
Molecular Modeling of Histamine Receptors—Recent Advances in Drug Discovery



Abstract

1. Introduction
1.1. Overall Structure of the Histamine Receptors
1.2. Recent Publications on Computational Studies Targeting HRs
2. HR-Targeted Ligands and Receptor Binding Site of Inactive/Active States of HRs
2.1. Computational Studies on Histamine H1 Receptor and Its ligands
2.1.1. Structural Aspects of Histamine H1 Receptor
2.1.2. H1R Targeted Ligands and Their Interactions in the Ligand-Binding Pocket
Receptor-Based in Silico Approaches Targeting H1R
Ligand-Based Computational Approaches in Search for Potential H1R Ligands
2.2. Computational Studies of H2R and Its Ligands
2.2.1. Homology Modeling of H2R
2.2.2. H2R-Targeted Ligands and Their Interactions at H2R Active Site
2.3. Computational Studies on H3R and Its Ligands
2.3.1. Homology Modeling and Structural Aspects of H3R
2.3.2. Ligands Targeting H3R and Their Interactions
2.3.3. Multi-Target H3R Ligands
2.4. Computational Studies on H4R and Its Ligands
2.4.1. Homology Modeling and Structural Aspects of H4R
2.4.2. Ligands Targeting H4R and Their Interactions
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Buchan, N.S.; Rajpal, D.K.; Webster, Y.; Alatorre, C.; Gudivada, R.C.; Zheng, C.; Sanseau, P.; Koehler, J. The role of translational bioinformatics in drug discovery. Drug Discov. Today 2011, 16, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Ferreira, R.S.; Irwin, J.J.; Shoichet, B.K. Docking and chemoinformatic screens for new ligands and targets. Curr. Opin. Biotechnol. 2009, 20, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y. Hierarchical virtual screening approaches in small molecule drug discovery. Methods 2015, 71, 26–37. [Google Scholar] [CrossRef]
- Tuccinardi, T. Docking-based virtual screening: Recent developments. Comb. Chem. High Throughput Screen. 2009, 12, 303–314. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Jespers, W.; van Vlijmen, H.W.; IJzerman,, A.P.; van Westen, G.J. Interacting with GPCRs: Using Interaction Fingerprints for Virtual Screening. J. Chem. Inf. Model. 2016, 56, 2053–2060. [Google Scholar] [CrossRef]
- Vass, M.; Schmidt, E.; Horti, F.; Keseru, G.M. Virtual fragment screening on GPCRs: A case study on dopamine D3 and histamine H4 receptors. Eur. J. Med. Chem. 2014, 77, 38–46. [Google Scholar] [CrossRef]
- Tian, S.; Wang, X.; Li, L.; Zhang, X.; Li, Y.; Zhu, F.; Huo, T.; Zhen, X. Discovery of Novel and Selective Adenosine A2A Receptor Antagonists for Treating Parkinson’s Disease through Comparative Structure-based Virtual Screening. J. Chem. Inf. Model. 2017, 57, 1474–1487. [Google Scholar] [CrossRef]
- Szollosi, E.; Bobok, A.; Kiss, L.; Vass, M.; Kurko, D.; Kolok, S.; Visegrady, A.; Gyorgy, M.; Keseru, G.M. Cell-based and virtual fragment screening for adrenergic α2C receptor agonists. Bioorg. Med. Chem. 2015, 23, 3991–3999. [Google Scholar] [CrossRef][Green Version]
- Kooistra, A.J.; Vischer, H.F.; McNaught-Flores, D.; Leurs, R.; de Esch, I.J.; de Graaf, C. Function-specific virtual screening for GPCR ligands using a combined scoring method. Sci. Rep. 2016, 6, 28288. [Google Scholar] [CrossRef] [PubMed]
- Shonberg, J.; Kling, R.C.; Gmeiner, P.; Lober, S. GPCR crystal structures: Medicinal chemistry in the pocket. Bioorg. Med. Chem. 2015, 23, 3880–3906. [Google Scholar] [CrossRef]
- Zaid, H.; Raiyn, J.; Osman, M.; Falah, M.; Srouji, S.; Rayan, A. In silico modeling techniques for predicting the tertiary structure of human H4 receptor. Front. Biosci. (Landmark Ed.) 2016, 21, 597–619. [Google Scholar] [CrossRef]
- Pappalardo, M.; Shachaf, N.; Basile, L.; Milardi, D.; Zeidan, M.; Raiyn, J.; Guccione, S.; Rayan, A. Sequential application of ligand and structure based modeling approaches to index chemicals for their hH4R antagonism. PLoS ONE 2014, 9, e109340. [Google Scholar] [CrossRef]
- Ehling, S.; Rossbach, K.; Dunston, S.M.; Stark, H.; Baumer, W. Allergic inflammation is augmented via histamine H4 receptor activation: The role of natural killer cells in vitro and in vivo. J. Dermatol. Sci. 2016, 83, 106–115. [Google Scholar] [CrossRef]
- Mommert, S.; Kleiner, S.; Gehring, M.; Eiz-Vesper, B.; Stark, H.; Gutzmer, R.; Werfel, T.; Raap, U. Human basophil chemotaxis and activation are regulated via the histamine H4 receptor. Allergy 2016, 71, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Nicoud, M.B.; Formoso, K.; Medina, V.A. Pathophysiological Role of Histamine H4 Receptor in Cancer: Therapeutic Implications. Front. Pharmacol. 2019, 10, 556. [Google Scholar] [CrossRef]
- Sanna, M.D.; Stark, H.; Lucarini, L.; Ghelardini, C.; Masini, E.; Galeotti, N. Histamine H4 receptor activation alleviates neuropathic pain through differential regulation of ERK, JNK, and P38 MAPK phosphorylation. Pain 2015, 156, 2492–2504. [Google Scholar] [CrossRef] [PubMed]
- de Esch, I.J.; Thurmond, R.L.; Jongejan, A.; Leurs, R. The histamine H4 receptor as a new therapeutic target for inflammation. Trends Pharmacol. Sci. 2005, 26, 462–469. [Google Scholar] [CrossRef]
- Salem, A.; Almahmoudi, R.; Listyarifah, D.; Siponen, M.; Maaninka, K.; Al-Samadi, A.; Salo, T.; Eklund, K.K. Histamine H4 receptor signalling in tongue cancer and its potential role in oral carcinogenesis—A short report. Cell. Oncol. 2017, 40, 621–630. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Zoheir, K.M.; Abdel-Hamied, H.E.; Alrashidi, I.; Attia, S.M.; Bakheet, S.A.; Ashour, A.E.; Abd-Allah, A.R. Role of a histamine 4 receptor as an anti-inflammatory target in carrageenan-induced pleurisy in mice. Immunology 2014, 142, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Ye, F.; Ijima, R.; Kachi, S.; Kato, S.; Nagaya, M.; Higuchi, A.; Terasaki, H. Histamine H4 receptor as a new therapeutic target for choroidal neovascularization in age-related macular degeneration. Br. J. Pharmacol. 2014, 171, 3754–3763. [Google Scholar] [CrossRef]
- Czerner, C.P.; Klos, A.; Seifert, R.; Neumann, D. Histamine induces chemotaxis and phagocytosis in murine bone marrow-derived macrophages and RAW 264.7 macrophage-like cells via histamine H4-receptor. Inflamm. Res. 2014, 63, 239–247. [Google Scholar] [CrossRef]
- Labeeuw, O.; Levoin, N.; Billot, X.; Danvy, D.; Calmels, T.; Krief, S.; Ligneau, X.; Berrebi-Bertrand, I.; Robert, P.; Lecomte, J.M.; et al. Synthesis and evaluation of a 2-benzothiazolylphenylmethyl ether class of histamine H4 receptor antagonists. Bioorg. Med. Chem. Lett. 2016, 26, 5263–5266. [Google Scholar] [CrossRef]
- Levoin, N.; Labeeuw, O.; Billot, X.; Calmels, T.; Danvy, D.; Krief, S.; Berrebi-Bertrand, I.; Lecomte, J.M.; Schwartz, J.C.; Capet, M. Discovery of nanomolar ligands with novel scaffolds for the histamine H4 receptor by virtual screening. Eur. J. Med. Chem. 2017, 125, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Lazewska, D.; Mogilski, S.; Hagenow, S.; Kuder, K.; Gluch-Lutwin, M.; Siwek, A.; Wiecek, M.; Kaleta, M.; Seibel, U.; Buschauer, A.; et al. Alkyl derivatives of 1,3,5-triazine as histamine H4 receptor ligands. Bioorg. Med. Chem. 2019, 27, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Jojart, B.; Schmidt, E.; Kiss, B.; Keseru, G.M. Identification of Novel Histamine H4 Ligands by Virtual Screening on Molecular Dynamics Ensembles. Mol. Inform. 2014, 33, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.; Kim, H.J.; Ho, P.S.; Lee, S.O.; Lee, J.E.; Min, C.R.; Kim, Y.C.; Yoon, J.H.; Park, E.J.; Kwon, Y.J.; et al. Discovery of a Novel Highly Selective Histamine H4 Receptor Antagonist for the Treatment of Atopic Dermatitis. J. Med. Chem. 2018, 61, 2949–2961. [Google Scholar] [CrossRef]
- Schultes, S.; Kooistra, A.J.; Vischer, H.F.; Nijmeijer, S.; Haaksma, E.E.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. Combinatorial Consensus Scoring for Ligand-Based Virtual Fragment Screening: A Comparative Case Study for Serotonin 5-HT3A, Histamine H1, and Histamine H4 Receptors. J. Chem. Inf. Model. 2015, 55, 1030–1044. [Google Scholar] [CrossRef]
- Lazewska, D.; Kuder, K.; Kononowicz, K. Monocyclic and Fused Azines and Azoles as Histamine H4 Receptor Ligands. Curr. Med. Chem. 2016, 23, 1870–1925. [Google Scholar] [CrossRef]
- Correa, M.F.; Barbosa, A.J.R.; Teixeira, L.B.; Duarte, D.A.; Simoes, S.C.; Parreiras, E.S.L.T.; Balbino, A.M.; Landgraf, R.G.; Bouvier, M.; Costa-Neto, C.M.; et al. Pharmacological Characterization of 5-Substituted 1-[(2,3-dihydro-1-benzofuran-2-yl)methyl]piperazines: Novel Antagonists for the Histamine H3 and H4 Receptors with Anti-inflammatory Potential. Front. Pharmacol. 2017, 8, 825. [Google Scholar] [CrossRef]
- Watanabe, M.; Kobayashi, T.; Ito, Y.; Fukuda, H.; Yamada, S.; Arisawa, M.; Shuto, S. Design and synthesis of histamine H3/H4 receptor ligands with a cyclopropane scaffold. Bioorg. Med. Chem. Lett. 2018, 28, 3630–3633. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.G.; Gobleder, S.; Naporra, F.; Wittmann, H.J.; Elz, S.; Heinrich, M.R.; Strasser, A. 2,4-Diaminopyrimidines as dual ligands at the histamine H1 and H4 receptor-H1/H4-receptor selectivity. Bioorg. Med. Chem. Lett. 2016, 26, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Bao, A.M.; Swaab, D.F. The human histaminergic system in neuropsychiatric disorders. Trends Neurosci. 2015, 38, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, M.F.; Fernandes, J.P.d.S. Histamine H4 receptor ligands: Future applications and state of art. Chem. Biol. Drug Des. 2015, 85, 461–480. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon, S.C., Ed.; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Zhu, Y.; Michalovich, D.; Wu, H.-L.; Tan, K.B.; Dytko, G.M.; Mannan, I.J.; Boyce, R.; Alston, J.; Tierney, L.A.; Li, X.; et al. Cloning, Expression, and Pharmacological Characterization of a Novel Human Histamine Receptor. Mol. Pharmacol. 2001, 59, 434–441. [Google Scholar] [CrossRef]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.; Stark, H.; Thurmond, R.L.; Haas, H.L. International union of basic and clinical pharmacology. XCVIII. Histamine receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef] [PubMed]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs—Theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef]
- Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.S. Construction of a sequence motif characteristic of aminergic G protein–coupled receptors. Protein Sci. 2003, 12, 1360–1367. [Google Scholar] [CrossRef]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef]
- Shimamura, T.; Shiroishi, M.; Weyand, S.; Tsujimoto, H.; Winter, G.; Katritch, V.; Abagyan, R.; Cherezov, V.; Liu, W.; Han, G.W.; et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. [Google Scholar] [CrossRef]
- Kooistra, A.J.; de Graaf, C.; Timmerman, H. The receptor concept in 3D: From hypothesis and metaphor to GPCR-ligand structures. Neurochem. Res. 2014, 39, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Tafi, A.; Desaubry, L.; Nebigil, C.G. Discovery of GPCR ligands for probing signal transduction pathways. Front. Pharmacol. 2014, 5, 255. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Dias, J.M.; Marshall, F.H. Structure-based drug design for G protein-coupled receptors. Prog. Med. Chem. 2014, 53, 1–63. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Leurs, R.; de Esch, I.J.; de Graaf, C. From three-dimensional GPCR structure to rational ligand discovery. Adv. Exp. Med. Biol. 2014, 796, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Zobayer, N.; Hossain, A.B.M.A. In silico Characterization and Homology Modeling of Histamine Receptors. J. Biol. Sci. 2018, 18, 178–191. [Google Scholar] [CrossRef]
- Pandy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsoe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef]
- Chovatiya, R.; Georrge, J.J. Identification of potential phytochemical inhibitors for the treatment of allergic asthma from the medicinal plants. In Proceedings of the 9th National Level Science Symposium, Rajkot, Gujarat, India, 14 February 2016; Volume 3, pp. 277–285. [Google Scholar]
- Lepailleur, A.; Freret, T.; Lemaitre, S.; Boulouard, M.; Dauphin, F.; Hinschberger, A.; Dulin, F.; Lesnard, A.; Bureau, R.; Rault, S. Dual histamine H3R/serotonin 5-HT4R ligands with antiamnesic properties: Pharmacophore-based virtual screening and polypharmacology. J. Chem. Inf. Model. 2014, 54, 1773–1784. [Google Scholar] [CrossRef]
- Frandsen, I.O.; Boesgaard, M.W.; Fidom, K.; Hauser, A.S.; Isberg, V.; Brauner-Osborne, H.; Wellendorph, P.; Gloriam, D.E. Identification of Histamine H3 Receptor Ligands Using a New Crystal Structure Fragment-based Method. Sci. Rep. 2017, 7, 4829. [Google Scholar] [CrossRef]
- Ghamari, N.; Zarei, O.; Reiner, D.; Dastmalchi, S.; Stark, H.; Hamzeh-Mivehroud, M. Histamine H3 receptor ligands by hybrid virtual screening, docking, molecular dynamics simulations, and investigation of their biological effects. Chem. Biol. Drug Des. 2019, 93, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Schaller, D.; Hagenow, S.; Stark, H.; Wolber, G. Ligand-guided homology modeling drives identification of novel histamine H3 receptor ligands. PLoS ONE 2019, 14, e0218820. [Google Scholar] [CrossRef]
- Istyastono, E.P.; Kooistra, A.J.; Vischer, H.F.; Kuijer, M.; Roumen, L.; Nijmeijer, S.; Smits, R.A.; de Esch, I.J.P.; Leurs, R.; de Graaf, C. Structure-based virtual screening for fragment-like ligands of the G protein-coupled histamine H4 receptor. MedChemComm 2015, 6, 1003–1017. [Google Scholar] [CrossRef]
- Herrera-Zuniga, L.D.; Moreno-Vargas, L.M.; Ballaud, L.; Correa-Basurto, J.; Prada-Gracia, D.; Pastre, D.; Curmi, P.A.; Arrang, J.M.; Maroun, R.C. A complex view of GPCR signaling: Multidimensional analysis of extended molecular dynamics simulations reveals the complexity of signal transduction by the histamine H3 membrane receptor. bioRxiv 2019. [Google Scholar] [CrossRef]
- Sader, S.; Cai, J.; Muller, A.C.G.; Wu, C. Can human allergy drug fexofenadine, an antagonist of histamine (H1) receptor, be used to treat dog and cat? Homology modeling, docking and molecular dynamic Simulation of three H1 receptors in complex with fexofenadine. J. Mol. Graph. Model. 2017, 75, 106–116. [Google Scholar] [CrossRef]
- Riza, Y.M.; Parves, M.R.; Tithi, F.A.; Alam, S. Quantum chemical calculation and binding modes of H1R; a combined study of molecular docking and DFT for suggesting therapeutically potent H1R antagonist. In Silico Pharmacol. 2019, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Darras, F.H.; Pockes, S.; Huang, G.; Wehle, S.; Strasser, A.; Wittmann, H.J.; Nimczick, M.; Sotriffer, C.A.; Decker, M. Synthesis, biological evaluation, and computational studies of Tri- and tetracyclic nitrogen-bridgehead compounds as potent dual-acting AChE inhibitors and hH3 receptor antagonists. ACS Chem. Neurosci. 2014, 5, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, H.J.; Seifert, R.; Strasser, A. Mathematical analysis of the sodium sensitivity of the human histamine H3 receptor. In Silico Pharmacol. 2014, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Al-Jomaily, M.; Arafat, T.; Al-Kaissi, E.; Ghattas, M.; Muhi-Eldeen, Z. Synthesis of amino acetylenic benzophenone derivatives as H3-antagonists. Int. J. Pharm. Pharm. Sci. 2015, 7, 174–179. [Google Scholar]
- Hauwert, N.J.; Mocking, T.A.M.; Da Costa Pereira, D.; Kooistra, A.J.; Wijnen, L.M.; Vreeker, G.C.M.; Verweij, E.W.E.; De Boer, A.H.; Smit, M.J.; De Graaf, C.; et al. Synthesis and Characterization of a Bidirectional Photoswitchable Antagonist Toolbox for Real-Time GPCR Photopharmacology. J. Am. Chem. Soc. 2018, 140, 4232–4243. [Google Scholar] [CrossRef]
- Szczepanska, K.; Karcz, T.; Siwek, A.; Kuder, K.J.; Latacz, G.; Bednarski, M.; Szafarz, M.; Hagenow, S.; Lubelska, A.; Olejarz-Maciej, A.; et al. Structural modifications and in vitro pharmacological evaluation of 4-pyridyl-piperazine derivatives as an active and selective histamine H3 receptor ligands. Bioorg. Chem. 2019, 91, 103071. [Google Scholar] [CrossRef]
- Kumar, A.; Pasam, V.R.; Thakur, R.K.; Singh, M.; Singh, K.; Shukla, M.; Yadav, A.; Dogra, S.; Sona, C.; Umrao, D.; et al. Novel Tetrahydroquinazolinamines as Selective Histamine 3 Receptor Antagonists for the Treatment of Obesity. J. Med. Chem. 2019, 62, 4638–4655. [Google Scholar] [CrossRef]
- Geyer, R.; Nordemann, U.; Strasser, A.; Wittmann, H.-J.; Buschauer, A. Conformational restriction and enantioseparation increase potency and selectivity of cyanoguanidine-type histamine H4 receptor agonists. J. Med. Chem. 2016, 59, 3452–3470. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Y.; Pan, Y.; Wang, J.; Lin, F.; Wang, C.; Zhang, S.; Yang, L. Computational Analysis of Structure-Based Interactions for Novel H(1)-Antihistamines. Int. J. Mol. Sci. 2016, 17, 129. [Google Scholar] [CrossRef] [PubMed]
- Siddalingamurthy, E.; Mahadevan, K.M.; Jagadeesh, N.M.; Kumara, M.N. Synthesis of Indolecarboxamides and Their Docking Studies with H1, 5HT and CCR2 Antagonist Receptors. Am. J. Pharm. Health Res. 2014, 2, 245–258. [Google Scholar]
- Saxena, M.; Bhunia, S.S.; Saxena, A.K. Molecular modelling studies on 2-substituted octahydropyrazinopyridoindoles for histamine H2 receptor antagonism. SAR QSAR Environ. Res. 2015, 26, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Szczepanska, K.; Karcz, T.; Mogilski, S.; Siwek, A.; Kuder, K.J.; Latacz, G.; Kubacka, M.; Hagenow, S.; Lubelska, A.; Olejarz, A.; et al. Synthesis and biological activity of novel tert-butyl and tert-pentylphenoxyalkyl piperazine derivatives as histamine H3R ligands. Eur. J. Med. Chem. 2018, 152, 223–234. [Google Scholar] [CrossRef]
- Jonczyk, J.; Lodarski, K.; Staszewski, M.; Godyń, J.; Zaręba, P.; Soukup, O.; Janockova, J.; Korabecny, J.; Sałat, K.; Malikowska-Racia, N.; et al. Search for multifunctional agents against Alzheimer’s disease among non-imidazole histamine H3 receptor ligands. In vitro and in vivo pharmacological evaluation and computational studies of piperazine derivatives. Bioorg. Chem. 2019, 90, 103084. [Google Scholar] [CrossRef]
- Siddalingamurthy, E.; Mahadevan, K.M.; Jagadeesh, N.M.; Kumara, M.N. Synthesis and docking study of 3-(N-Alkyl/Aryl piperidyl) indoles with serotonin-5HT, H1 and CCR2 antagonist receptors. Int. J. Pharm. Pharm. Sci. 2014, 6, 475–482. [Google Scholar]
- Elbayaa, R.Y. Computer-aided Design, Synthesis, and Biological Evaluation of 5-Substituted Aminomethylenepyrimidine-2, 4, 6-Triones as H1 Antihistaminic Agents (Part2). Med. Chem. 2014, 10, 66–73. [Google Scholar] [CrossRef]
- Hariono, M.; Wahab, H.A. In Silico study of N1-alkyltheobromine as histamine-H1 receptor antagonist. Int. J. Life Sci. Biotech. Pharma Res. 2015, 4, 108. [Google Scholar]
- Kuhne, S.; Kooistra, A.J.; Bosma, R.; Bortolato, A.; Wijtmans, M.; Vischer, H.F.; Mason, J.S.; de Graaf, C.; de Esch, I.J.; Leurs, R. Identification of Ligand Binding Hot Spots of the Histamine H1 Receptor following Structure-Based Fragment Optimization. J. Med. Chem. 2016, 59, 9047–9061. [Google Scholar] [CrossRef]
- Sureshkumar, K.; Maheshwaran, V.; Dharma Rao, T.; Themmila, K.; Ponnuswamy, M.N.; Kadhirvel, S.; Dhandayutham, S. Synthesis, characterization, crystal structure, in-vitro anti-inflammatory and molecular docking studies of 5-mercapto-1-substituted tetrazole incorporated quinoline derivative. J. Mol. Struct. 2017, 1146, 314–323. [Google Scholar] [CrossRef]
- Stoddart, L.A.; Vernall, A.J.; Bouzo-Lorenzo, M.; Bosma, R.; Kooistra, A.J.; de Graaf, C.; Vischer, H.F.; Leurs, R.; Briddon, S.J.; Kellam, B.; et al. Development of novel fluorescent histamine H1-receptor antagonists to study ligand-binding kinetics in living cells. Sci. Rep. 2018, 8, 1572. [Google Scholar] [CrossRef]
- Soldner, C.A.; Horn, A.H.C.; Sticht, H. Binding of histamine to the H1 receptor—A molecular dynamics study. J. Mol. Model. 2018, 24, 346. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Hage-Melim, L.I.; Poiani, J.G.C.; de Paula da Silva, C.H.T.; Boylan, F. In silico study of the mechanism of action, pharmacokinetic and toxicological properties of some N-methylanthranilates and their analogs. Food Chem. Toxicol. 2019, 131, 110556. [Google Scholar] [CrossRef]
- Pockes, S.; Wifling, D.; Keller, M.; Buschauer, A.; Elz, S. Highly Potent, Stable, and Selective Dimeric Hetarylpropylguanidine-Type Histamine H2 Receptor Agonists. ACS Omega 2018, 3, 2865–2882. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, O.M.; Hagenow, S.; Palomino-Antolin, A.; Farre-Alins, V.; Ismaili, L.; Joffrin, P.L.; Jimeno, M.L.; Soukup, O.; Janockova, J.; Kalinowsky, L.; et al. Multitarget-Directed Ligands Combining Cholinesterase and Monoamine Oxidase Inhibition with Histamine H3 R Antagonism for Neurodegenerative Diseases. Angew. Chem. Int. Ed. Engl. 2017, 56, 12765–12769. [Google Scholar] [CrossRef]
- Wen, G.; Liu, Q.; Hu, H.; Wang, D.; Wu, S. Design, synthesis, biological evaluation, and molecular docking of novel flavones as H3R inhibitors. Chem. Biol. Drug Des. 2017, 90, 580–589. [Google Scholar] [CrossRef]
- Jonczyk, J.; Malawska, B.; Bajda, M. Hybrid approach to structure modeling of the histamine H3 receptor: Multi-level assessment as a tool for model verification. PLoS ONE 2017, 12, e0186108. [Google Scholar] [CrossRef]
- Wagner, G.; Mocking, T.A.M.; Arimont, M.; Provensi, G.; Rani, B.; Silva-Marques, B.; Latacz, G.; Da Costa Pereira, D.; Karatzidou, C.; Vischer, H.F.; et al. 4-(3-Aminoazetidin-1-yl)pyrimidin-2-amines as High-Affinity Non-imidazole Histamine H3 Receptor Agonists with in Vivo Central Nervous System Activity. J. Med. Chem. 2019, 62, 10848–10866. [Google Scholar] [CrossRef] [PubMed]
- Wifling, D.; Löffel, K.; Nordemann, U.; Strasser, A.; Bernhardt, G.; Dove, S.; Seifert, R.; Buschauer, A. Molecular determinants for the high constitutive activity of the human histamine H4 receptor: Functional studies on orthologues and mutants. Br. J. Pharmacol. 2015, 172, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Shobana, S. Virtual Screening, Pharmacophore Modeling, and Quantitative Structure Activity Relationship Studies on Histamine 4 Receptor. Asian J. Pharm. Clin. Res. 2017, 10, 150–154. [Google Scholar] [CrossRef][Green Version]
- Jakubik, J.; El-Fakahany, E.E.; Dolezal, V. Towards predictive docking at aminergic G-protein coupled receptors. J. Mol. Model. 2015, 21, 284. [Google Scholar] [CrossRef]
- Krzan, M.; Vianello, R.; Marsavelski, A.; Repic, M.; Zaksek, M.; Kotnik, K.; Fijan, E.; Mavri, J. The Quantum Nature of Drug-Receptor Interactions: Deuteration Changes Binding Affinities for Histamine Receptor Ligands. PLoS ONE 2016, 11, e0154002. [Google Scholar] [CrossRef] [PubMed]
- Correa, M.F.; Balico-Silva, A.L.; Kiss, D.J.; Fernandes, G.A.B.; Maraschin, J.C.; Parreiras, E.S.L.T.; Varela, M.T.; Simoes, S.C.; Bouvier, M.; Keseru, G.M.; et al. Novel potent (dihydro)benzofuranyl piperazines as human histamine receptor ligands—Functional characterization and modeling studies on H3 and H4 receptors. Bioorg Med Chem 2021, 30, 115924. [Google Scholar] [CrossRef]
- Shankaran, K.S.; Ganai, S.A.; Arun, K.P.; Brindha, P.; Mahadevan, V. In silico and In vitro evaluation of the anti-inflammatory potential of Centratherum punctatum Cass-A. J. Biomol. Struct. Dyn. 2017, 35, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Kuder, K.J.; Lazewska, D.; Kaleta, M.; Latacz, G.; Kottke, T.; Olejarz, A.; Karcz, T.; Fruzinski, A.; Szczepanska, K.; Karolak-Wojciechowska, J.; et al. Synthesis and biological activity of novel tert-amylphenoxyalkyl (homo)piperidine derivatives as histamine H3R ligands. Bioorg. Med. Chem. 2017, 25, 2701–2712. [Google Scholar] [CrossRef] [PubMed]
- Szczepanska, K.; Karcz, T.; Kotanska, M.; Siwek, A.; Kuder, K.J.; Latacz, G.; Mogilski, S.; Hagenow, S.; Lubelska, A.; Sobolewski, M.; et al. Optimization and preclinical evaluation of novel histamine H3 receptor ligands: Acetyl and propionyl phenoxyalkyl piperazine derivatives. Bioorg. Med. Chem. 2018, 26, 6056–6066. [Google Scholar] [CrossRef]
- Xin, J.; Hu, M.; Liu, Q.; Zhang, T.T.; Wang, D.M.; Wu, S. Design, synthesis, and biological evaluation of novel iso-flavones derivatives as H3R antagonists. J. Enzyme Inhib. Med. Chem. 2018, 33, 1545–1553. [Google Scholar] [CrossRef]
- Hamzeh-Mivehroud, M.; Khoshravan-Azar, Z.; Dastmalchi, S. QSAR and Molecular Docking Studies on Non-Imidazole-Based Histamine H3 Receptor Antagonists. Pharm. Sci. 2020, 26, 165–174. [Google Scholar] [CrossRef]
- Kaminska, K.; Ziemba, J.; Ner, J.; Schwed, J.S.; Lazewska, D.; Wiecek, M.; Karcz, T.; Olejarz, A.; Latacz, G.; Kuder, K.; et al. (2-Arylethenyl)-1,3,5-triazin-2-amines as a novel histamine H4 receptor ligands. Eur. J. Med. Chem. 2015, 103, 238–251. [Google Scholar] [CrossRef]
- Correa, M.F.; Dos Santos Fernandes, J.P. QSAR Modeling of Histamine H3R Antagonists/inverse Agonists as Future Drugs for Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 749–757. [Google Scholar] [CrossRef]
- Lazewska, D.; Jonczyk, J.; Bajda, M.; Szalaj, N.; Wieckowska, A.; Panek, D.; Moore, C.; Kuder, K.; Malawska, B.; Kiec-Kononowicz, K. Cholinesterase inhibitory activity of chlorophenoxy derivatives-Histamine H3 receptor ligands. Bioorg. Med. Chem. Lett. 2016, 26, 4140–4145. [Google Scholar] [CrossRef] [PubMed]
- Kuder, K.; Lazewska, D.; Latacz, G.; Schwed, J.S.; Karcz, T.; Stark, H.; Karolak-Wojciechowska, J.; Kiec-Kononowicz, K. Chlorophenoxy aminoalkyl derivatives as histamine H(3)R ligands and antiseizure agents. Bioorg. Med. Chem. 2016, 24, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Wifling, D.; Bernhardt, G.; Dove, S.; Buschauer, A. The Extracellular Loop 2 (ECL2) of the Human Histamine H4 Receptor Substantially Contributes to Ligand Binding and Constitutive Activity. PLoS ONE 2015, 10, e0117185. [Google Scholar] [CrossRef]
- Karthic, V.M.; Poongodi, B.; Shanmugapriya, P.; Sivaraman, D. In-silico molecular docking analysis of potential phytotherapeutics from the medicinal herb Corallocarpus Epigaeus for treating urticaria. Int. J. Trans. Res. Ind. Med. 2019, 1, 5–12. [Google Scholar]
- Bosma, R.; Wang, Z.; Kooistra, A.J.; Bushby, N.; Kuhne, S.; van den Bor, J.; Waring, M.J.; de Graaf, C.; de Esch, I.J.; Vischer, H.F.; et al. Route to Prolonged Residence Time at the Histamine H1 Receptor: Growing from Desloratadine to Rupatadine. J. Med. Chem. 2019, 62, 6630–6644. [Google Scholar] [CrossRef]
- Soldner, C.A.; Horn, A.H.C.; Sticht, H. A Metadynamics-Based Protocol for the Determination of GPCR-Ligand Binding Modes. Int. J. Mol. Sci. 2019, 20, 1970. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Che, D.; Wei, D.; Wang, C.; Xie, Y.; Zhang, K.; Cao, J.; Fu, J.; Zhou, N.; He, H. Phenothiazine antipsychotics exhibit dual properties in pseudo-allergic reactions: Activating MRGPRX2 and inhibiting the H1 receptor. Mol. Immunol. 2019, 111, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Snyder, G.L.; Vanover, K.E. Dopamine Targeting Drugs for the Treatment of Schizophrenia: Past, Present and Future. Curr. Top. Med. Chem. 2016, 16, 3385–3403. [Google Scholar] [CrossRef] [PubMed]
- Stepnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [PubMed]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L. Harnessing Polypharmacology with Medicinal Chemistry. ACS Med. Chem. Lett. 2019, 10, 273–275. [Google Scholar] [CrossRef]
- Mota, F.V.B.; de Araujo Neta, M.S.; de Souza Franco, E.; Bastos, I.; da Araujo, L.C.C.; da Silva, S.C.; de Oliveira, T.B.; Souza, E.K.; de Almeida, V.M.; Ximenes, R.M.; et al. Evaluation of anti-inflammatory activity and molecular docking study of new aza-bicyclic isoxazoline acylhydrazone derivatives. MedChemComm 2019, 10, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Sumathi, M.; Shanmugapriya, C.; Meenakumari, R. Evaluation of anti-inflammatory, anti-allergic and immune modulatory potential of siddha formulation Oma Legium by computational docking analysis. Eur. J. Biomed. Pharm. Sci. 2017, 4, 549–556. [Google Scholar]
- Daddam, J.R.; Sreenivasulu, B.; Peddanna, K.; Umamahesh, K. Designing, docking and molecular dynamics simulation studies of novel cloperastine analogues as anti-allergic agents: Homology modeling and active site prediction for the human histamine H1 receptor. RSC Adv. 2020, 10, 4745–4754. [Google Scholar] [CrossRef]
- Gurjar, V.K.; Pal, D. Design, in silico studies, and synthesis of new 1,8-naphthyridine-3-carboxylic acid analogues and evaluation of their H1R antagonism effects. RSC Adv. 2020, 10, 13907–13921. [Google Scholar] [CrossRef]
- Mobarec, J.C.; Filizola, M. Advances in the Development and Application of Computational Methodologies for Structural Modeling of G-Protein Coupled Receptors. Expert. Opin. Drug Discov. 2008, 3, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, R.; Heim, A.J.; Li, Z. Improving homology modeling of G-protein coupled receptors through multiple-template derived conserved inter-residue interactions. J. Comput. Aided Mol. Des. 2015, 29, 413–420. [Google Scholar] [CrossRef]
- Costanzi, S.; Wang, K. The GPCR crystallography boom: Providing an invaluable source of structural information and expanding the scope of homology modeling. Adv. Exp. Med. Biol. 2014, 796, 3–13. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2017, 1654, 39–54. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Chaudhary, A.; Yadav, B.S.; Singh, S.; Maurya, P.K.; Mishra, A.; Srivastva, S.; Varadwaj, P.K.; Singh, N.K.; Mani, A. Docking-based Screening of Ficus religiosa Phytochemicals as Inhibitors of Human Histamine H2 Receptor. Pharmacogn. Mag. 2017, 13, S706–S714. [Google Scholar] [CrossRef]
- Singh, V.; Gohil, N.; Ramirez-Garcia, R. New insight into the control of peptic ulcer by targeting the histamine H2 receptor. J. Cell. Biochem. 2018, 119, 2003–2011. [Google Scholar] [CrossRef]
- Boddupally, S.; Gunda, S.K.; Naga Harini, P.; Shaik, M. In silico predictive studies of histamine H2 receptor protein binding using homology modelling and molecular docking. Res. J. Life Sci. Bioinf. Pharm. Chem. Sci. 2019, 5, 500–514. [Google Scholar] [CrossRef]
- Lazewska, D.; Bajda, M.; Kaleta, M.; Zareba, P.; Doroz-Plonka, A.; Siwek, A.; Alachkar, A.; Mogilski, S.; Saad, A.; Kuder, K.; et al. Rational design of new multitarget histamine H3 receptor ligands as potential candidates for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2020, 207, 112743. [Google Scholar] [CrossRef]
- Bajda, M.; Lazewska, D.; Godyn, J.; Zareba, P.; Kuder, K.; Hagenow, S.; Latka, K.; Stawarska, E.; Stark, H.; Kiec-Kononowicz, K.; et al. Search for new multi-target compounds against Alzheimer’s disease among histamine H3 receptor ligands. Eur. J. Med. Chem. 2020, 185, 111785. [Google Scholar] [CrossRef]
- Yuan, S.; Filipek, S.; Palczewski, K.; Vogel, H. Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nat. Commun. 2014, 5, 4733. [Google Scholar] [CrossRef]
- Yuan, S.; Hu, Z.; Filipek, S.; Vogel, H. W246(6.48) opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew. Chem. Int. Ed. 2015, 54, 556–559. [Google Scholar] [CrossRef]
- Kim, S.K.; Fristrup, P.; Abrol, R.; Goddard, W.A., 3rd. Structure-based prediction of subtype selectivity of histamine H3 receptor selective antagonists in clinical trials. J. Chem. Inf. Model. 2011, 51, 3262–3274. [Google Scholar] [CrossRef]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Ghamari, N.; Dastmalchi, S.; Zarei, O.; Arias-Montaño, J.A.; Reiner, D.; Ustun-Alkan, F. In silico and in vitro studies of two non-imidazole multiple targeting agents at histamine H3 receptors and cholinesterase enzymes. Chem. Biol. Drug Des. 2020, 95, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Sadek, B.; Stark, H. Cherry-picked ligands at histamine receptor subtypes. Neuropharmacology 2016, 106, 56–73. [Google Scholar] [CrossRef]
- Jayaraju, A.; Sreeramulu, J. Synthesis, insilico studies and biological evaluation of pramepexole carbamodithiolate metal complexes. J. Chem. Pharm. Res. 2016, 8, 884–890. [Google Scholar]
- Miszta, P.; Pasznik, P.; Jakowiecki, J.; Sztyler, A.; Latek, D.; Filipek, S. GPCRM: A homology modeling web service with triple membrane-fitted quality assessment of GPCR models. Nucleic Acids Res. 2018, 46, W387–W395. [Google Scholar] [CrossRef] [PubMed]
- Schihada, H.; Ma, X.; Zabel, U.; Vischer, H.F.; Schulte, G.; Leurs, R. Development of a Conformational Histamine H3 Receptor Biosensor for the Synchronous Screening of Agonists and Inverse Agonists. ACS Sens. 2020, 5, 1734–1742. [Google Scholar] [CrossRef]
- Song, M.; Yan, R.; Zhang, Y.; Guo, D.; Zhou, N.; Deng, X. Design, synthesis, and anticonvulsant effects evaluation of nonimidazole histamine H3 receptor antagonists/inverse agonists containing triazole moiety. J. Enzyme Inhib. Med. Chem. 2020, 35, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.D.; de Graaf, C.; Jiang, W.; Sadek, P.; McGovern, P.M.; Istyastono, E.P.; Bakker, R.A.; de Esch, I.J.P.; Thurmond, R.L.; Leurs, R. Molecular determinants of ligand binding to H4R species variants. Mol. Pharmacol. 2010, 77, 734–743. [Google Scholar] [CrossRef]
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers—Aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. [Google Scholar] [CrossRef]
- Mehta, P.; Miszta, P.; Rzodkiewicz, P.; Michalak, O.; Krzeczyński, P.; Filipek, S. Enigmatic Histamine Receptor H4 for Potential Treatment of Multiple Inflammatory, Autoimmune, and Related Diseases. Life (Basel) 2020, 10, 50. [Google Scholar] [CrossRef]
- Woolley, M.J.; Conner, A.C. Understanding the common themes and diverse roles of the second extracellular loop (ECL2) of the GPCR super-family. Mol. Cell. Endocrinol. 2017, 449, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Mogilski, S.; Kubacka, M.; Łażewska, D.; Więcek, M.; Głuch-Lutwin, M.; Tyszka-Czochara, M.; Bukowska-Strakova, K.; Filipek, B.; Kieć-Kononowicz, K. Aryl-1,3,5-triazine ligands of histamine H4 receptor attenuate inflammatory and nociceptive response to carrageen, zymosan and lipopolysaccharide. Inflamm. Res. 2016, 66, 79–95. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Templates | Receptor State | Strategy | Hit Rate | Reference |
|---|---|---|---|---|---|
| H1R | β2-adrenergic (2R4R) | Inactive | SBVS of Phytochemical inhibitors | 5 hits | [48] |
| H1R | 3RZE | Inactive | IFP-based SBVS | 60.6% (20/33) | [9] |
| PLANTS-based SBVS | 45.5% (15/33) | ||||
| Combined IFP and PLANTS scoring based SBVS | 73.1% (19/26) | ||||
| H3R | H1R (3RZE) | Inactive | Pharmacophore-based virtual screening | 100% (5/5) | [49] |
| H3R | H1R (3RZE) | Inactive | Prospective crystal structure-based pharmacophore virtual screening | 8% (6/76) | [50] |
| H3R | M3 muscarinic receptor (4DAJ) | Inactive | FP2 fingerprint/Electroshape / Spectrophores/LBVS | 50% (2/4) | [51] |
| Hybrid VS | 100% (1/1) | ||||
| H3R | H1R (3RZE), M2 muscarinic (3UON), M3 muscarinic (4U15) | Inactive | Pharmacophore screening and redocking. | 25% (2/8) | [52] |
| H4R | H1R (3RZE) | Inactive | Single Structure | 22% (11/50) | [6] |
| Ensemble Docking | 16% (8/50) | ||||
| Overlap hits | 27% (4/15) | ||||
| H4R | H1R (3RZE) | Inactive | Ligand-based chemoinformatics: Intelligent Learning Engine/Iterative Stochastic Elimination/Extended connectivity fingerprint (ECFP4). | 11 hits | [12] |
| H4R | Bovine rhodopsin (1F88) | Inactive | Ensemble docking | 5.3% (4/75) | [25] |
| Ensemble docking followed by consensus scoring. | 15.4% (2/13) | ||||
| H4R | β2R (2RH1) | Inactive | Prospective SBVS. | 26% (6/23) | [53] |
| H1R (3RZE) | Inactive | Prospective SBVS. | 21.4% (3/14) | ||
| H4R | Bovine rhodopsin (1L9H) | Inactive | Homology model refined by “scout screening”, VS using ECFP_4 fingerprint. | 23% (28/120) | [22,23] |
| H4R | H1R (3RZE) | Inactive | Pharmacophore-based virtual screening (Tanimoto similarity coefficient ≥0.9). | 1% (3/291) | [26] |
| Residue | Residues | Reference | |||
|---|---|---|---|---|---|
| H1R | H2R | H3R | H4R | ||
| 2.50 | D80 | [49,54] | |||
| 2.61 | N84 | S75 | Y91 | Y72 | [26,55,56,57,58,59,60,61,62,63,64] |
| 2.62 | I85 | [31] | |||
| 2.64 | Y87 | Y78 | Y94 | H75 | [55,60,63] |
| 2.65 | L88 | [55] | |||
| ECL1 | W93 | [65] | |||
| 3.28 | W103 | Y94 | W110 | W90 | [54,55,59,62,63,66,67,68] |
| 3.29 | L104 | T95 | L111 | [55,57,62,66] | |
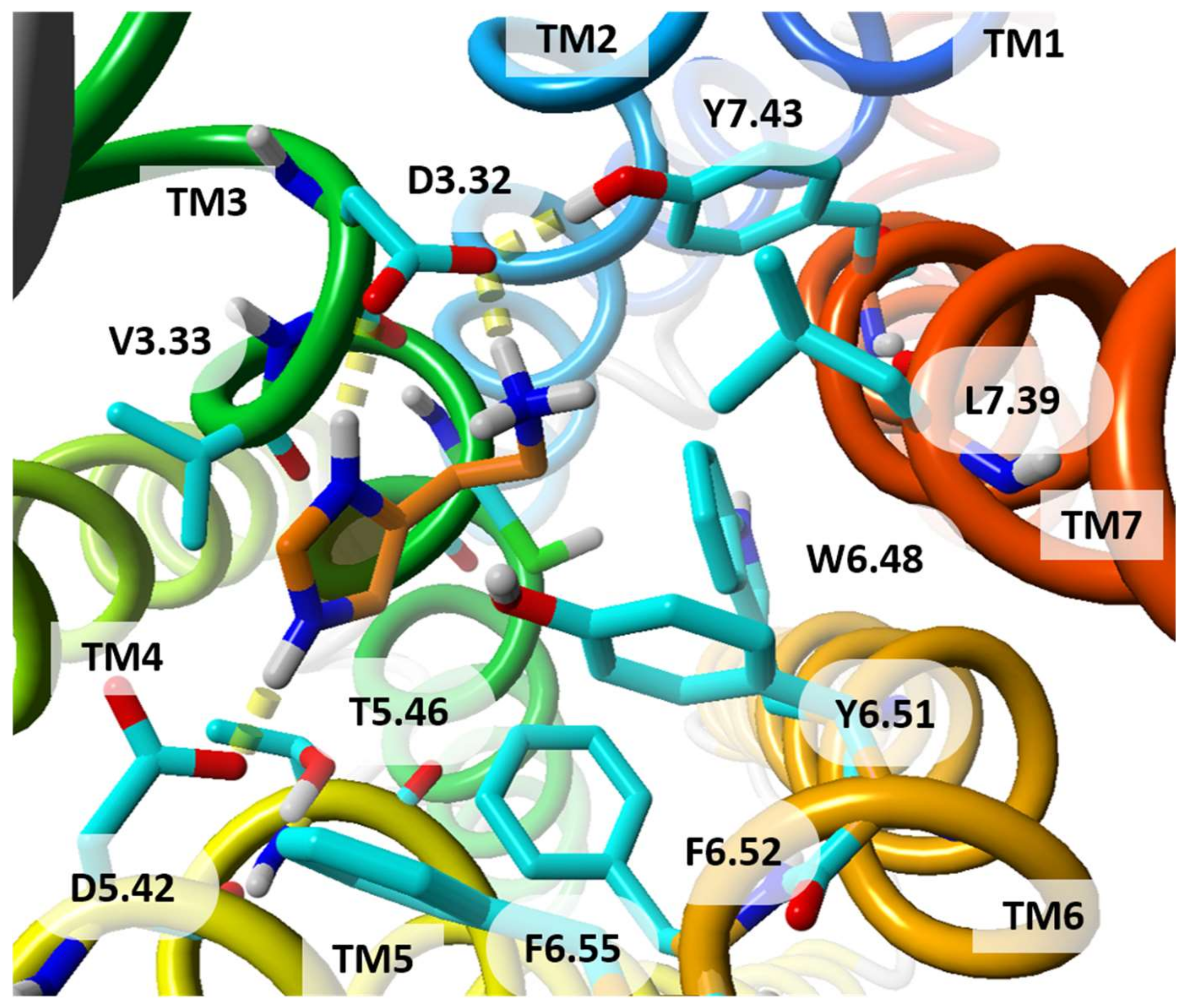
| 3.32 | D107 | D98 | D114 | D94 | [6,9,12,22,23,24,25,26,31,48,49,51,52,53,54,55,56,57,58,59,60,62,63,64,66,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86] |
| 3.33 | Y108 | V99 | Y115 | Y95 | [6,12,22,24,26,31,51,52,55,56,57,59,60,62,63,64,66,68,69,70,71,72,75,76,77,78,79,80,82,83,84,86,87,88,89,90,91,92] |
| 3.36 | S111 | C102 | C118 | C98 | [6,12,31,48,57,64,66,76,79,88,90] |
| 3.37 | T112 | T119 | T99 | [12,31,51,57,64,73,78,88] | |
| 3.40 | I115 | I106 | A122 | V102 | [31,55,57,64,76,78,88,89] |
| 3.41 | F116 | [31] | |||
| 4.56 | W158 | L149 | L166 | V146 | [9,31,52,55,56,57,63,64,66,72,73,76,78,90] |
| 4.57 | Y167 | N147 | [6,12,53,93] | ||
| 4.60 | S153 | M150 | [6,12,77] | ||
| 4.61 | I154 | [66] | |||
| ECL2 | N159 | E175 | [49,66] | ||
| ECL2 | H167 | [55] | |||
| ECL2 | R175 | [65] | |||
| ECL2 | R176 | [55,65,69] | |||
| ECL2 | T171 | [77] | |||
| ECL2 | D178 | S162 | [12,55,83] | ||
| ECL2 | K179 | H187 | E163 | [12,55,56,58,59,65,66,69,77,82,83] | |
| ECL2 | C180 | C174 | C164 | [12,55,66] | |
| ECL2 | E181 | K175 | Y189 | E165 | [12,51,55,66,67,68,80,85,88,91,94] |
| ECL2 | T182 | V176 | A190 | P166 | [12,48,55,62,66,77,80,94,95] |
| ECL2 | D183 | Q177 | E191 | [48,49,54,70,77,94,95] | |
| ECL2 | F184 | V178 | F192 | F168 | [6,12,26,31,53,54,66,77,78,80,82,96] |
| ECL2 | Y185 | F193 | F169 | [12,31,55,57,58,59,62,68,69,77,80,82,83,89] | |
| ECL2 | Y194 | [62,94,95] | |||
| ECL2 | W196 | [62,94,95] | |||
| 5.38 | F190 | Y182 | F198 | I174 | [6,12,31,51,57,59,63,66,88,90,94] |
| 5.39 | K191 | G183 | L199 | L175 | [6,12,26,31,53,55,56,57,62,63,66,69,70,71,87] |
| 5.42 | T194 | D186 | A202 | T178 | [6,12,56,57,63,64,66,75,77,79,82,85,87,90] |
| 5.43 | A195 | G187 | S203 | S179 | [12,26,31,55,57,63,64,66,82,83,87] |
| 5.46 | N198 | T190 | E206 | E182 | [6,12,23,24,25,26,31,49,50,51,52,53,57,58,59,60,61,63,64,65,66,67,68,69,75,76,77,79,80,81,82,83,85,86,87,88,89,90,91,92,93,94,95] |
| 5.47 | F199 | F207 | F183 | [9,12,26,31,55,64,67,72,76,90] | |
| 6.44 | F424 | F367 | V184 | [66,72,76,82,90] | |
| 6.48 | W428 | W247 | W371 | W316 | [9,12,24,26,31,49,51,52,53,55,56,57,58,61,63,66,67,72,73,75,76,79,80,84,87,89,90,91,94,97] |
| 6.51 | Y431 | Y250 | Y374 | Y319 | [6,12,22,24,31,48,49,51,52,53,55,56,57,59,60,61,63,65,66,67,69,70,71,72,73,75,76,77,79,80,81,82,83,85,86,87,88,89,91,92,94,95,98] |
| 6.52 | F432 | F251 | T375 | S320 | [9,12,31,55,56,63,66,72,73,75,76,83,84,87] |
| 6.55 | F435 | F254 | M378 | T323 | [9,12,31,53,55,56,57,66,72,75,76,83,84] |
| 6.58 | I438 | R381 | L326 | [12,55,61,62,65,80,89] | |
| 6.59 | A439 | S327 | [12,55] | ||
| ECL3 | K442 | S330 | [12,55] | ||
| ECL3 | N443 | S331 | [12,55,65,69,83] | ||
| 7.35 | H450 | E270 | Y394 | Y340 | [48,55,56,60,62,67,68,69,73,74,77,83,89] |
| 7.39 | I454 | L274 | F398 | F344 | [6,23,24,51,52,53,55,57,59,60,62,63,66,68,72,76,84] |
| 7.42 | G457 | G277 | L401 | Q347 | [6,23,24,26,31,52,53,57,58,63,79,80,82,86] |
| 7.43 | Y458 | Y278 | W402 | W348 | [23,24,26,31,54,55,56,58,63,66,71,72,80,87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, P.; Miszta, P.; Filipek, S. Molecular Modeling of Histamine Receptors—Recent Advances in Drug Discovery. Molecules 2021, 26, 1778. https://doi.org/10.3390/molecules26061778
Mehta P, Miszta P, Filipek S. Molecular Modeling of Histamine Receptors—Recent Advances in Drug Discovery. Molecules. 2021; 26(6):1778. https://doi.org/10.3390/molecules26061778
Chicago/Turabian StyleMehta, Pakhuri, Przemysław Miszta, and Sławomir Filipek. 2021. "Molecular Modeling of Histamine Receptors—Recent Advances in Drug Discovery" Molecules 26, no. 6: 1778. https://doi.org/10.3390/molecules26061778
APA StyleMehta, P., Miszta, P., & Filipek, S. (2021). Molecular Modeling of Histamine Receptors—Recent Advances in Drug Discovery. Molecules, 26(6), 1778. https://doi.org/10.3390/molecules26061778