
Prediction Models for Brain Distribution of Drugs Based on Biomimetic Chromatographic Data


Abstract
:1. Introduction
2. Results and Discussion
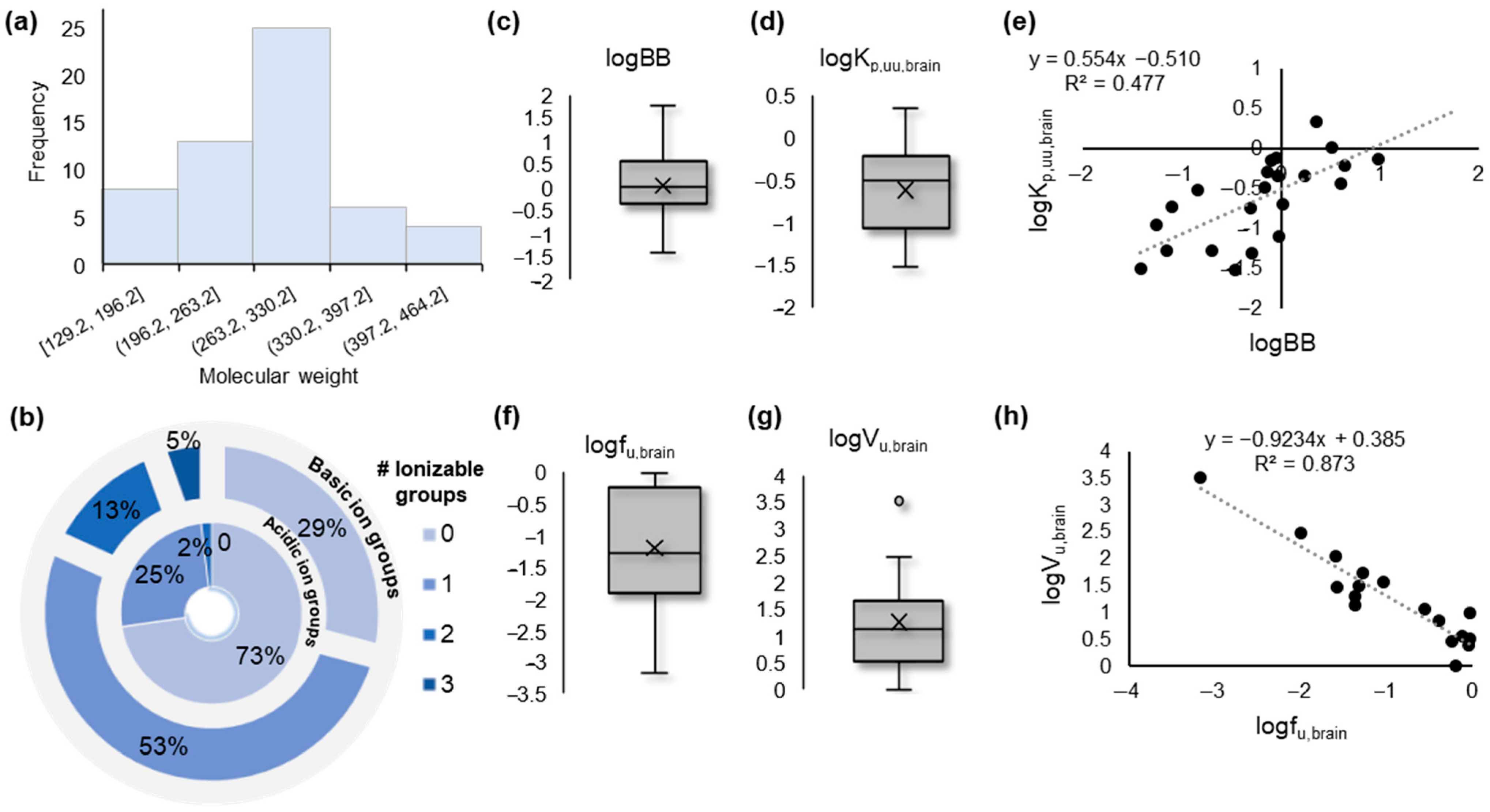
2.1. Data Overview with Unsupervised Data Analysis
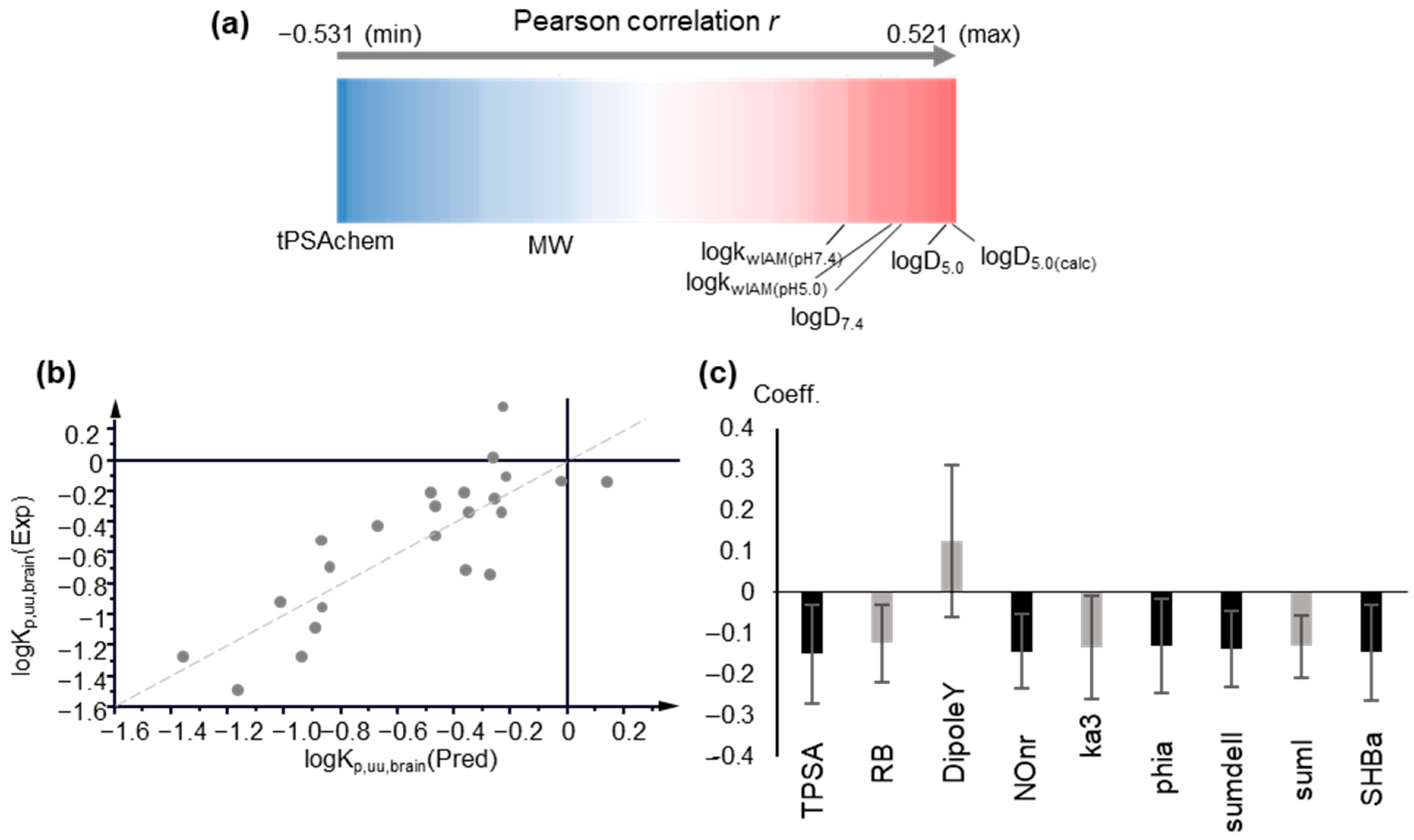
2.2. Modeling logKp,uu,brain
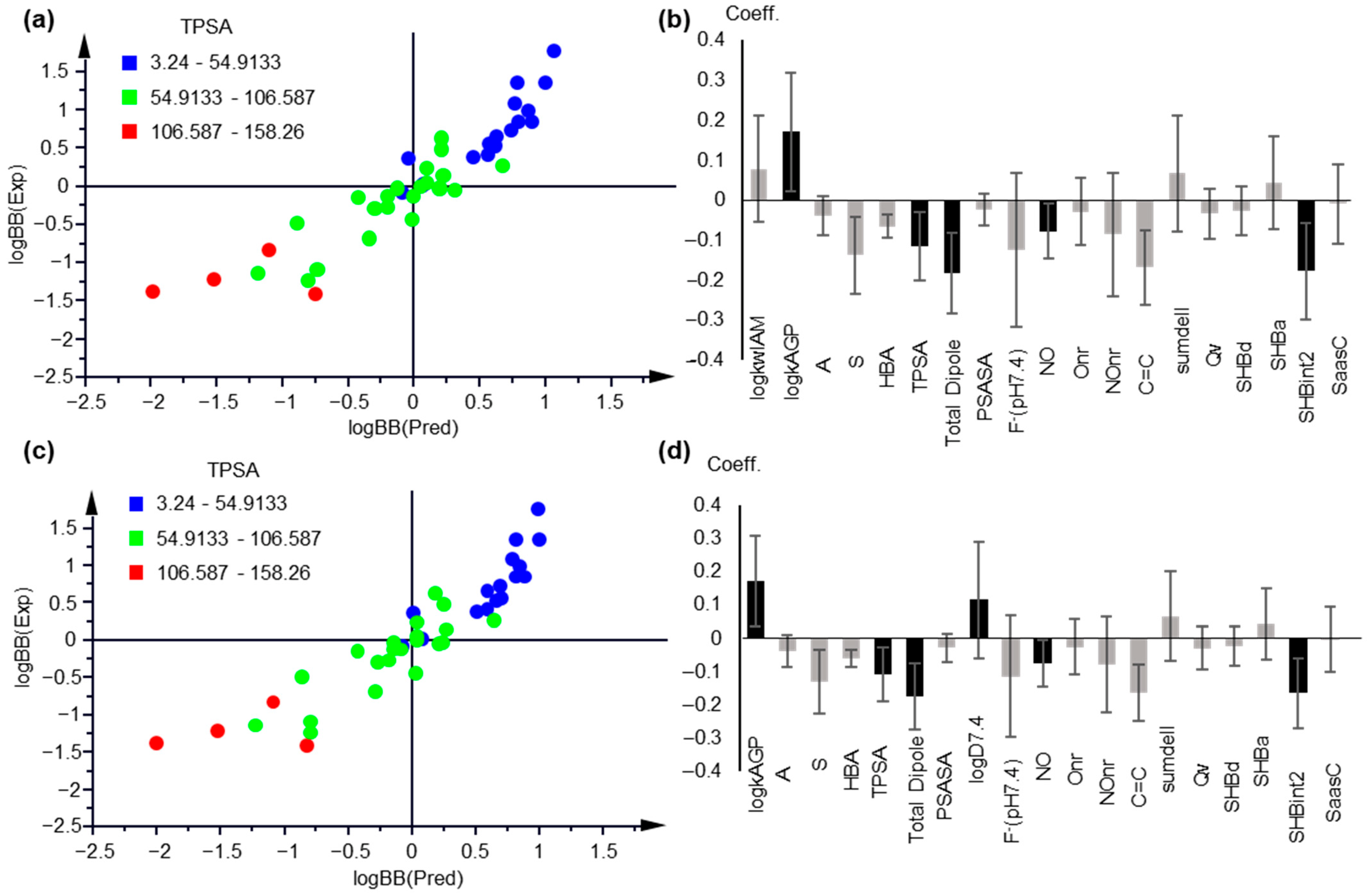
2.3. Modeling logBB
2.3.1. Multiple Linear Regression Models
(* Piracetam did not have a logkwIAM measured value).
2.3.2. Partial Least Squares Models
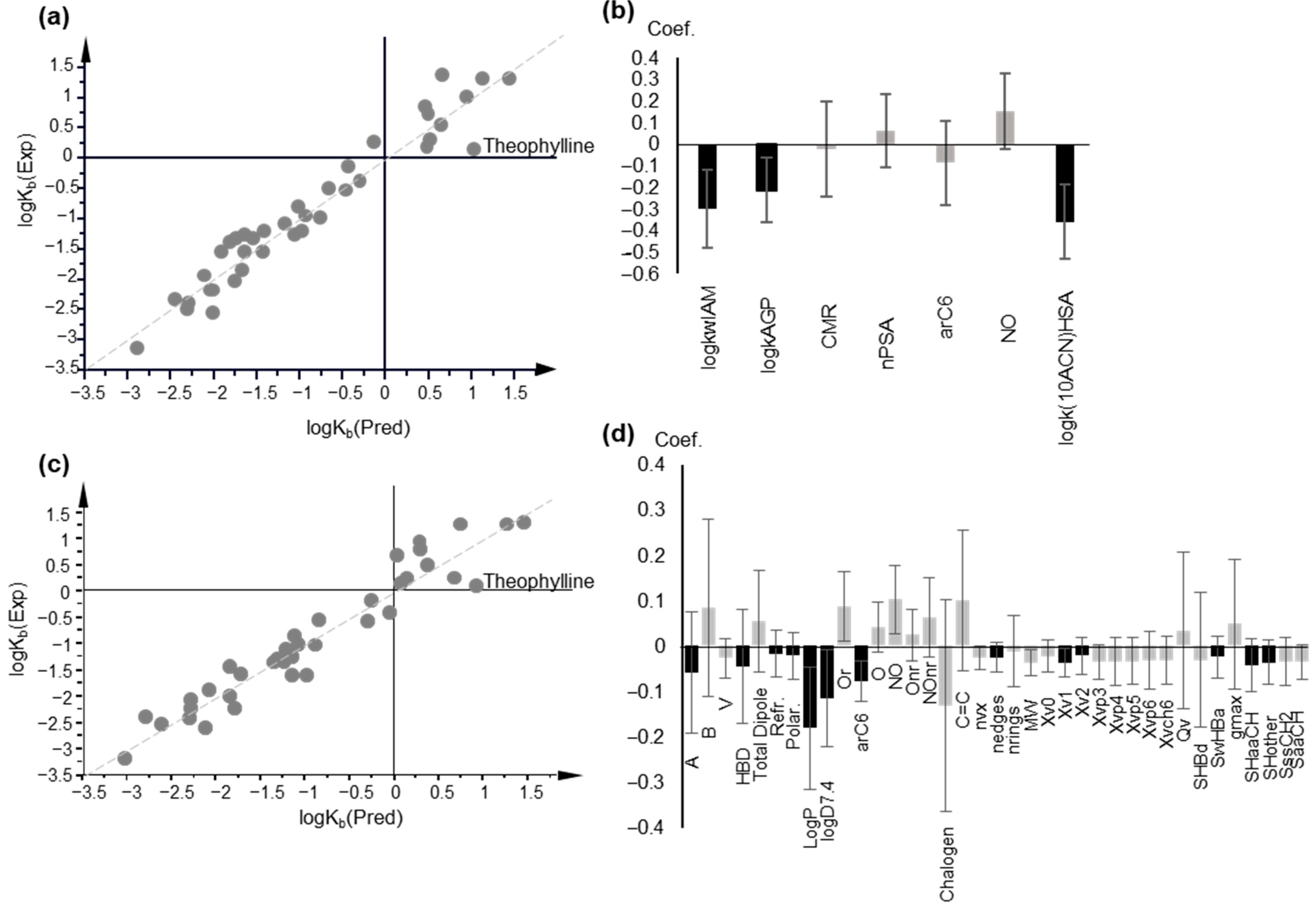
2.4. Modeling logKb
2.4.1. Multiple Linear Regression
2.4.2. Partial Least Squares Models
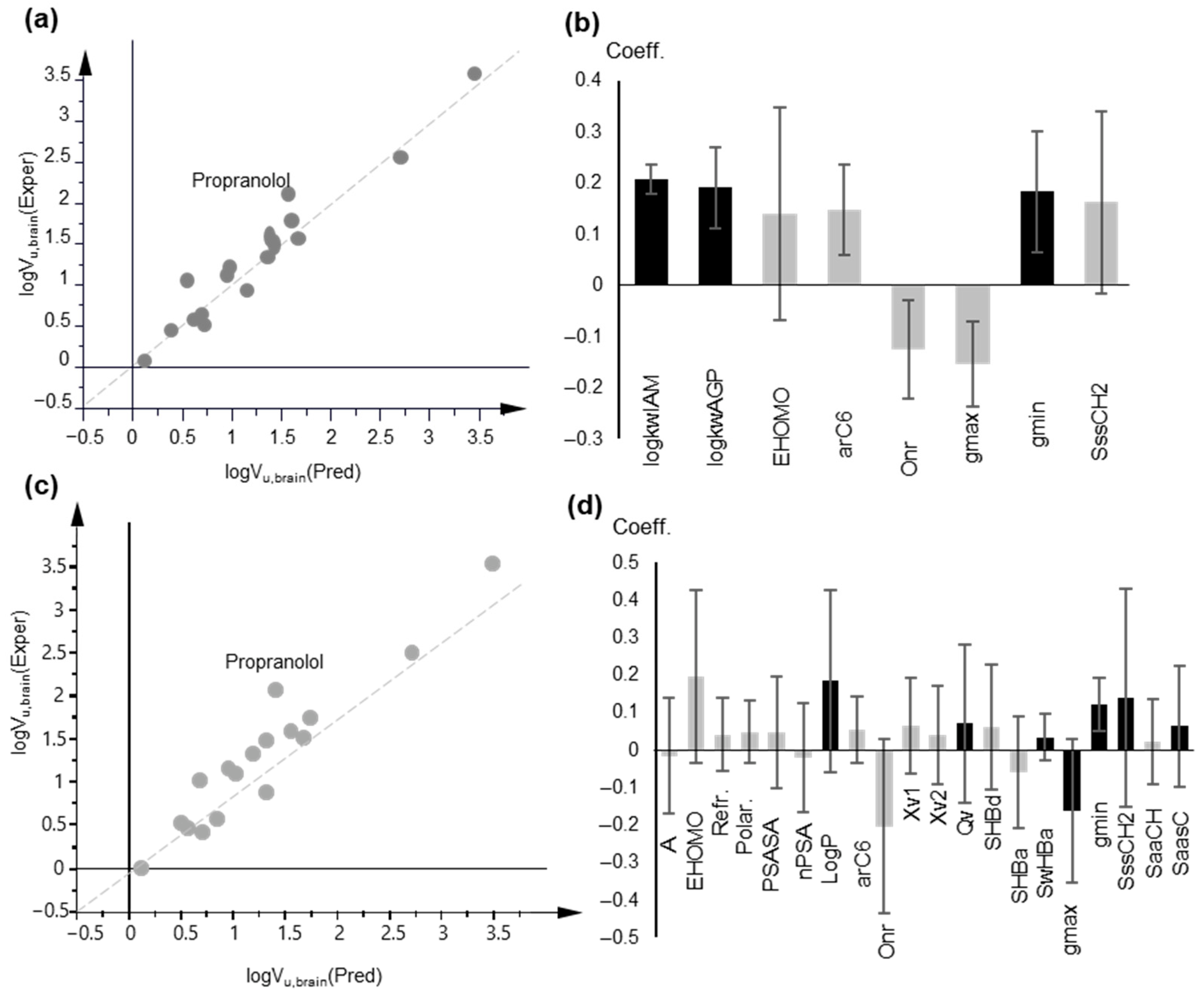
2.5. Unbound Volume of Distribution, Vu, Brain
2.5.1. Correlation between fraction unbound and unbound volume of distribution in the brain
2.5.2. Multiple Linear Regression
2.5.3. Partial Least Squares (PLS) Models
3. Materials and Methods
3.1. Dataset and Chromatographic Data
3.2. Brain Disposition Data
3.3. Physicochemical and Molecular Descriptors
3.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef]
- Choi, D.W.; Armitage, R.; Brady, L.S.; Coetzee, T.; Fisher, W.; Hyman, S.; Pande, A.; Paul, S.; Potter, W.; Roin, B.; et al. Medicines for the mind: Policy-based “pull” incentives for creating breakthrough CNS drugs. Neuron 2014, 84, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease. Epilepsia 2012, 53 (Suppl. 6), 1–6. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef]
- Eyal, S.; Hsiao, P.; Unadkat, J.D. Drug interactions at the blood-brain barrier: Fact or fantasy? Pharmacol. Ther. 2009, 123, 80–104. [Google Scholar] [CrossRef] [Green Version]
- Loryan, I.; Sinha, V.; Mackie, C.; Van Peer, A.; Drinkenburg, W.; Vermeulen, A.; Morrison, D.; Monshouwer, M.; Heald, D.; Hammarlund-Udenaes, M. Mechanistic understanding of brain drug disposition to optimize the selection of potential neurotherapeutics in drug discovery. Pharm. Res. 2014, 31, 2203–2219. [Google Scholar] [CrossRef]
- Hammarlund-Udenaes, M.; Bredberg, U.; Friden, M. Methodologies to assess brain drug delivery in lead optimization. Curr. Top. Med. Chem. 2009, 9, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund-Udenaes, M.; Friden, M.; Syvanen, S.; Gupta, A. On the rate and extent of drug delivery to the brain. Pharm. Res. 2008, 25, 1737–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friden, M.; Bergstrom, F.; Wan, H.; Rehngren, M.; Ahlin, G.; Hammarlund-Udenaes, M.; Bredberg, U. Measurement of unbound drug exposure in brain: Modeling of pH partitioning explains diverging results between the brain slice and brain homogenate methods. Drug Metab. Dispos. 2011, 39, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Friden, M.; Winiwarter, S.; Jerndal, G.; Bengtsson, O.; Wan, H.; Bredberg, U.; Hammarlund-Udenaes, M.; Antonsson, M. Structure-brain exposure relationships in rat and human using a novel data set of unbound drug concentrations in brain interstitial and cerebrospinal fluids. J. Med. Chem. 2009, 52, 6233–6243. [Google Scholar] [CrossRef]
- Friden, M.; Ducrozet, F.; Middleton, B.; Antonsson, M.; Bredberg, U.; Hammarlund-Udenaes, M. Development of a high-throughput brain slice method for studying drug distribution in the central nervous system. Drug Metab. Dispos. 2009, 37, 1226–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luptáková, D.; Vallianatou, T.; Nilsson, A.; Shariatgorji, R.; Hammarlund-Udenaes, M.; Loryan, I.; Andrén, P.E. Neuropharmacokinetic visualization of regional and subregional unbound antipsychotic drug transport across the blood-brain barrier. Mol. Psychiatry 2021, 26, 7732–7745. [Google Scholar] [CrossRef]
- Loryan, I.; Sinha, V.; Mackie, C.; Van Peer, A.; Drinkenburg, W.H.; Vermeulen, A.; Heald, D.; Hammarlund-Udenaes, M.; Wassvik, C.M. Molecular properties determining unbound intracellular and extracellular brain exposure of CNS drug candidates. Mol. Pharm. 2015, 12, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Prediction of blood-brain barrier permeation in drug discovery from in vivo, in vitro and in silico models. Drug Discov. Today Technol. 2004, 1, 407–416. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, H.; Wu, Z.; Wang, T.; Li, W.; Tang, Y.; Liu, G. In Silico Prediction of Blood-Brain Barrier Permeability of Compounds by Machine Learning and Resampling Methods. ChemMedChem 2018, 13, 2189–2201. [Google Scholar] [CrossRef]
- Clark, D.E. In silico prediction of blood-brain barrier permeation. Drug Discov. Today 2003, 8, 927–933. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, H.; Oprea, T.I.; Golbraikh, A.; Tropsha, A. QSAR modeling of the blood-brain barrier permeability for diverse organic compounds. Pharm. Res. 2008, 25, 1902–1914. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, C.A.; Charman, S.A.; Nicolazzo, J.A. Computational prediction of CNS drug exposure based on a novel in vivo dataset. Pharm. Res. 2012, 29, 3131–3142. [Google Scholar] [CrossRef]
- Varadharajan, S.; Winiwarter, S.; Carlsson, L.; Engkvist, O.; Anantha, A.; Kogej, T.; Friden, M.; Stalring, J.; Chen, H. Exploring in silico prediction of the unbound brain-to-plasma drug concentration ratio: Model validation, renewal, and interpretation. J. Pharm. Sci. 2015, 104, 1197–1206. [Google Scholar] [CrossRef]
- Chrysanthakopoulos, M.; Kolletsou, A.; Nicolaou, I.; Demopoulos, V.J.; Tsantili-Kakoullidou, A. Lipophilicity Studies on Pyrrolyl-Acetic Acid Derivatives. Experimental Versus Predicted logP Values in Relationship with Aldose Reductase Inhibitory Activity. QSAR Comb. Sci. 2009, 28, 551–560. [Google Scholar] [CrossRef]
- Lanevskij, K.; Dapkunas, J.; Juska, L.; Japertas, P.; Didziapetris, R. QSAR analysis of blood-brain distribution: The influence of plasma and brain tissue binding. J. Pharm. Sci. 2011, 100, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Vrakas, D.; Giaginis, C.; Tsantili-Kakoulidou, A. Different retention behavior of structurally diverse basic and neutral drugs in immobilized artificial membrane and reversed-phase high performance liquid chromatography: Comparison with octanol-water partitioning. J. Chromatogr. A 2006, 1116, 158–164. [Google Scholar] [CrossRef]
- Vrakas, D.; Giaginis, C.; Tsantili-Kakoulidou, A. Electrostatic interactions and ionization effect in immobilized artificial membrane retention. A comparative study with octanol-water partitioning. J. Chromatogr. A 2008, 1187, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Valkó, K.L. Lipophilicity and biomimetic properties measured by HPLC to support drug discovery. J. Pharm. Biomed. Anal. 2016, 130, 35–54. [Google Scholar] [CrossRef]
- Valko, K.; Nunhuck, S.; Bevan, C.; Abraham, M.H.; Reynolds, D.P. Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. J. Pharm. Sci. 2003, 92, 2236–2248. [Google Scholar] [CrossRef]
- Tsopelas, F.; Vallianatou, T.; Tsantili-Kakoulidou, A. The potential of immobilized artificial membrane chromatography to predict human oral absorption. Eur. J. Pharm. Sci. 2016, 81, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Tsopelas, F.; Vallianatou, T.; Tsantili-Kakoulidou, A. Advances in immobilized artificial membrane (IAM) chromatography for novel drug discovery. Expert Opin. Drug Discov. 2016, 11, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Tsopelas, F.; Malaki, N.; Vallianatou, T.; Chrysanthakopoulos, M.; Vrakas, D.; Ochsenkuhn-Petropoulou, M.; Tsantili-Kakoulidou, A. Insight into the retention mechanism on immobilized artificial membrane chromatography using two stationary phases. J. Chromatogr. A 2015, 1396, 25–33. [Google Scholar] [CrossRef]
- Grumetto, L.; Russo, G.; Barbato, F. Indexes of polar interactions between ionizable drugs and membrane phospholipids measured by IAM-HPLC: Their relationships with data of Blood-Brain Barrier passage. Eur. J. Pharm. Sci. 2014, 65, 139–146. [Google Scholar] [CrossRef]
- Grumetto, L.; Carpentiero, C.; Di Vaio, P.; Frecentese, F.; Barbato, F. Lipophilic and polar interaction forces between acidic drugs and membrane phospholipids encoded in IAM-HPLC indexes: Their role in membrane partition and relationships with BBB permeation data. J. Pharm. Biomed. Anal. 2013, 75, 165–172. [Google Scholar] [CrossRef]
- Grumetto, L.; Carpentiero, C.; Barbato, F. Lipophilic and electrostatic forces encoded in IAM-HPLC indexes of basic drugs: Their role in membrane partition and their relationships with BBB passage data. Eur. J. Pharm. Sci. 2012, 45, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Tsantili-Kakoulidou, A. Alternative measures of lipophilicity: From octanol-water partitioning to IAM retention. J. Pharm. Sci. 2008, 97, 2984–3004. [Google Scholar] [CrossRef]
- Verzele, D.; Lynen, F.; De Vrieze, M.; Wright, A.G.; Hanna-Brown, M.; Sandra, P. Development of the first sphingomyelin biomimetic stationary phase for immobilized artificial membrane (IAM) chromatography. Chem. Commun. 2012, 48, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Salminen, T.; Pulli, A.; Taskinen, J. Relationship between immobilised artificial membrane chromatographic retention and the brain penetration of structurally diverse drugs. J. Pharm. Biomed. Anal. 1997, 15, 469–477. [Google Scholar] [CrossRef]
- Reichel, A.; Begley, D.J. Potential of immobilized artificial membranes for predicting drug penetration across the blood-brain barrier. Pharm. Res. 1998, 15, 1270–1274. [Google Scholar] [CrossRef]
- Norinder, U.; Osterberg, T. Theoretical calculation and prediction of drug transport processes using simple parameters and partial least squares projections to latent structures (PLS) statistics. The use of electrotopological state indices. J. Pharm. Sci. 2001, 90, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, E.; Ràfols, C.; Abraham, M.H.; Rosés, M. Chromatographic estimation of drug disposition properties by means of immobilized artificial membranes (IAM) and C18 columns. J. Med. Chem. 2006, 49, 4861–4870. [Google Scholar] [CrossRef]
- Hollósy, F.; Valkó, K.; Hersey, A.; Nunhuck, S.; Kéri, G.; Bevan, C. Estimation of volume of distribution in humans from high throughput HPLC-based measurements of human serum albumin binding and immobilized artificial membrane partitioning. J. Med. Chem. 2006, 49, 6958–6971. [Google Scholar] [CrossRef]
- De Vrieze, M.; Lynen, F.; Chen, K.; Szucs, R.; Sandra, P. Predicting drug penetration across the blood-brain barrier: Comparison of micellar liquid chromatography and immobilized artificial membrane liquid chromatography. Anal. Bioanal. Chem. 2013, 405, 6029–6041. [Google Scholar] [CrossRef]
- Dash, A.K.; Elmquist, W.F. Separation methods that are capable of revealing blood-brain barrier permeability. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 797, 241–254. [Google Scholar] [CrossRef]
- Valko, K.; Rava, S.; Bunally, S.; Anderson, S. Revisiting the application of Immobilized Artificial Membrane (IAM) chromatography to estimate in vivo distribution properties of drug discovery compounds based on the model of marketed drugs. ADMET DMPK 2020, 8, 78–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, C.H.; Kim, S.J.; Shin, B.S.; Lee, K.C.; Yoo, S.D. Rapid screening of blood-brain barrier penetration of drugs using the immobilized artificial membrane phosphatidylcholine column chromatography. J. Biomol. Screen 2006, 11, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Chrysanthakopoulos, M.; Vallianatou, T.; Giaginis, C.; Tsantili-Kakoulidou, A. Investigation of the retention behavior of structurally diverse drugs on alpha(1) acid glycoprotein column: Insight on the molecular factors involved and correlation with protein binding data. Eur. J. Pharm. Sci. 2014, 60, 24–31. [Google Scholar] [CrossRef]
- Chrysanthakopoulos, M.; Giaginis, C.; Tsantili-Kakoulidou, A. Retention of structurally diverse drugs in human serum albumin chromatography and its potential to simulate plasma protein binding. J. Chromatogr. A 2010, 1217, 5761–5768. [Google Scholar] [CrossRef] [PubMed]
- Bteich, M. An overview of albumin and alpha-1-acid glycoprotein main characteristics: Highlighting the roles of amino acids in binding kinetics and molecular interactions. Heliyon 2019, 5, e02879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrinidis, G.; Vallianatou, T.; Tsantili-Kakoulidou, A. In vitro, in silico and integrated strategies for the estimation of plasma protein binding. A review. Adv. Drug Deliv. Rev. 2015, 86, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Ahman, M.; Holmen, A.G. Relationship between brain tissue partitioning and microemulsion retention factors of CNS drugs. J. Med. Chem. 2009, 52, 1693–1700. [Google Scholar] [CrossRef]
- Wan, H.; Rehngren, M.; Giordanetto, F.; Bergstrom, F.; Tunek, A. High-throughput screening of drug-brain tissue binding and in silico prediction for assessment of central nervous system drug delivery. J. Med. Chem. 2007, 50, 4606–4615. [Google Scholar] [CrossRef]
- Summerfield, S.G.; Stevens, A.J.; Cutler, L.; del Carmen Osuna, M.; Hammond, B.; Tang, S.P.; Hersey, A.; Spalding, D.J.; Jeffrey, P. Improving the in vitro prediction of in vivo central nervous system penetration: Integrating permeability, P-glycoprotein efflux, and free fractions in blood and brain. J. Pharmacol. Exp. Ther. 2006, 316, 1282–1290. [Google Scholar] [CrossRef] [Green Version]
- Mateus, A.; Matsson, P.; Artursson, P. Rapid measurement of intracellular unbound drug concentrations. Mol. Pharm. 2013, 10, 2467–2478. [Google Scholar] [CrossRef]
- Longhi, R.; Corbioli, S.; Fontana, S.; Vinco, F.; Braggio, S.; Helmdach, L.; Schiller, J.; Boriss, H. Brain tissue binding of drugs: Evaluation and validation of solid supported porcine brain membrane vesicles (TRANSIL) as a novel high-throughput method. Drug Metab. Dispos. 2011, 39, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Kalvass, J.C.; Maurer, T.S. Influence of nonspecific brain and plasma binding on CNS exposure: Implications for rational drug discovery. Biopharm. Drug Dispos. 2002, 23, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Ball, K.; Bouzom, F.; Scherrmann, J.M.; Walther, B.; Decleves, X. Physiologically based pharmacokinetic modelling of drug penetration across the blood-brain barrier--towards a mechanistic IVIVE-based approach. AAPS J. 2013, 15, 913–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanevskij, K.; Japertas, P.; Didziapetris, R.; Petrauskas, A. Ionization-specific prediction of blood-brain permeability. J. Pharm. Sci. 2009, 98, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Toth, A.E.; Nielsen, S.S.E.; Tomaka, W.; Abbott, N.J.; Nielsen, M.S. The endo-lysosomal system of bEnd.3 and hCMEC/D3 brain endothelial cells. Fluids Barriers CNS 2019, 16, 14. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [Green Version]
- Karalis, V.; Tsantili-Kakoulidou, A.; Macheras, P. Multivariate statistics of disposition pharmacokinetic parameters for structurally unrelated drugs used in therapeutics. Pharm. Res. 2002, 19, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Daniel, W.A.; Wojcikowski, J. Lysosomal trapping as an important mechanism involved in the cellular distribution of perazine and in pharmacokinetic interaction with antidepressants. Eur. Neuropsychopharmacol. 1999, 9, 483–491. [Google Scholar] [CrossRef]
- Friden, M.; Gupta, A.; Antonsson, M.; Bredberg, U.; Hammarlund-Udenaes, M. In vitro methods for estimating unbound drug concentrations in the brain interstitial and intracellular fluids. Drug Metab. Dispos. 2007, 35, 1711–1719. [Google Scholar] [CrossRef] [Green Version]
- Friden, M.; Ljungqvist, H.; Middleton, B.; Bredberg, U.; Hammarlund-Udenaes, M. Improved measurement of drug exposure in the brain using drug-specific correction for residual blood. J. Cereb. Blood Flow Metab. 2010, 30, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Winiwarter, S.; Friden, M.; Antonsson, M.; Engkvist, O. In silico prediction of unbound brain-to-plasma concentration ratio using machine learning algorithms. J. Mol. Graph. Model. 2011, 29, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, M.; Jacobson, M.P. In silico prediction of brain exposure: Drug free fraction, unbound brain to plasma concentration ratio and equilibrium half-life. Curr. Top. Med. Chem. 2013, 13, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Kier, L.B.; Hall, L.H. An electrotopological-state index for atoms in molecules. Pharm. Res. 1990, 7, 801–807. [Google Scholar] [CrossRef]
- Gramatica, P. On the development and validation of QSAR models. Methods Mol. Biol. 2013, 930, 499–526. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validated PLS Model: 4 Different Test Sets with n = 6 | R2train/Q2train | RMSEE | R2test | RMSEP |
|---|---|---|---|---|
| PLS Model 4 (based on computational descriptors) | 0.687–0.831/0.597–0.736 | 0.212–0.276 | 0.212 *–0.824 | 0.229–0.407 * |
| Validated MLR Models: 6 Different Test Sets with n = 7 or n = 6 | R2train | strain | R2test | stest |
|---|---|---|---|---|
| Equation (1) (based on IAM retention) | 0.626–0.705 | 0.388–0.499 | 0.583–0.932 | 0.167–0.669 |
| Equation (2) (based on lipophilicity) | 0.639–692 | 0.416–0.472 | 0.532–0.848 | 0.244–0.510 |
| Validated PLS Models: 5 Different Test Sets with n = 8 or n = 9 | R2train/Q2train | RMSEE | R2test | RMSEP |
| PLS Model 5 (based on IAM retention) | 0.850–0.863/ 0.724–0.793 | 0.272–0.316 | 0.457 *–0.936 | 0.302–0.525 * |
| PLS Model 6 (based on lipophilicity) | 0.850–0.865/ 0.725–0.791 | 0.272–0.328 | 0.600 **–0.919 | 0.317–0.516 ** |
| Validated MLR Models: 5 Different Test Sets with n = 8 or n = 9 | R2train | strain | R2test | stest |
|---|---|---|---|---|
| Equation (7) (IAM retention) | 0.924–0.937 | 0.306–0.341 | 0.823–0.966 | 0.270–0.377 |
| Equation (11) (lipophilicity) | 0.848–0.890 | 0.403–0.462 | 0.797–0.955 | 0.310–0.664 |
| Validated PLS Models: 5 different test sets with n = 8 or = 9 | R2train/Q2train | RMSEE | R2test | RMSEP |
| PLS Model 8 (based on IAM retention) | 0.919–0.946/ 0.851–0.927 | 0.286–0.353 | 0.822–0.988 | 0.277–0.519 |
| PLS Model 9 (based on lipophilicity) | 0.751–0.931/ 0.669–0.857 | 0.358–0.610 | 0.711–0.939 | 0.402–0.676 |
| Validated MLR Model: 3 Different Test Sets with n = 4–6 | R2train | Strain | R2test | stest |
|---|---|---|---|---|
| Equation (15) (IAM retention) | 0.686–0.923 | 0.262–0.376 | 0.433 *–0.920 | 0.257–0.434 |
| Equation (17) (lipophilicity) | 0.526 *–0.852 | 0.364–0.485 | 0.437 *–0. 0.950 | 0.132–0.429 |
| Validated PLS Model: 2 different test sets with n = 8–9 | R2train/Q2train | RMSEE | R2test | RMSEP |
| PLS Model 10 (based on IAM retention) | 0.927, 0.962/ 0.690, 0.922 | 0.160, 0.268 | 0.718, 0.956 | 0.316, 0.656 |
| PLS Model 11 (based on lipophilicity) | 0.747, 0.962/ 0.747, 0.880 | 0.160, 0.251 | 0.718, 0.782 | 0.650, 0.656 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallianatou, T.; Tsopelas, F.; Tsantili-Kakoulidou, A. Prediction Models for Brain Distribution of Drugs Based on Biomimetic Chromatographic Data. Molecules 2022, 27, 3668. https://doi.org/10.3390/molecules27123668
Vallianatou T, Tsopelas F, Tsantili-Kakoulidou A. Prediction Models for Brain Distribution of Drugs Based on Biomimetic Chromatographic Data. Molecules. 2022; 27(12):3668. https://doi.org/10.3390/molecules27123668
Chicago/Turabian StyleVallianatou, Theodosia, Fotios Tsopelas, and Anna Tsantili-Kakoulidou. 2022. "Prediction Models for Brain Distribution of Drugs Based on Biomimetic Chromatographic Data" Molecules 27, no. 12: 3668. https://doi.org/10.3390/molecules27123668
APA StyleVallianatou, T., Tsopelas, F., & Tsantili-Kakoulidou, A. (2022). Prediction Models for Brain Distribution of Drugs Based on Biomimetic Chromatographic Data. Molecules, 27(12), 3668. https://doi.org/10.3390/molecules27123668