
Stretching of Bombyx mori Silk Protein in Flow



Abstract
1. Introduction
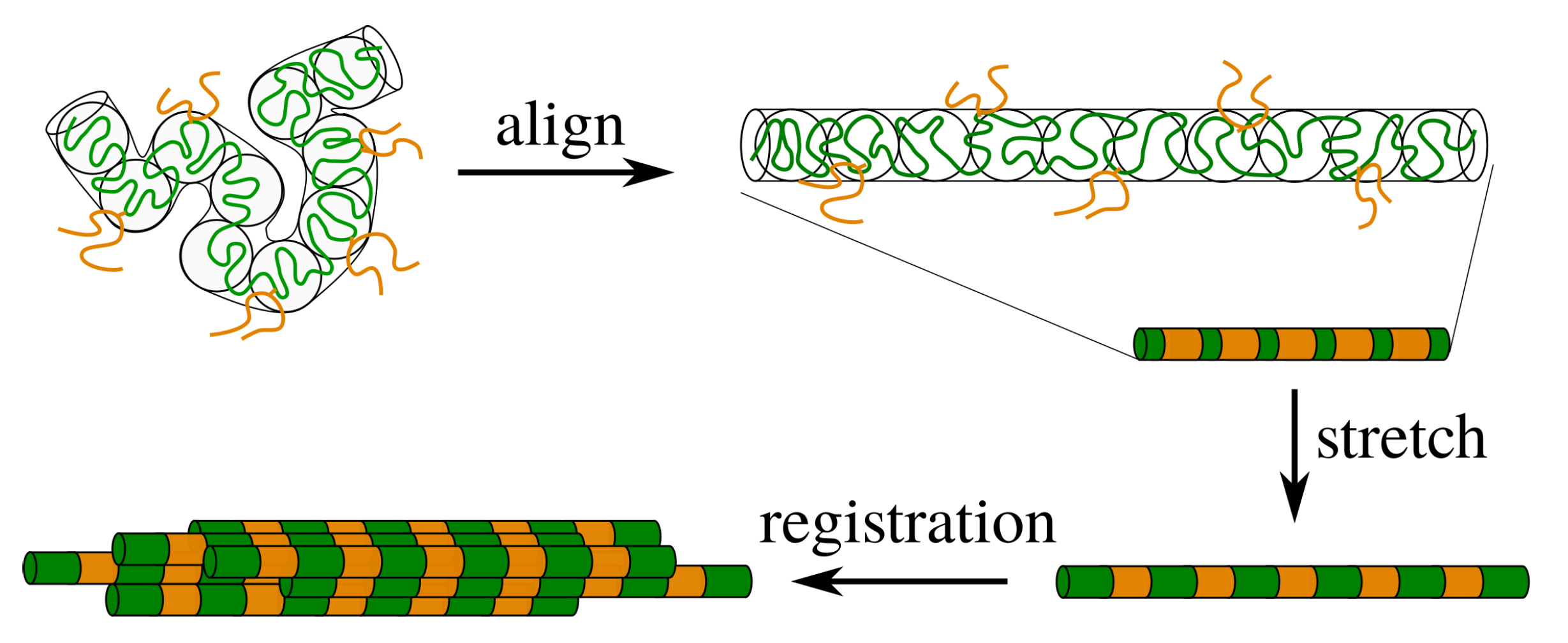
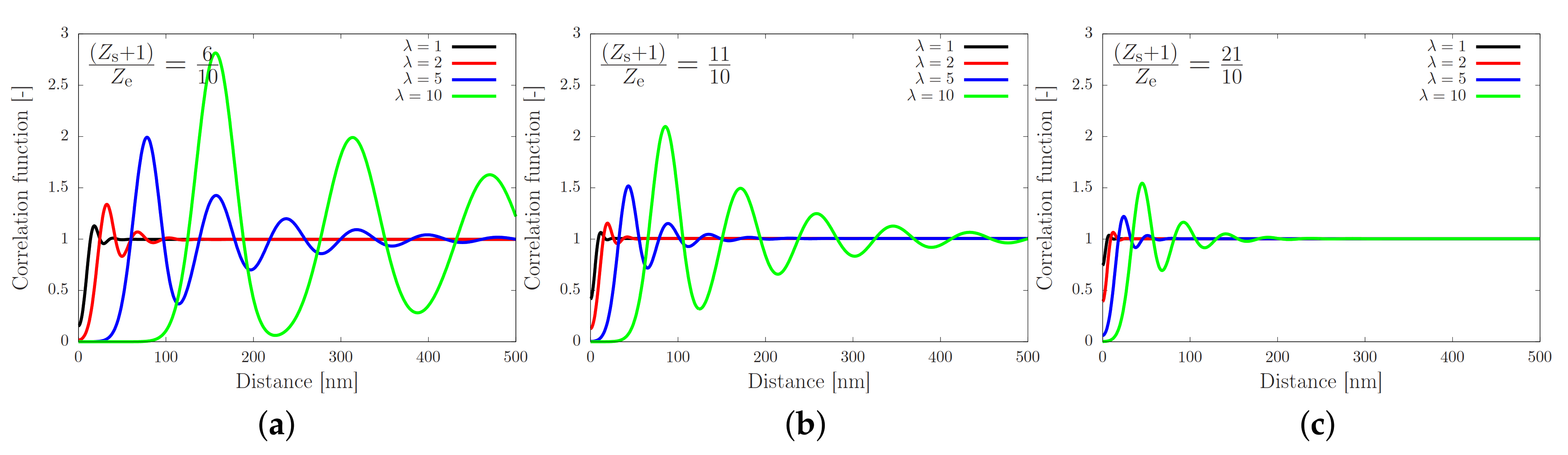
2. Theory for Registration of Stretched Chains
3. Experimental and Modelling Methods
3.1. Experimental Methods
3.2. Tube Model: Brownian Dynamics
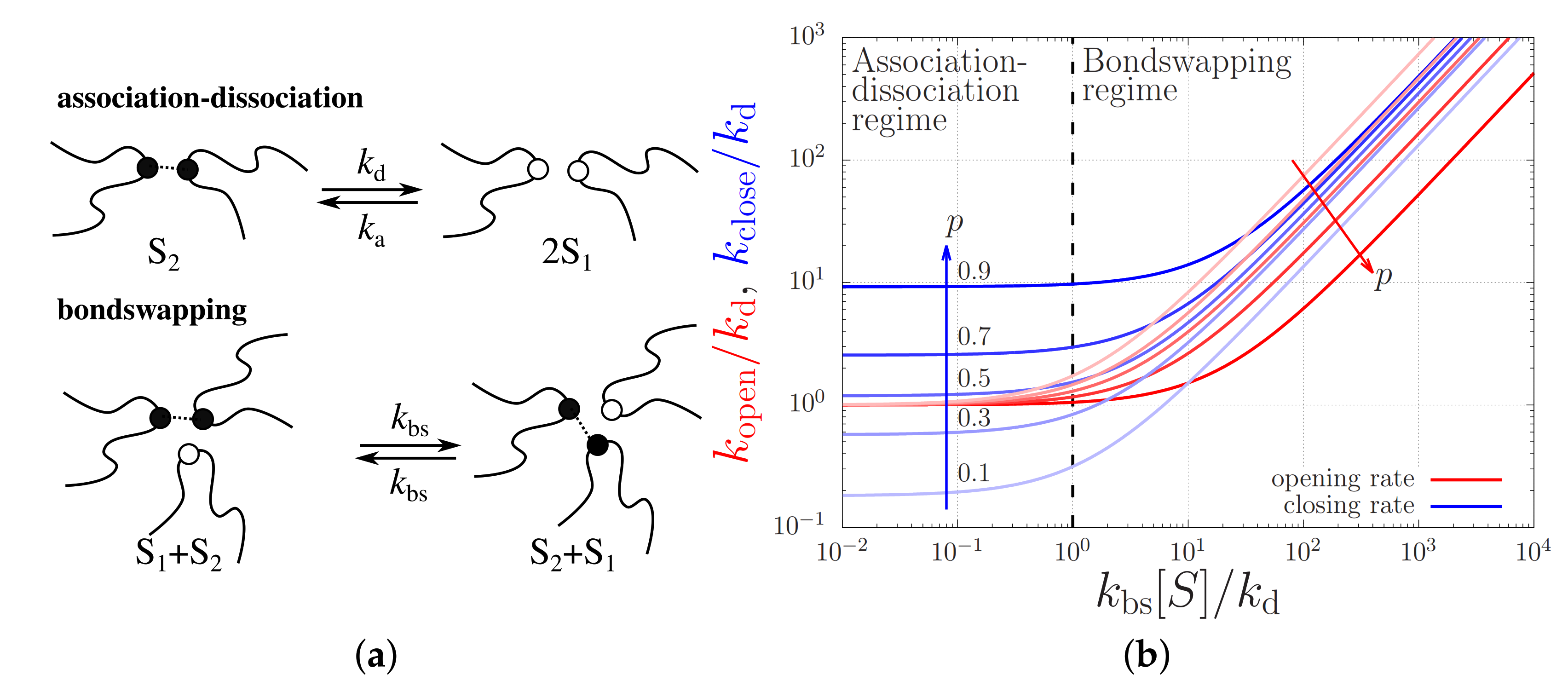
3.3. Physical Chemistry of the Stickers
3.4. Simulation Method
4. Results
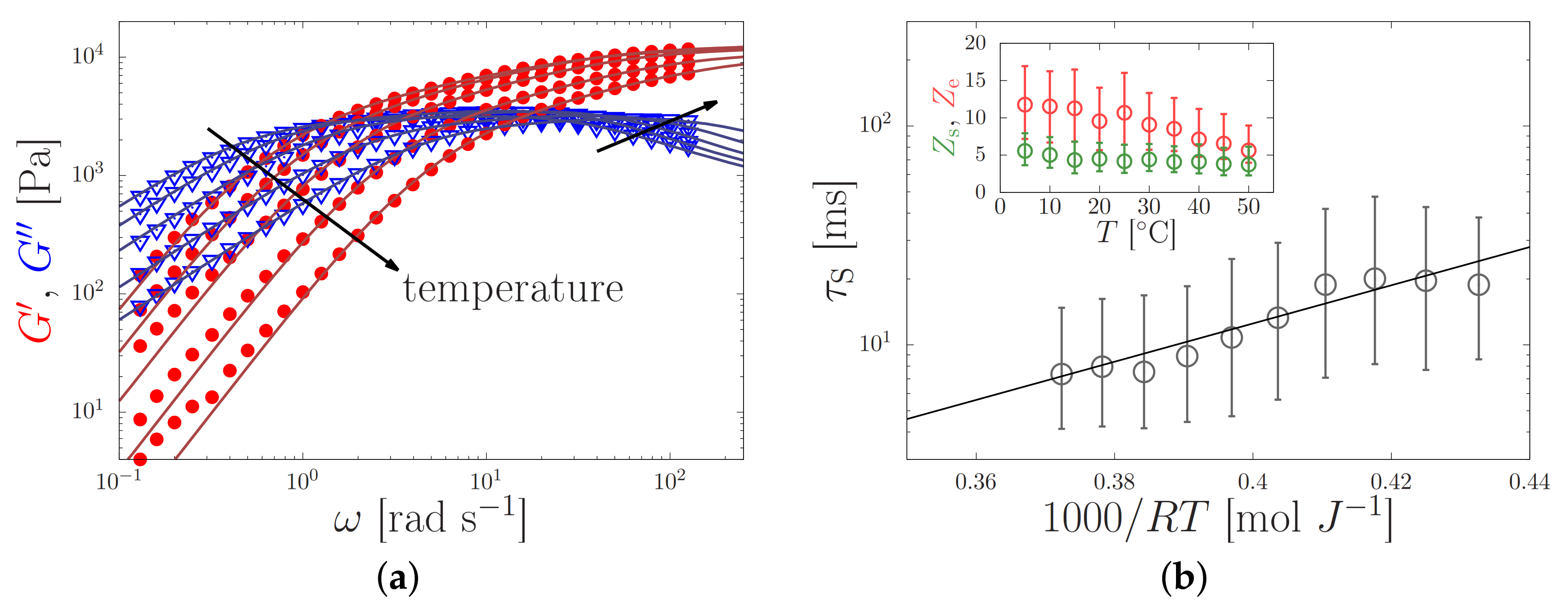
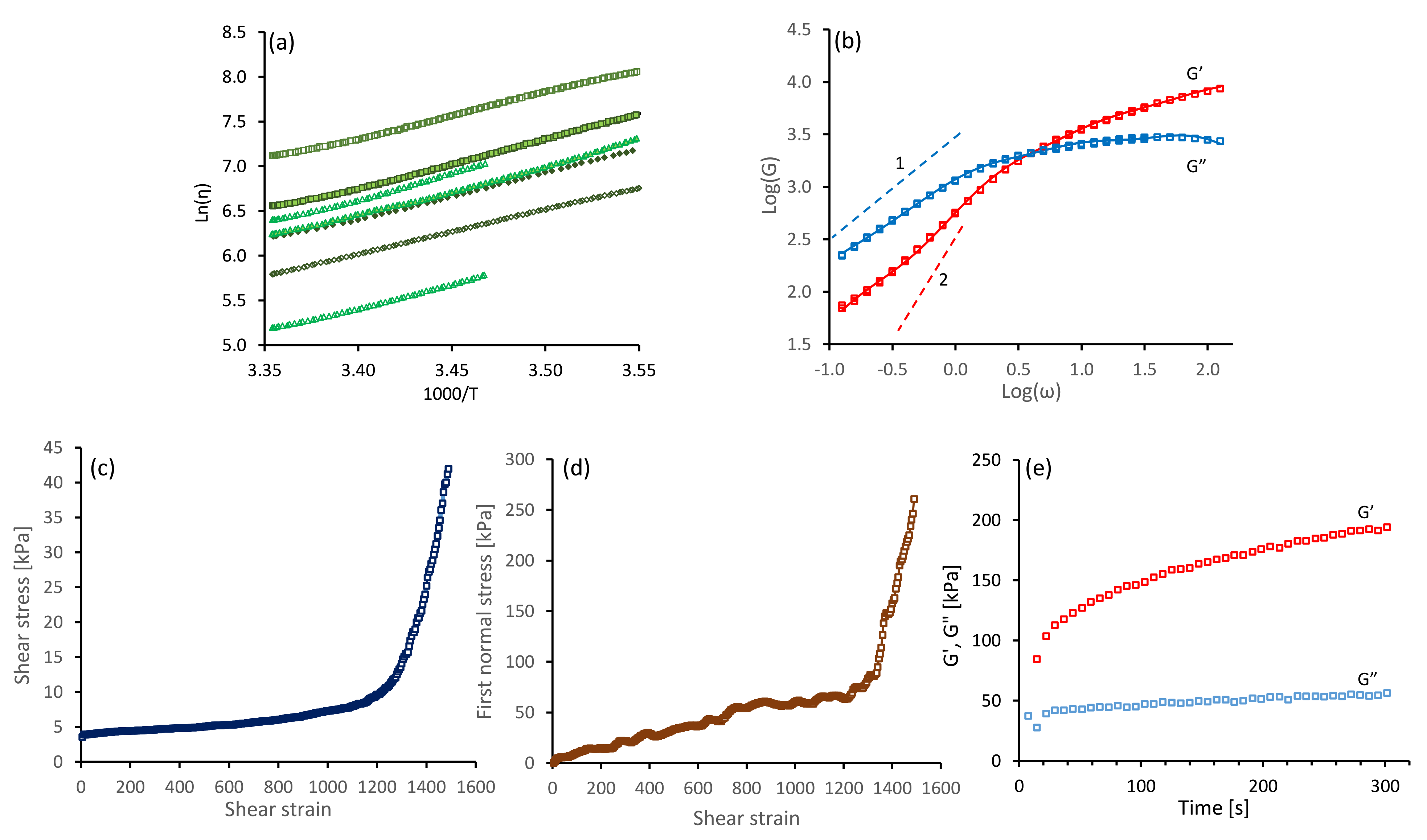
4.1. Silk Gelation under Constant Shear

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode Number, i | Modulus Contribution, [Pa] | Relaxation Time, [s] |
|---|---|---|
| 1 | ||
| 2 | ||
| 3 | ||
| 4 |
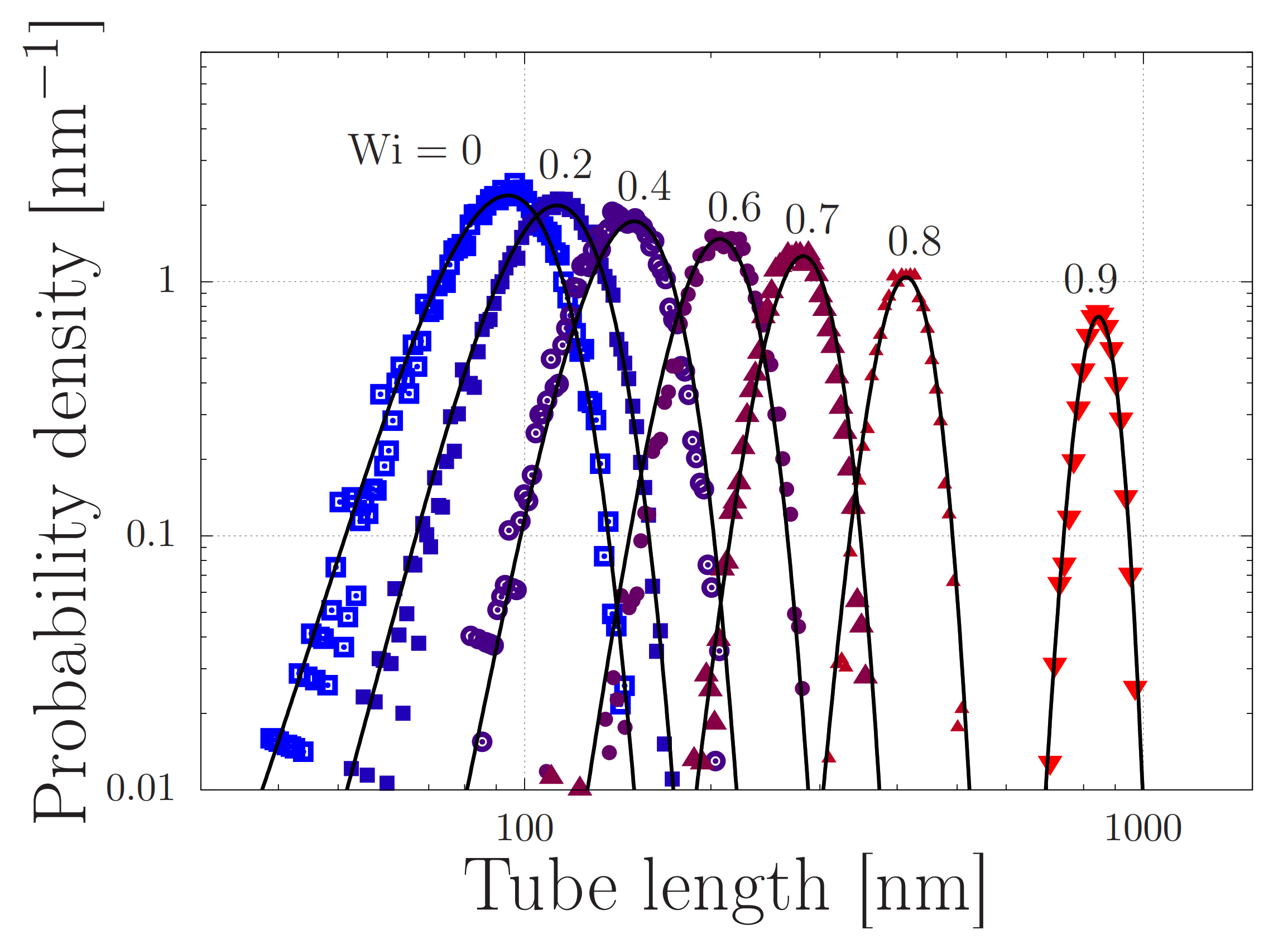
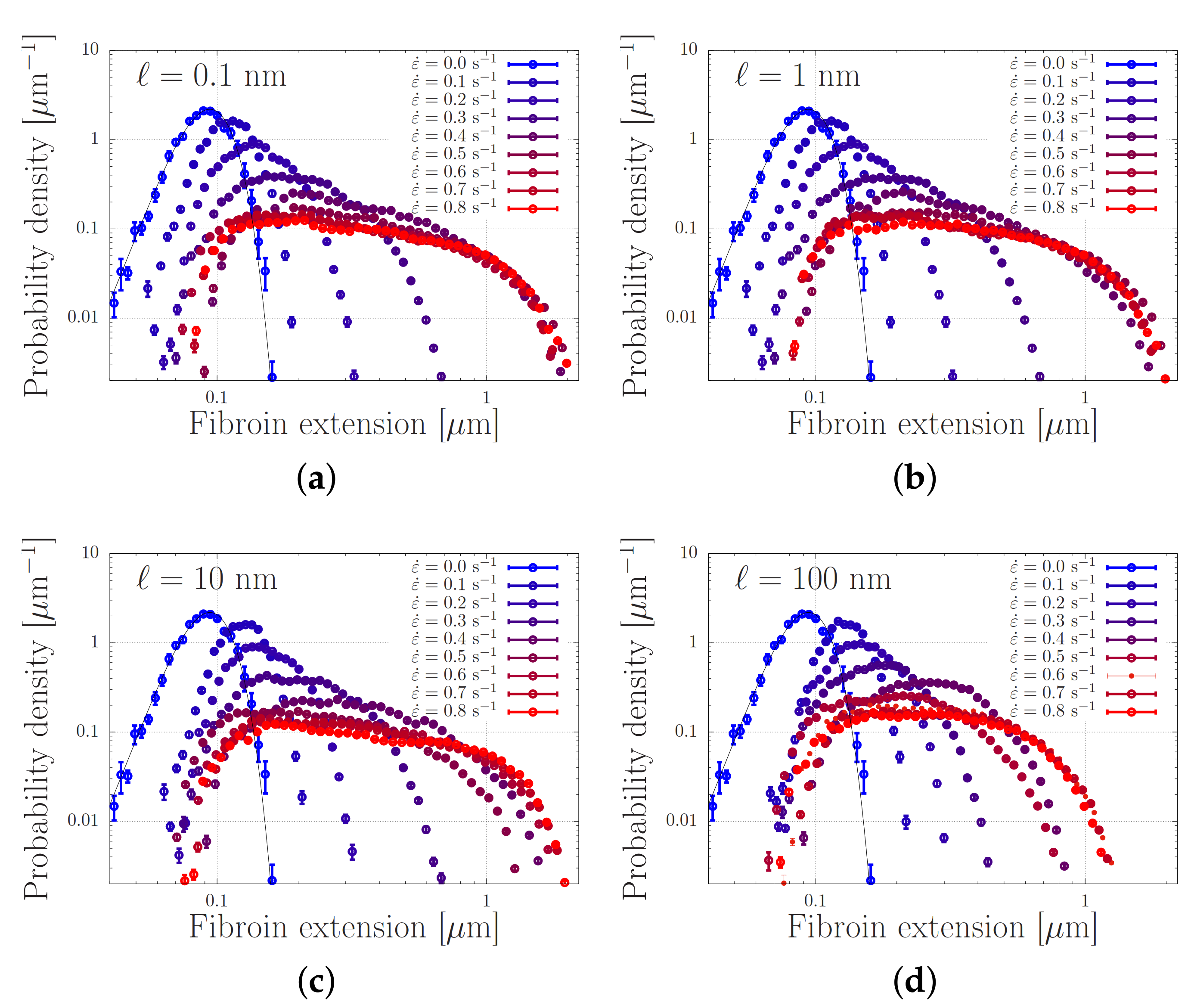
4.2. Modelling of Dispersed Chain Stretching in Extensional Flow
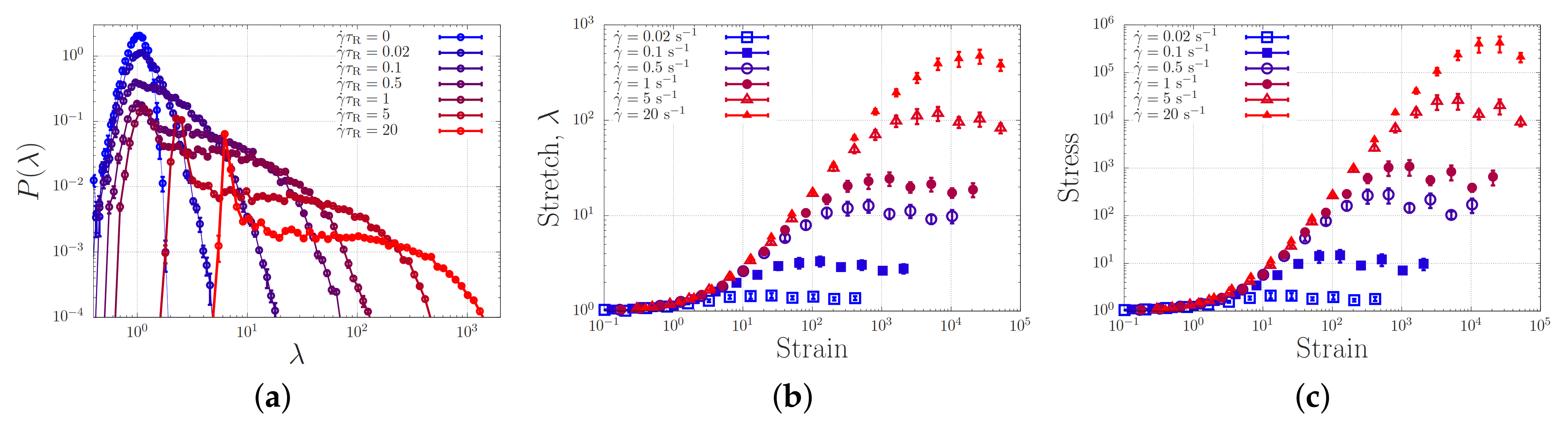
4.3. Toy-Model Simulations of Associating Polymers in Shear Flow
5. Discussion
- 1.
- The broad stretch distribution may set a probability by which proteins with a similar stretch may ‘register’ or associate and eventually stretch further or give rise to nucleation events; in this case, correctly parameterised single-chain simulations will enable to calculate nucleation rates. Within this mechanism, the finite extensibility of the chains and the tension-dependence of the sticker stability compress the high end of the stretch distribution, and are expected to be important control parameters of the nucleation rate.
- 2.
- At small strains, registration may develop swiftly, so that the stretch of a protein correlates with the stretch of its surrounding proteins, which in turn leads to collective stretching of the entire network.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Wegst, U.G.K.; Bai, H.; Saiz, E.; Tomsia, A.P.; Ritchie, R.O. Bioinspired structural materials. Nat. Mater. 2015, 14, 23–36. [Google Scholar] [CrossRef]
- Pennisi, E. Untangling spider biology. Science 2017, 358, 288–291. [Google Scholar] [CrossRef]
- Service, R.F. Silken promises. Science 2017, 358, 293–294. [Google Scholar] [CrossRef]
- Holland, C.; Vollrath, F.; Ryan, A.J.; Mykhaylyk, O.O. Silk and synthetic polymers: Reconciling 100 degrees of separation. Adv. Mater. 2012, 24, 105–109. [Google Scholar] [CrossRef]
- Laity, P.R.; Baldwin, E.; Holland, C. Changes in silk feedstock rheology during cocoon construction: The role of calcium and potassium ions. Macromol. Biosci. 2018, 19, 1800188. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.S.; Olmsted, P.D. Coarse-Grained Simulations of Flow-Induced Nucleation in Semicrystalline Polymers. Phys. Rev. Lett. 2009, 103, 115702. [Google Scholar] [CrossRef]
- Troise, E.M.; Caelers, H.J.M.; Peters, G.W.M. Full Characterization of Multiphase, Multimorphological Kinetics in Flow-Induced Crystallization of IPP at Elevated Pressure. Macromolecules 2017, 50, 3868–3882. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.A.; Rutledge, G.C. An assessment of models for flow-enhanced nucleation in an n-alkane melt by molecular simulation. J. Rheol. 2019, 63, 465–475. [Google Scholar] [CrossRef]
- Moghadamand, S.; Dalal, I.S.; Larson, R.G. Unraveling Dynamics of Entangled Polymers in Strong Extensional Flows. Macromolecules 2019, 52, 1296–1307. [Google Scholar] [CrossRef]
- Read, D.J.; McIlroy, C.; Das, C.; Harlen, O.G.; Graham, R.S. PolySTRAND Model of Flow-Induced Nucleation in Polymers. Phys. Rev. Lett. 2020, 124, 147802. [Google Scholar] [CrossRef]
- Dunderdale, G.J.; Davidson, S.J.; Ryan, A.J.; Mykhaylyk, O.O. Flow-induced crystallisation of polymers from aqueous solution. Nat. Comm. 2020, 11, 3372. [Google Scholar] [CrossRef]
- Schaefer, C.; Laity, P.R.; Holland, C.; McLeish, T.C.B. Silk Protein Solution: A Natural Example of Sticky Reptation. Macromolecules 2020, 53, 2669–2676. [Google Scholar] [CrossRef] [PubMed]
- Cranford, S.W. Increasing silk fibre strength through heterogeneity of bundled fibrils. J. R. Soc. Interface 2014, 10, 0130148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ling, S.; Yao, Q.; Li, Q.; Hu, D.; Dai, Q.; Weitz, D.A.; Kaplan, D.L.; Bueler, M.J.; Zhang, Y. Observations of 3 nm Silk Nanofibrils Exfoliated from Natural Silkworm Silk Fibers. ACS Mater. Lett. 2020, 2, 153–160. [Google Scholar] [CrossRef]
- Schaefer, C.; McLeish, T.C.B. Power-law stretching of Associating Polymers in Steady-State Extensional Flow. Phys. Rev. Lett. 2021, 126, 057801. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Suzuki, H.; Watanabe, Y. Conformational characterization of silk fibroin in intact Bombyx mori and Philosamia cynthia ricini silkworms by 13C NMR spectroscopy. Macromolecules 1983, 16, 1024–1026. [Google Scholar] [CrossRef]
- Asakura, T.; Watanabe, Y.; Uchida, A.; Minagawa, H. NMR of silk fibroin. 2. 13C NMR study of the chain dynamics and solution structure of Bombyx mori silk fibroin. Macromolecules 1984, 17, 1075–1081. [Google Scholar] [CrossRef]
- Zhao, C.; Asakura, T. Structure of silk studied with NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2001, 39, 301–352. [Google Scholar] [CrossRef]
- Asakura, T.; Okushita, K.; Williamson, M.P. Analysis of the structure of bombyx mori silk fibroin by NMR. Macromolecules 2015, 48, 2345–2357. [Google Scholar] [CrossRef]
- Laity, P.R.; Gilks, S.E.; Holland, C. Rheological behaviour of native silk feedstocks. Polymer 2015, 67, 28–39. [Google Scholar] [CrossRef]
- Laity, P.R.; Holland, C. Native silk feedstock as a model biopolymer: A arheological perspective. Biomacromolecules 2016, 17, 2662–2671. [Google Scholar] [CrossRef] [PubMed]
- Leibler, L.; Rubinstein, M.; Colby, R.H. Dynamics of reversible networks. Macromolecules 1991, 24, 4701–4707. [Google Scholar] [CrossRef]
- Kramer, O. Biological and Synthetic Polymer Networks; Elsevier Applied Science: New York, NY, USA, 1988. [Google Scholar]
- Seiffert, S.; Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev. 2012, 41, 909–930. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Q.; Colby, R.H. Dynamics of associative polymers. Soft Matter 2018, 14, 2961–2977. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. USA 1951, 37, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Ainaverapu, S.R.K.; Brujić, J.; Huang, H.H.; Wiita, A.P.; Liu, H.; Li, L.; Walther, K.A.; Carrion-Vazques, M.; Li, H.; Fernandez, J.M. Contour length and refolding rate of a small protein controlled by engineered disulfide bonds. Biophys. J. 2007, 92, 225–233. [Google Scholar] [CrossRef]
- Müller-Späth, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Rüegger, S.; Reymond, L.; Nettels, D.; Schuler, B. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, R.; Hofmann, H.; Nettels, D.; Borgia, M.B.; Mittal, J.; Best, R.B.; Schuler, B. Temperature-dependent solvation modulates the dimensions of disordered proteins. Proc. Natl. Acad. Sci. USA 2014, 11, 5213–5218. [Google Scholar] [CrossRef]
- Greving, I.; Dicko, C.; Terry, A.; Callowc, P.; Vollrath, F. Small angle neutron scattering of native and reconstituted silk fibroin. Soft Matter 2010, 6, 4389–4395. [Google Scholar] [CrossRef]
- Tang, S.; Wang, M.; Olsen, B.D. Anomalous Self-Diffusion and Sticky Rouse Dynamics in Associative Protein Hydrogels. Proc. Natl. Acad. Sci. USA 2015, 137, 3946–3957. [Google Scholar] [CrossRef]
- Rubinstein, M.; Semenov, A.N. Dynamics of Entangled Solutions of Associating Polymers. Macromolecules 2001, 34, 1058–1068. [Google Scholar] [CrossRef]
- Graham, R.S.; Likhtman, A.E.; McLeish, T.C.B.; Milner, S.T. Microscopic theory of linear, entangled polymer chains under rapid deformation including chain stretch and convective constraint release. J. Rheol. 2003, 47, 1171–1200. [Google Scholar] [CrossRef]
- Laity, P.R.; Holland, C. Thermo-rheological behaviour of native silk feedstocks. Eur. Polym. J. 2017, 87, 519–534. [Google Scholar] [CrossRef]
- Koeppel, A.; Laity, P.R.; Holland, C. The influence of metal ions on native silk rheology. Acta Biomater. 2020, 117, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Holland, C.; Urbach, J.; Blair, D.L. Direct visualization of shear dependent silk fibrillogenesis. Soft Matter 2012, 8, 2590–2594. [Google Scholar] [CrossRef]
- Boulet-Audet, M.; Terry, A.E.; Vollrath, F.; Holland, C. Silk protein aggregation kinetics revealed by Rheo-IR. Acta Biomater. 2014, 10, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Laity, P.R.; Holland, C. The Rheology behind Stress-Induced Solidification in Native Silk Feedstocks. Int. J. Mol. Sci. 2016, 17, 1812. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, Z.; Colby, R.H. Viscoelasticity of entangled random polystyrene ionomers. J. Rheol. 2016, 60, 1031–1040. [Google Scholar] [CrossRef]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon: Oxford, UK, 1986. [Google Scholar]
- Likhtman, A.E.; McLeish, T.C.B. Quantitative theory for linear dynamics of linear entangled polymers. Macromolecules 2002, 35, 6332–6343. [Google Scholar] [CrossRef]
- Likhtman, A.E.; Graham, R.S. Simple constitutive equation for linear polymer melts derived from molecular theory: Rolie–Poly equation. J. Non–Newton. Fluid Mech. 2003, 114, 1–12. [Google Scholar] [CrossRef]
- Boudara, V.A.H.; Read, D.J. Stochastic and preaveraged nonlinear rheology models for entangled telechelic starpolymers. J. Rheol. 2017, 61, 339–362. [Google Scholar] [CrossRef][Green Version]
- Boudara, V.A.H.; Read, D.J.; Ramírez, J. RepTate rheology software: Toolkit for the analysis of theories and experiments. J. Rheol. 2020, 64, 709–722. [Google Scholar] [CrossRef]
- Collis, M.W.; Lele, A.K.; Mackley, M.R.; Graham, R.S.; Groves, D.J.; Likhtman, A.E.; Nicholsona, T.M.; Harlen, O.G.; McLeish, T.C.B.; Hutchings, L.R.; et al. Constriction flows of monodisperse linear entangled polymers: Multiscale modeling and flow visualization. J. Rheol. 2005, 49, 501–522. [Google Scholar] [CrossRef]
- Doi, M. Explanation for the 3.4-Power Law for Viscosity of Polymeric Liquids on the Basis of the Tube Model. J. Polym. Phys. 1983, 21, 667–684. [Google Scholar] [CrossRef]
- Auhl, D.; Chambon, P.; McLeish, T.C.B.; Read, D.J. Elongational Flow of Blends of Long and Short Polymers: Effective Stretch Relaxation Time. Phys. Rev. Lett. 2009, 103, 136001. [Google Scholar] [CrossRef]
- Cohen, A. A Padé approximant to the inverse Langevin function. Rheol. Acta 1991, 30, 270–273. [Google Scholar] [CrossRef]
- Tomkovic, T.; Hatzikiriakos, S.G. Nonlinear rheology of poly(ethylene-co-methacrylic acid) ionomers. J. Rheol. 2018, 62, 1319–1329. [Google Scholar] [CrossRef]
- Tomkovic, T.; Mitsoulis, E.; Hatzikiriakos, S.G. Contraction flow of ionomers and their corresponding copolymers: Ionic and hydrogen bonding effects. Phys. Fluids 2019, 31, 033102. [Google Scholar] [CrossRef]
- Zuliki, M.; Zhang, S.; Nyamajaro, K.; Tomkovic, T.; Hatzikiriakos, S.G. Rheology of sodium and zinc ionomers: Effects of neutralization and valency. Phys. Fluids 2020, 32, 023104. [Google Scholar] [CrossRef]
- Bell, G.I. Models for the specific adhesion of cells to cells. Science 1978, 200, 618–627. [Google Scholar] [CrossRef]
- Huang, Q.; Ahn, J.; Parisi, D.; Chang, T.; Hassager, O.; Panyukov, S.; Rubinstein, M.; Vlassopoulos, D. Unexpected Stretching of Entangled Ring Macromolecules. Phys. Rev. Lett. 2019, 122, 208001. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.C.; Ge, T.; Rubinstein, M.; Grest, G.S. Topological Linking Drives Anomalous Thickening of Ring Polymers in Weak Extensional Flows. Phys. Rev. Lett. 2020, 124, 027801. [Google Scholar] [CrossRef] [PubMed]
- Mohottalalage, S.; Senanayake, M.; O’Connor, T.; Grest, G.; Perahia, D. Nonlinear Elongation Flows Effects on Aggregation in Associating Polymer Melts. Presented at the APS March Meeting, Virtual, 15–19 April 2021. V04.00008. [Google Scholar]








| Property | Symbol | Value |
|---|---|---|
| Number of monomers per chain | N | 5524 |
| Kuhn length | b | nm |
| Number of entanglements | 10 | |
| Number of stickers | 5 | |
| Entanglement relaxation time | s | |
| † Quiescent sticker lifetime | 10 ms | |
| † Quiescent fraction of closed stickers | p | |
| Sticker activation energy | ||
| Sticker dissociation length | ℓ | 0.1–100 nm |
| Maximum stretch ratio |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaefer, C.; Laity, P.R.; Holland, C.; McLeish, T.C.B. Stretching of Bombyx mori Silk Protein in Flow. Molecules 2021, 26, 1663. https://doi.org/10.3390/molecules26061663
Schaefer C, Laity PR, Holland C, McLeish TCB. Stretching of Bombyx mori Silk Protein in Flow. Molecules. 2021; 26(6):1663. https://doi.org/10.3390/molecules26061663
Chicago/Turabian StyleSchaefer, Charley, Peter R. Laity, Chris Holland, and Tom C. B. McLeish. 2021. "Stretching of Bombyx mori Silk Protein in Flow" Molecules 26, no. 6: 1663. https://doi.org/10.3390/molecules26061663
APA StyleSchaefer, C., Laity, P. R., Holland, C., & McLeish, T. C. B. (2021). Stretching of Bombyx mori Silk Protein in Flow. Molecules, 26(6), 1663. https://doi.org/10.3390/molecules26061663