
Na+/K+-ATPase Revisited: On Its Mechanism of Action, Role in Cancer, and Activity Modulation



Abstract

1. Introduction
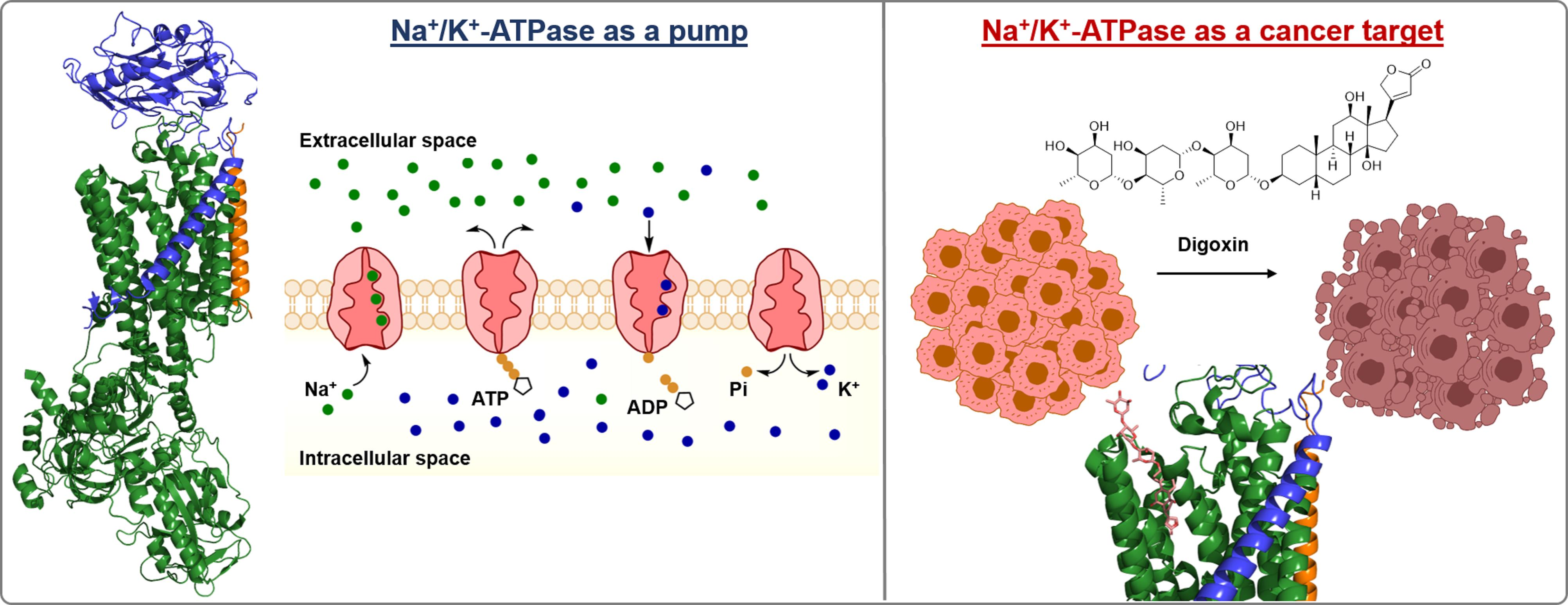
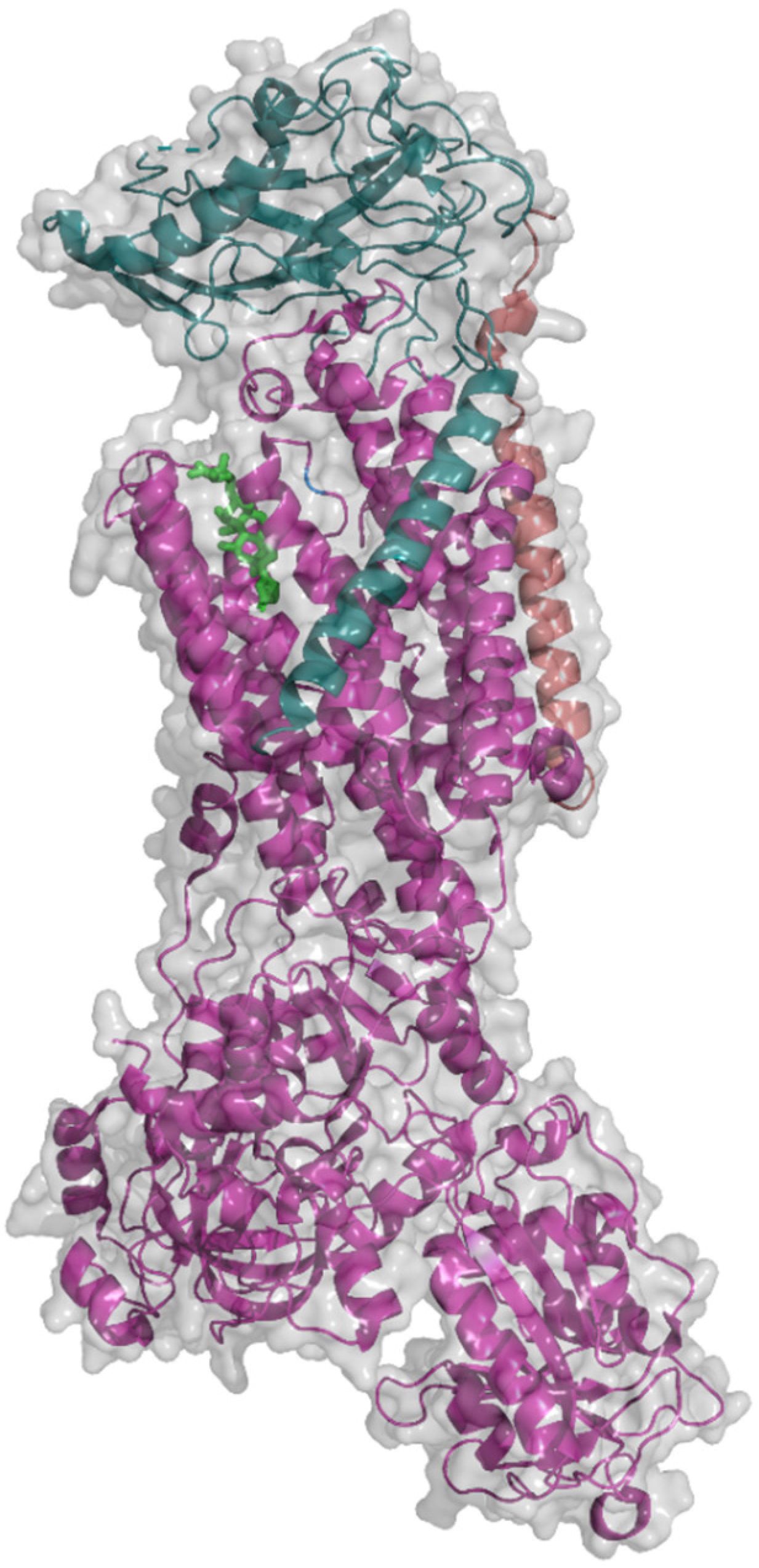
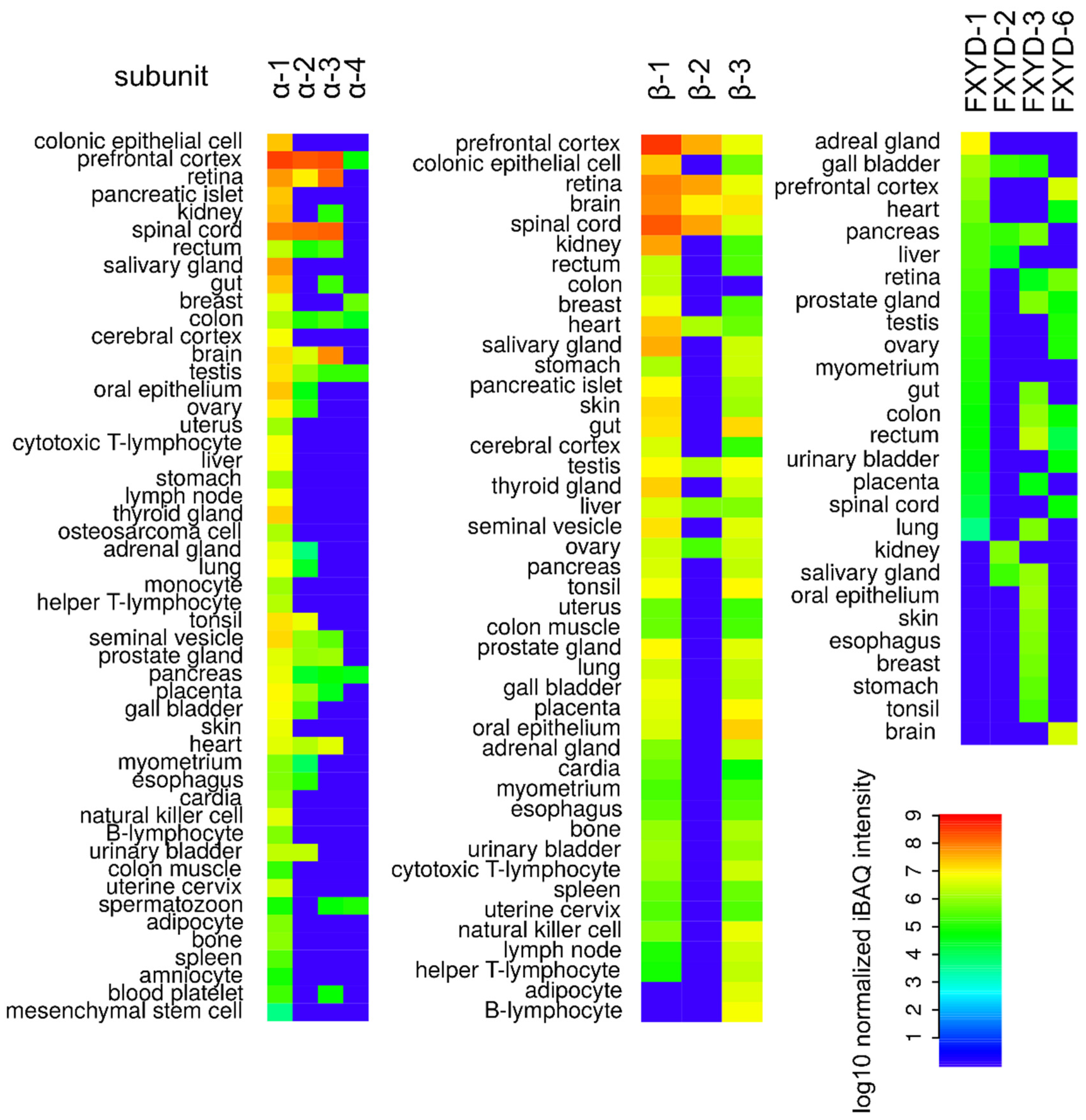
2. Na+/K+-ATPase Structure
3. Catalytic Cycle of Na+/K+-ATPase
- In the E1 state with bound ATP, the Na+ binding site in NKA is opened to the intracellular space and NKA has a high affinity for Na+ in this state.
- NKA phosphorylation occurs only when all Na+ binding sites are occupied since binding of the third Na+ ion causes a conformational change in the transmembrane domain, which is subsequently transferred to the nucleotide-binding domain [72].
- Then, after NKA phosphorylation, another conformational change takes place leading to the opening of the NKA cavity to the extracellular space, i.e., to the E2 state, and a release of Na+ [73].
4. Na+/K+-ATPase Functions and Anticancer Potential of Cardiac Steroids
5. Regulation of Na+/K+-ATPase Activity
5.1. Exogenous NKA Modulators
5.2. Endogenous NKA Modulators
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABL1 | Abelson tyrosine-protein kinase 1 |
| Akt | Protein kinase B |
| ATP | Adenosine triphosphate |
| BAD | Bcl2 associated agonist of cell death |
| Bcl-XL | B-cell lymphoma-extra large |
| cAMP | Cyclic adenosine monophosphate |
| cGMP | Cyclic guanosine monophosphate |
| CSs | Cardiac steroids |
| EPAC | Exchange of protein directly activated by cAMP |
| FAK | Focal adhesion kinase |
| GTP | Guanosine triphosphate |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| IC50 | Half maximal inhibitory concentration |
| IP3 | 1,4,5-Triphosphate |
| K-Ras | Kirsten sarcoma virus protein |
| MAPK | p38-mitogen-activated protein kinase |
| mTOR | Mammalian target of rapamycin |
| NKA | Sodium-potassium-ATPase |
| p21cip1 | Cyclin-dependent kinase inhibitor 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PGE2 | Prostaglandin E2 |
| ROS | Reactive oxygen species |
| SERCA | Sarco-/endoplasmic reticular Ca2+-ATPase |
| siRNA | Small interfering ribonucleic acid |
| SrcK | Non-receptor tyrosine kinase |
| VEGF | Vascular endothelial growth factor |
| VRAC | Volume-regulated anion channels |
References
- Castagnetti, F.; Gugliotta, G.; Breccia, M.; Stagno, F.; Iurlo, A.; Albano, F.; Abruzzese, E.; Martino, B.; Levato, L.; Intermesoli, T.; et al. Long-term outcome of chronic myeloid leukemia patients treated frontline with imatinib. Leukemia 2015, 29, 1823–1831. [Google Scholar] [CrossRef]
- Zhang, H.; Berel, D.; Wang, Y.; Li, P.; Bhowmick, N.A.; Figlin, R.A.; Kim, H.L. A comparison of Ku0063794, a dual mTORC1 and mTORC2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PLoS ONE 2013, 8, e54918. [Google Scholar] [CrossRef]
- FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-pi3k-inhibitor-breast-cancer (accessed on 13 October 2020).
- McCarthy, M.J.; Pagba, C.V.; Prakash, P.; Naji, A.K.; van der Hoeven, D.; Liang, H.; Gupta, A.K.; Zhou, Y.; Cho, K.J.; Hancock, J.F. Discovery of high-affinity noncovalent allosteric KRAS inhibitors that disrupt effector binding. ACS Omega 2019, 4, 2921–2930. [Google Scholar] [CrossRef]
- Guimarães, I.S.; Daltoé, R.D.; Herlinger, A.L.; Madeira, K.P.; Ladislau, T.; Valadão, J.C.; Lyra, P.C.M., Jr.; Teixeira, S.F.; Amorim, G.M.; dos Santos, D.Z.; et al. Conventional cancer treatment. In Cancer Treatment—Conventional and Innovative Approaches; Rangel, L., Ed.; IntechOpen: London, UK, 2013; pp. 3–35. [Google Scholar] [CrossRef]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.P.; De Giorgis, D.; Basilio, D.; Gadsby, D.C.; Rosenthal, J.J.C.; Latorre, R.; Holmgren, M.; Bezanilla, F. Energy landscape of the reactions governing the Na+ deeply occluded state of the Na+/K+-ATPase in the giant axon of the humboldt squid. Proc. Natl. Acad. Sci. USA 2011, 108, 20556–20561. [Google Scholar] [CrossRef]
- Blom, H.; Bernhem, K.; Brismara, H. Sodium pump organization in dendritic spines. Neurophotonics 2016, 3, 041803. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ferrer-Martinez, A.; Casado, F.J.; Felipe, A.; Pastor-Anglada, M. Regulation of Na+,K(+)-ATPase and the Na+/K+/Cl- co-transporter in the renal epithelial cell line NBL-1 ender osmotic stress. Biochem. J. 1996, 319, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Wu, W.Y.; Tsai, S.C.; Yoshinaga, T.; Lee, T.H. Elevated Na+/K+-ATPase responses and its potential role in triggering ion reabsorption in kidneys for homeostasis of marine euryhaline milkfish (Chanos chanos) when acclimated to hypotonic fresh water. J. Comp. Physiol. B 2010, 180, 813–824. [Google Scholar] [CrossRef]
- Wong, M.K.S.; Pipil, S.; Ozaki, H.; Suzuki, Y.; Iwasaki, W.; Takei, Y. Flexible selection of diversified Na+/K+-ATPase α-subunit isoforms for osmoregulation in teleosts. Zool. Lett. 2016, 2, 15. [Google Scholar] [CrossRef]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J.C. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2021, 276, 46605–46611. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jiang, P.; Li, J.; Xie, Z.; Xu, Y.; Qu, W.; Feng, F.; Liu, W. Periplocin promotes wound healing through the activation of Src/ERK and PI3K/Akt pathways mediated by Na/K-ATPase. Phytomedicine 2019, 57, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Prassas, I.; Karagiannis, G.S.; Batruch, I.; Dimitromanolakis, A.; Datti, A.; Diamandis, E.P. Digitoxin-induced cytotoxicity in cancer cells is mediated through distinct kinase and interferon signaling networks. Mol. Cancer Ther. 2011, 10, 2083–2093. [Google Scholar] [CrossRef]
- Khajah, M.A.; Mathew, P.M.; Luqmani, Y.A. Na+/K+ ATPase activity promotes invasion of endocrine resistant breast cancer cells. PLoS ONE 2018, 13, e0193779. [Google Scholar] [CrossRef]
- Edwards, I.A.; Bruce, G.; Lawrenson, C.; Howe, L.; Clapcote, S.J.; Deuchars, S.A.; Deuchars, J. Na+/K+ ATPase α1 and α3 isoforms are differentially expressed in α- and γ-motoneurons. J. Neurosci. 2013, 33, 9913–9919. [Google Scholar] [CrossRef]
- Radzyukevich, T.L.; Neumann, J.C.; Rindler, T.N.; Oshiro, N.; Goldhamer, D.J.; Lingrel, J.B.; Heiny, J.A. Tissue-specific role of the Na,K-ATPase α2 isozyme in skeletal muscle. J. Biol. Chem. 2013, 288, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.; Nguyen, A.N.T.; Timmerberg, B.; Tash, J.S.; Blanco, G. The Na,K-ATPase alpha4 isoform from humans has distinct enzymatic properties and is important for sperm motility. Mol. Hum. Reprod. 2006, 12, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Hundal, H.S.; Marette, A.; Ramlal, T.; Liu, Z.; Klip, A. Expression of beta subunit isoforms of the Na+,K(+)-ATPase is muscle type-specific. FEBS Lett. 1993, 328, 253–258. [Google Scholar] [CrossRef]
- Arystarkhova, E.; Sweadner, K.J. Tissue-specific expression of the Na,K-ATPase beta3 subunit. The presence of beta3 in lung and liver addresses the problem of the missing subunit. J. Biol. Chem. 1997, 272, 22405–22408. [Google Scholar] [CrossRef]
- PDB. Available online: https://www.rcsb.org/structure/3A3Y (accessed on 2 February 2021).
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/endoplasmic reticulum calcium ATPase inhibitors: Beyond anticancer perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef]
- Morth, J.P.; Pedersen, B.P.; Toustrup-Jensen, M.S.; Sørensen, T.L.; Petersen, J.; Andersen, J.P.; Vilsen, B.; Nissen, P. Crystal structure of the sodium-potassium pump. Nature 2007, 450, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Hilbers, F.; Kopec, W.; Isaksen, T.J.; Holm, T.H.; Lykke-Hartmann, K.; Nissen, P.; Khandelia, H.; Poulsen, H. Tuning of the Na,K-ATPase by the beta subunit. Sci. Rep. 2016, 6, 20442. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Gopal, J.; Willis, D.; Espineda, C.; Twiss, J.L.; Rajasekaran, A.K. Na,K-ATPase β1-subunit increases the translation efficiency of the α1-Subunit in MSV-MDCK cells. Mol. Biol. Cell 2004, 15, 3005–3508. [Google Scholar] [CrossRef]
- Miller, R.P.; Farley, R.A. All three potential N-glycosylation sites of the dog kidney (Na+ + K+)-ATPase beta-subunit contain oligosaccharide. Biochim. Biophys. Acta 1988, 954, 50–57. [Google Scholar] [CrossRef]
- Beggah, A.T.; Jaunin, P.; Geering, K. Role of glycosylation and disulfide bond formation in the beta subunit in the folding and functional expression of Na,K-ATPase. J. Biol. Chem. 1997, 272, 10318–10326. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, M.; Matsumoto, K.; Iwasaki, N.; Hayashi, Y.; Yamaguchi, Y. Structural analysis of N-glycans attached to pig kidney Na+/K+-ATPase. J. Glyc. Lip. 2013, S5, 005. [Google Scholar] [CrossRef]
- Tokhtaeva, E.; Munson, K.; Sachs, G.; Vagin, O. N-Glycan-dependent quality control of the Na,K-ATPase β2 subunit. Biochemistry 2010, 49, 3116–3128. [Google Scholar] [CrossRef][Green Version]
- Béguin, P.; Hasler, U.; Staub, O.; Geering, K. Endoplasmic reticulum quality control of oligomeric membrane proteins: Topogenic determinants involved in the degradation of the unassembled Na,K-ATPase alpha subunit and in its stabilization by beta subunit assembly. Mol. Biol. Cell. 2000, 11, 1657–1672. [Google Scholar] [CrossRef]
- Tokhtaeva, E.; Sachs, G.; Vagin, O. Assembly with the Na,K-ATPase α1 subunit is required for export of β1 and β2 subunits from the endoplasmic reticulum. Biochemistry 2009, 48, 11421–11431. [Google Scholar] [CrossRef]
- Lian, W.N.; Wu, T.W.; Dao, R.L.; Chen, Y.J.; Lin, C.H. Deglycosylation of Na+/K+-ATPase causes the basolateral protein to undergo apical targeting in polarized hepatic cells. J. Cell Sci. 2006, 119, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Vagin, O.; Tokhtaeva, E.; Sachs, G. The role of the β1 subunit of the Na,K-ATPase and its glycosylation in cell-cell adhesion. J. Cell. Biol. 2006, 281, 39573–39587. [Google Scholar] [CrossRef]
- Wang, P.J.; Lin, C.H.; Hwang, H.H.; Lee, T.H. Branchial FXYD protein expression in response to salinity change and its interaction with Na+/K+-ATPase of the euryhaline teleost Tetraodon nigroviridis. J. Exp. Biol. 2008, 211, 3750–3758. [Google Scholar] [CrossRef][Green Version]
- Wang, L.J.; Li, Q.J.; Le, Y.; Ouyang, H.Y.; He, M.K.; Yu, Z.S.; Zhang, Y.F.; Shi, M. Prognostic significance of sodium-potassium ATPase regulator, FXYD3, in human hepatocellular. Carcinoma Oncol. Lett. 2018, 15, 3024–3030. [Google Scholar] [CrossRef]
- Widegren, E.; Önnesjö, S.; Arbman, G.; Kayed, H.; Zentgraf, H.; Kleeff, J.; Zhang, H.; Sun, X.-F. Expression of FXYD3 protein in relation to biological and clinicopathological variables in colorectal cancers. Chemotherapy 2009, 55, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pang, S.T.; Kasper, K.A.; Luan, C.; Wondergem, B.; Lin, F.; Chuang, C.K.; Teh, B.T.; Yang, X.J. FXYD3: A promising biomarker for urothelial carcinoma. Biomark Insights 2011, 6, 17–26. [Google Scholar] [CrossRef]
- Herrmann, P.; Aronica, S.M. Estrogen and tamoxifen up-regulate FXYD3 on breast cancer cells: Assessing the differential roles of ER α and ZEB1. Springerplus 2015, 4, 245. [Google Scholar] [CrossRef] [PubMed]
- Kayed, H.; Kleeff, J.; Kolb, A.; Ketterer, K.; Keleg, S.; Felix, K.; Giese, T.; Penzel, R.; Zentgraf, H.; Büchler, M.W.; et al. FXYD3 is overexpressed in pancreatic ductal adenocarcinoma and influences pancreatic cancer cell growth. Int. J. Cancer 2006, 118, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Uniprot. Available online: https://www.uniprot.org/uniprot/O00168 (accessed on 10 October 2020).
- Uniprot. Available online: https://www.uniprot.org/uniprot/P54710 (accessed on 10 October 2020).
- Uniprot. Available online: https://www.uniprot.org/uniprot/Q14802-5 (accessed on 10 October 2020).
- Uniprot. Available online: https://www.uniprot.org/uniprot/P59646 (accessed on 10 October 2020).
- Uniprot. Available online: https://www.uniprot.org/uniprot/Q96DB9 (accessed on 10 October 2020).
- Uniprot. Available online: https://www.uniprot.org/uniprot/Q9H0Q3 (accessed on 10 October 2020).
- Uniprot. Available online: https://www.uniprot.org/uniprot/P58549 (accessed on 10 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#human/proteinDetails/P05023/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/56274/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/52946/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/59668/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/51770/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/53052/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/56697/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/47759/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/56698/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/60188/expression (accessed on 6 October 2020).
- Proteomics DB. Available online: https://www.proteomicsdb.org/proteomicsdb/#protein/proteinDetails/80311/expression (accessed on 6 October 2020).
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Mishra, N.K.; Peleg, Y.; Cirri, E.; Belogus, T.; Lifshitz, Y.; Voelker, D.R.; Apell, H.J.; Garty, H.; Karlish, S.J.D. FXYD proteins stabilize Na,K-ATPase: Amplification of specific phosphatidylserine-protein interactions. J. Biol. Chem. 2011, 286, 9699–9712. [Google Scholar] [CrossRef] [PubMed]
- Tokhtaeva, E.; Sun, H.; Deiss-Yehiely, N.; Wen, Y.; Soni, P.N.; Gabrielli, N.M.; Marcus, E.A.; Ridge, K.M.; Sachs, G.; Vazquez-Levin, M.; et al. The O-glycosylated ectodomain of FXYD5 impairs adhesion by disrupting cell-cell trans-dimerization of Na,K-ATPase β1 subunits. J. Cell Sci. 2016, 129, 2394–2406. [Google Scholar] [CrossRef] [PubMed]
- Cirri, E.; Katz, A.; Mishra, N.K.; Belogus, T.; Lifshitz, Y.; Garty, H.; Karlish, S.J.D.; Apell, H.J. Phospholemman (FXYD1) raises the affinity of the human α1β1 isoform of Na,K-ATPase for Na ions. Biochemistry 2011, 50, 3736–3748. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Despa, S.; Bossuyt, J.; Han, F.; Ginsburg, K.S.; Jia, L.G.; Kutchai, H.; Tucker, A.L.; Bers, D.M. Phospholemman-phosphorylation mediates the β-adrenergic effects on Na/K pump function in cardiac myocytes. Circ. Res. 2005, 97, 252–259. [Google Scholar] [CrossRef]
- Silva, E.C.C.; Masui, D.C.; Furriel, R.P.; McNamara, J.C.; Barrabina, H.; Scofano, H.M.; Perales, J.; Teixeira-Ferreira, A.; Leone, F.A.; Fontesa, C.F.L. Identification of a crab gill FXYD2 protein and regulation of crab microsomal Na,K-ATPase activity by mammalian FXYD2 peptide. Biochim. Biophys. Acta-Biomembr. 2012, 1818, 2588–2597. [Google Scholar] [CrossRef] [PubMed]
- Bibert, S.; Aebischer, D.; Desgranges, F.; Roy, S.; Schaer, D.; Kharoubi-Hess, S.; Horisberger, J.D.; Geering, K. A link between FXYD3 (Mat-8)-mediated Na,K-ATPase regulation and differentiation of Caco-2 intestinal epithelial cells. Mol. Biol. Cell 2009, 20, 1132–1140. [Google Scholar] [CrossRef]
- Béguin, P.; Crambert, G.; Guennoun, S.; Garty, H.; Horisberger, J.D. CHIF, a member of the FXYD protein family, is a regulator of Na,K-ATPase distinct from the gamma-subunit. EMBO J. 2001, 20, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.J.; Davis, P.B. FXYD5 modulates Na+ absorption and is increased in cystic fibrosis airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L654–L664. [Google Scholar] [CrossRef]
- Delprat, B.; Schaer, D.; Roy, S.; Wang, J.; Puel, J.L.; Geering, K. FXYD6 is a novel regulator of Na,K-ATPase expressed in the inner ear. J. Biol. Chem. 2007, 282, 7450–7456. [Google Scholar] [CrossRef]
- Béguin, P.; Crambert, G.; Monnet-Tschudi, F.; Uldry, M.; Horisberger, J.D.; Garty, H.; Geering, K. FXYD7 is a brain-specific regulator of Na,K-ATPase alpha 1-beta isozymes. EMBO J. 2002, 21, 3264–3273. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, J.L.; Mattle, D.; Fedosova, N.U.; Nissena, P.; Reinhard, L. Isolation, crystallization and crystal structure determination of bovine kidney Na+,K+-ATPase. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72, 282–287. [Google Scholar] [CrossRef]
- Sørensen, L.M.T.; Møller, J.V.; Nissen, P. Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science 2004, 304, 1672–1675. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, A.; Apell, H.J. Ion selectivity of the cytoplasmic binding sites of the Na,K-ATPase: I. sodium binding is associated with a conformational rearrangement. J. Membr. Biol. 1999, 168, 221–228. [Google Scholar] [CrossRef]
- Stolz, M.; Lewitzki, E.; Bergbauer, R.; Mäntele, W.; Grell, E.; Barth, A. Structural changes in the catalytic cycle of the Na+,K+-ATPase studied by infrared spectroscopy. Biophys. J. 2009, 96, 3433–3442. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Castillo, J.P.; Rui, H.; Basilio, D.; Das, A.; Roux, B.; Latorre, R.; Bezanilla, F.; Holmgren, M. Mechanism of potassium ion uptake by the Na+/K+-ATPase. Nat. Commun. 2015, 6, 7622. [Google Scholar] [CrossRef]
- Leone, F.A.; Furriel, R.P.M.; McNamara, J.C.; Horisberger, J.D.; Borin, I.A. Cation transport coupled to ATP hydrolysis by the (Na, K)-ATPase: An integrated, animated model. Biochem. Mol. Biol. Educ. 2010, 38, 276–279. [Google Scholar] [CrossRef]
- Rui, H.; Artigas, P.; Roux, B. The selectivity of the Na+/K+-pump is controlled by binding site protonation and self-correcting occlusion. eLife 2016, 5, e16616. [Google Scholar] [CrossRef] [PubMed]
- Grycova, L.; Sklenovsky, P.; Lansky, Z.; Janovska, M.; Otyepka, M.; Amlerd, E.; Teisinger, J.; Kubalac, M. ATP and magnesium drive conformational changes of the Na+/K+-ATPase cytoplasmic headpiece. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 1081–1091. [Google Scholar] [CrossRef]
- Tejral, G.; Sopko, B.; Necas, A.; Schoner, W.; Amler, E. Computer modelling reveals new conformers of the ATP binding loop of Na+/K+-ATPase involved in the transphosphorylation process of the sodium pump. PeerJ 2017, 5, e3087. [Google Scholar] [CrossRef] [PubMed]
- Razavi, A.M.; Delemotte, L.; Berlin, J.R.; Carnevale, V.; Voelz, V.A. Molecular simulations and free-energy calculations suggest conformation-dependent anion binding to a cytoplasmic site as a mechanism for Na+/K+-ATPase ion selectivity. J. Biol. Chem. 2017, 292, 12412–12423. [Google Scholar] [CrossRef]
- Apell, H.J.; Benz, G.; Sauerbrunn, D. Proton diet for the sodium pump. Biochemistry 2011, 50, 409–418. [Google Scholar] [CrossRef][Green Version]
- el-Masri, M.A.; Clark, B.J.; Qazzaz, H.M.; Valdes, R. Human adrenal cells in culture produce both ouabain-like and dihydroouabain-like factors. Jr. Clin. Chem. 2002, 48, 1720–1730. [Google Scholar] [CrossRef]
- Gao, J.; Wymore, R.S.; Wang, Y.; Gaudette, G.R.; Krukenkamp, I.B.; Cohen, I.S.; Mathias, R.T. Isoform-specific stimulation of cardiac Na/K pumps by nanomolar concentrations of glycosides. J. Gen. Physiol. 2002, 119, 297–312. [Google Scholar] [CrossRef]
- Khundmiri, S.J.; Amin, V.; Henson, J.; Lewis, J.; Ameen, M.; Rane, M.J.; Delamere, N.A. Ouabain stimulates protein kinase B (Akt) phosphorylation in opossum kidney proximal tubule cells through an ERK-dependent pathway. Am. J. Physiol. Cell Physiol. 2007, 293, C1171–C1180. [Google Scholar] [CrossRef]
- Khundmiri, S.J.; Metzler, M.A.; Ameen, M.; Amin, V.; Rane, M.J.; Delamere, N.A. Ouabain induces cell proliferation through calcium-dependent phosphorylation of Akt (protein kinase B) in opossum kidney proximal tubule cells. Am. J. Physiol. Cell Physiol. 2006, 291, C1247–C1257. [Google Scholar] [CrossRef]
- Manunta, P.; Messaggio, E.; Ballabeni, C.; Sciarrone, M.T.; Lanzani, C.; Ferrandi, M.; Hamlyn, J.M.; Cusi, D.; Galletti, F.; Bianchi, G. Salt sensitivity study group of the Italian society of hypertension. Plasma ouabain-like factor during acute and chronic changes in sodium balance in essential hypertension. Hypertension 2001, 38, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Stella, P.; Rivera, R.; Ciurlino, D.; Cusi, D.; Ferrandi, M.; Hamlyn, J.M.; Bianchi, G. Left ventricular mass, stroke volume, and ouabain-like factor in essential hypertension. Hypertension 1999, 34, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Pierdomenico, S.D.; Bucci, A.; Manunta, P.; Rivera, R.; Ferrandi, M.; Hamlyn, J.M.; Lapenna, D.; Cuccurullo, F.; Mezzetti, A. Endogenous ouabain and hemodynamic and left ventricular geometric patterns in essential hypertension. Am. J. Hyper. 2001, 14, 44–50. [Google Scholar] [CrossRef]
- Wang, H.; Haas, M.; Liang, M.; Cai, T.; Tian, J.; Li, S.; Xie, Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J. Biol. Chem. 2004, 279, 17250–17259. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.J. Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.N.T.; Jansson, K.; Sánchez, G.; Sharma, M.; Reif, G.A.; Blanco, G. Ouabain activates the Na-K-ATPase signalosome to induce autosomal dominant polycystic kidney disease cell proliferation. Am. J. Physiol. Renal. Physiol. 2011, 301, F897–F906. [Google Scholar] [CrossRef]
- Tverskoi, A.M.; Sidorenko, S.V.; Klimanova, E.A.; Akimova, O.A.; Smolyaninova, L.V.; Lopina, O.D.; Orlov, S.N. Effects of ouabain on proliferation of human endothelial cells correlate with Na,K-ATPase activity and intracellular ratio of Na and K. Biochemistry 2016, 81, 876–883. [Google Scholar] [CrossRef]
- Banerjee, G.M.; Cui, X.; Li, Z.; Yu, H.; Cai, L.; Jia, X.; He, D.; Wang, C.; Gao, T.; Xie, Z. Na/K-ATPase Y260 phosphorylation–mediated Src regulation in control of aerobic glycolysis and tumor. Sci. Rep. 2018, 8, 12322. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Nichols, A.; Mallick, A.; Klug, R.L.; Liu, J.; Wang, X.; Srikanthan, K.; Goguet-Rubio, P.; Nawab, A.; Pratt, R.; et al. The Na/K-ATPase oxidant amplifcation loop regulates aging. Sci. Rep. 2018, 8, 9721. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Srikanthan, K.; Goguet-Rubio, P.; Nichols, A.; Mallick, A.; Nawab, A.; Martin, R.; Shah, P.T.; Chaudhry, M.; Sigdel, S.; et al. pNaKtide attenuates steatohepatitis and atherosclerosis by blocking Na/K-ATPase/ROS amplification in C57Bl6 and ApoE knockout mice fed a western diet. Sci. Rep. 2017, 7, 193. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, Q.; Liu, C.; Xie, J.X.; Yan, Y.; Lai, F.; Duan, Q.; Li, X.; Tian, J.; Xie, Z. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 2014, 71, 415–426. [Google Scholar] [CrossRef]
- Yan, Y.; Shapiro, A.P.; Mopidevi, B.R.; Chaudhry, M.A.; Maxwell, K.; Haller, S.T.; Drummond, C.A.; Kennedy, D.J.; Tian, J.; Malhotra, D.; et al. Protein carbonylation of an amino acid residue of the Na/K-ATPase α1 subunit determines Na/K-ATPase signaling and sodium transport in renal proximal tubular cells. J. Am. Heart Assoc. 2016, 5, e003675. [Google Scholar] [CrossRef]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, D.; Xie, Z.; Abraham, N.G.; et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef]
- Yu, H.; Cui, X.; Zhang, J.; Xie, J.X.; Banerjee, M.; Pierre, S.V.; Xie, Z. Heterogeneity of signal transduction by Na-K-ATPase α-isoforms: Role of Src interaction. Am. J. Physiol. Cell Physiol. 2018, 314, C202–C210. [Google Scholar] [CrossRef]
- Miyakawa-Naito, A.; Uhlén, P.; Lal, M.; Aizman, O.; Mikoshiba, K.; Brismar, H.; Zelenin, S.; Aperia, A. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J. Biol. Chem. 2003, 278, 50355–50361. [Google Scholar] [CrossRef]
- Yuan, Z.; Cai, T.; Tian, J.; Ivanov, A.V.; Giovannucci, D.R.; Xie, Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol. Biol. Cell 2005, 16, 4034–4045. [Google Scholar] [CrossRef]
- Burlaka, I.; Liu, X.L.; Rebetz, J.; Arvidsson, I.; Yang, L.; Brismar, H.; Karpman, D.; Aperia, A. Ouabain protects against shiga toxin–triggered apoptosis by reversing the imbalance between Bax and Bcl-xL. J. Am. Soc. Nephrol. 2013, 24, 1413–1423. [Google Scholar] [CrossRef]
- Panizza, E.; Zhang, L.; Fontana, J.M.; Hamada, K.; Svensson, D.; Akkuratov, E.E.; Scott, L.; Mikoshiba, K.; Brismar, H.; Lehtiö, J.; et al. Ouabain-regulated phosphoproteome reveals molecular mechanisms for Na+, K+-ATPase control of cell adhesion, proliferation, and survival. FASEB J. 2019, 33, 10193–10206. [Google Scholar] [CrossRef] [PubMed]
- Barwe, S.P.; Anilkumar, G.; Moon, S.Y.; Zheng, Y.; Whitelegge, J.P.; Rajasekaran, S.A.; Rajasekaran, A.K. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 2005, 16, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Vilchis-Nestor, C.A.; Roldán, M.L.; Leonardi, A.; Navea, J.G.; Padilla-Benavides, T.; Shoshani, L. Ouabain enhances cell-cell adhesion mediated by β 1 subunits of the Na+,K+-ATPase in CHO fibroblasts. Int. J. Mol. Sci. 2019, 20, 2111. [Google Scholar] [CrossRef] [PubMed]
- del Toro, A.O.; Jimenez, L.; Hinojosa, L.; Martínez-Rendón, J.; Castillo, A.; Cereijido, M.; Ponce, A. Influence of endogenous cardiac glycosides, digoxin, and marinobufagenin in the physiology of epithelial cells. Cardiol. Res. Pract. 2019, 2019, 8646787. [Google Scholar] [CrossRef]
- Verdejo-Torres, O.; Flores-Maldonado, C.; Padilla-Benavides, T.; Campos-Blázquez, J.P.; Larré, I.; Lara-Lemus, R.; Perez Salazar, E.; Cereijido, M.; Contreras, R.G. Ouabain accelerates collective cell migration through a cSrc and ERK1/2 sensitive metalloproteinase activity. J. Membr. Biol. 2019, 252, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Reuter, H.; Henderson, S.A.; Han, T.; Ross, R.S.; Goldhaber, J.I.; Philipson, K.D. The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ. Res. 2002, 90, 305–308. [Google Scholar] [CrossRef]
- Winnicka, K.; Bielawski, K.; Bielawska, A.; Miltyk, W. Dual effects of ouabain, digoxin and proscillaridin A on the regulation of apoptosis in human fibroblasts. Nat. Prod. Res. 2010, 24, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhang, Y.; Zhao, W.; Zhou, X.; Wang, C.; Deng, F. The cardiac glycoside oleandrin induces apoptosis in human colon cancer cells via the mitochondrial pathway. Cancer Chemother. Pharmacol. 2017, 80, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Kometiani, P.; Liu, L.; Askari, A. Digitalis-induced signaling by Na+/K+-ATPase in human breast cancer cells. Mol. Pharmacol. 2005, 67, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Chang, H.H.; Lai, Y.H.; Lin, C.H.; Chen, M.H.; Chang, G.C.; Tsai, M.F.; Chen, J.J.W. Digoxin suppresses tumor malignancy through inhibiting multiple Src-related signaling pathways in non-small cell lung cancer. PLoS ONE 2015, 10, e0123305. [Google Scholar] [CrossRef]
- Shin, H.K.; Ryu, B.J.; Choi, S.W.; Kim, S.H.; Le, K. Inactivation of Src-to-ezrin pathway: A possible mechanism in the ouabain-mediated inhibition of A549 cell migration. Biomed. Res. Int. 2015, 2015, 537136. [Google Scholar] [CrossRef]
- Pongrakhananon, V.; Chunhacha, P.; Chanvorachote, P. Ouabain suppresses the migratory behavior of lung cancer cells. PLoS ONE 2013, 8, e68623. [Google Scholar] [CrossRef]
- Kornberg, L.J.; Shaw, L.C.; Spoerri, P.E.; Caballero, S.; Grant, M.B. Focal adhesion kinase overexpression induces enhanced pathological retinal angiogenesis. Inv. Ophthalmol. Vis. Sci. 2004, 45, 4463–4469. [Google Scholar] [CrossRef]
- Cabrita, M.A.; Jones, L.M.; Quizi, J.L.; Sabourin, L.A.; McKay, B.C.; Addison, C.L. Focal adhesion kinase inhibitors are potent anti-angiogenic agents. Mol. Oncol. 2011, 5, 517–526. [Google Scholar] [CrossRef]
- Pedrosa, A.R.; Bodrug, N.; Gomez-Escudero, J.; Carter, E.P.; Reynolds, L.E.; Georgiou, P.N.; Fernandez, I.; Lees, D.M.; Kostourou, V.; Alexopoulou, A.N.; et al. Tumor angiogenesis is differentially regulated by phosphorylation of endothelial cell focal adhesion kinase tyrosines-397 and -861. Cancer Res. 2019, 79, 4371–4386. [Google Scholar] [CrossRef]
- Trenti, A.; Zulato, E.; Pasqualini, L.; Indraccolo, S.; Bolego, C.; Trevisi, L. Therapeutic concentrations of digitoxin inhibit endothelial focal adhesion kinase and angiogenesis induced by different growth factors. Br. J. Pharmacol. 2017, 174, 3094–3106. [Google Scholar] [CrossRef]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ. Res. 2003, 93, 664–667. [Google Scholar] [CrossRef]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.A.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef]
- Lee, D.H.; Oh, S.C.; Giles, A.J.; Jung, J.; Gilbert, M.R.; Park, D.M. Cardiac glycosides suppress the maintenance of stemness and malignancy via inhibiting HIF-1α in human glioma stem cells. Oncotarget 2017, 8, 40233–40245. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.S.; Xu, Z.W.; Yi, T.L.; Xu, R.C.; Li, J.; Zhang, W.B.; Zhang, S.; Sun, H.T.; Yu, Z.Q.; Xu, H.X.; et al. Ouabain suppresses the growth and migration abilities of glioma U-87MG cells through inhibiting the Akt/mTOR signaling pathway and downregulating the expression of HIF-1α. Mol. Med. Rep. 2018, 17, 5595–5600. [Google Scholar] [CrossRef]
- Fujii, T.; Shimizu, T.; Yamamoto, S.; Funayama, K.; Fujita, K.; Tabuchi, Y.; Ikari, A.; Takeshima, H.; Sakai, H. Crosstalk between Na+,K+-ATPase and a volume-regulated anion channel in membrane microdomains of human cancer cells. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 3792–3804. [Google Scholar] [CrossRef]
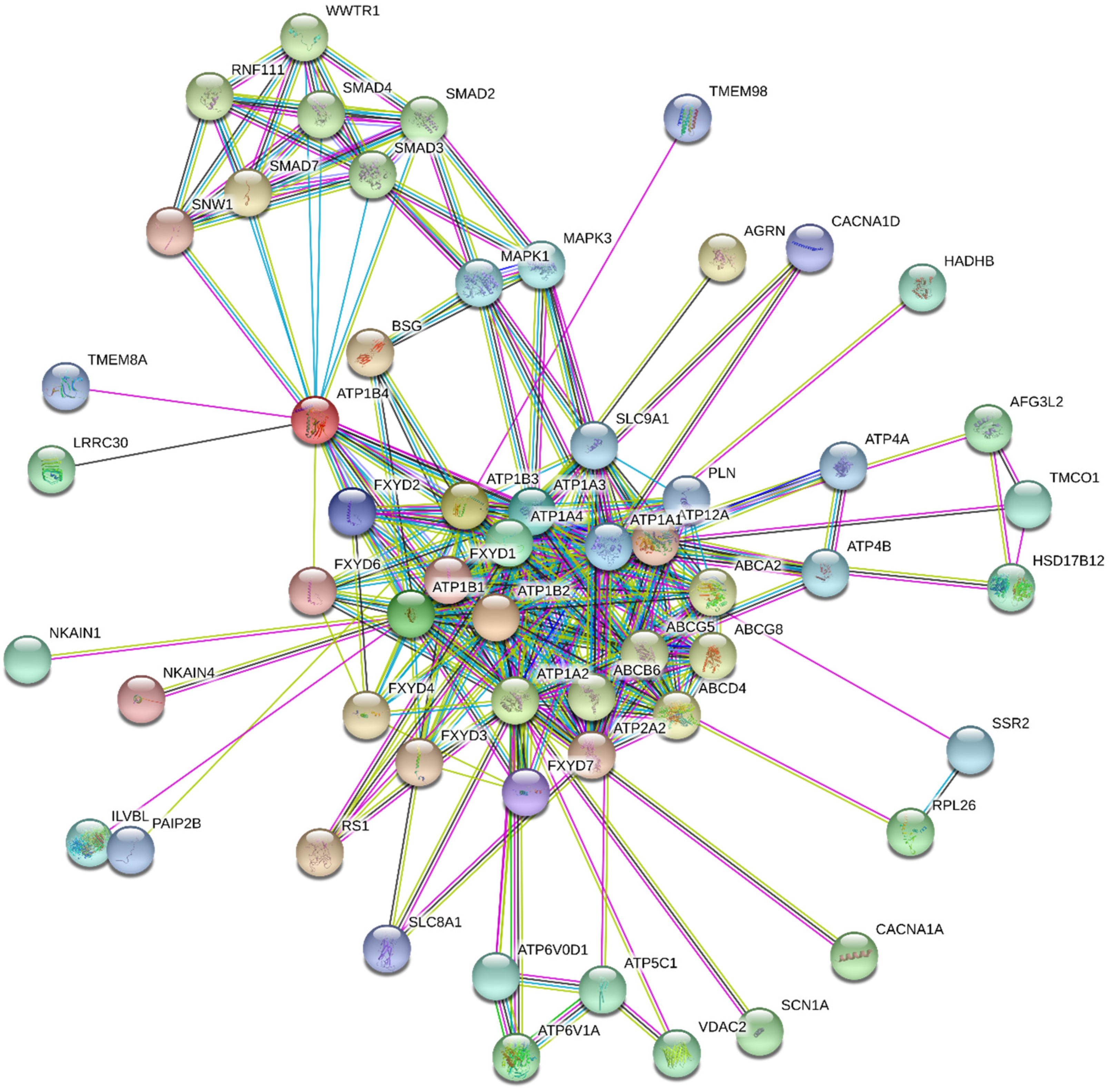
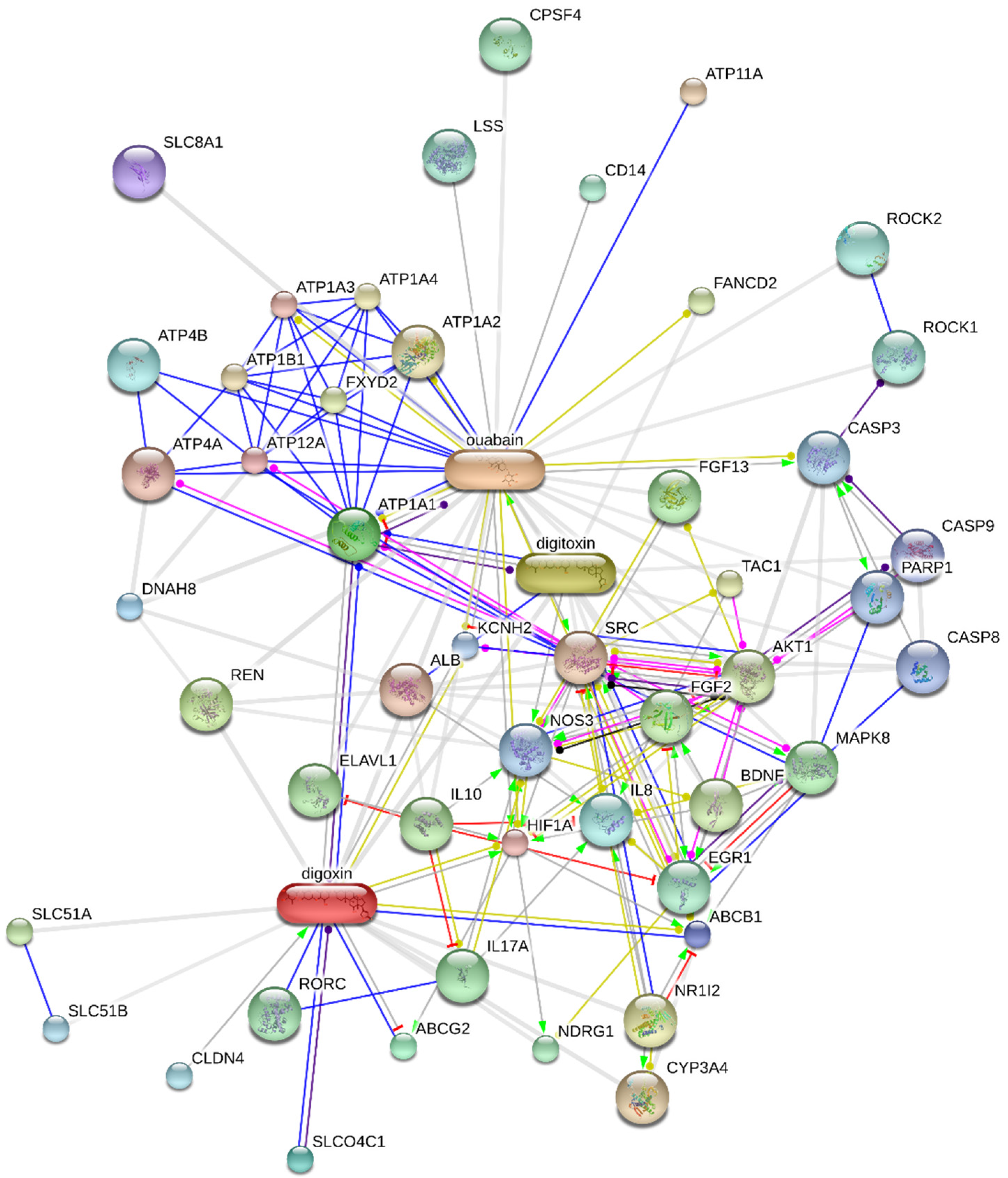
- STRING. Available online: http://string.embl.de/ (accessed on 14 August 2020).
- Laursen, M.; Gregersen, J.L.; Yatime, L.; Nissen, P.; Fedosova, N.U. Structures and characterization of digoxin- and bufalin-bound Na+,K+-ATPase compared with the ouabain-bound complex. Proc. Natl. Acad. Sci. USA 2015, 112, 1755–1760. [Google Scholar] [CrossRef]
- Laursen, M.; Yatime, L.; Nissen, P.; Fedosova, N.U. Crystal structure of the high-affinity Na+K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Proc. Natl. Acad. Sci. USA 2013, 110, 10958–10963. [Google Scholar] [CrossRef]
- Canfield, V.; Emanuel, J.R.; Spickofsky, N.; Levenson, R.; Margolskee, R. Ouabain-resistant mutants of the rat Na,K-ATPase x2 isoform identified by using an episomal expression vector. Mol. Cell. Biol. 1990, 10, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, W.J.; Wallick, E.T.; Lingrel, J.B. Amino acid residues of the Na,K-ATPase involved in ouabain sensitivity do not bind the sugar moiety of cardiac glycosides. J. Biol. Chem. 1993, 268, 7707–7712. [Google Scholar] [CrossRef]
- Croyle, M.L.; Woo, A.L.; Lingrel, J.B. Extensive random mutagenesis analysis of the Na+/K+-ATPase alpha subunit identifies known and previously unidentified amino acid residues that alter ouabain sensitivity—Implications for ouabain binding. Eur. J. Biochem. 1997, 248, 488–495. [Google Scholar] [CrossRef]
- Dalla, S.; Swarts, H.G.P.; Koenderink, J.B.; Dobler, S. Amino acid substitutions of Na,K-ATPase conferring decreased sensitivity to cardenolides in insects compared to mammals insect. Biochem. Mol. Biol. 2013, 43, 1109–1115. [Google Scholar] [CrossRef]
- STITCH. Available online: https://stitch-db.org/ (accessed on 18 August 2020).
- Cornelius, F.; Kanai, R.; Toyoshima, C. A structural view on the functional importance of the sugar moiety and steroid hydroxyls of cardiotonic steroids in binding to Na,K-ATPase. J. Biol. Chem. 2013, 288, 6602–6616. [Google Scholar] [CrossRef]
- Magpusao, A.N.; Omolloh, G.; Johnson, J.; Gascón, J.; Peczuh, M.W.; Fenteany, G. Cardiac glycoside activities link Na+/K+ ATPase ion-transport to breast cancer cell migration via correlative SAR. ACS Chem. Biol. 2015, 10, 561–569. [Google Scholar] [CrossRef]
- Manunta, P.; Hamilton, B.P.; Hamlyn, J.M. Structure-activity relationships for the hypertensinogenic activity of ouabain: Role of the sugar and lactone ring. Hypertension 2001, 37, 472–477. [Google Scholar] [CrossRef]
- Ren, Y.; Ribas, H.T.; Heath, K.; Wu, S.; Ren, J.; Shriwas, P.; Chen, X.; Johnson, M.E.; Cheng, X.; Burdette, J.E.; et al. Na+/K+-ATPase-targeted cytotoxicity of (+)-digoxin and several semisynthetic derivatives. J. Nat. Prod. 2020, 83, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.C.; Pessoa, M.T.C.; Neves, L.D.R.; Alves, S.L.G.; Silva, L.M.; Santos, H.L.; Oliveira, S.M.F.; Taranto, A.G.; Comar, M.; Gomes, I.V.; et al. 21-Benzylidene digoxin: A proapoptotic cardenolide of cancer cells that up-regulates Na,K-ATPase and epithelial tight junctions. PLoS ONE 2014, 9, e108776. [Google Scholar] [CrossRef]
- Alves, S.L.G.; Paixão, N.; Ferreira, L.G.R.; Santos, F.R.S.; Neves, L.D.R.; Oliveira, G.C.; Cortes, V.F.; Salomé, K.S.; Barison, A.; Santos, F.V.; et al. 9γ-Benzylidene digoxin derivatives synthesis and molecular modeling: Evaluation of anticancer and the Na,K-ATPase activity effect. Bioorg. Med. Chem. 2015, 23, 4397–4404. [Google Scholar] [CrossRef] [PubMed]
- Pessôa, M.T.C.; Alves, S.L.G.; Taranto, A.G.; Villar, J.A.F.P.; Blanco, G.; Barbosa, L.A. Selectivity analyses of γ-benzylidene digoxin derivatives to different Na,K-ATPase α isoforms: A molecular docking approach. J. Enzyme Inhib. Med. Chem. 2018, 33, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Syeda, S.S.; Sánchez, G.; Hong, K.H.; Hawkinson, J.E.; Georg, G.I.; Blanco, G. Design, synthesis, and in vitro and in vivo evaluation of ouabain analogues as potent and selective Na,K-ATPase α4 isoform inhibitors for male contraception. J. Med. Chem. 2018, 61, 1800–1820. [Google Scholar] [CrossRef]
- Iyer, A.K.V.; Zhou, M.; Azad, N.; Elbaz, H.; Wang, L.; Rogalsky, D.K.; Rojanasakul, Y.; O’Doherty, G.A.; Langenhan, J.M. A direct comparison of the anticancer activities of digitoxin MeON-neoglycosides and O-glycosides. ACS Med. Chem. Lett. 2010, 1, 326–330. [Google Scholar] [CrossRef]
- Elbaz, H.A.; Stueckle, T.A.; Wang, H.Y.L.; O’Doherty, G.A.; Lowry, D.T.; Sargent, L.M.; Wang, L.; Dinu, C.Z.; Rojanasakula, Y. Digitoxin and a synthetic monosaccharide analog inhibit cell viability in lung cancer cells. Toxicol. Appl. Pharmacol. 2012, 258, 51–60. [Google Scholar] [CrossRef]
- Katz, A.; Lifshitz, Y.; Bab-Dinitz, E.; Kapri-Pardes, E.; Goldshleger, R.; Tal, D.M.; Karlish, S.J.D. Selectivity of digitalis glycosides for isoforms of human Na,K-ATPase. J. Biol. Chem. 2010, 285, 19582–19592. [Google Scholar] [CrossRef]
- Reddy, D.; Kumavath, R.; Barh, D.; Azevedo, V.; Ghosh, P. Anticancer and antiviral properties of cardiac glycosides: A review to explore the mechanism of actions. Molecules 2020, 25, 3596. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials. Available online: https://www.clinicaltrials.gov/ (accessed on 15 March 2021).
- Clinical Trials. Available online: https://clinicaltrials.gov/ct2/show/NCT04141995 (accessed on 15 March 2021).
- Clinical Trials. Available online: https://clinicaltrials.gov/ct2/show/NCT01765569 (accessed on 15 March 2021).
- Clinical Trials. Available online: https://www.clinicaltrials.gov/ct2/results?recrs=&cond=cancer&term=digoxin&cntry=&state=&city=&dist= (accessed on 15 March 2021).
- Liang, G.; Chung, T.; Guo, J.; Zhang, R.; Xü, W.; Tzen, J.T.C.; Jiang, R. Novel cinobufagin oxime ether derivatives as potential Na+/K+-ATPase inhibitors: Synthesis, biological screening and molecular docking. Chem. Res. Chin. Univ. 2017, 33, 378–383. [Google Scholar] [CrossRef]
- Morita, Y.; Matsumura, E.; Tsujibo, H.; Yasuda, M.; Sakagami, Y.; Okabe, T.; Ishida, N.; Inamori, Y. Biological activity of alpha-thujaplicin, the minor component of Thujopsis dolabrata SIEB. et ZUCC. var. hondai MAKINO. Biol. Pharm. Bull. 2001, 24, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.; Yang, W.Y.; Park, J.; Lee, S.; Mar, W.; Oh, K.B.; Shin, J. In vitro Na+/K+-ATPase inhibitory activity and antimicrobial activity of sesquiterpenes isolated from Thujopsis dolabrata. Arch. Pharm. Res. 2011, 34, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, P.; Tu, G.; Li, M.; Pan, B.; Guo, Y.; Zhai, J.; Fu, H. Synthesis and evaluation of panaxatriol derivatives as Na+, K+-ATPase inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 2885–2889. [Google Scholar] [CrossRef] [PubMed]
- De Munari, S.; Cerri, A.; Gobbini, M.; Almirante, N.; Banfi, L.; Carzana, G.; Ferrari, P.; Marazzi, G.; Micheletti, R.; Schiavone, A.; et al. Structure-based design and synthesis of novel potent Na+,K+-ATPase inhibitors derived from a 5α,14α-androstane scaffold as positive inotropic compounds. J. Med. Chem. 2003, 46, 3644–3654. [Google Scholar] [CrossRef]
- Alevizopoulos, K.; Dimas, K.; Papadopoulou, N.; Schmidt, E.M.; Tsapara, A.; Alkahtani, S.; Honisch, S.; Prousis, K.C.; Alarifi, S.; Calogeropoulou, T.; et al. Functional characterization and anti-cancer action of the clinical phase II cardiac Na+/K+ ATPase inhibitor istaroxime: In vitro and in vivo properties and cross talk with the membrane androgen receptor. Oncotarget 2016, 7, 24415–24428. [Google Scholar] [CrossRef]
- Gobbini, M.; Armaroli, S.; Banfi, L.; Benicchio, A.; Carzana, G.; Ferrari, P.; Giacalone, G.; Marazzi, G.; Moro, B.; Micheletti, R.; et al. Novel analogues of Istaroxime, a potent inhibitor of Na(+),K(+)-ATPase: Synthesis, structure-activity relationship and 3D-quantitative structure-activity relationship of derivatives at position 6 on the androstane scaffold. Bioorg. Med. Chem. 2010, 18, 4275–4299. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Z.; Tian, J.; Jiang, W.; Wang, Y.; Zhang, X.; Li, Z.; You, Q.; Shapiro, J.I.; Si, S.; et al. Identification of hydroxyxanthones as Na/K-ATPase ligands. Mol. Pharmacol. 2010, 77, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.N.; Liou, S.J.; Lee, T.H.; Chuang, Y.C.; Won, S.J. Xanthone derivatives as potential anti-cancer drugs. J. Pharm. Pharm. 1996, 48, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Alvarez, I.; Lee, L.; Jensen, R.T. Cyclic AMP-dependent protein kinase A and EPAC mediate VIP and secretin stimulation of PAK4 and activation of Na+,K+-ATPase in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G263–G277. [Google Scholar] [CrossRef]
- Moussawi, L.E.; Mohamed, C.; Kreydiyyeh, S.I. Epinephrine modulates Na+/K+ ATPase activity in Caco-2 cells via Src, p38MAPK, ERK and PGE2. PLoS ONE 2018, 13, e0193139. [Google Scholar] [CrossRef]
- Moussawi, L.E.; Mohamed, C.; Kreydiyyeh, S. The epinephrine-induced PGE2 reduces Na+/K+ ATPase activity in Caco-2 cells via PKC, NF-κB and NO. PLoS ONE 2019, 14, e0220987. [Google Scholar] [CrossRef]
- Shahidullah, M.; Mandal, A.; Delamere, N.A. Src family kinase links insulin signaling to short term regulation of Na,K-ATPase in nonpigmented ciliary epithelium. J. Cell. Physiol. 2017, 232, 1489–1500. [Google Scholar] [CrossRef]
- Alharbi, Y.; Kapur, A.; Felder, M.; Barroilhet, L.; Stein, T.; Pattnaik, B.R.; Patankar, M.S. Plumbagin-induced oxidative stress leads to inhibition of Na+/K+-ATPase (NKA) in canine cancer cells. Sci. Rep. 2019, 9, 11471. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-ATPase catalytic α subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [PubMed]
- Juel, C. Oxidative stress (glutathionylation) and Na,K-ATPase activity in rat skeletal muscle. PLoS ONE 2014, 13, e110514. [Google Scholar] [CrossRef] [PubMed]
- Bibert, S.; Liu, C.C.; Figtree, G.A.; Garcia, A.; Hamilton, E.J.; Marassi, F.M.; Sweadner, K.J.; Cornelius, F.; Geering, K.; Rasmussen, H.H. FXYD proteins reverse inhibition of the Na+-K+ pump mediated by glutathionylation of its beta1 subunit. J. Biol. Chem. 2011, 286, 18562–18572. [Google Scholar] [CrossRef] [PubMed]
- Dada, L.A.; Chandel, N.S.; Ridge, K.M.; Pedemonte, C.; Bertorello, A.M.; Sznajder, J.I. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J. Clin. Investig. 2003, 111, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Comellas, A.P.; Dada, L.A.; Lecuona, E.; Pesce, L.M.; Chandel, N.S.; Quesada, N.; Budinger, G.R.S.; Strous, G.J.; Ciechanover, A.; Sznajder, J.I. Hypoxia-mediated degradation of Na,K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-conjugating system. Circ. Res. 2006, 98, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Krmar, R.T.; Dada, L.; Efendiev, R.; Leibiger, I.B.; Pedemonte, C.H.; Katz, A.I.; Sznajder, J.I.; Bertorello, A.M. Phosphorylation of adaptor protein-2 mu2 is essential for Na+,K+-ATPase endocytosis in response to either G protein-coupled receptor or reactive oxygen species. Am. J. Respir. Cell Mol. Biol. 2006, 35, 127–132. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoform | Affinity for Ions | References | |
|---|---|---|---|
| Na+ | K+ | ||
| 1 a | +/− | n.e. b | [62,63] |
| 2 | n.e. | + | [64] |
| 3 | - | - | [65] |
| 4 | + | - | [66] |
| 5 | + | - | [67] |
| 6 c | +/− | +/− | [68] |
| 7 c | +/− | n.e. | [69] |
| Clinical Trial Identifier | Study Title | Condition or Disease | First Posted | Status | Phase | Intervention/ Treatment |
|---|---|---|---|---|---|---|
| NCT02906800 | Potentiation of cisplatin-based chemotherapy by digoxin in advanced unresectable head and neck cancer patients | Head and neck cancer | 20 September 2016 | Unknown | I, II | Digoxin |
| NCT04094519 | A study to evaluate the effect of multiple doses of enzalutamide on the pharmacokinetics of substrates of P-glycoprotein (digoxin) and breast cancer resistant protein (rosuvastatin) in male subjects with prostate cancer | Prostate cancer | 19 September 2019 | Active, not recruiting | I | Enzultamide, enzultamide placebo, digoxin, rosuvastatin |
| NCT01763931 | DIG-HIF-1 pharmacodynamic trial in newly diagnosed operable breast cancer | Breast cancer | 9 January 2013 | Completed | II | Digoxin |
| NCT01162135 | Digoxin for recurrent prostate cancer | Prostate cancer | 14 July 2010 | Completed | II | Digoxin |
| NCT01887288 | Capecitabine with digoxin for metastatic breast cancer | Metastatic breast cancer | 26 June 2013 | Terminated | II | Capetabine, digoxin |
| NCT00650910 | Study to examine the effects of lapatinib on the pharmacokinetics of digoxin in subjects w/ErbB2 positive breast cancer | Neoplasm, breast | 2 April 2008 | Completed | I | Lapatinib, digoxin |
| NCT03889795 | Phase IB metformin, digoxin, simvastatin in solid tumors | Advanced pancreatic cancer, advanced solid tumor | 26 March 2019 | Recruiting | I | Metformin, simvastatin, digoxin |
| NCT03928210 | Digoxin induced dissolution of CTC clusters | Breast cancer, circulating tumor cells (CTCs) | 26 April 2019 | Not yet recruiting | I | Digoxin |
| NCT04141995 | FOLFIRINOX with digoxin in patients with resectable pancreatic cancer | Pancreas cancer, adenocarcinoma of the pancreas | 28October 2019 | Not yet recruiting | II | Digoxin, 5-fluorouracil, calcium leucovorin, irinotecan, oxaliplatin |
| NCT02106845 | Effect of regorafenib on digoxin and rosuvastatin in patients with advanced solid malignant tumors | Neoplasms | 8 April 2014 | Completed | I | Digoxin, rosuvastatin, regorafenib |
| NCT04322552 | A pharmacokinetic interaction study between apatinib mesylate and transporter Pgp substrate digoxin in advanced solid tumor subjects | Advanced solid tumor | 26 March 2020 | Recruiting | I | Apatinib mesylate, digoxin |
| NCT01517399 | Drug-drug interaction study of tivantinib (ARQ 197) with omeprazole, S-warfarin, caffeine, midazolam, and digoxin in cancer subjects | Solid tumors | 25 January 2012 | Completed | I | Tivantinib, omeprazole, s-warfarin, caffeine, vitamin K (dietary supplement), digoxin, midazolam |
| NCT02626234 | A drug-drug interaction (DDI) study to assess the effect of INC280 on the pharmacokinetics of digoxin and rosuvastatin in patients with cMET-dysregulated advanced solid tumors | cMET-dysregulated Advanced Solid Tumors | 10 December 2015 | Completed | I | INC280, digoxin, rosuvastatin |
| NCT00281021 | Second line erlotinib (Tarceva) plus eigoxin in non-small cell lung cancer | Carcinoma, non-small cell lung | 24 January 2006 | Terminated | II | Erlotinib, digoxin |
| NCT01765569 | A pharmacokinetic study to investigate the effect of vemurafenib on Digoxin in Patients With BRAFV600 mutation-positive Metastatic Melanoma | Malignant melanoma, neoplasms | 10 January 2013 | Completed | I | Digoxin, vemurafenib |
| NCT02740712 | Pharmacokinetic drug-drug interaction study of rucaparib (DDI) | Neoplasms | 15 April 2016 | Completed | I | Caffeine, warfarin, omeprazole, midazolam, digoxin, vitamin K, rucaparib |
| NCT02212639 | Phase II multicentric study of digoxin per os in classic or endemic Kaposi’s sarcoma (KADIG 01) | Classic and endemic Kaposi’s sarcoma, lymph angio proliferations | 8 August 2014 | Unknown | II | Digoxin |
| NCT03720366 | A study of perpetrator drug interactions of enasidenib in AML patients | Leukemia, myeloid, acute | 25 October 2018 | Not yet recruiting | I | Caffeine, dextromethorphan, flurbiprofen, midazolam, omeprazole, digoxin, rosuvastatin, pioglitazone |
| NCT03684772 | Topical ionic contra-viral therapy in actinic keratosis | Actinic keratosis | 26 September 2018 | Recruiting | II | ICVT topical gel, furosemide topical, digoxin topical gel, vehicle topical gel |
| NCT02138292 | A phase 1B clinical trial of trametinib plus digoxin in patients with unresectable or metastatic BRAF wild-type melanoma | Melanoma | 14 May 2014 | Completed | I | Trametinib, digoxin |
| NCT02915666 | A clinical trial of patients with melanoma | Melanoma | 27 September 2016 | Withdrawn | I | Digoxin combination, dabrafenib, trametinib |
| NCT02732275 | DS-3201b in participants with lymphomas | Lymphoma, malignant, non-Hodgkin lymphoma | 8 April 2016 | Recruiting | I | DS-3201b, midazolam, digoxin |
| NCT02333643 | A phase 2 efficacy study of CLS003 ICVT in subjects with cutaneous warts | Cutaneous warts | 7 January 2015 | Completed | II | CLS003, furosemide, digoxin, vehicle topical |
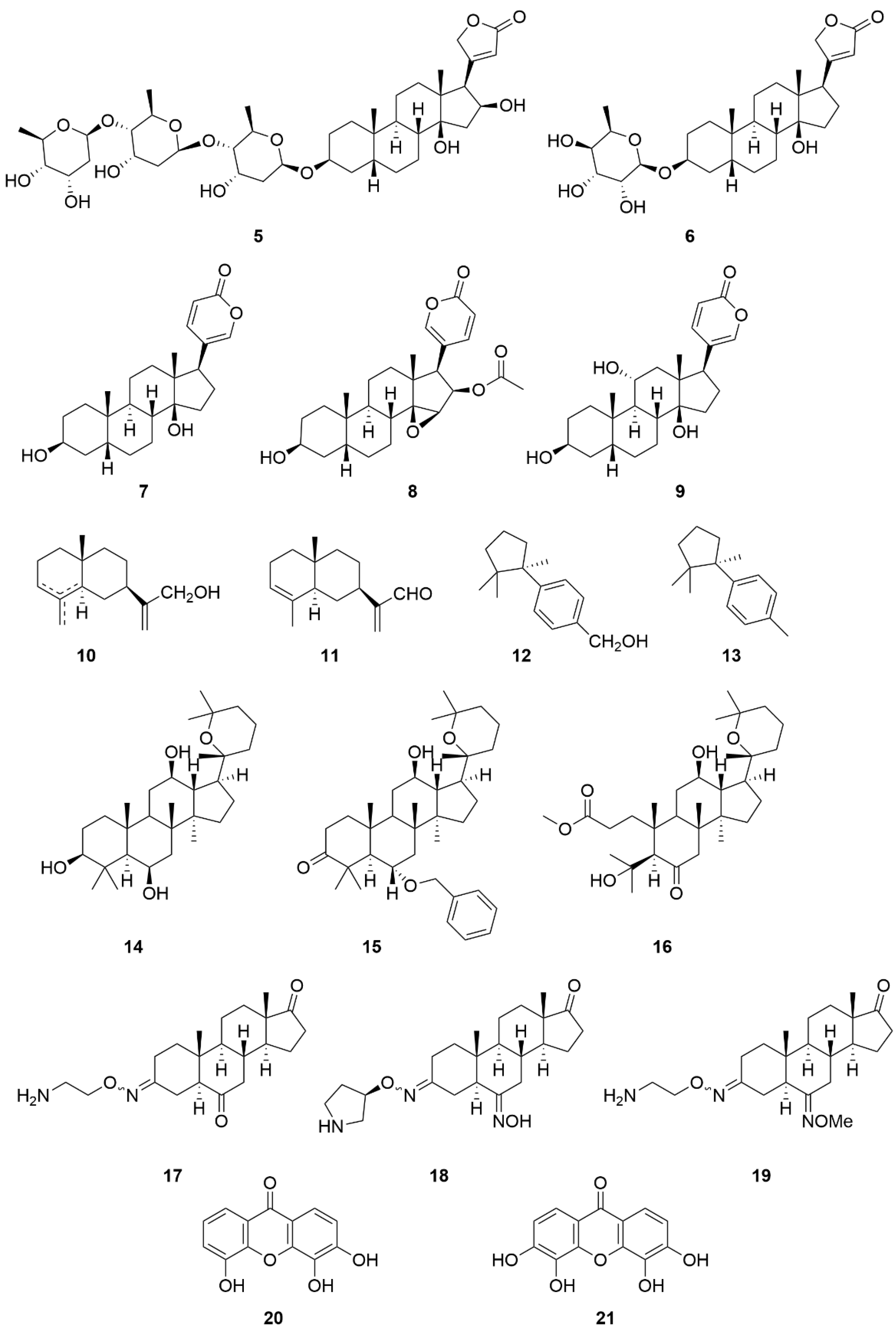
| Compound Name (Code) | Group of Compounds | Ki or IC50 [μM] | Isoform/Source | Ref. |
|---|---|---|---|---|
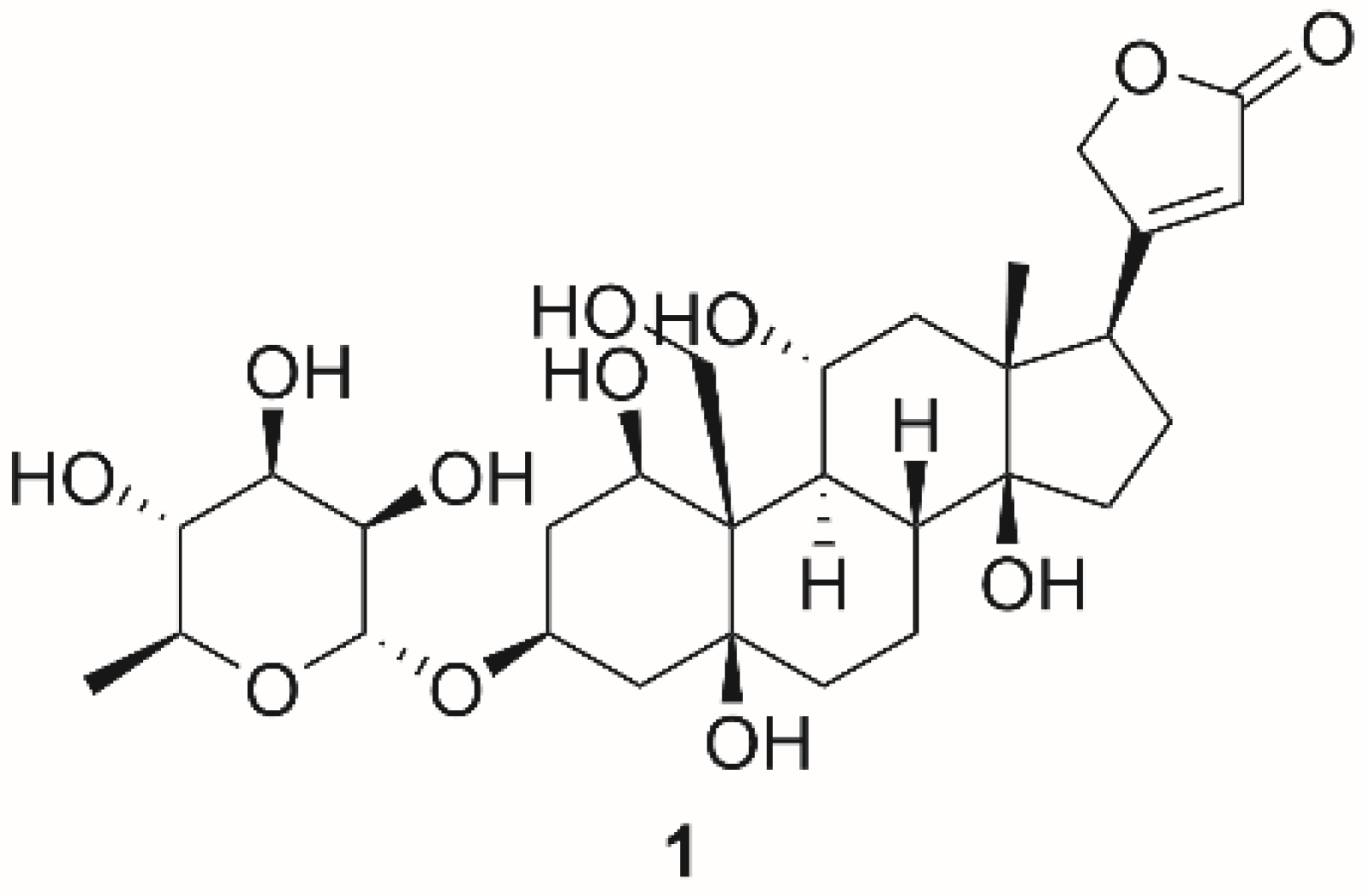
| Ouabain (1) | Cardiac steroids | 0.09 ± 0.01 | Shark (rectal gland microsomes, α3) | [136] |
| Digoxin (2) | 0.13 ± 0.02 | |||
| Digitoxin (3) | 0.18 ± 0.01 | |||
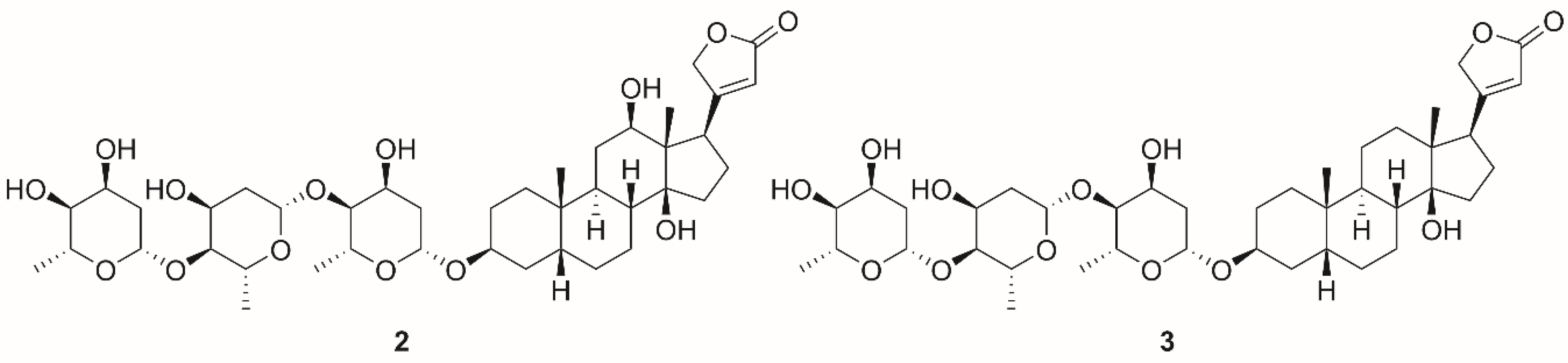
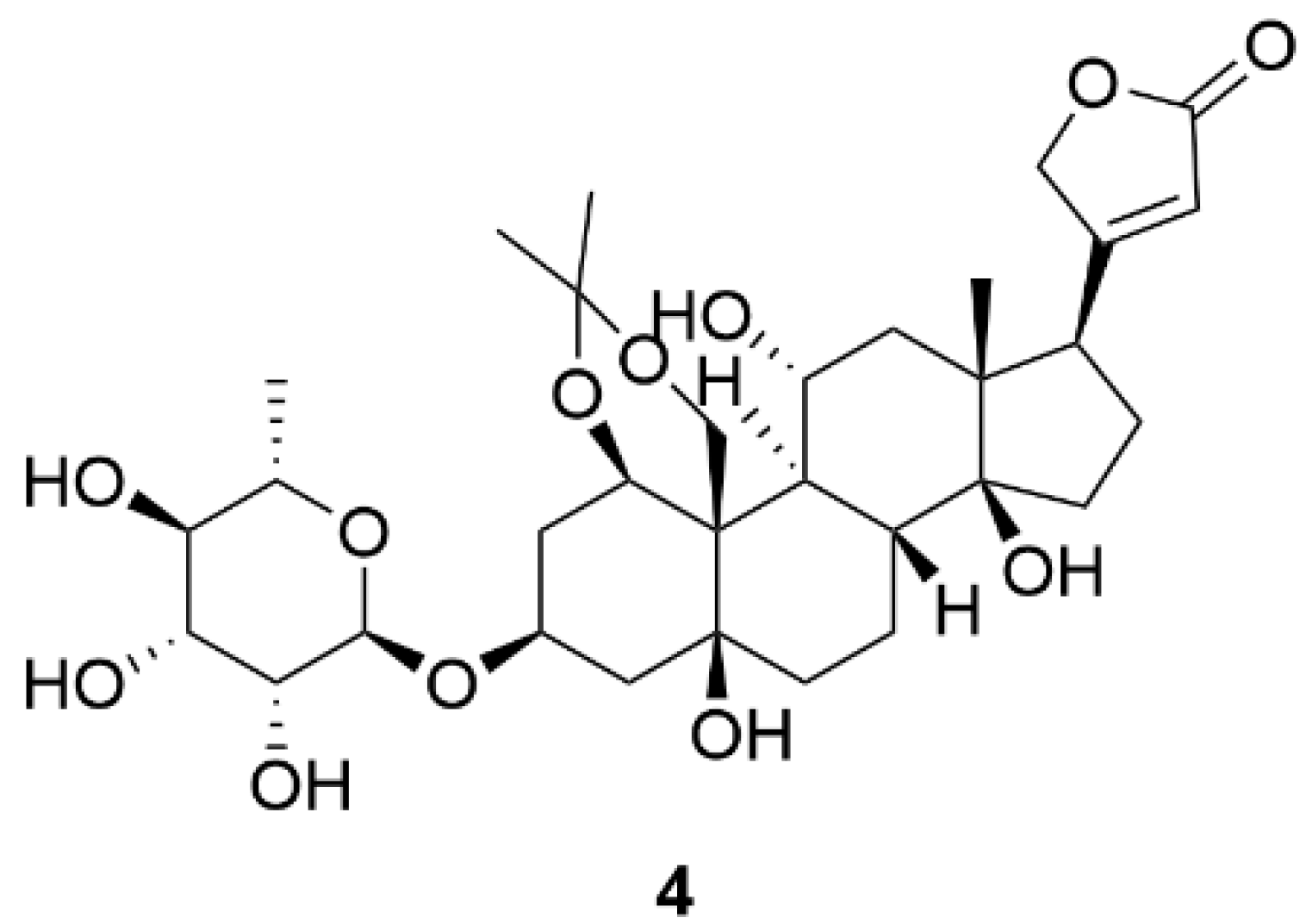
| 4 | 12.4 * | Porcine cerebral cortex | [137] | |
| Gitoxin (5) | 0.16 ± 0.04 | Shark (rectal gland microsomes, α3) | [136] | |
| Evomonoside (6) | 0.11 ± 0.01 | |||
| Bufalin (7) | 0.13 ± 0.00 | |||
| Cinobufagin (8) | 0.68 | Pig kidney | [152] | |
| Gamabufotalin (9) | 0.16 ± 0.02 | Shark (rectal gland microsomes, α3) | [136] | |
| 10 | Sesquiterpenes | 55.62 ± 0.41 | Porcine cerebral cortex | [154] |
| 11 | 212.0 ± 1.92 | |||
| 12 | 108.09 ± 2.01 | |||
| 13 | >494.22 | |||
| Panaxatriol (14) | Triterpenes | 1.09 ± 0.11 | Human Na+/K+-ATPase | [155] |
| 15 | 0.33 ± 0.03 | |||
| 16 | 0.26 ± 0.03 | |||
| Istaroxime (17) | Steroids | 0.11 | Dog kidney | [158] |
| 18 | 0.02 | |||
| 19 | 0.02 | |||
| 3,4,5-trihydroxyxanthone (20) | Hydroxyxanthones | 10.0 | Dog kidney | [159] |
| 3,4,5,6-tetrahydroxyxanthone (21) | 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bejček, J.; Spiwok, V.; Kmoníčková, E.; Rimpelová, S. Na+/K+-ATPase Revisited: On Its Mechanism of Action, Role in Cancer, and Activity Modulation. Molecules 2021, 26, 1905. https://doi.org/10.3390/molecules26071905
Bejček J, Spiwok V, Kmoníčková E, Rimpelová S. Na+/K+-ATPase Revisited: On Its Mechanism of Action, Role in Cancer, and Activity Modulation. Molecules. 2021; 26(7):1905. https://doi.org/10.3390/molecules26071905
Chicago/Turabian StyleBejček, Jiří, Vojtěch Spiwok, Eva Kmoníčková, and Silvie Rimpelová. 2021. "Na+/K+-ATPase Revisited: On Its Mechanism of Action, Role in Cancer, and Activity Modulation" Molecules 26, no. 7: 1905. https://doi.org/10.3390/molecules26071905
APA StyleBejček, J., Spiwok, V., Kmoníčková, E., & Rimpelová, S. (2021). Na+/K+-ATPase Revisited: On Its Mechanism of Action, Role in Cancer, and Activity Modulation. Molecules, 26(7), 1905. https://doi.org/10.3390/molecules26071905