at 110 °C overnight. After cooling to room temperature, the mixture was evaporated under reduced pressure, and the yellow powder obtained was washed with diethyl ether (5 × 5 mL) to provide quinolinone (S)-5 (158 mg, quantitative) as a yellow solid, which was used in the next step without further purification. HRMS (ESI) m/z 314.9981 [M+H]+ (calculated for [C11H12IN2O]+ 314.9988).
- ((R)-3-(1-aminoethyl)-6-iodoquinolin-2(1H)-one hydrochloride salt ((R)-5). Using the same procedure as described for compound (S)-5, the enantiomer (R)-5 was obtained as a yellow solid. Yield 95%. HRMS (ESI) m/z 314.9983 [M+H]+ (calculated for [C11H12IN2O]+ 314.9989).
- ((S)-3-(1-aminoethyl)-6-fluoroquinolin-2(1H)-one hydrochloride salt ((S)-6). Using the same procedure as described for compound (S)-5, the fluorinated analogue (S)-6 was obtained as a yellow solid. Yield 80%. HRMS (ESI) m/z 207.0925 [M+H]+ (calculated for [C11H12FN2O]+ 207.0928).
- ((R)-3-(1-aminoethyl)-6-fluoroquinolin-2(1H)-one hydrochloride salt ((R)-6). Using the same procedure as described for compound (S)-5, the compound (R)-6 was obtained as a yellow solid. Yield 88%. HRMS (ESI) m/z 207.0926 [M+H]+ (calculated for [C11H12FN2O]+ 207.0928).
- (S)-5-{[1-(6-iodo-2-oxo-1,2-dihydroquinolin-3-yl)ethyl]-amino}-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile ((S)-2). DIPEA (750 µL, 4.28 mmol) was added to a solution of 4 (254 mg, 1.67 mmol) and amine (S)-5 (500 mg, 1.43 mmol) in dimethylsulfoxyde (DMSO, 11 mL). The resulting solution was stirred at 110 °C for 15 h. After cooling to room temperature, deionised water (150 mL) was added, and the resulting solution was extracted with EtOAc (3 × 100 mL). The organic extracts were combined, washed with deionised water (200 mL) to eliminate residual traces of DMSO, dried on MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by reversed-phase flash chromatography (flow 30 mL.min−1; water/ACN: 95/5, v/v for 2 min; 95/5 to 0/100, v/v for 13 min; and 0/100, v/v for 5 min) to provide end-product (S)-2 (341 mg, 0.764 mmol, 54%) as a yellow powder. Rf (SiO2, cyclohexane/EtOAc, 3/7, v/v) 0.35. IR (ATR, cm−1) 2208, 1647, 1591, 1544, 809. Mp 172 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 12.36 (s, 1H, NH), 7.86 (d, 1H,4J = 1.7 Hz, H-5), 7.76 (dd, 1H, 3J = 8.6 Hz,4J = 1.8 Hz, H-7), 7.59 (s, 1H, H-4), 7.21 (d, 1H,3J = 8.6 Hz, H-8), 6.68 (d, 1H,3J = 7.8 Hz, H-3′), 6.29 (br.s, 1H, NH), 5.90 (d, 1H, 3J = 7.9 Hz, H-4′), 4.85 (q, 1H,3J = 6.6 Hz, CHCH3), 3.75 (s, 3H, NCH3), 1.66 (d, 3H, 3J = 6.7 Hz, CHCH3). 13C NMR (126 MHz, CDCl3) δ 163.06 (1C, C-2), 156.94 (1C, C-6′), 141.31 (1C, C-5′ or C-2′), 139.09 (1C, C-7), 137.03 (1C, C-8a or C-4a), 136.45 (1C, C-5), 134.25 (1C, C-3), 134.16 (1C, C-4), 121.96 (1C, C-8a or C-4a), 119.42 (1C, C-3′), 117.70 (1C, C-8), 114.81 (1C, CN), 106.11 (1C, C-5′ or C-2′), 104.60 (1C, C-4′), 85.89 (1C, C-6), 47.91 (1C, CHCH3), 34.81 (1C, NCH3), 21.53 (1C, CHCH3). HRMS (ESI) m/z 447.0302 [M+H]+ (calculated for [C18H16IN4O2]+ 447.0312). Analytical HPLC characterisation: Rt: 12.49 min, 95.2% purity at 254 and 360 nm.
- (R)-5-{[1-(6-iodo-2-oxo-1,2-dihydroquinolin-3-yl)ethyl]-amino}-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile ((R)-2). DIPEA (150 µL, 0.861 mmol) was added to a solution of 4 (52 mg, 0.343 mmol) and amine (R)-5 (100 mg, 0.285 mmol) in DMSO (2.25 mL). The resulting solution was stirred at 110 °C for 22 h. After cooling to room temperature, deionised water (30 mL) was added, and the resulting solution was extracted with EtOAc (3 × 20 mL). The organic extracts were combined, dried on MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified first by silica column chromatography (SiO2, cyclohexane/EtOAc, 4/6, v/v) and then by alumina column chromatography (Al2O3, cyclohexane/EtOAc, EtOAc gradient varying from 80 to 100%, and then methanol gradient in EtOAc from 1 to 5%) to provide derivative (R)-2 (55 mg, 0.123 mmol, 43%) as a green powder. Rf (SiO2, cyclohexane/EtOAc, 3/7, v/v) 0.49. IR (ATR, cm−1) 2209, 1633, 1589, 1544, 811. Mp 257 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 11.92 (s, 1H, NH), 7.85 (d, 1H,4J = 1.8 Hz, H-5), 7.76 (dd, 1H, 3J = 8.6 Hz,4J = 1.8 Hz, H-7), 7.57 (s, 1H, H-4), 7.17 (d, 1H,3J = 8.6 Hz, H-8), 6.68 (d, 1H,3J = 7.8 Hz, H-3′), 6.26 (br.d, 1H, NH), 5.90 (d, 1H, 3J = 7.9 Hz, H-4′), 4.88-4.81 (m, 1H, CHCH3), 3.75 (s, 3H, NCH3), 1.65 (d, 3H, 3J = 6.7 Hz, CHCH3). 13C NMR (126 MHz, CDCl3) δ 163.12 (1C, C-2), 156.93 (1C, C-6′), 141.31 (1C, C-5′ or C-2′), 139.09 (1C, C-7), 137.03 (1C, C-8a or C-4a), 136.45 (1C, C-5), 134.20 (1C, C-3), 134.17 (1C, C-4), 121.93 (1C, C-8a or C-4a), 119.42 (1C, C-3′), 117.69 (1C, C-8), 114.81 (1C, CN), 106.11 (1C, C-5′ or C-2′), 104.59 (1C, C-4′), 85.81 (1C, C-6), 47.92 (1C, CHCH3), 34.80 (1C, NCH3), 21.53 (1C, CHCH3). HRMS (ESI) m/z 447.0303 [M+H]+ (calculated for [C18H16IN4O2]+ 447.0312). Analytical HPLC characterisation: Rt: 12.52 min, 95.3% and 96.5% purity at 254 and 360 nm, respectively.
- (S)-5-{[1-(6-fluoro-2-oxo-1,2-dihydroquinolin-3-yl)ethyl]-amino}-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile ((S)-3). DIPEA (206 µL, 1.18 mmol) was added to a solution of 4 (71 mg, 0.467 mmol) and amine (S)-6 (96 mg, 0.396 mmol) in DMSO (3 mL). The resulting solution was stirred at 110 °C during 19 h. After cooling to room temperature, deionised water (30 mL) was added, and the aqueous layer was extracted with EtOAc (3 × 20 mL). The organic extracts were combined, dried on MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (Al2O3, cyclohexane/EtOAc, EtOAc gradient varying from 80 to 100%, and then methanol gradient in EtOAc from 1 to 5%) to provide derivative (S)-3 (56 mg, 0.166 mmol, 42%) as a brown powder. Rf (Al2O3, MeOH 3% in EtOAc) 0.34. IR (ATR, cm−1) 2209, 1652, 1627, 1594, 1544, 1227, 814. Mp 171 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 12.90 (s, 1H, NH), 7.65 (s, 1H, H-4), 7.47 (dd, 1H, 3JH-H = 8.9 Hz,4JH-F = 4.5 Hz, H-8), 7.27 (td, 3JH-F = 3JH-H = 8.6 Hz, 4JH-H = 2.6 Hz, 1H, H-7), 7.19 (dd, 1H,3JH-F = 8.5 Hz, 4JH-H = 2.6 Hz, H-5), 6.68 (d, 1H,3JH-H = 7.9 Hz, H-3′), 6.38 (br.s, 1H, NH), 5.94 (d, 1H, 3JH-H = 7.9 Hz, H-4′), 4.92–4.84 (m, 1H, CHCH3), 3.75 (s, 3H, NCH3), 1.67 (d, 3H, 3JH-H = 6.7 Hz, CHCH3). 13C NMR (126 MHz, CDCl3) δ 163.25 (1C, C-2), 158.31 (d, 1C, 1JC-F= 243 Hz, C-6), 156.95 (1C, C-6′), 141.39 (1C, C-5′ or C-2′), 134.65 (d, 1C, 4JC-F= 3 Hz, C-4), 134.39 (d, 1C, 4JC-F= 4 Hz, C-8a), 134.38 (1C, C-3), 120.52 (d, 1C, 3JC-F= 9 Hz, C-4a), 119.47 (1C, C-3′), 118.97 (d, 1C, 2JC-F= 25 Hz, C-7), 117.60 (d, 1C, 3JC-F= 8 Hz, C-8), 114.81 (1C, CN), 112.64 (d, 1C, 2JC-F= 23 Hz, C-5), 105.93 (1C, C-5′ or C-2′), 104.53 (1C, C-4′), 47.97 (1C, CHCH3), 34.76 (1C, NCH3), 21.50 (1C, CHCH3). 19F NMR (470 MHz, CDCl3) δ—119.21. HRMS (ESI) m/z 339.1245 [M+H]+ (calculated for [C18H16FN4O2]+ 339.1252). Analytical HPLC characterisation: Rt: 11.27 min, 97.2% and 96.4% purity at 254 and 360 nm, respectively.
- (R)-5-{[1-(6-fluoro-2-oxo-1,2-dihydroquinolin-3-yl)ethyl]-amino}-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile ((R)-3). DIPEA (150 µL, 0.861 mmol) was added to a solution of 4 (53 mg, 0.346 mmol) and amine (R)-6 (71 mg, 0.291 mmol) in DMSO (3 mL). The resulting solution was stirred at 110 °C during 19 h. After cooling to room temperature, deionised water (25 mL) was added, and the aqueous layer was extracted with EtOAc (3 × 20 mL). The organic extracts were combined, dried on MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (SiO2, cyclohexane/EtOAc, 2/8, v/v) to provide derivative (R)-3 (39 mg, 0.115 mmol, 40%) as a brown powder. Rf (SiO2, cyclohexane/EtOAc, 2/8, v/v) 0.33. IR (ATR, cm−1) 2208, 1652, 1626, 1592, 1544, 1227, 815. Mp 166 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 12.69 (br.s, 1H, NH), 7.65 (s, 1H, H-4), 7.46 (dd, 1H, 3JH-H = 8.9 Hz,4JH-F = 4.5 Hz, H-8), 7.27 (td, 1H, 3JH-F = 3JH-H = 8.4 Hz, 4JH-H = 2.8 Hz, H-7), 7.19 (dd, 1H,3JH-F = 8.5 Hz, 4JH-H = 2.7 Hz, H-5), 6.68 (d, 1H,3JH-H = 7.9 Hz, H-3′), 6,34 (br.s, 1H, NH), 5.93 (d, 1H, 3JH-H = 7.9 Hz, H-4′), 4.87 (q, 1H, 3JH-H = 6.7 Hz, CHCH3), 3.75 (s, 3H, NCH3), 1.67 (d, 3H, 3JH-H = 6.7 Hz, CHCH3). 13C NMR (126 MHz, CDCl3) δ 163.12 (1C, C-2), 158.35 (d, 1C, 1JC-F = 243 Hz, C-6), 156.96 (1C, C-6′), 141.39 (1C, C-5′ or C-2′), 134.69 (d, 1C, 4JC-F = 3 Hz, C-4), 134.44 (1C, C-3), 134.32 (s, 1C, C-8a), 120.56 (d, 1C, 3JC-F = 9 Hz, C-4a), 119.47 (1C, C-3′), 119.02 (d, 1C, 2JC-F = 25 Hz, C-7), 117.59 (d, 1C, 3JC-F = 8 Hz, C-8), 114.82 (1C, CN), 112.69 (d, 1C, 2JC-F = 23 Hz, C-5), 105.98 (1C, C-5′ or C-2′), 104.55 (1C, C-4′), 47.97 (1C, CHCH3), 34.77 (1C, NCH3), 21.53 (1C, CHCH3). 19F NMR (470 MHz, CDCl3) δ—119.10. HRMS (ESI) m/z 339.1245 [M+H]+ (calculated for [C18H16FN4O2]+ 339.1252). Analytical HPLC characterisation: Rt: 12.26 min, 95.6% and 95.9% purity at 254 and 360 nm, respectively.
- 5-((S)-1-(6-(tributylstannyl)-1,2-dihydro-2-oxoquinolin-3-yl)ethylamino)-1,6-dihydro-1-methyl-6-oxopyridine-2-carbonitrile ((S)-15). To a solution of iodinated compound (S)-2 (150 mg, 0.336 mmol) in anhydrous propan-2-ol (17 mL), beforehand degassed under argon flow, were successively added, under stirring, tris(dibenzylidenacetone)palladium(0) (7.7 mg, 8.4 µmol), hexabutylditin (187 µL, 0.370 mmol), and DIPEA (146 µL, 0.84 mmol). The reaction mixture was then stirred at room temperature for 21 h before further addition of tris(dibenzylidenacetone)palladium(0) (7.7 mg, 8.4 µmol). After 2 h stirring at room temperature, the mixture was filtered on Celite 545 and washed with EtOAc (3 × 50 mL). After evaporation of the filtrate under reduced pressure, the crude residue was purified by column chromatography (SiO2, cyclohexane/EtOAc, 7/3, v/v and then cyclohexane/EtOAc, 3/7, v/v) to provide derivative (S)-15 (111 mg, 0.182 mmol, 54%) as a light yellow solid to be stored at 4 °C under argon atmosphere and protected from light. Rf (SiO2, cyclohexane/EtOAc, 5/5, v/v) 0.23. IR (ATR, cm−1) 2955, 2921, 2870, 1850, 2209, 1650, 1635, 1594, 1548, 815. Mp 82 ± 1 °C. 1H NMR (500 MHz, CDCl3) δ 1H NMR (500 MHz, CDCl3) δ 10.94 (s, 1H, NH), 7.67 (s, 1H, H-4), 7.61–7.48 (m, 2H, H-5, H-7), 7.29 (d, 1H,3J = 8.1 Hz, H-8), 6.69 (d, 1H,3J = 7.8 Hz, H-3′), 6.26 (d, 1H, 3J = 6.6 Hz, NH), 5.96 (d, 1H, 3J = 7.8 Hz, H-4′), 4.88 (qt, 1H, 3J = 6.7 Hz, CHCH3), 3.76 (s, 3H, NCH3), 1.64 (d, 3H, 3J = 6.6 Hz, CHCH3), 1.60–1.45 (m, 6H, Sn(CH2CH2CH2CH3)3, 1.39–1.26 (m, 6H, Sn(CH2CH2CH2CH3)3), 1.15–0.98 (m, 6H, Sn(CH2CH2CH2CH3)3), 0.95–0.84 (m, 9H, Sn(CH2CH2CH2CH3)3). 13C NMR (126 MHz, CDCl3) δ 162.61 (1C, C-2),157.05 (1C, C-6′), 141.50 (1C, C-5′ or C-2′), 138.28 (1C, C-7), 137.52 (1C, C-8a or C-4a), 136.08 (1C, C-5), 135.27 (1C, C-3), 133.16 (1C, C-4), 130.64 (1C, C-6), 120.01 (1C, C-8a or C-4a), 119.67, 119.59 (2C, C-3′, C-8), 114.88 (1C, CN), 105.82 (1C, C-5′ or C-2′), 104.68 (1C, C-4′), 47.67 (1C, CHCH3), 34.77 (1C, NCH3), 21.81 (1C, CHCH3), 27.49 (3C, 3J119Sn/117Sn-C = 62 Hz, Sn(CH2CH2CH2CH3)3), 29,20 (3C, Sn(CH2CH2CH2CH3)3), 13.82 (3C, Sn(CH2CH2CH2CH3)3), 9.87 (3C, Sn(CH2CH2CH2CH3)3). HRMS (ESI) m/z 611.2399 [M+H]+ (calculated for [C30H43N4O2120Sn]+ 611.2403).
3.2. Biological Evaluation
3.2.1. Mutant IDH1 Enzyme Assay for Determination of the Inhibitory Potency
3.2.2. wt IDH Enzyme Assay for Determination of Inhibitory Potency
3.3. Radiochemistry with Iodine-125
3.3.1. General Information
3.3.2. Radiolabelling with Iodine-125
3.4. Radiochemistry with Fluorine-18
3.4.1. General Information
3.4.2. Radiolabelling with Fluorine-18
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Khallaf, H. Isocitrate Dehydrogenases in Physiology and Cancer: Biochemical and Molecular Insight. Cell Biosci. 2017, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. Oncogenic Isocitrate Dehydrogenase Mutations: Mechanisms, Models, and Clinical Opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef]
- Dang, L.; Yen, K.; Attar, E.C. IDH Mutations in Cancer and Progress toward Development of Targeted Therapeutics. Ann. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Krell, D.; Assoku, M.; Galloway, M.; Mulholland, P.; Tomlinson, I.; Bardella, C. Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH Mutations in Glioblastoma. PLoS ONE 2011, 6, e19868. [Google Scholar] [CrossRef] [PubMed]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [PubMed]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 Codon 132 Mutation in Brain Tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; McLendon, R.; Kos, I.; Riggins, G.J.; Reardon, D.; Velculescu, V.E.; Bigner, D.D. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, E.; Wilding, C.; Engelman, B.; Huang, P.; Jones, R.L. Is the IDH Mutation a Good Target for Chondrosarcoma Treatment? Curr. Mol. Bio. Rep. 2020, 6, 1–9. [Google Scholar] [CrossRef]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 Mutations Are Frequent Events in Central Chondrosarcoma and Central and Periosteal Chondromas but Not in Other Mesenchymal Tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, C.J.; Yan, H. The Implications of IDH Mutations for Cancer Development and Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Juratli, T.A.; Peitzsch, M.; Geiger, K.; Schackert, G.; Eisenhofer, G.; Krex, D. Accumulation of 2-Hydroxyglutarate Is Not a Biomarker for Malignant Progression in IDH-Mutated Low-Grade Gliomas. Neuro-oncology 2013, 15, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [PubMed]
- FDA-Approves-Ivosidenib-First-Line-Treatment-Aml-Idh1-Mutation. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ivosidenib-first-line-treatment-aml-idh1-mutation (accessed on 9 April 2022).
- FDA-Granted-Regular-Approval-Enasidenib-Treatment-Relapsed-or-Refractory-Aml. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-granted-regular-approval-enasidenib-treatment-relapsed-or-refractory-aml (accessed on 9 April 2022).
- FDA-Approves-Ivosidenib-Advanced-or-Metastatic-Cholangiocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ivosidenib-advanced-or-metastatic-cholangiocarcinoma (accessed on 9 April 2022).
- Zarei, M.; Hue, J.J.; Hajihassani, O.; Graor, H.J.; Katayama, E.S.; Loftus, A.W.; Bajor, D.; Rothermel, L.D.; Vaziri-Gohar, A.; Winter, J.M. Clinical Development of IDH1 Inhibitors for Cancer Therapy. Cancer Treat. Rev. 2022, 103, 102334. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Gori, S.S.; Tsukamoto, T.; Rais, R.; Slusher, B.S. Clinical Development of Metabolic Inhibitors for Oncology. J. Clin. Invest. 2022, 132, e148550. [Google Scholar] [CrossRef] [PubMed]
- Chitneni, S.K.; Yan, H.; Zalutsky, M.R. Synthesis and Evaluation of a 18F-Labeled Triazinediamine Analogue for Imaging Mutant IDH1 Expression in Gliomas by PET. ACS Med. Chem. Lett. 2018, 9, 606–611. [Google Scholar] [CrossRef]
- Chitneni, S.K.; Reitman, Z.J.; Spicehandler, R.; Gooden, D.M.; Yan, H.; Zalutsky, M.R. Synthesis and Evaluation of Radiolabeled AGI-5198 Analogues as Candidate Radiotracers for Imaging Mutant IDH1 Expression in Tumors. Bioorg. Med. Chem. Lett. 2018, 28, 694–699. [Google Scholar] [CrossRef]
- Chitneni, S.K.; Reitman, Z.J.; Gooden, D.M.; Yan, H.; Zalutsky, M.R. Radiolabeled Inhibitors as Probes for Imaging Mutant IDH1 Expression in Gliomas: Synthesis and Preliminary Evaluation of Labeled Butyl-Phenyl Sulfonamide Analogs. Eur. J. Med. Chem. 2016, 119, 218–230. [Google Scholar] [CrossRef]
- Wang, T.; Lin, Q.; Zhang, Y.; Xu, Z.; Shi, D.; Cheng, Y.; Fu, Z.; Tan, H.; Cheng, D.; Shi, H. Synthesis and Biological Evaluation of Novel PET Tracers [18F]AG120 & [18F]AG135 for Imaging Mutant Isocitrate Dehydrogenase 1 Expression. Bioorg. Med. Chem. 2022, 53, 116525. [Google Scholar] [CrossRef]
- Caravella, J.A.; Lin, J.; Diebold, R.B.; Campbell, A.-M.; Ericsson, A.; Gustafson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gotur, D.; et al. Structure-Based Design and Identification of FT-2102 (Olutasidenib), a Potent Mutant-Selective IDH1 Inhibitor. J. Med. Chem. 2020, 63, 1612–1623. [Google Scholar] [CrossRef]
- Lin, J.; Lu, W.; Caravella, J.A.; Campbell, A.M.; Diebold, R.B.; Ericsson, A.; Fritzen, E.; Gustafson, G.R.; Lancia, D.R.; Shelekhin, T.; et al. Discovery and Optimization of Quinolinone Derivatives as Potent, Selective, and Orally Bioavailable Mutant Isocitrate Dehydrogenase 1 (MIDH1) Inhibitors. J. Med. Chem. 2019, 62, 6575–6596. [Google Scholar] [CrossRef] [PubMed]
- Salifu, E.Y.; Agoni, C.; Soliman, M.E.S. Highlighting the Mechanistic Role of Olutasidenib (FT-2102) in the Selective Inhibition of Mutated Isocitrate Dehydrogenase 1 (mIDH1) in Cancer Therapy. Inf. Med. Unlocked 2022, 28, 100829. [Google Scholar] [CrossRef]
- Ashwell, S.; Campbell, A.-M.; Caravella, J.A.; Diebold, R.B.; Ericsson, A.; Gustafson, G.; Lancia, D.R.; Lin, J.; Lu, W.; Wang, Z. Phenyl Quinolinone Derivatives as Mutant-Isocitrate Dehydrogenase Inhibitors. U.S. Patent WO/2016/044782, 23 April 2016. [Google Scholar]
- Meth-Cohn, O.; Narine, B.; Tarnowski, B. A Versatile New Synthesis of Quinolines and Related Fused Pyridines, Part 5. The Synthesis of 2-Chloroquinoline-3-Carbaldehydes. J. Chem. Soc. Perkin Trans. 1 1981, 1520–1530. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Narine, B.; Tarnowski, B. A Versatile New Synthesis of Quinolines and Related Fused Pyridines. Part II. Tetrahedron Lett. 1979, 20, 3111–3114. [Google Scholar] [CrossRef]
- Ali, M.M.; Sana, S.; Tasneem; Rajanna, K.C.; Saiprakash, P.K. Ultrasonically Accelerated Vilsmeier Haack Cyclisation and Formylation Reactions. Synth. Commun. 2002, 32, 1351–1356. [Google Scholar] [CrossRef]
- Ali, M.M.; Tasneem, K.C.; Rajanna, P.K.; Prakash, P.K.S. An Efficient and Facile Synthesis of 2-Chloro-3-Formyl Quinolines from Acetanilides in Micellar Media by Vilsmeier-Haack Cyclisation. Synlett 2001, 2001, 0251–0253. [Google Scholar] [CrossRef]
- Pickett, J.E.; Váradi, A.; Palmer, T.C.; Grinnell, S.G.; Schrock, J.M.; Pasternak, G.W.; Karimov, R.R.; Majumdar, S. Mild, Pd-Catalyzed Stannylation of Radioiodination Targets. Bioorg. Med. Chem. Lett. 2015, 25, 1761–1764. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.N.; Chezal, J.-M.; Debiton, E.; Canitrot, D.; Witkowski, T.; Levesque, S.; Degoul, F.; Tarrit, S.; Wenzel, B.; Miot-Noirault, E.; et al. A Convenient Route to New (Radio)Fluorinated and (Radio)Iodinated Cyclic Tyrosine Analogs. Pharmaceuticals 2022, 15, 162. [Google Scholar] [CrossRef]
- Chen, Z.; Destro, G.; Guibbal, F.; Chan, C.Y.; Cornelissen, B.; Gouverneur, V. Copper-Mediated Radiosynthesis of [18F]Rucaparib. Org. Lett. 2021, 23, 7290–7294. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Schäfer, D.; Urusova, E.A.; Guliyev, M.; Bannykh, O.; Endepols, H.; Neumaier, B. Discovery of 7-[18F]Fluorotryptophan as a Novel Positron Emission Tomography (PET) Probe for the Visualization of Tryptophan Metabolism in Vivo. J. Med. Chem. 2018, 61, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Elsevier: Amsterdam, The Netherlands; Butterworth-Heinemann: Oxford, UK, 2009; ISBN 978-1-85617-567-8. [Google Scholar]
- Eliel, E.L. Infelicitous Stereochemical Nomenclature. Chirality 1997, 9, 428–430. [Google Scholar] [CrossRef]
- Bergmann, E.D.; Bentov, M. Sandmeyer reactions of monoacyl arylenediamines. J. Org. Chem. 1955, 20, 1654–1656. [Google Scholar] [CrossRef]
- Stavber, S.; Zupan, M. Fluorination with Cesium Fluoroxysulfate. Room Temperature Fluorination of Benzene and Naphthalene Derivatives. J. Org. Chem. 1985, 50, 3609–3612. [Google Scholar] [CrossRef]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.-F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.-X.; et al. Mutations in Isocitrate Dehydrogenase 1 and 2 Occur Frequently in Intrahepatic Cholangiocarcinomas and Share Hypermethylation Targets with Glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| IC50 (nM) a | |||||
| Compound | R | IDH1 R132H | IDH1 R132C | wt IDH1 | wt IDH2 |
| (S)-FT-2102 | Cl | 4.88 ± 1.63 | 178 ± 70 | >10,000 | >10,000 |
| (S)-2 | I | 5.23 ± 0.75 | 160 ± 24 | >10,000 | >10,000 |
| (R)-2 | I | 323 ± 83 | >10,000 | >10,000 | >10,000 |
| (S)-3 | F | 22.7 ± 5.2 | 579 ± 110 | >10,000 | >10,000 |
| (R)-3 | F | 698 ± 145 | >10,000 | >10,000 | >10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, V.; Arnaud, L.; Dukic-Stefanovic, S.; Wenzel, B.; Roux, V.; Chezal, J.-M.; Lai, T.-H.; Teodoro, R.; Kopka, K.; Miot-Noirault, E.; et al. Novel Radioiodinated and Radiofluorinated Analogues of FT-2102 for SPECT or PET Imaging of mIDH1 Mutant Tumours. Molecules 2022, 27, 3766. https://doi.org/10.3390/molecules27123766
Weber V, Arnaud L, Dukic-Stefanovic S, Wenzel B, Roux V, Chezal J-M, Lai T-H, Teodoro R, Kopka K, Miot-Noirault E, et al. Novel Radioiodinated and Radiofluorinated Analogues of FT-2102 for SPECT or PET Imaging of mIDH1 Mutant Tumours. Molecules. 2022; 27(12):3766. https://doi.org/10.3390/molecules27123766
Chicago/Turabian StyleWeber, Valérie, Lucie Arnaud, Sladjana Dukic-Stefanovic, Barbara Wenzel, Valérie Roux, Jean-Michel Chezal, Thu-Hang Lai, Rodrigo Teodoro, Klaus Kopka, Elisabeth Miot-Noirault, and et al. 2022. "Novel Radioiodinated and Radiofluorinated Analogues of FT-2102 for SPECT or PET Imaging of mIDH1 Mutant Tumours" Molecules 27, no. 12: 3766. https://doi.org/10.3390/molecules27123766
APA StyleWeber, V., Arnaud, L., Dukic-Stefanovic, S., Wenzel, B., Roux, V., Chezal, J.-M., Lai, T.-H., Teodoro, R., Kopka, K., Miot-Noirault, E., Deuther-Conrad, W., & Maisonial-Besset, A. (2022). Novel Radioiodinated and Radiofluorinated Analogues of FT-2102 for SPECT or PET Imaging of mIDH1 Mutant Tumours. Molecules, 27(12), 3766. https://doi.org/10.3390/molecules27123766