
Bioinformatic Analysis of Genome-Predicted Bat Cathelicidins

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
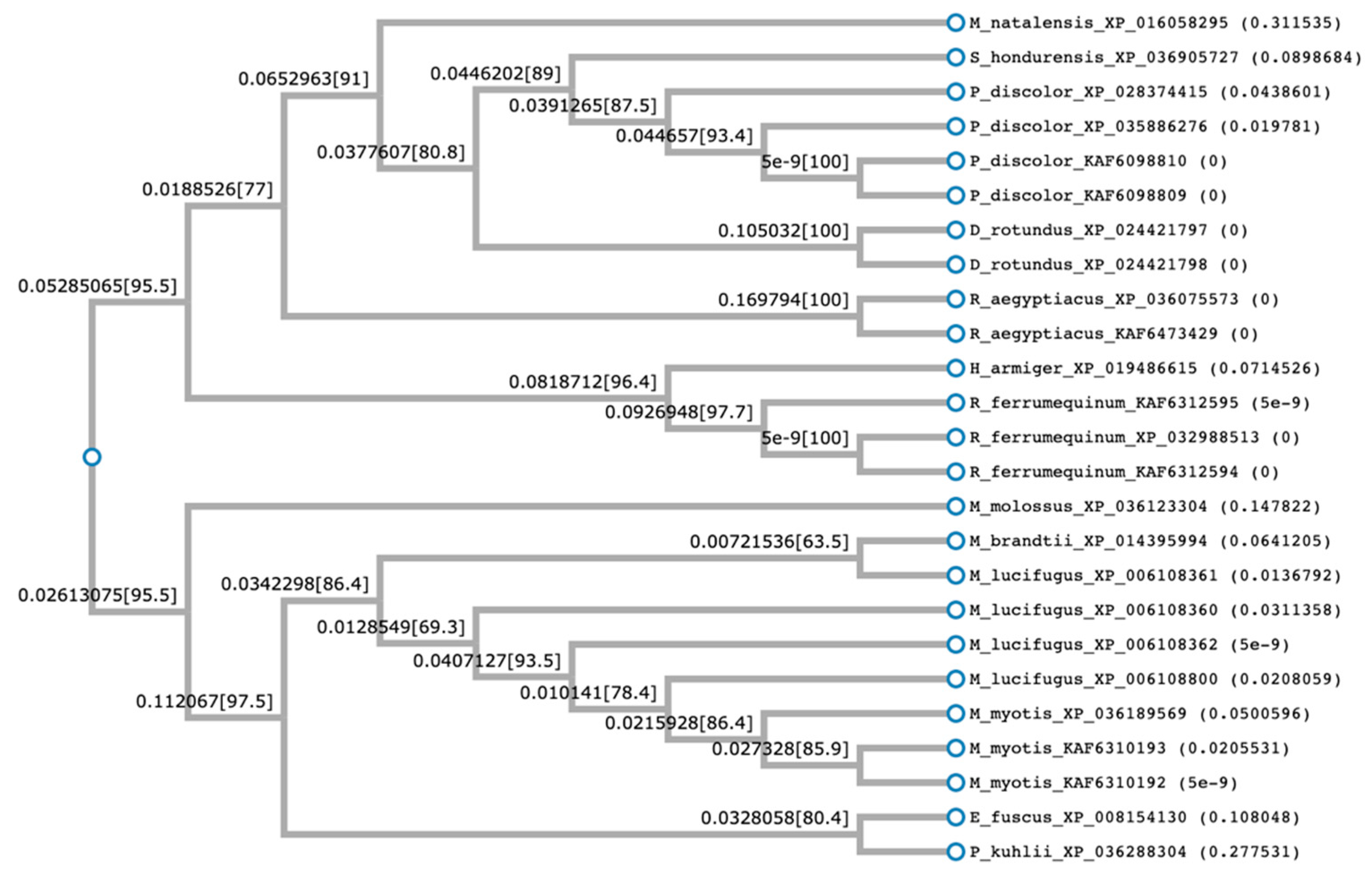
2.1. Prediction of Cathelicidin Active Peptides
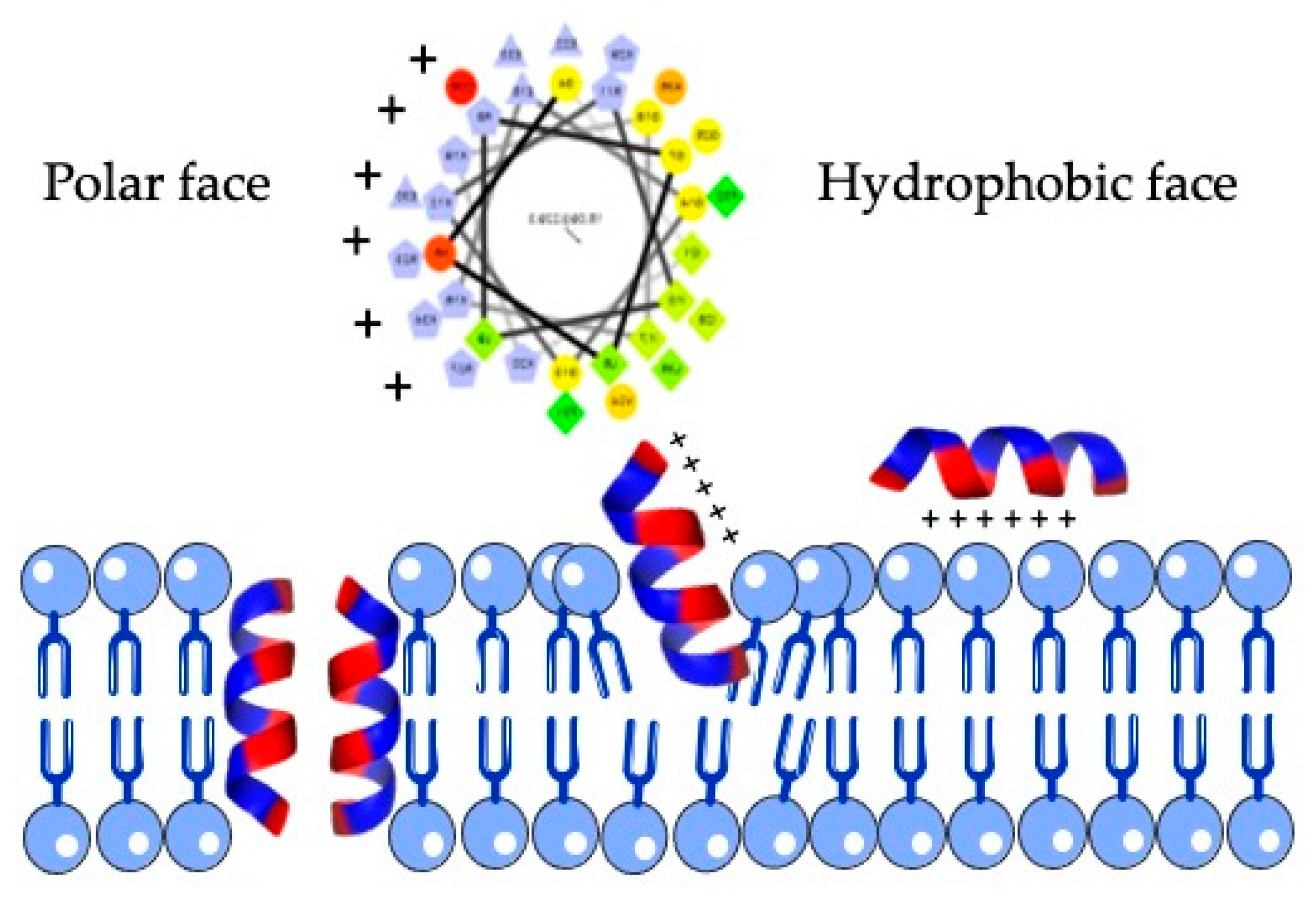
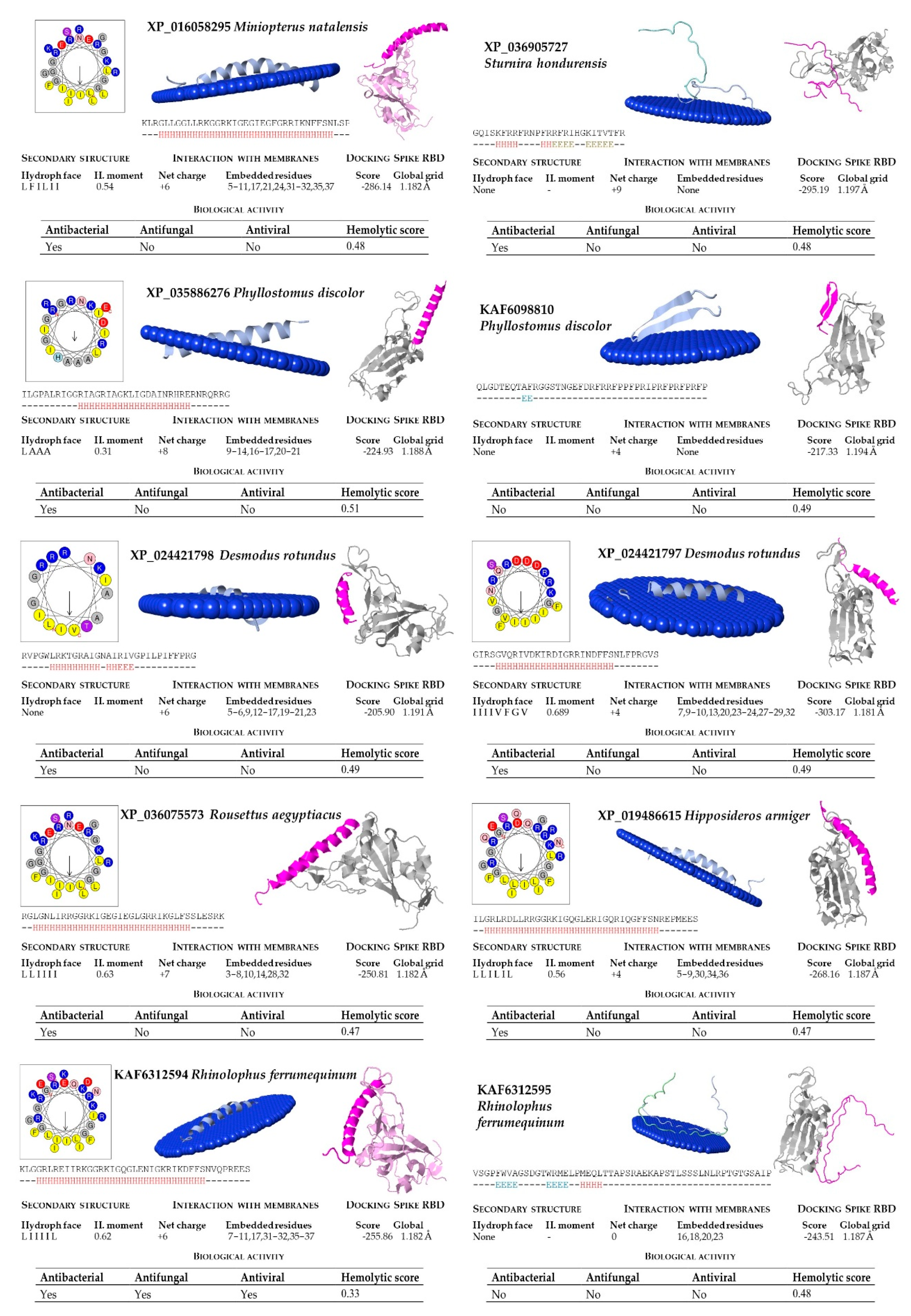
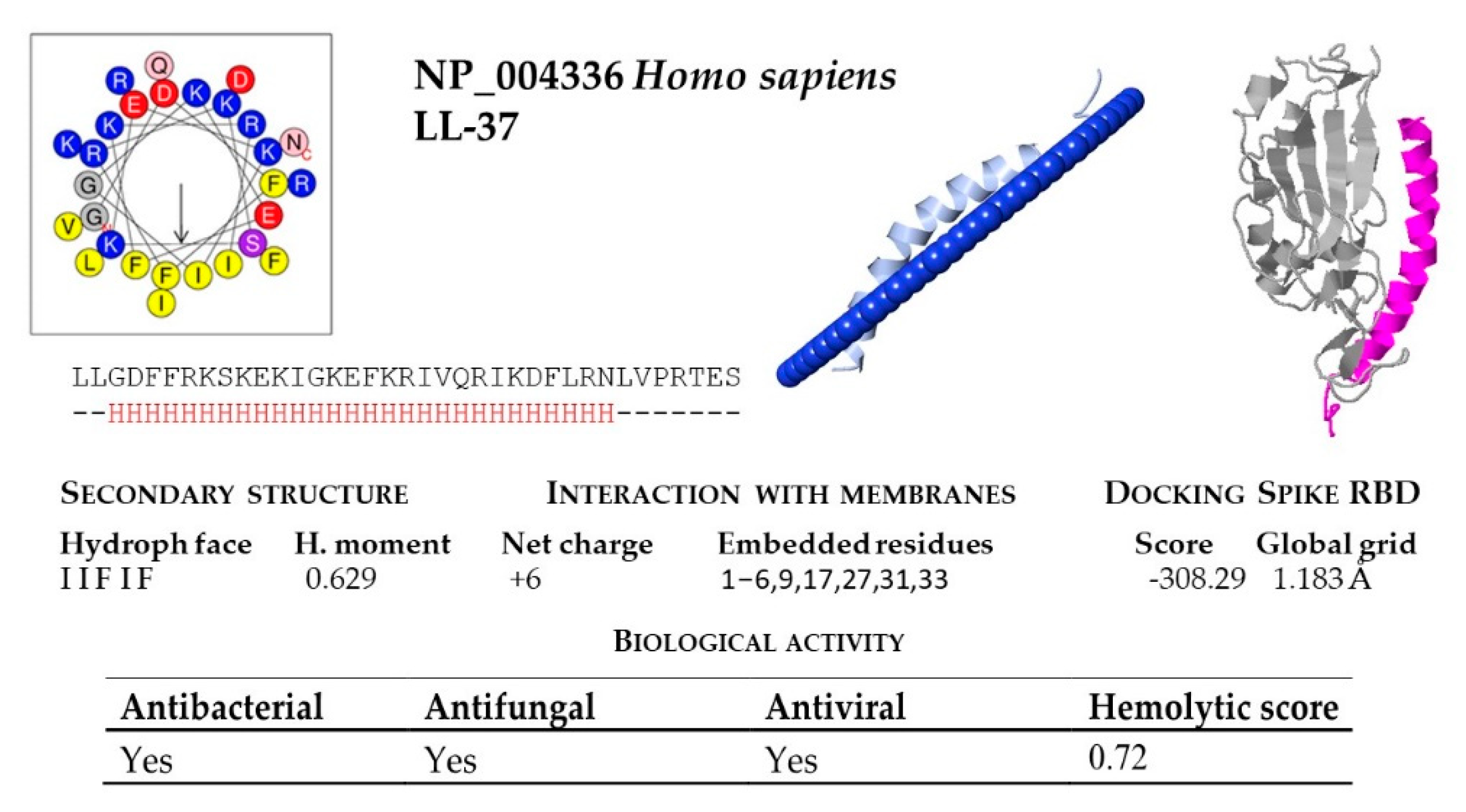
2.2. Structural and Functional Prediction of Bat Cathelicidins
3. Discussion
4. Materials and Methods
4.1. Retrieval of Bat Cathelicidin Sequences
4.2. Structural Analysis
4.3. Multiple Alignment and Phylogenetic Tree
4.4. In silico Analysis of Biological Activity
4.5. Structural Analysis of Peptides
4.6. Modeling of Peptides
4.7. Molecular Docking studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020, 1–14. [Google Scholar] [CrossRef]
- Olival, K.J.; Cryan, P.M.; Amman, B.R.; Baric, R.S.; Blehert, D.S.; Brook, C.E.; Calisher, C.H.; Castle, K.T.; Coleman, J.T.H.; Daszak, P.; et al. Possibility for reverse zoonotic transmission of SARS-CoV-2 to free-ranging wildlife: A case study of bats. PLOS Pathog. 2020, 16, e1008758. [Google Scholar] [CrossRef]
- Brunet-Rossinni, A.K.; Austad, S.N. Ageing studies on bats: A review. Biogerontology 2004, 5, 211–222. [Google Scholar] [CrossRef]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Subudhi, S.; Rapin, N.; Misra, V. Immune System Modulation and Viral Persistence in Bats: Understanding Viral Spillover. Viruses 2019, 11, 192. [Google Scholar] [CrossRef]
- Wang, L.F.; Shi, Z.; Zhang, S.; Field, H.; Daszak, P.; Eaton, B.T. Review of bats and SARS. Emerg. Infect. Dis. 2006, 12, 1834–1840. [Google Scholar] [CrossRef]
- Baker, M.L.; Schountz, T.; Wang, L.F. Antiviral Immune Responses of Bats: A Review. Zoonoses Public Health 2013, 60, 104–116. [Google Scholar] [CrossRef]
- Teeling, E.C.; Vernes, S.C.; Dávalos, L.M.; Ray, D.A.; Gilbert, M.T.P.; Myers, E. Bat Biology, Genomes, and the Bat1K Project: To Generate Chromosome-Level Genomes for All Living Bat Species. Annu. Rev. Anim. Biosci. 2018, 6, 23–46. [Google Scholar] [CrossRef]
- Tan, C.P.Y.T. Molecular Cloning and Characterization of Defensin beta 103a, Hepcidin and Cathelicidin Antimicrobial peptides in Eonycteris speleae and Pteropus Alecto, 2016. Nanyang Technological University. Available online: http://hdl.handle.net/10356/68531 (accessed on 15 December 2020).
- Boto, A.; Pérez de la Lastra, J.; González, C. The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules 2018, 23, 311. [Google Scholar] [CrossRef] [PubMed]
- Kościuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.; Krzyżewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef]
- Roth, A.; Lütke, S.; Meinberger, D.; Hermes, G.; Sengle, G.; Koch, M.; Streichert, T.; Klatt, A.R. LL-37 fights SARS-CoV-2: The Vitamin D-Inducible Peptide LL-37 Inhibits Binding of SARS-CoV-2 Spike Protein to its Cellular Receptor Angiotensin Converting Enzyme 2 In Vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tomasinsig, L.; Zanetti, M. The Cathelicidins—Structure, Function and Evolution. Curr. Protein Pept. Sci. 2005, 6, 23–34. [Google Scholar] [CrossRef]
- Pérez de la Lastra, J.M.; Garrido-Orduña, C.; Borges, A.A.; Jiménez-Arias, D.; García-Machado, F.J.; Hernández, M.; González, C.; Boto, A. Bioinformatics Discovery of Vertebrate Cathelicidins from the Mining of Available Genomes. In Drug Discovery—Concepts to Market; Bobbarala, V., Ed.; IntechOpen: London, UK, 2018; pp. 165–181. [Google Scholar]
- Pérez De La Lastra, J.M.; Garrido-Orduña, C.; Borges, C.L.; Borges, A.A.; Boto, A. Antimicrobial activity of cathelicidins of mammals from avian, aquatic and terrestrial environments. In Antimacribial Research: Novel Bioknowledge and Educational Programs; Mendez-Vilas, A., Ed.; Formatex: Badajoz, Spain, 2017; p. 682. [Google Scholar]
- Zanetti, M. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 2004, 75, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R. The evolution of flight and echolocation in bats: Another leap in the dark. Mamm. Rev. 2001, 31, 111–130. [Google Scholar] [CrossRef]
- Xiao, Y.; Cai, Y.; Bommineni, Y.R.; Fernando, S.C.; Prakash, O.; Gilliland, S.E.; Zhang, G. Identification and Functional Characterization of Three Chicken Cathelicidins with Potent Antimicrobial Activity. J. Biol. Chem. 2006, 281, 2858–2867. [Google Scholar] [CrossRef]
- Hao, X.; Yang, H.; Wei, L.; Yang, S.; Zhu, W.; Ma, D.; Yu, H.; Lai, R. Amphibian cathelicidin fills the evolutionary gap of cathelicidin in vertebrate. Amino Acids 2012, 43, 677–685. [Google Scholar] [CrossRef]
- Van der Does, A.M.; Bergman, P.; Agerberth, B.; Lindbom, L. Induction of the human cathelicidin LL-37 as a novel treatment against bacterial infections. J. Leukoc. Biol. 2012, 92, 735–742. [Google Scholar] [CrossRef]
- Gennaro, R.; Zanetti, M. Structural features and biological activities of the cathelicidin-derived antimicrobial peptides. Biopolym. Pept. Sci. Sect. 2000, 55, 31–49. [Google Scholar] [CrossRef]
- Cole, A.M.; Shi, J.; Ceccarelli, A.; Kim, Y.-H.; Park, A.; Ganz, T. Inhibition of neutrophil elastase prevents cathelicidin activation and impairs clearance of bacteria from wounds. Blood 2001, 97, 297–304. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Zanetti, M.; Gennaro, R.; Romeo, D. Cathelicidins: A novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995, 374, 1–5. [Google Scholar] [CrossRef]
- Schneider, V.A.F.; Coorens, M.; Ordonez, S.R.; Tjeerdsma-Van Bokhoven, J.L.M.; Posthuma, G.; Van Dijk, A.; Haagsman, H.P.; Veldhuizen, E.J.A. Imaging the antimicrobial mechanism(s) of cathelicidin-2. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Vaello, E.; Gil-Carton, D.; François, P.; Bonetti, E.J.; Kreir, M.; Pothula, K.R.; Kleinekathöfer, U.; Zeth, K. The structure of the antimicrobial human cathelicidin LL-37 shows oligomerization and channel formation in the presence of membrane mimics. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Wang, J.; Gao, H.; Wang, Z.; Dong, N.; Ma, Q.; Shan, A. Antimicrobial Properties and Membrane-Active Mechanism of a Potential α-Helical Antimicrobial Derived from Cathelicidin PMAP-36. PLoS ONE 2014, 9, e86364. [Google Scholar] [CrossRef] [PubMed]
- Sevcsik, E.; Pabst, G.; Richter, W.; Danner, S.; Amenitsch, H.; Lohner, K. Interaction of LL-37 with Model Membrane Systems of Different Complexity: Influence of the Lipid Matrix. Biophys. J. 2008, 94, 4688–4699. [Google Scholar] [CrossRef] [PubMed]
- Dobson, A.P. VIROLOGY: What Links Bats to Emerging Infectious Diseases? Science 2005, 310, 628–629. [Google Scholar] [CrossRef]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and Coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef]
- Mandl, J.N.; Ahmed, R.; Barreiro, L.B.; Daszak, P.; Epstein, J.H.; Virgin, H.W.; Feinberg, M.B. Reservoir Host Immune Responses to Emerging Zoonotic Viruses. Cell 2015, 160, 20–35. [Google Scholar] [CrossRef]
- Lokhande, K.B.; Banerjee, T.; Swamy, K.V.; Deshpande, M. An in Silico Scientific Basis for LL-37 as a Therapeutic and Vitamin D as Preventive for Covid-19. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Irving, A.T.; Ahn, M.; Goh, G.; Anderson, D.E.; Wang, L.-F. Lessons from the host defences of bats, a unique viral reservoir. Nature 2021, 589, 363. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-J.; Wen, H.; Zhou, C.-M.; Chen, F.-F.; Luo, L.-M.; Liu, J.; Yu, X.-J. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 2015, 205, 1–6. [Google Scholar] [CrossRef]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important Reservoir Hosts of Emerging Viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Timmons, P.B.; Hewage, C.M. HAPPENN is a novel tool for hemolytic activity prediction for therapeutic peptides which employs neural networks. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Kumar, R.; Singh, S.; Tuknait, A.; Gautam, A.; Mathur, D.; Anand, P.; Varshney, G.C.; Raghava, G.P.S. A Web Server and Mobile App for Computing Hemolytic Potency of Peptides. Nat. Publ. Gr. 2016. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. IAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Wang, F.; Zhang, J.; Wang, F.; Chang, S. CoDockPP: A Multistage Approach for Global and Site-Specific Protein-Protein Docking. J. Chem. Inf. Model. 2019, 59, 3556–3564. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bat Species | Genome ID | Length (Mb) | Proteins | Cathelicidins |
|---|---|---|---|---|
| Artibeus jamaicensis | 12026 | 2316.20 | 42,600 | 1 |
| Desmodus rotundus | 15041 | 2063.80 | 29,845 | 2 |
| Eptesicus fuscus | 11703 | 2026.63 | 49,822 | 1 |
| Hipposideros armiger | 15002 | 2236.58 | 45,831 | 1 |
| Miniopterus natalensis | 44094 | 1803.10 | 29,787 | 1 |
| Molossus molossus | 93829 | 2315.57 | 53,797 | 1 |
| Myotis brandtii | 18281 | 2107.24 | 40,808 | 2 |
| Myotis davidii | 14635 | 2059.80 | 33,106 | 2 |
| Myotis lucifugus | 614 | 2034.58 | 43,106 | 4 |
| Myotis myotis | 43810 | 2148.60 | 61,156 | 3 |
| Phyllostomus discolor | 75334 | 2080.20 | 46,999 | 4 |
| Pipistrellus kuhlii | 93828 | 1775.69 | 39,923 | 1 |
| Pteropus alecto | 12056 | 1985.96 | 39,693 | 1 |
| Pteropus vampyrus | 757 | 2198.28 | 43,630 | 1 |
| Rhinolophus ferrumequinum | 10960 | 2072.56 | 45,117 | 3 |
| Rousettus aegyptiacus | 7672 | 1904.62 | 61,105 | 2 |
| Sturnira hondurensis | 95481 | 2098.36 | 43,530 | 2 |
| Bat Species | Accession No. | Interval | Cysteines | Peptide Length |
|---|---|---|---|---|
| A. jamaicensis | XP_036984419 | 30–104 | 2 | 14 |
| S. hondurensis | XP_036905728 | 30–127 | 4 | 10 |
| M. brandtii | XP_005867268 | 33–131 | 4 | 3 |
| M. davidii | ELK24988 | 39–82 | 3 | 43 |
| M. davidii | ELK24989 | 32–126 | 4 | 7 |
| P. alecto | XP_006914980 | 23–102 | 3 | 67 |
| P. vampyrus | XP_011373748 | 23–94 | 2 | 64 |
| Bat Species | Accession | Sequence of Active Peptide |
|---|---|---|
| D. rotundus | XP_024421798 | RVPGWLRKTGRAIGNAIRIVGPILPIFFPRG |
| D. rotundus | XP_024421797 | GIRSGVQRIVDKIRDIGRRINDFFSNLFPRGVS |
| E. fuscus | XP_008154130 | KFNARKLGELIRRGGEGFGRKVEKIGRRIKEFFTNLAPREEEA |
| H. armiger | XP_019486615 | ILGRLRDLLRRGGRKIGQGLERIGQRIQGFFSNREPMEES |
| M. brandtii | XP_014395994 | ELNIENLGERIKNAKKKVWEKIKSFGRRIKDFFRKPSPEVEP |
| M. lucifugus | XP_006108362 | RFNYDRLSNIIKRGGYKLGEGLEIVGGILRRS |
| M. lucifugus | XP_006108800 | GLILWGWRPPGALGRLWDRIRYRVRRPRDVSENLSP |
| M. lucifugus | XP_006108360 | LNPLIKAGIFILKHRRPIGRGIEITGRGIKKFFSK |
| M. lucifugus | XP_006108361 | LNPWIIGGALAWKHRRPIGRGLEKAGSGIKRFFSKRSPEQEP |
| M. molossus | XP_036123304 | SLGGLLKKGGQIIGKKIEKIGKRIKDFFTNTESMEEAKSV |
| M. myotis | XP_036189569 | LNPDTPKPVSFTLKETVCPRTTRQPPEECDFKENGLVKVCGGTVTLDQDTDYYDVHCEEIKDVAIRPLVSGALFLWKNRRPIGWGIEKTGRGIKRIFSKRSPEQEP |
| M. myotis | KAF6310192 | AIRPLVSGALFLWKNRRPIGWGIEKTGRGIKRIFSKRSPEQEP |
| M. myotis | KAF6310193 | AIRPLVSGALFLWKNRRPIGWGIEKTGRGIKRIFSKRSPEQEP |
| M. natalensis | XP_016058295 | KLRGLLGGLLRKGGRKIGEGIEGFGRRIKNFFSNLSPREES |
| P. discolor | KAF6098810 | QLGDTEQTAFRGGSTNGEFDRFRRFPPFPRIPRFPRFPRFP |
| P. discolor | KAF6098809 | QLGDTEQTAFRGGSTNGEFDRFRRFPPFPRIPRFPRFPRFP |
| P. discolor | XP_035886276 | ILGPALRIGGRIAGRIAGKLIGDAINRHRERNRQRRG |
| P. discolor | XP_028374415 | ILGPALRIGGRIAGRIAGKLIGDAINRHRERNRQRRG |
| P. kuhlii | XP_036288304 | NLDNLIQKGREKLGRLRELFRKGGQKVGKLLQKGGQKLGEIGQRIRDFFSNLRPREEGPQPRGEGPQPPEGGPQLPEEDTQPQEES |
| R. aegyptiacus | XP_036075573 | RGLGNLIRRGGRKIGEGIEGLGRRIKGLFSSLESRK |
| R. aegyptiacus | KAF6473429 | RGLGNLIRRGGRKIGEGIEGLGRRIKGLFSSLESRK |
| R. ferrumequinum | KAF6312594 | KLGGRLREIIRKGGRKIGQGLENIGKRIKDFFSNVQPREES |
| R. ferrumequinum | XP_032988513 | KLGGRLREIIRKGGRKIGQGLENIGKRIKDFFSNVQPREES |
| R. ferrumequinum | KAF6312595 | SGPFWVAGSDGTWRMELPMEQLTTAPSRAEKAPSTLSSSLNLRPTGTGSAIP |
| S. hondurensis | XP_036905727 | GQISKFRRFRNPFRRFRIHGKITVTFR |
| * H. sapiens | NP_004336 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez de la Lastra, J.M.; Asensio-Calavia, P.; González-Acosta, S.; Baca-González, V.; Morales-delaNuez, A. Bioinformatic Analysis of Genome-Predicted Bat Cathelicidins. Molecules 2021, 26, 1811. https://doi.org/10.3390/molecules26061811
Pérez de la Lastra JM, Asensio-Calavia P, González-Acosta S, Baca-González V, Morales-delaNuez A. Bioinformatic Analysis of Genome-Predicted Bat Cathelicidins. Molecules. 2021; 26(6):1811. https://doi.org/10.3390/molecules26061811
Chicago/Turabian StylePérez de la Lastra, José Manuel, Patricia Asensio-Calavia, Sergio González-Acosta, Victoria Baca-González, and Antonio Morales-delaNuez. 2021. "Bioinformatic Analysis of Genome-Predicted Bat Cathelicidins" Molecules 26, no. 6: 1811. https://doi.org/10.3390/molecules26061811
APA StylePérez de la Lastra, J. M., Asensio-Calavia, P., González-Acosta, S., Baca-González, V., & Morales-delaNuez, A. (2021). Bioinformatic Analysis of Genome-Predicted Bat Cathelicidins. Molecules, 26(6), 1811. https://doi.org/10.3390/molecules26061811