
Aha1 Exhibits Distinctive Dynamics Behavior and Chaperone-Like Activity

 ,
,
Abstract
1. Introduction
2. Results
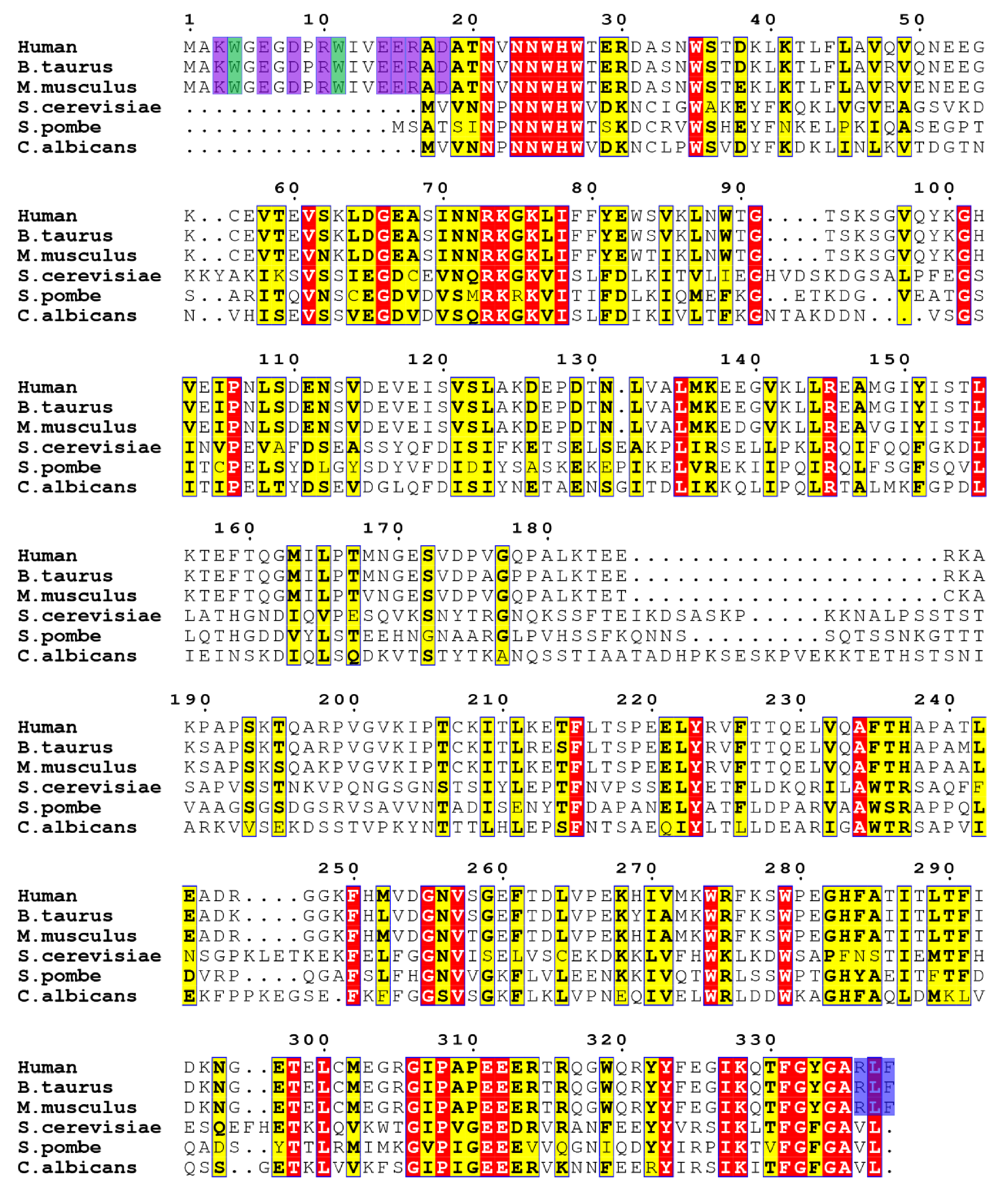
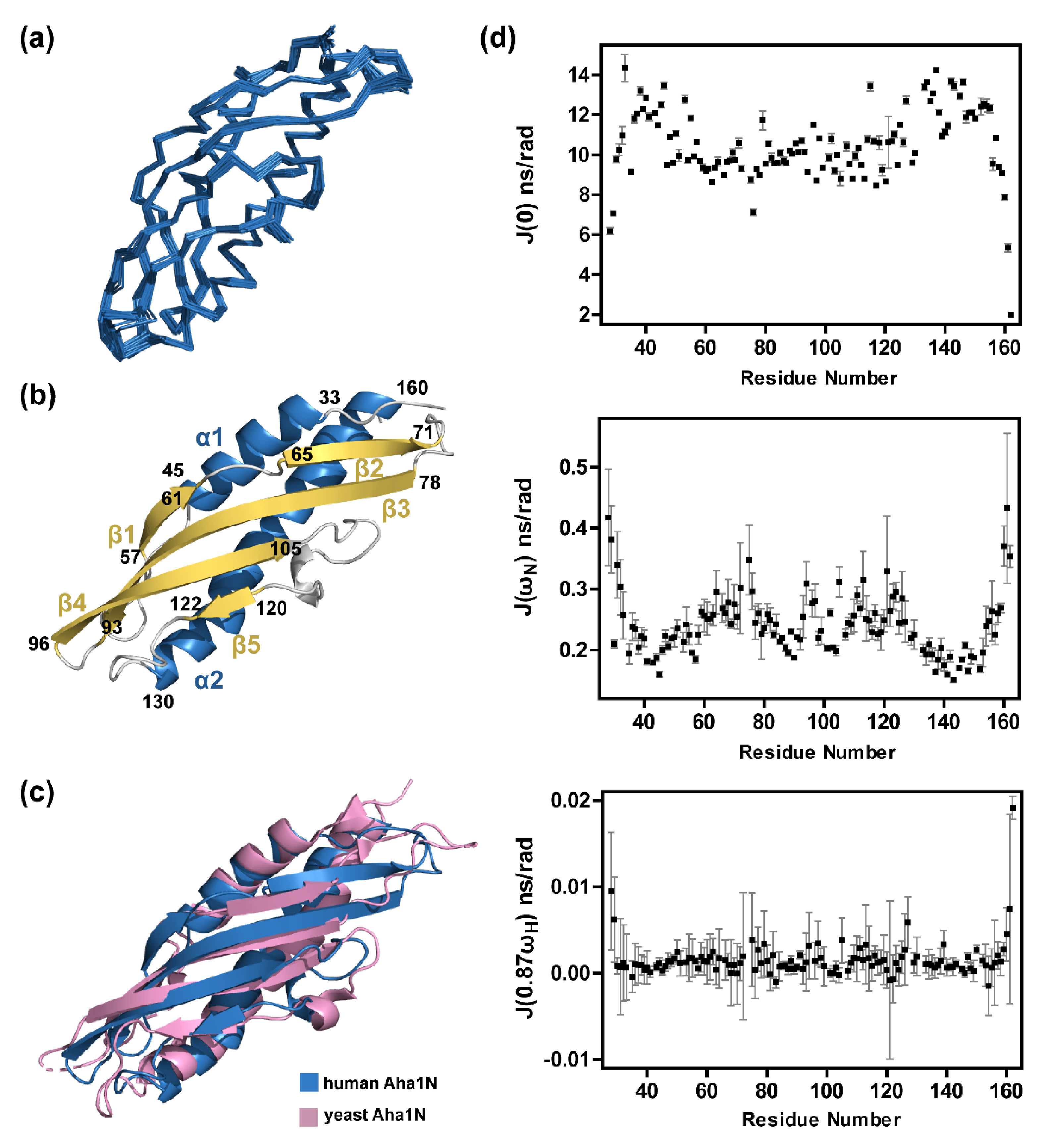
2.1. Solution Structure Determination of Human Aha1’s N-Terminal Domain
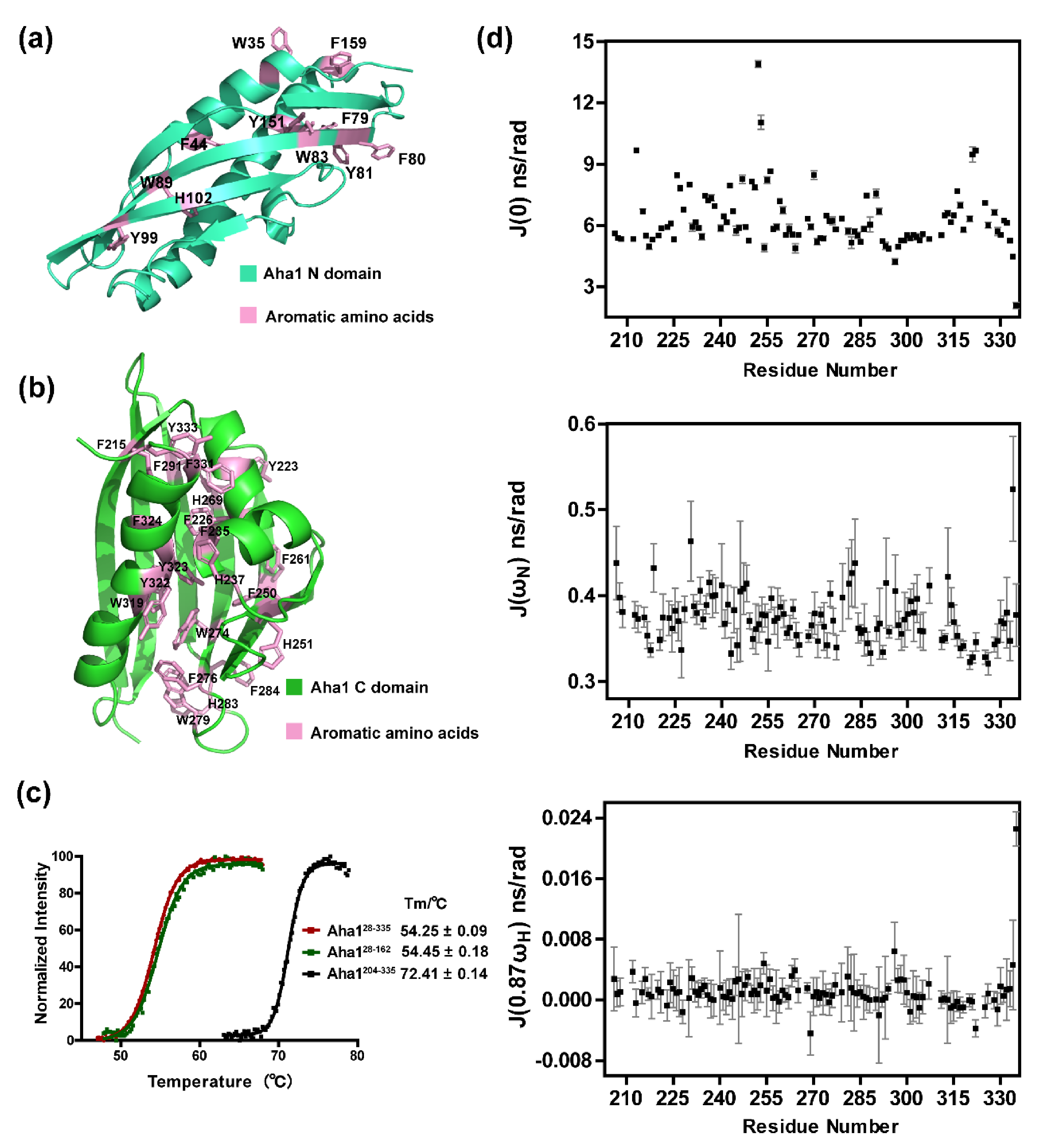
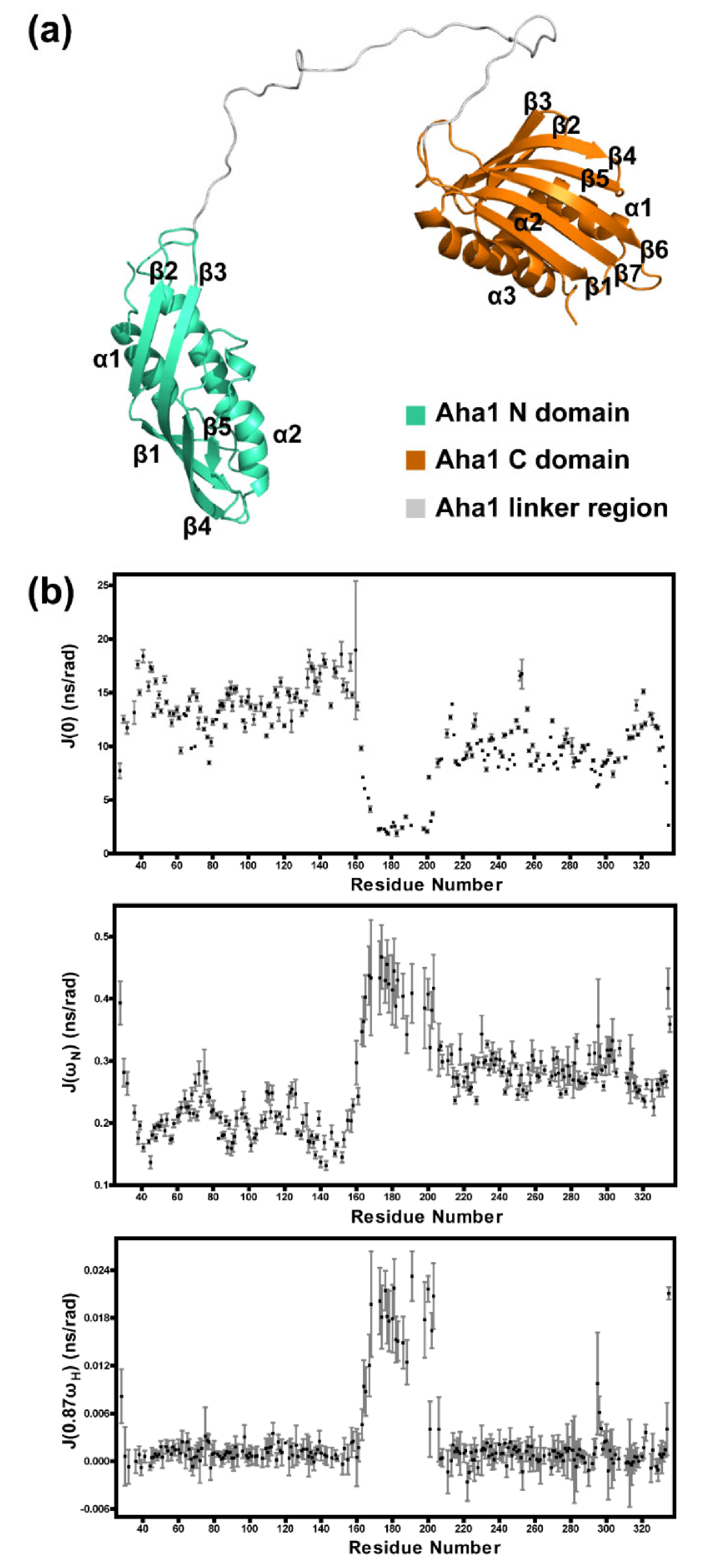
2.2. Aha1’s N-Terminal Domain Presents Unique Dynamics Behavior and Structural Features Different from Aha1’s C-Terminal Domain
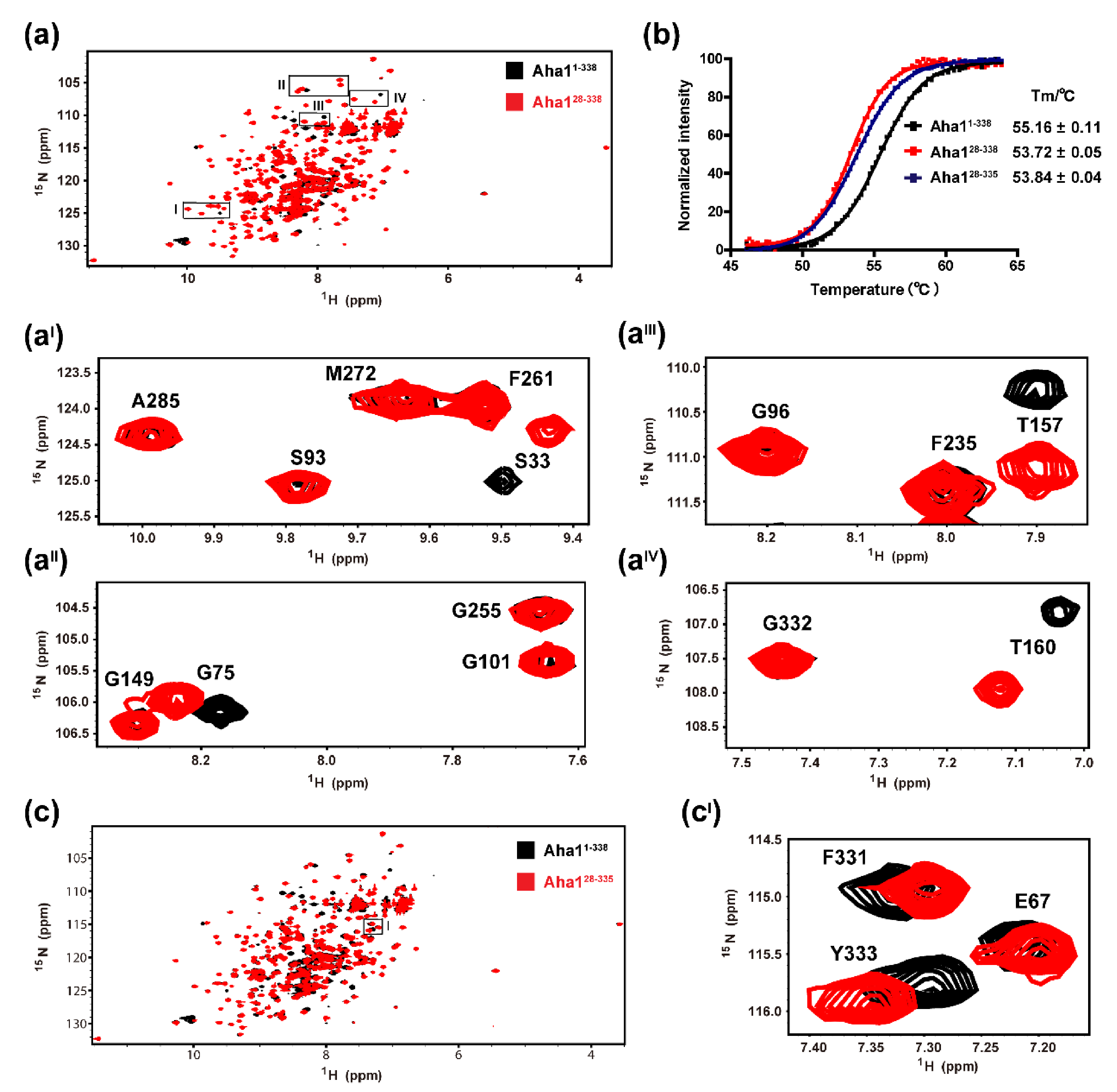
2.3. The Very N-Terminal Region and the C-Terminal RLF Motif of Aha1 Show No Significant Effects on the Global Fold of the Protein
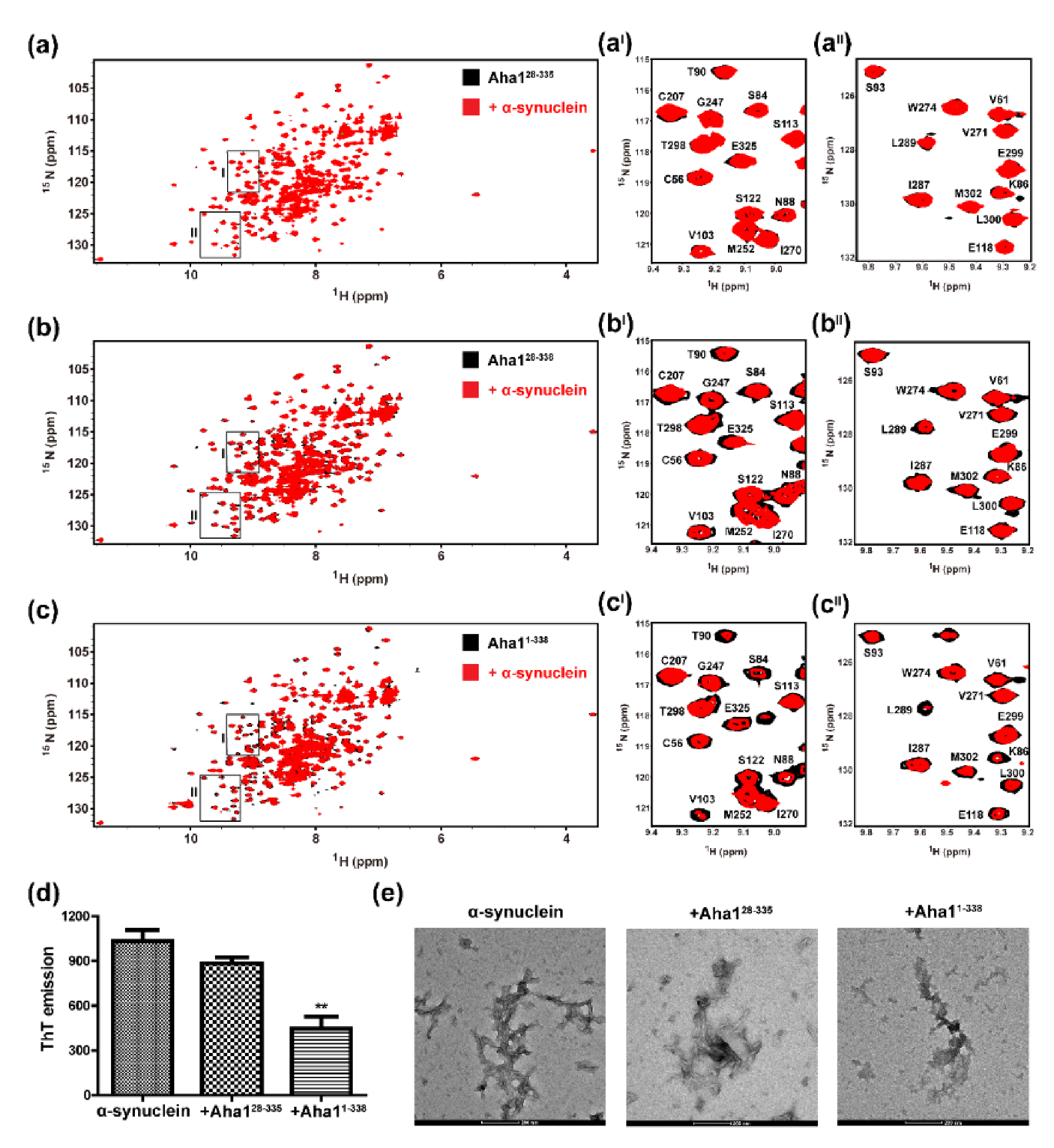
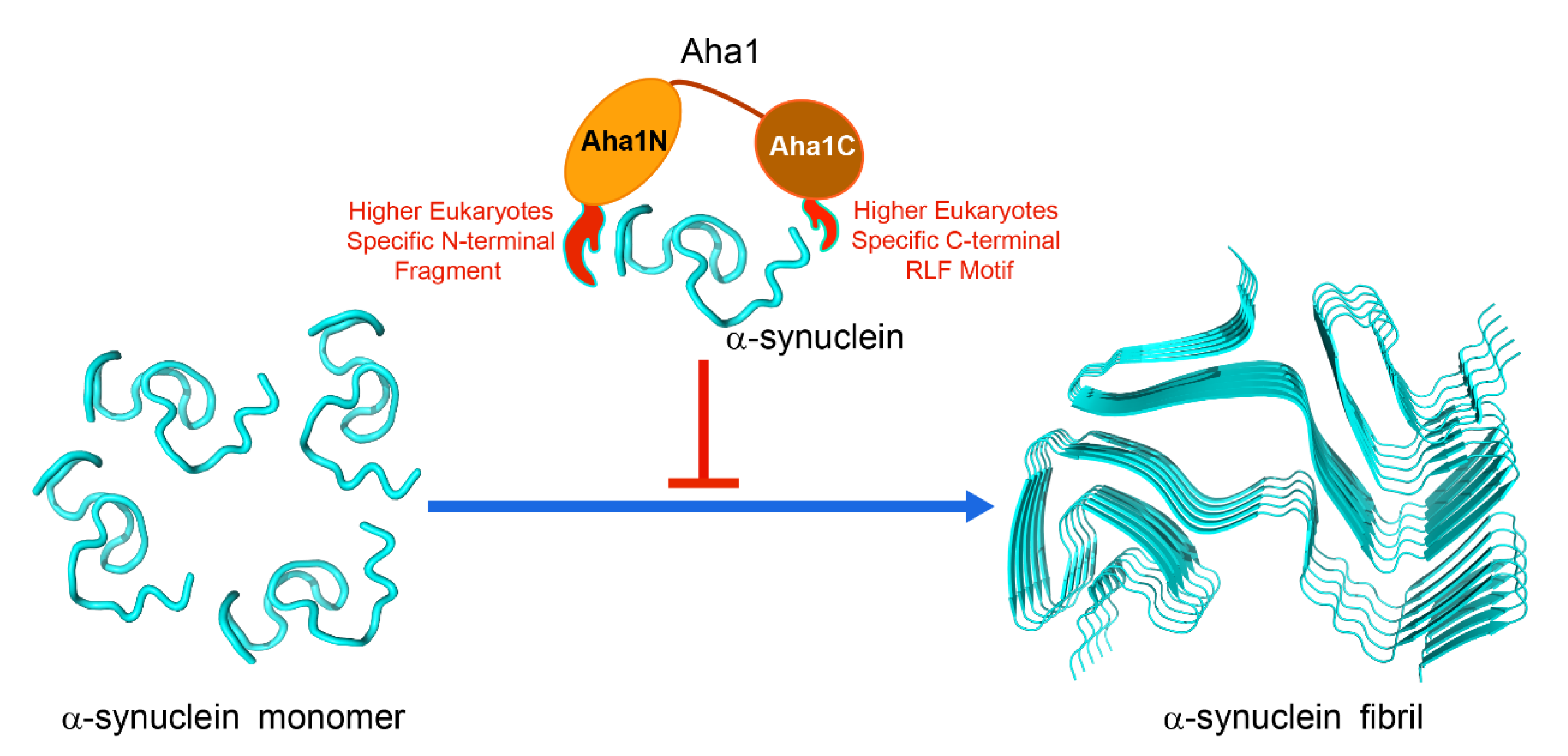
2.4. Both the N-Terminal Fragment M1-W27 and the C-Terminal RLF Motif of Aha1 Contribute to the Recognition of α-Synuclein
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Protein Expression and Purification
4.3. NMR Spectroscopy
4.4. Structure Calculation
4.5. NMR Relaxation Experiments
4.6. Dynamics Behavior Analysis
4.7. NMR Titration Experiments
4.8. Thermal Shift Assay
4.9. In Vitro α-Synuclein Fibril Formation Assay
4.10. Thioflavin T Fluorescence Assay (ThT)
4.11. Electron Microscopy Experiments
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dobson, C.M.; Sali, A.; Karplus, M. Protein folding: A perspective from theory and experiment. Angew. Chem. Int. Ed. 1998, 37, 868–893. [Google Scholar] [CrossRef]
- Bartlett, A.I.; Radford, S.E. An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 582–588. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Horwich, A.L. Protein folding in the cell: An inside story. Nat. Med. 2011, 17, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Saio, T.; Guan, X.; Rossi, P.; Economou, A.; Kalodimos, C.G. Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 2014, 344, 1250494. [Google Scholar] [CrossRef]
- Burmann, B.M.; Hiller, S. Chaperones and chaperone-substrate complexes: Dynamic playgrounds for NMR spectroscopists. Prog. Nucl. Magn. Reason. Spectrosc. 2015, 86, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Prodromou, C. Structure and in vivo function of Hsp90. Curr. Opin. Struct. Biol. 2000, 10, 46–51. [Google Scholar] [CrossRef]
- Pearl, L.H.; Prodromou, C. Structure, function, and mechanism of the Hsp90 molecular chaperone. Adv. Protein Chem. 2001, 59, 157–186. [Google Scholar]
- Pearl, L.H.; Prodromou, C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 2006, 75, 271–294. [Google Scholar] [CrossRef]
- Prodromou, C.; Panaretou, B.; Chohan, S.; Siligardi, G.; O’Brien, R.; Ladbury, J.E.; Roe, S.M.; Piper, P.W.; Pearl, L.H. The ATPase cycle of Hsp90 drives a molecular ’clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000, 19, 4383–4392. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C.; Pearl, L.H. Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 2003, 3, 301–323. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C. The ’active life’ of Hsp90 complexes. Biochim. Biophys. Acta 2012, 1823, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Panaretou, B.; Siligardi, G.; Meyer, P.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.H.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone Aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar] [CrossRef]
- Meyer, P.; Prodromou, C.; Liao, C.; Hu, B.; Mark Roe, S.; Vaughan, C.K.; Vlasic, I.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 2004, 23, 511–519. [Google Scholar] [CrossRef]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef]
- Prodromou, C. Regulatory Mechanisms of Hsp90. J. Biochem. Mol. Biol. 2017, 3, 2. [Google Scholar] [CrossRef]
- Zuehlke, A.; Johnson, J.L. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 2010, 93, 211–217. [Google Scholar] [CrossRef]
- Biebl, M.M.; Buchner, J. Structure, function, and regulation of the Hsp90 machinery. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Retzlaff, M.; Hagn, F.; Mitschke, L.; Hessling, M.; Gugel, F.; Kessler, H.; Richter, K.; Buchner, J. Asymmetric activation of the hsp90 dimer by its cochaperone Aha1. Mol. Cell 2010, 37, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Blair, L.J.; Zweckstetter, M. Dynamic Aha1 co-chaperone binding to human Hsp90. Protein Sci. 2019, 28, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Mercier, R.; Wolmarans, A.; Schubert, J.; Neuweiler, H.; Johnson, J.L.; LaPointe, P. The conserved NxNNWHW motif in Aha-type co-chaperones modulates the kinetics of Hsp90 ATPase stimulation. Nat. Commun. 2019, 10, 1273. [Google Scholar] [CrossRef]
- Tripathi, V.; Darnauer, S.; Hartwig, N.R.; Obermann, W.M.J. Aha1 can act as an autonomous chaperone to prevent aggregation of stressed proteins. J. Biol. Chem. 2014, 289, 36220–36228. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Koulov, A.V.; LaPointe, P.; Lu, B.W.; Razvi, A.; Coppinger, J.; Dong, M.Q.; Matteson, J.; Laister, R.; Arrowsmith, C.; Yates, J.R.; et al. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 2010, 21, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef]
- Dunn, D.M.; Woodford, M.R.; Truman, A.W.; Jensen, S.M.; Schulman, J.; Caza, T.; Remillard, T.C.; Loiselle, D.; Wolfgeher, D.; Blagg, B.S.; et al. c-Abl mediated tyrosine phosphorylation of Aha1 activates its co-chaperone function in cancer cells. Cell Rep. 2015, 12, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, P.; Rohrberg, J.; Biebl, M.M.; Rutz, D.A.; Buchner, J. The plasticity of the Hsp90 co-chaperone system. Mol. Cell 2017, 67, 947. [Google Scholar] [CrossRef]
- Horvat, N.K.; Armstrong, H.; Lee, B.L.; Mercier, R.; Wolmarans, A.; Knowles, J.; Spyracopoulos, L.; LaPointe, P. A mutation in the catalytic loop of Hsp90 specifically impairs ATPase stimulation by Aha1p, but not Hch1p. J. Mol. Biol. 2014, 426, 2379–2392. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal-W–improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Peng, J.W.; Wagner, G. Mapping of the spectral densities of N-H bond motions in Eglin c using heteronuclear relaxation experiments. Biochemistry 1992, 31, 8571–8586. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.W.; Wagner, G. Mapping of spectral density-Functions using heteronuclear NMR relaxation measurements. J. Magn. Reason. 1992, 98, 308–332. [Google Scholar] [CrossRef][Green Version]
- Lefevre, J.F.; Dayie, K.T.; Peng, J.W.; Wagner, G. Internal mobility in the partially folded DNA binding and dimerization domains of GAL4: NMR analysis of the N-H spectral density functions. Biochemistry 1996, 35, 2674–2686. [Google Scholar] [CrossRef] [PubMed]
- Farrow, N.A.; Zhang, O.; Szabo, A.; Torchia, D.A.; Kay, L.E. Spectral density function mapping using 15N relaxation data exclusively. J. Biomol. NMR 1995, 6, 153–162. [Google Scholar] [CrossRef]
- Jarymowycz, V.A.; Stone, M.J. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem. Rev. 2006, 106, 1624–1671. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Didenko, T.; Duarte, A.M.; Karagoz, G.E.; Rudiger, S.G. Hsp90 structure and function studied by NMR spectroscopy. Biochim. Biophys. Acta 2012, 1823, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.M.; Ali, M.M.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef]
- Vaughan, C.K.; Gohlke, U.; Sobott, F.; Good, V.M.; Ali, M.M.; Prodromou, C.; Robinson, C.V.; Saibil, H.R.; Pearl, L.H. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 2006, 23, 697–707. [Google Scholar] [CrossRef]
- Zhang, M.; Kadota, Y.; Prodromou, C.; Shirasu, K.; Pearl, L.H. Structural basis for assembly of Hsp90-Sgt1-CHORD protein complexes: Implications for chaperoning of NLR innate immunity receptors. Mol. Cell 2010, 39, 269–281. [Google Scholar] [CrossRef]
- Karagoz, G.E.; Duarte, A.M.; Ippel, H.; Uetrecht, C.; Sinnige, T.; van Rosmalen, M.; Hausmann, J.; Heck, A.J.; Boelens, R.; Rudiger, S.G. N-terminal domain of human Hsp90 triggers binding to the cochaperone p23. Proc. Natl. Acad. Sci. USA 2011, 108, 580–585. [Google Scholar] [CrossRef]
- Prodromou, C.; Siligardi, G.; O’Brien, R.; Woolfson, D.N.; Regan, L.; Panaretou, B.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J. 1999, 18, 754–762. [Google Scholar] [CrossRef]
- Siligardi, G.; Hu, B.; Panaretou, B.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle. J. Biol. Chem. 2004, 279, 51989–51998. [Google Scholar] [CrossRef]
- Woodford, M.R.; Sager, R.A.; Marris, E.; Dunn, D.M.; Blanden, A.R.; Murphy, R.L.; Rensing, N.; Shapiro, O.; Panaretou, B.; Prodromou, C.; et al. Tumor suppressor Tsc1 is a new Hsp90 co-chaperone that facilitates folding of kinase and non-kinase clients. EMBO J. 2017, 36, 3650–3665. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.R.; Arakawa, A.; Liu, Y.X.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Lanzarotti, E.; Biekofsky, R.R.; Estrin, D.A.; Marti, M.A.; Turjanski, A.G. Aromatic-aromatic interactions in proteins: Beyond the dimer. J. Chem. Inf. Model. 2011, 51, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Rooman, M. Physical and molecular bases of protein thermal stability and cold adaptation. Curr. Opin. Struc. Biol. 2017, 42, 117–128. [Google Scholar] [CrossRef]
- Karshikoff, A.; Nilsson, L.; Ladenstein, R. Rigidity versus flexibility: The dilemma of understanding protein thermal stability. FEBS J. 2015, 282, 3899–3917. [Google Scholar] [CrossRef]
- Siddiqui, K.S. Defying the activity-stability trade-off in enzymes: Taking advantage of entropy to enhance activity and thermostability. Crit. Rev. Biotechnol. 2017, 37, 309–322. [Google Scholar] [CrossRef]
- He, L.C.; Sharpe, T.; Mazur, A.; Hiller, S. A molecular mechanism of chaperone-client recognition. Sci. Adv. 2016, 2. [Google Scholar] [CrossRef]
- Wen, Y.; Lin, D.-H. Protein dynamics studied by NMR spin relaxation. Am. Chem. Soc. 2012, 29, 288–306. [Google Scholar]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of alpha-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Weikl, T.; Bugl, H.; Buchner, J. Chaperone function of Hsp90-associated proteins. Science 1996, 274, 1715–1717. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.C.; Toft, D.O.; Morimoto, R.I. Molecular chaperone machines: Chaperone activities of the cyclophilin Cyp-40 and the steroid aporeceptor-associated protein p23. Science 1996, 274, 1718–1720. [Google Scholar] [CrossRef] [PubMed]
- Pirkl, F.; Buchner, J. Functional analysis of the Hsp90-associated human peptidyl prolyl cis/trans isomerases FKBP51, FKBP52 and Cyp40. J. Mol. Biol. 2001, 308, 795–806. [Google Scholar] [CrossRef]
- Shelton, L.B.; Baker, J.D.; Zheng, D.L.; Sullivan, L.E.; Solanki, P.K.; Webster, J.M.; Sun, Z.Y.; Sabbagh, J.J.; Nordhues, B.A.; Koren, J.; et al. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. USA 2017, 114, 9707–9712. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Keller, R.L.J. The Computer Aided Resonance Assignment Tutorial; Cantina Verlag: Goldau, Switzerland, 2004. [Google Scholar]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. Talos plus: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Ottiger, M.; Delaglio, F.; Bax, A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reason. 1998, 131, 373–378. [Google Scholar] [CrossRef]
- Bryce, D.L.; Bax, A. Application of correlated residual dipolar couplings to the determination of the molecular alignment tensor magnitude of oriented proteins and nucleic acids. J. Biomol. NMR 2004, 29, 219. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.S.; Ying, J.F.; Bax, A. Improved accuracy of N-15-H-1 scalar and residual dipolar couplings from gradient-enhanced IPAP-HSQC experiments on protonated proteins. J. Biomol. NMR 2009, 43, 161–170. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Kneller, D.G.; Kuntz, I.D. Ucsf Sparky—An Nmr display, annotation and assignment tool. J. Cell Biochem. 1993, 53, 254. [Google Scholar]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M.Y. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aha128−162 | Aha128−335 | ||
|---|---|---|---|
| NOE Distance Restraints | |||
| Total NOE | 2210 | 6180 | |
| Intra-residual | 728 | 1449 | |
| Sequential (|i-j| =1) | 467 | 1375 | |
| Medium range (2≤ |i-j| ≤4) | 425 | 1061 | |
| Long range (|i-j| >4) | 590 | 2295 | |
| Hydrogen Bonds | 143 | 136 | |
| RDCs | 129 | 248 | |
| Dihedral Angle Restraints | |||
| Total | 238 | 452 | |
| Φ | 119 | 226 | |
| Ψ | 119 | 226 | |
| Ramachandran Statistics | |||
| Most favored region (%) | 81.9 | 81.5 | |
| Additionally allowed region (%) | 14.1 | 16.5 | |
| Generously allowed region (%) | 3.7 | 1.8 | |
| Disallowed region (%) | 0.3 | 0.2 | |
| RMSD from Mean Structure (Å) | S33-T160 | S33-T160 | C207-F331 |
| Backbone atoms | 0.62 ± 0.11 | 0.67 ± 0.14 | 0.16 ± 0.04 |
| Heavy atoms. | 1.41 ± 0.23 | 1.55 ± 0.24 | 0.78 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Wang, Q.; Du, J.; Liu, Z.; Ding, Y.; Xue, H.; Zhou, C.; Feng, L.; Zhang, N. Aha1 Exhibits Distinctive Dynamics Behavior and Chaperone-Like Activity. Molecules 2021, 26, 1943. https://doi.org/10.3390/molecules26071943
Hu H, Wang Q, Du J, Liu Z, Ding Y, Xue H, Zhou C, Feng L, Zhang N. Aha1 Exhibits Distinctive Dynamics Behavior and Chaperone-Like Activity. Molecules. 2021; 26(7):1943. https://doi.org/10.3390/molecules26071943
Chicago/Turabian StyleHu, Huifang, Qing Wang, Jingwen Du, Zhijun Liu, Yiluan Ding, Hongjuan Xue, Chen Zhou, Linyin Feng, and Naixia Zhang. 2021. "Aha1 Exhibits Distinctive Dynamics Behavior and Chaperone-Like Activity" Molecules 26, no. 7: 1943. https://doi.org/10.3390/molecules26071943
APA StyleHu, H., Wang, Q., Du, J., Liu, Z., Ding, Y., Xue, H., Zhou, C., Feng, L., & Zhang, N. (2021). Aha1 Exhibits Distinctive Dynamics Behavior and Chaperone-Like Activity. Molecules, 26(7), 1943. https://doi.org/10.3390/molecules26071943