
Minimal Active Space for Diradicals Using Multistate Density Functional Theory


Abstract
:1. Introduction
2. Theoretical Background
3. Computational Details
4. Results and Discussion
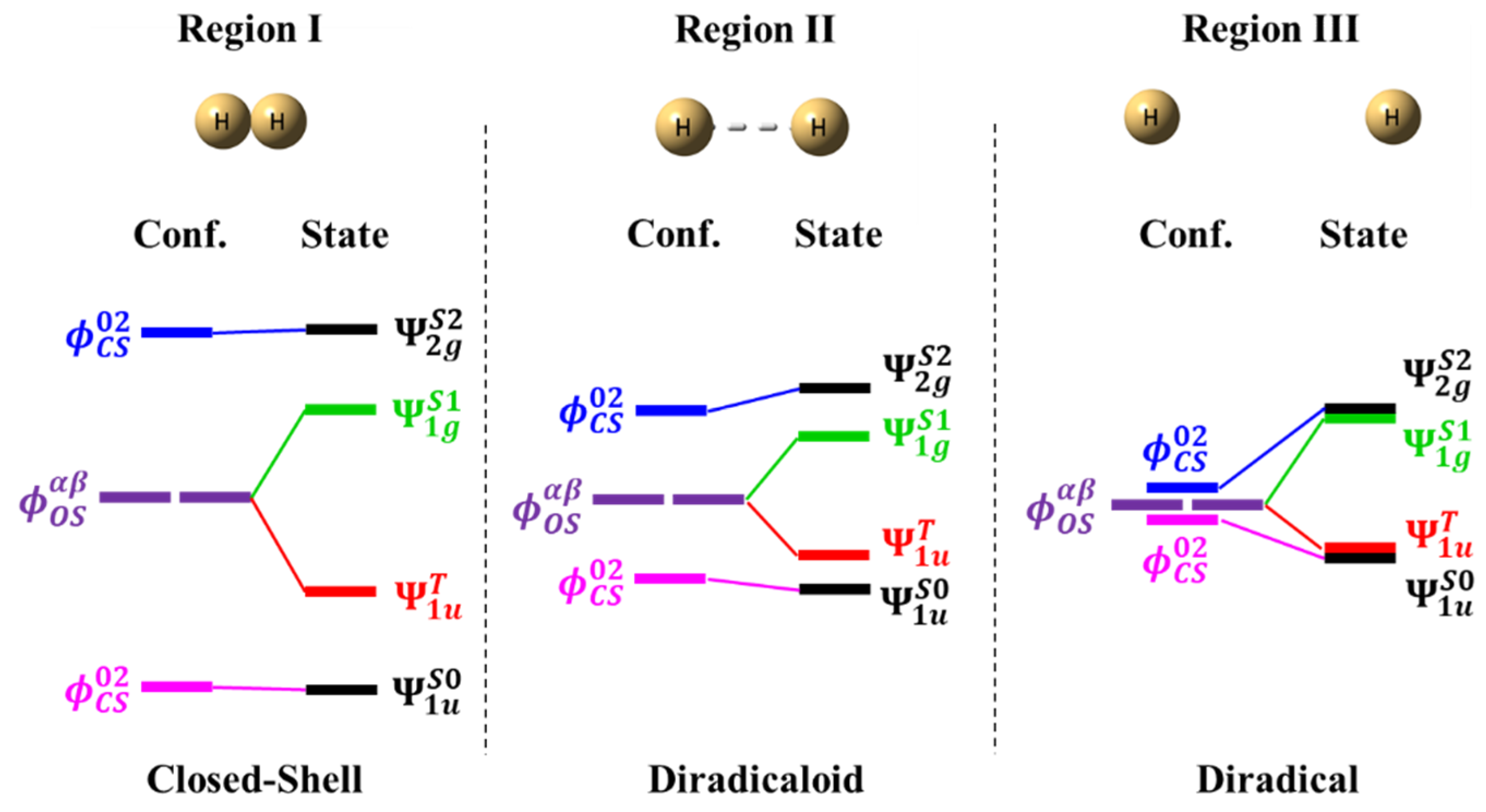
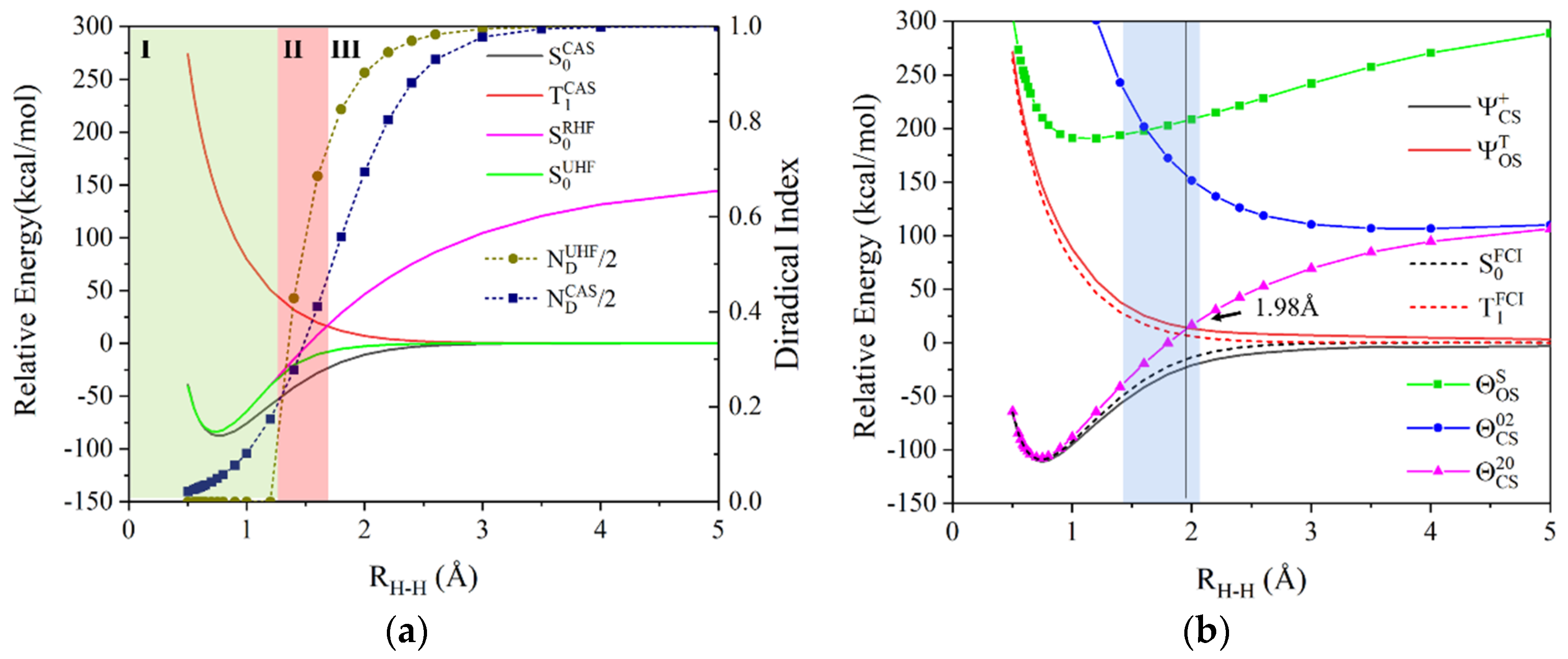
4.1. Potential Energy Curves of Hydrogen Molecule
4.2. Singlet–Triplet-Energy Gap
4.2.1. Benzyne Isomers
4.2.2. Cyclobutadiene
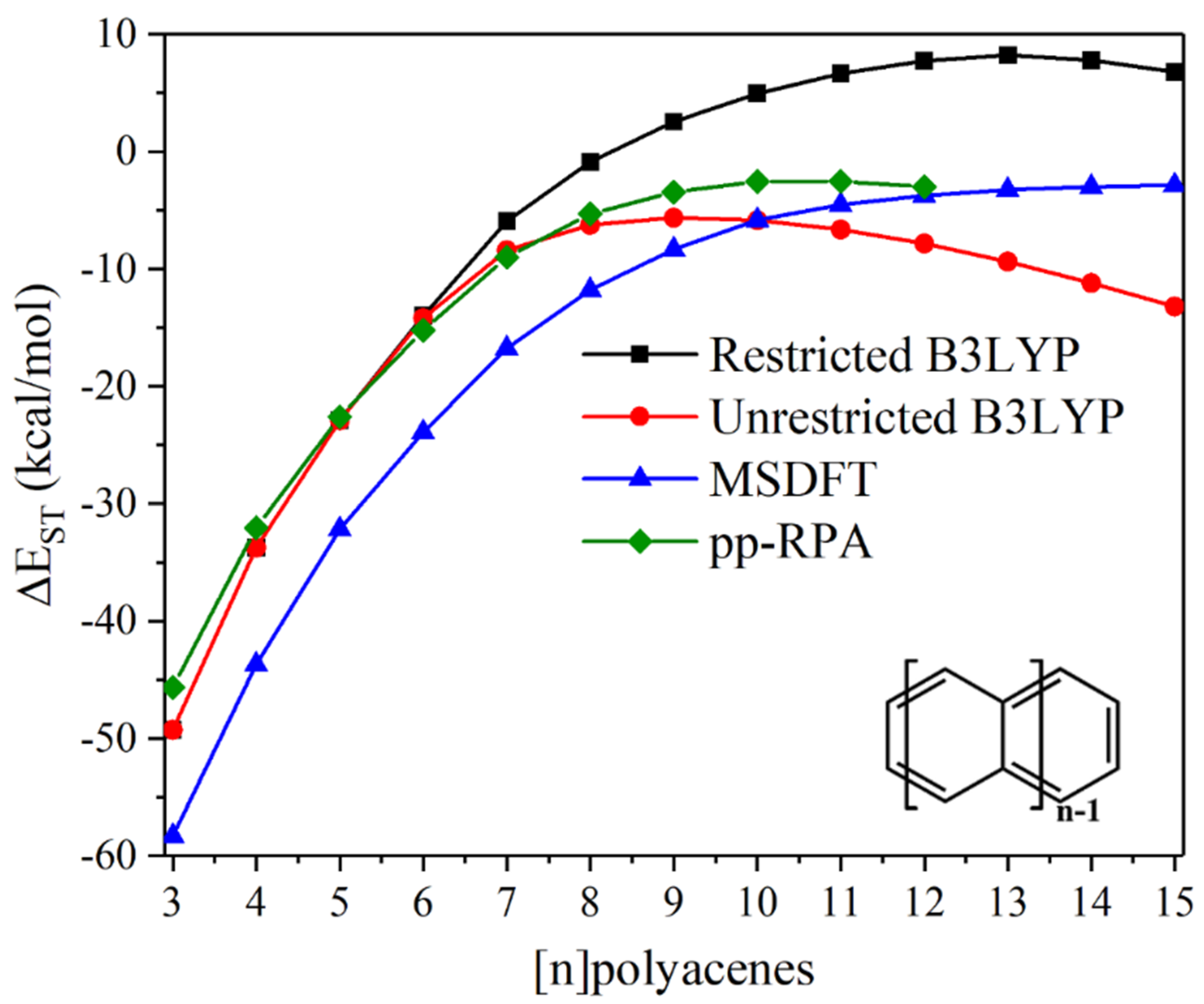
4.2.3. Polyacenes
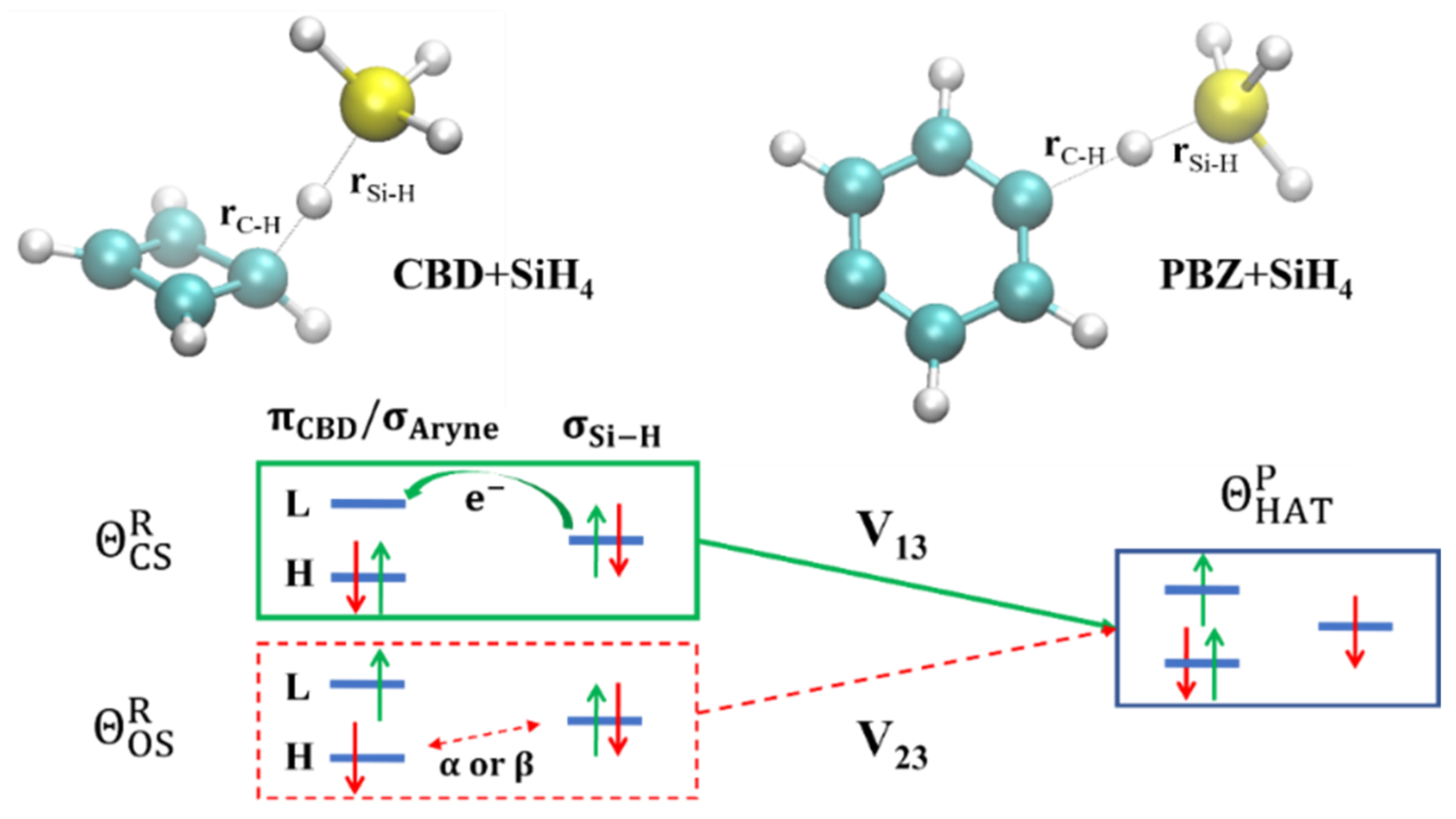
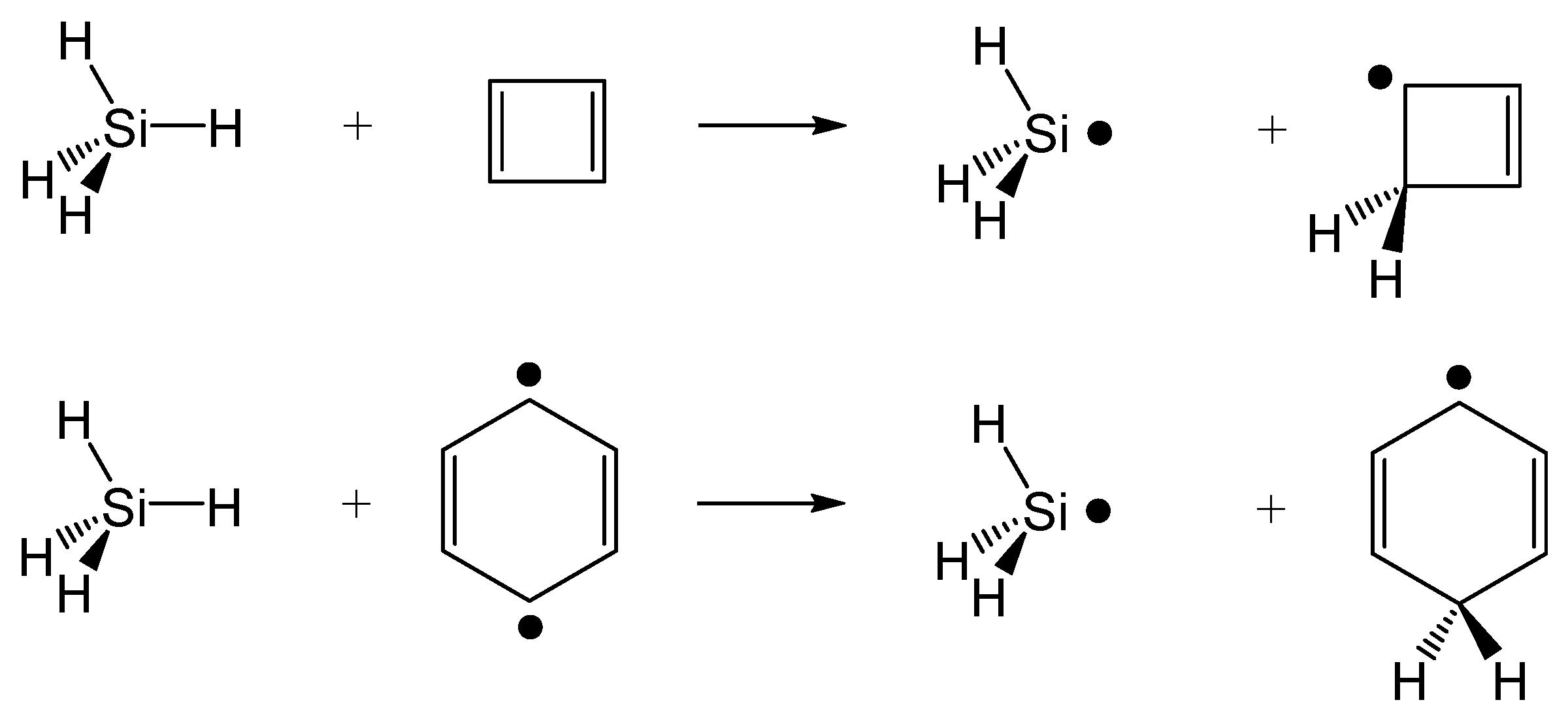
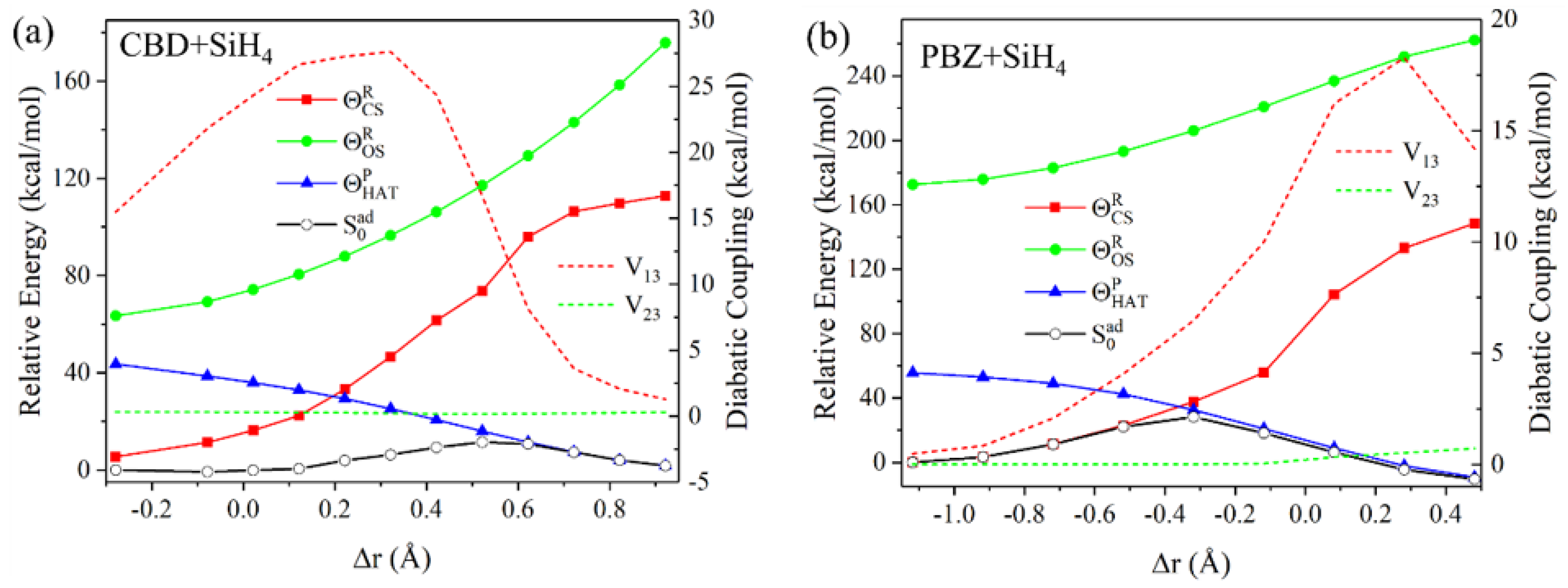
4.3. Hydrogen-Atom Transfer Reactions
- The diagonal elements of are directly determined as the BLKS-DFT energies of the corresponding determinants. The two reactant diabatic states can be separately obtained as the lower root of a 2 × 2 NOSI diagonalization of the two states in Equations (16) and (17) for illustration in Figure 6, although it is not needed to determine the potential energy surface of the adiabatic ground state. The four determinants in Equations (16) and (17) involve block-local excitations, for which the optimization has been detailed in reference [41]. For the product state, only ground-state BLKS optimization is sufficient;
- The spin-coupling matrix element between the spin-pair determinants in Equation (18) is evaluated using Equations (1) and (5), which requires a separate BLKS calculation of the triplet state with MS = +1;
- For all other off-diagonal matrix elements of , we used the DFT energy-scaled nonorthogonal determinant value to approximate (Equation (6));
- To examine the variations of state interactions as the HAT occurs, we also computed the effective diabatic coupling values between the reactant (Equations (16) and (17)) and product (Equation (18)) states, denoted as and according to , where R = 1 and 2, P = 3, and is the adiabatic ground-state energy.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Stuyver, T.; Chen, B.; Zeng, T.; Geerlings, P.; De Proft, F.; Hoffmann, R. Do Diradicals Behave Like Radicals? Chem. Rev. 2019, 119, 11291–11351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Truhlar, D.G. Spin Splitting Energy of Transition Metals: A New, More Affordable Wave Function Benchmark Method and Its Use to Test Density Functional Theory. J. Chem. Theory Comput. 2020, 16, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Rajca, A. Organic Diradicals and Polyradicals: From Spin Coupling to Magnetism? Chem. Rev. 1994, 94, 871–893. [Google Scholar] [CrossRef]
- Abe, M. Diradicals. Chem. Rev. 2013, 113, 7011–7088. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, G.; Teufel, E.; Hopf, H. First Spectroscopic Detection of Diradicals in photocycloreversions. Angew. Chem. Int. Ed. 1979, 18, 215–217. [Google Scholar] [CrossRef]
- Borden, W.T.; Davidson, E.R. Effects of electron repulsion in conjugated hydrocarbon diradicals. J. Am. Chem. Soc. 1977, 99, 4587–4594. [Google Scholar] [CrossRef]
- Fukui, K.; Tanaka, K. A Theoretical Study on Biradicals. I. Theoretical Characteristics of Biradicals. Bull. Chem. Soc. Jpn. 2006, 50, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Salem, L.; Rowland, C. The Electronic Properties of Diradicals. Angew. Chem. Int. Ed. 1972, 11, 92–111. [Google Scholar] [CrossRef]
- Nakano, M. Electronic Structure of Open-Shell Singlet Molecules: Diradical Character Viewpoint. Top. Curr. Chem. 2017, 375, 47. [Google Scholar] [CrossRef]
- Scheschkewitz, D.; Amii, H.; Gornitzka, H.; Schoeller, W.W.; Bourissou, D.; Bertrand, G. Singlet diradicals: From transition states to crystalline compounds. Science 2002, 295, 1880–1881. [Google Scholar] [CrossRef]
- Yamaguchi, K. The electronic structures of biradicals in the unrestricted Hartree-Fock approximation. Chem. Phys. Lett. 1975, 33, 330–335. [Google Scholar] [CrossRef]
- Nakano, M. Open-Shell-Character-Based Molecular Design Principles: Applications to Nonlinear Optics and Singlet Fission. Chem. Rec. 2016, 17, 27–62. [Google Scholar] [CrossRef] [PubMed]
- Rivero, P.; Jiménez-Hoyos, C.A.; Scuseria, G.E. Entanglement and Polyradical Character of Polycyclic Aromatic Hydrocarbons Predicted by Projected Hartree–Fock Theory. J. Phys. Chem. B 2013, 117, 12750–12758. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wu, J. Open-shell polycyclic aromatic hydrocarbons. J. Mater. Chem. 2011, 22, 4151–4160. [Google Scholar] [CrossRef]
- Nagai, H.; Nakano, M.; Yoneda, K.; Kishi, R.; Takahashi, H.; Shimizu, A.; Kubo, T.; Kamada, K.; Ohta, K.; Botek, E.; et al. Signature of multiradical character in second hyperpolarizabilities of rectangular graphene nanoflakes. Chem. Phys. Lett. 2010, 489, 212–218. [Google Scholar] [CrossRef]
- Hajgató, B.; Szieberth, D.; Geerlings, P.; De Proft, F.; Deleuze, M. A benchmark theoretical study of the electronic ground state and of the singlet-triplet split of benzene and linear acenes. J. Chem. Phys. 2009, 131, 224321. [Google Scholar] [CrossRef]
- Hachmann, J.; Dorando, J.J.; Avilés, M.; Chan, G.K.-L. The radical character of the acenes: A density matrix renormalization group study. J. Chem. Phys. 2007, 127, 134309. [Google Scholar] [CrossRef] [Green Version]
- Cembran, A.; Song, L.; Mo, Y.; Gao, J. Block-Localized Density Functional Theory (BLDFT), Diabatic Coupling, and Their Use in Valence Bond Theory for Representing Reactive Potential Energy Surfaces. J. Chem. Theory Comput. 2009, 5, 2702–2716. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Provorse, M.R.; Bao, P.; Qu, Z.; Gao, J. Multistate Density Functional Theory for Effective Diabatic Electronic Coupling. J. Phys. Chem. Lett. 2016, 7, 2286–2293. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Grofe, A.; Ren, H.; Bao, P. Beyond Kohn–Sham Approximation: Hybrid Multistate Wave Function and Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 5143–5149. [Google Scholar] [CrossRef] [Green Version]
- Mo, Y.; Gao, J.; Peyerimhoff, S.D. Energy decomposition analysis of intermolecular interactions using a block-localized wave function approach. J. Chem. Phys. 2000, 112, 5530–5538. [Google Scholar] [CrossRef]
- Mo, Y.; Bao, P.; Gao, J. Energy decomposition analysis based on a block-localized wavefunction and multistate density functional theory. Phys. Chem. Chem. Phys. 2011, 13, 6760–6775. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, X.; Grofe, A.; Gao, J. Diabatic States at Construction (DAC) through Generalized Singular Value Decomposition. J. Phys. Chem. Lett. 2018, 9, 6038–6046. [Google Scholar] [CrossRef] [PubMed]
- Grofe, A.; Zhao, R.; Wildman, A.; Stetina, T.F.; Li, X.; Bao, P.; Gao, J. Generalization of Block-Localized Wave Function for Constrained Optimization of Excited Determinants. J. Chem. Theory Comput. 2021, 17, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Grofe, A.; Wang, Z.; Bao, P.; Chen, X.; Liu, W.; Gao, J. Dynamic-then-Static Approach for Core Excitations of Open-Shell Molecules. J. Phys. Chem. Lett. 2021, 12, 7409–7417. [Google Scholar] [CrossRef] [PubMed]
- Grofe, A.; Chen, X.; Liu, W.; Gao, J. Spin-Multiplet Components and Energy Splittings by Multistate Density Functional Theory. J. Phys. Chem. Lett. 2017, 8, 4838–4845. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Hettich, C.P.; Chen, X.; Gao, J. Minimal-active-space multistate density functional theory for excitation energy involving local and charge transfer states. Npj Comput. Mater. 2021, 7, 148. [Google Scholar] [CrossRef]
- Liu, W.; Hoffmann, M.R. SDS: The ‘static–dynamic–static’ framework for strongly correlated electrons. Theor. Chem. Acc. 2014, 133, 1481. [Google Scholar] [CrossRef]
- Yang, L.; Grofe, A.; Reimers, J.; Gao, J. Multistate density functional theory applied with 3 unpaired electrons in 3 orbitals: The singdoublet and tripdoublet states of the ethylene cation. Chem. Phys. Lett. 2019, 736, 136803. [Google Scholar] [CrossRef]
- Cembran, A.; Provorse, M.R.; Wang, C.; Wu, W.; Gao, J. The Third Dimension of a More O’Ferrall-Jencks Diagram for Hydrogen Atom Transfer in the Isoelectronic Hydrogen Exchange Reactions of (PhX)(2)H(*) with X = O, NH, and CH(2). J. Chem. Theory Comput. 2012, 8, 4347–4358. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.; Swaminathan, S.; Odell, B.G.; Gleiter, R. Potential surface for a nonconcerted reaction. Tetramethylene. J. Am. Chem. Soc. 1970, 92, 7091–7097. [Google Scholar] [CrossRef]
- Sirjean, B.; Glaude, P.A.; Ruiz-Lopez, A.M.F.; Fournet, R. Detailed Kinetic Study of the Ring Opening of Cycloalkanes by CBS-QB3 Calculations. J. Phys. Chem. A 2006, 110, 12693–12704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaque, P.; Toro-Labbé, A.; Geerlings, P.; De Proft, F. Theoretical Study of the Regioselectivity of [2 + 2] Photocycloaddition Reactions of Acrolein with Olefins. J. Phys. Chem. A 2009, 113, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, C.; Freccero, M.; Gandolfi, R.; Rastelli, A. Concerted vs. Stepwise Mechanism in 1,3-Dipolar Cycloaddition of Nitrone to Ethene, Cyclobutadiene, and Benzocyclobutadiene. A Computational Study. J. Org. Chem. 2000, 65, 6112–6120. [Google Scholar] [CrossRef]
- Bai, S.; Barbatti, M. Mechanism of Spin-Exchange Internal Conversion: Practical Proxies for Diabatic and Nonadiabatic Couplings. J. Chem. Theory Comput. 2019, 15, 1503–1513. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z. Reactivities of singlet oxygen: Open-shell or closed-shell? Phys. Chem. Chem. Phys. 2020, 22, 13373–13377. [Google Scholar] [CrossRef]
- Grofe, A.; Qu, Z.; Truhlar, D.G.; Li, H.; Gao, J. Diabatic-At-Construction Method for Diabatic and Adiabatic Ground and Excited States Based on Multistate Density Functional Theory. J. Chem. Theory Comput. 2017, 13, 1176–1187. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision A. 1; Gaussian Inc.: Wallingford, CT, USA, 2009; Volume 27, p. 34. [Google Scholar]
- Werner, H.; Knowles, P.; Amos, R.; Bernhardsson, A.; Berning, A.; Celani, P.; Cooper, D.; Deegan, M.; Dobbyn, A.; Eckert, F. MolPro2012; University College Cardiff Consultants Ltd.: Wales, UK, 2012. [Google Scholar]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Bao, P.; Hettich, C.P.; Shi, Q.; Gao, J. Block-Localized Excitation for Excimer Complex and Diabatic Coupling. J. Chem. Theory Comput. 2021, 17, 240–254. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Nguyen, N.A.; Csonka, G.I.; Kolossváry, I. Simple tests for density functional methods. J. Comput. Chem. 1997, 18, 1534–1545. [Google Scholar] [CrossRef]
- Johnson, B.G.; Gonzales, C.A.; Gill, P.M.W.; Pople, J.A. A density functional study of the simplest hydrogen abstraction reaction. Effect of self-interaction correction. Chem. Phys. Lett. 1994, 221, 100–108. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A.; Baerends, E.J. On the Calculation of Multiplet Energies by the Hartree-Fock-Slater Method. Theor. Chim. Acta 1977, 43, 261–271. [Google Scholar] [CrossRef]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Su, Y.; Wang, X.; Wang, L.; Zhang, Z.; Wang, X.; Song, Y.; Power, P.P. Thermally controlling the singlet–triplet energy gap of a diradical in the solid state. Chem. Sci. 2016, 7, 6514–6518. [Google Scholar] [CrossRef] [Green Version]
- Hajgató, B.; Huzak, M.; Deleuze, M.S. Focal Point Analysis of the Singlet–Triplet Energy Gap of Octacene and Larger Acenes. J. Phys. Chem. A 2011, 115, 9282–9293. [Google Scholar] [CrossRef]
- Goldberg, A.H.; Dougherty, D.A. Effects of through-bond and through-space interactions on singlet-triplet energy gaps in localized biradicals. J. Am. Chem. Soc. 1983, 105, 284–290. [Google Scholar] [CrossRef]
- Wei, H.; Hrovat, D.A.; Mo, Y.; Hoffmann, R.; Borden, W.T. The Contributions of Through-Bond Interactions to the Singlet-Triplet Energy Difference in 1,3-Dehydrobenzene. J. Phys. Chem. A 2009, 113, 10351–10358. [Google Scholar] [CrossRef]
- Ana-Maria, Y.S.; Cristian, C.; Krylov, A.I. Bonding Patterns in Benzene Triradicals from Structural, Spectroscopic, and Thermochemical Perspectives. J. Phys. Chem. A 2004, 108, 6581–6588. [Google Scholar]
- Wenthold, P.G.; Squires, R.R.; Lineberger, W.C. Ultraviolet Photoelectron Spectroscopy of the o-, m-, and p-Benzyne Negative Ions. Electron Affinities and Singlet−Triplet Splittings for o-, m-, and p-Benzyne. J. Am. Chem. Soc. 1998, 120, 5279–5290. [Google Scholar] [CrossRef]
- Borden, W.T.; Davidson, E.R.; Hart, P. The potential surfaces for the lowest singlet and triplet states of cyclobutadiene. J. Am. Chem. Soc. 1978, 100, 388–392. [Google Scholar] [CrossRef]
- Wu, J.I.-C.; Mo, Y.; Evangelista, F.A.; Schleyer, P.V.R. Is cyclobutadiene really highly destabilized by antiaromaticity? Chem. Commun. 2012, 48, 8437–8439. [Google Scholar] [CrossRef] [PubMed]
- Karadakov, P.B. Ground- and Excited-State Aromaticity and Antiaromaticity in Benzene and Cyclobutadiene. J. Phys. Chem. A 2008, 112, 7303–7309. [Google Scholar] [CrossRef] [PubMed]
- Eckert-Maksić, M.; Vazdar, M.; Barbatti, M.; Lischka, H.; Maksić, Z. Automerization reaction of cyclobutadiene and its barrier height: An ab initio benchmark multireference average-quadratic coupled cluster study. J. Chem. Phys. 2006, 125, 64310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levchenko, S.; Krylov, A.I. Equation-of-motion spin-flip coupled-cluster model with single and double substitutions: Theory and application to cyclobutadiene. J. Chem. Phys. 2004, 120, 175. [Google Scholar] [CrossRef]
- Kollmar, H.; Staemmler, V. Violation of Hund’s rule by spin polarization in molecules. Theor. Chim. Acta 1978, 48, 223–239. [Google Scholar] [CrossRef]
- Thatcher, B.W. Can a square or effectively square singlet be the ground state of cyclobutadiene. J. Am. Chem. Soc. 1976, 7, 5969. [Google Scholar]
- Hrovat, D.A.; Borden, W.T. Violations of Hund’s rule in molecules—Where to look for them and how to identify them. J. Mol. Struc.-Theochem. 1997, 398–399, 211–220. [Google Scholar] [CrossRef]
- Tofanelli, M.A.; Salorinne, K.; Ni, T.W.; Malola, S.; Newell, B.; Phillips, B.; Hakkinen, H.; Ackerson, C.J. Jahn-Teller Effects in Au25(SR)18. Chem. Sci. 2016, 7, 1882–1890. [Google Scholar] [CrossRef] [Green Version]
- Tachikawa, H. Jahn–Teller Effect of the Benzene Radical Cation: A Direct ab Initio Molecular Dynamics Study. J. Phys. Chem. A 2018, 122, 4121–4129. [Google Scholar] [CrossRef]
- Tachikawa, H.; Lund, A. Structures and electronic states of trimer radical cations of coronene: DFT–ESR simulation study. Phys. Chem. Chem. Phys. 2022, 24, 10318–10324. [Google Scholar] [CrossRef] [PubMed]
- Senn, F.; Krykunov, M. Excited State Studies of Polyacenes Using the All-Order Constricted Variational Density Functional Theory with Orbital Relaxation. J. Phys. Chem. A 2015, 119, 10575–10581. [Google Scholar] [CrossRef] [PubMed]
- Bendikov, M.; Duong, H.M.; Starkey, K.; Houk, K.N.; Carter, A.E.A.; Wudl, F. Oligoacenes: Theoretical Prediction of Open-Shell Singlet Diradical Ground States. J. Am. Chem. Soc. 2004, 126, 7416–7417. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhang, D.; Liu, C.; Jiang, Y. Open-Shell Ground State of Polyacenes: A Valence Bond Study. J. Phys. Chem. A 2009, 113, 7909–7914. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Davidson, E.R.; Yang, W. Nature of ground and electronic excited states of higher acenes. Proc. Natl. Acad. Sci. USA 2016, 113, E5098–E5107. [Google Scholar] [CrossRef] [Green Version]
- Houk, K.N.; Lee, P.S.; Nendel, M. Polyacene and Cyclacene Geometries and Electronic Structures: Bond Equalization, Vanishing Band Gaps, and Triplet Ground States Contrast with Polyacetylene. J. Org. Chem. 2001, 66, 5517–5521. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Cooper, D.L.; Gerratt, J.; Raimondi, M. Applications of spin-coupled valence bond theory. Chem. Rev. 1991, 91, 929–964. [Google Scholar] [CrossRef]
- Mo, Y.; Gao, J. An Ab Initio Molecular Orbital−Valence Bond (MOVB) Method for Simulating Chemical Reactions in Solution. J. Phys. Chem. A 2000, 104, 3012–3020. [Google Scholar] [CrossRef]
- Song, L.; Mo, Y.; Gao, J. An Effective Hamiltonian Molecular Orbital-Valence Bond (MOVB) Approach for Chemical Reactions as Applied to the Nucleophilic Substitution Reaction of Hydrosulfide Ion and Chloromethane. J. Chem. Theory Comput. 2009, 5, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Gao, J. On the Construction of Diabatic and Adiabatic Potential Energy Surfaces Based on Ab Initio Valence Bond Theory. J. Phys. Chem. A 2008, 112, 12925–12935. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | p-Benzyne | m-Benzyne | o-Benzyne | MUE |
|---|---|---|---|---|
| RDFT | 41.7 | 19.3 | −37.3 | 28.7 |
| UDFT | −1.9 | −4.0 | −37.3 | 6.4 |
| CASSCF | −0.7 | −12.9 | −28.5 | 6.8 |
| CASPT2 | −3.6 | −19.7 | −35.0 | 1.3 |
| SF-CCSD(UHF) a | −3.9 | −19.2 | −37.6 | 0.7 |
| MSDFT | −4.7 | −19.1 | −36.9 | 0.5 |
| Exp. b | −3.8 | −21.0 | −37.5 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Zhao, R.; Guo, Y.; Qu, Z.; Gao, J. Minimal Active Space for Diradicals Using Multistate Density Functional Theory. Molecules 2022, 27, 3466. https://doi.org/10.3390/molecules27113466
Han J, Zhao R, Guo Y, Qu Z, Gao J. Minimal Active Space for Diradicals Using Multistate Density Functional Theory. Molecules. 2022; 27(11):3466. https://doi.org/10.3390/molecules27113466
Chicago/Turabian StyleHan, Jingting, Ruoqi Zhao, Yujie Guo, Zexing Qu, and Jiali Gao. 2022. "Minimal Active Space for Diradicals Using Multistate Density Functional Theory" Molecules 27, no. 11: 3466. https://doi.org/10.3390/molecules27113466
APA StyleHan, J., Zhao, R., Guo, Y., Qu, Z., & Gao, J. (2022). Minimal Active Space for Diradicals Using Multistate Density Functional Theory. Molecules, 27(11), 3466. https://doi.org/10.3390/molecules27113466