3.5. Synthesis and Characterization of Compounds
3.5.1. ((1R,3S)-1,2,2-trimethylcyclopentane-1,3-diyl)dimethanol (2)
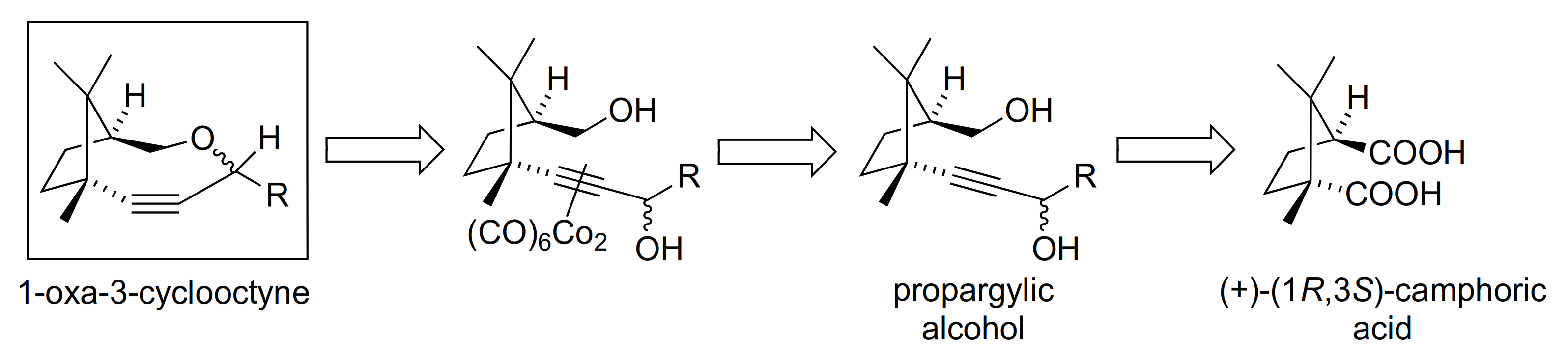
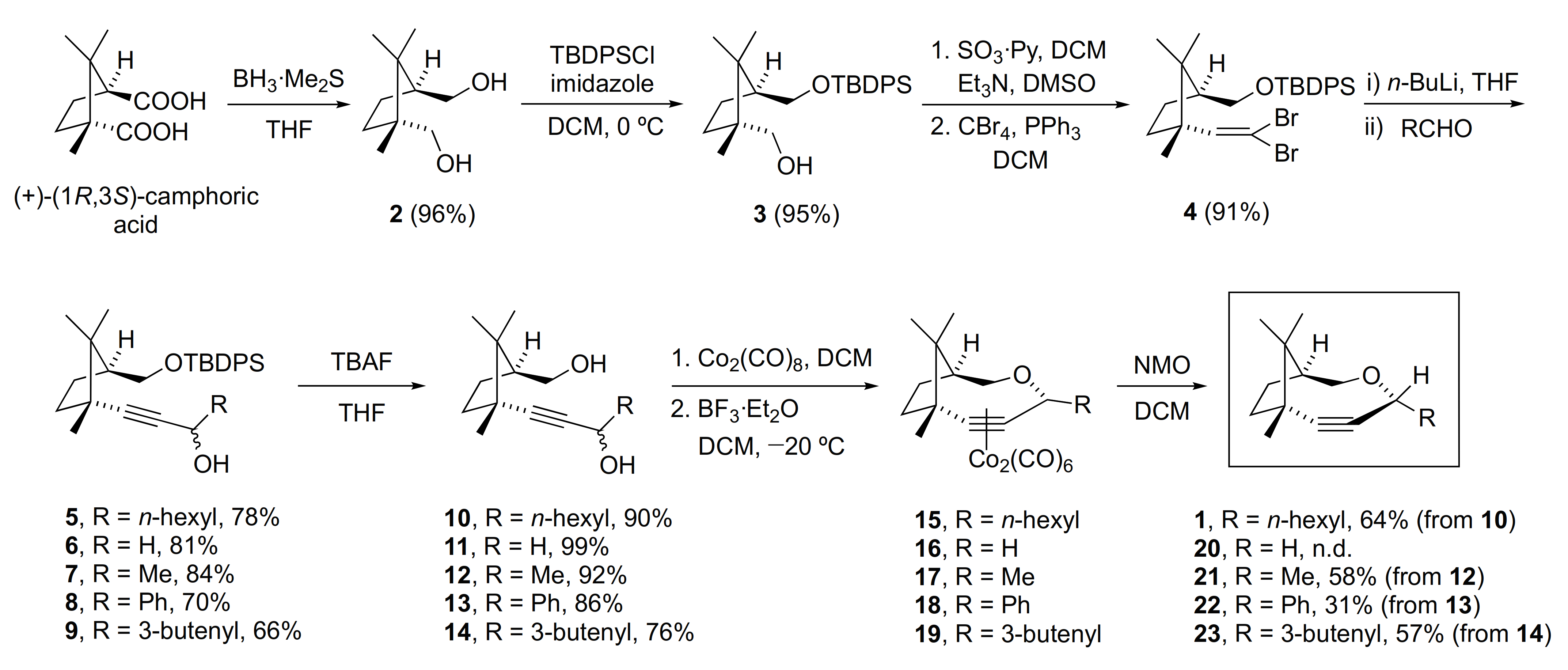
To a solution of camphoric acid (7.1 g, 35.0 mmol) in 250 mL THF at 0 °C under inert atmosphere, we added 46 mL of BH3·SMe2 (2.0 M in THF) dropwise, and the reaction mixture was stirred for 23 h at RT. The reaction was quenched by adding 17 mL of MeOH dropwise at 0 °C. Then, the solvent was evaporated under reduced pressure. The crude was purified by flash chromatography (silica gel, gradient n-hexane/EtOAc 60:40 to 40:60) to obtain 5.8 g (96% yield) of 2 as a white solid.
1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 3.73 (dd, J = 5.3, 10.3 Hz, 1H), 3.58 (d, J = 10.8 Hz, 1H), 3.51 (dd, J = 8.3, 10.3 Hz, 1H), 3.47 (d, J = 10.8 Hz, 1H), 2.08 (ddd, J = 5.3, 9.3, 17.8 Hz, 1H), 1.99–1.91 (m, 1H), 1.63–1.56 (m, 1H), 1.41–1.32 (m, 2H), 1.25 (br, 2H), 1.02 (s, 3H), 1.01 (s, 3H), 0.79 (s, 3H).
13C-NMR (125 MHz, CDCl3, 298 K): δ ppm 69.2 (t), 65.0 (t), 50.5 (d), 48.8 (s), 44.0 (s), 33.7 (t), 25.5 (t), 24.2 (q), 20.4 (q), 18.5 (q).
HRMS (ESI-negative): m/z: calculated for C10H19O [M − H+]: 171.1385, found: 171.1391.
Melting point: 130–132 °C.
[α]25D = + 54.1 (c 2.19, CHCl3).
3.5.2. ((1R,3S)-3-(((tert-butyldiphenylsilyl)oxy)methyl)-1,2,2-trimethylcyclopentyl)methanol (3)
The diol 2 (8.4 g, 48.8 mmol) was dissolved in 500 mL of DCM (0.1 M) under inert atmosphere (Ar). The solution was then cooled to 0 °C and imidazole (10.0 g, 146.3 mmol, 3 equiv) was added. Finally, TBDPSCl (13 mL, 48.8 mmol, 1.0 equiv) was added dropwise and the mixture was stirred for 3–4 h. The reaction was quenched with 800 mL of water and the 2 phases were separated. The aqueous layer was extracted with DCM (2 × 400 mL). The organic layers were collected, dried over anhydrous MgSO4, and filtered, and the solvent was removed under reduced pressure. The crude was purified by flash chromatography (silica gel, gradient n-hexane/EtOAc, 90:10 to 80:20) to obtain 19.1 g (95% yield) of the compound 3 as a colorless oil.
1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 7.69–7.66 (m, 4H), 7.45–7.36 (m, 6H), 3.70 (dd, J = 6.4, 10.1 Hz, 1H), 3.57–3.52 (m, 2H), 3.45 (d, J = 10.8 Hz, 1H), 2.14 (ddd, J = 7.2, 9.6, 16.4 Hz, 1H), 1.92–1.83 (m, 1H), 1.58–1.50 (m, 1H), 1.36–1.24 (m, 2H), 1.04 (s, 9H), 0.99 (s, 3H), 0.97 (s, 3H), 0.75 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 135.6 (d), 134.1 (s), 134.0 (s), 129.5 (d), 129.5 (d), 127.6 (d), 69.3 (t), 65.5 (t), 50.1 (d), 48.9 (s), 43.9 (s), 33.7 (t), 26.9 (q), 25.2 (t), 24.4 (q), 20.4 (q), 19.2 (s), 18.4 (q).
HRMS (ESI): m/z: calculated for C26H38O2Si [M + Na+]: 433.2539, found: 433.2536
[α]25D = + 20.5 (c 2.12, CHCl3).
3.5.3. tert-Butyl(((1S,3S)-3-(2,2-dibromovinyl)-2,2,3-trimethylcyclopentyl)methoxy)diphenylsilane (4)
To a solution of 3 (10.1 g, 24.59 mmol) in 82 mL of DCM (0.3 M) under inert atmosphere (Ar) at 0 °C, we sequentially added DMSO (16.2 mL, 0.66 mL/mmol) triethylamine (17.3 mL, 122.97 mmol, 5 equiv) and finally the SO3·Py complex (11.7 g, 3 equiv). The mixture was stirred for 2 h. After that, the reaction was quenched with 100 mL of water, and then the organic layer was separated and the aqueous phase was extracted with DCM (2 × 100 mL). The organic extracts were collected, dried over anhydrous MgSO4, and filtered, and the solvent was evaporated under reduced pressure. The crude was purified by flash chromatography (silica gel, n-hexane/EtOAc, 95:5) to obtain 9.3 g (93% yield) of the corresponding aldehyde, which was immediately used in the next step.
Intermediate aldehyde: 1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 9.63 (s, 1H) 7.68–7.65 (m, 4H), 7.45–7.37 (m, 6H), 3.6 (dd, J = 6.7, 10.2 Hz, 1H), 3.57 (dd, J = 6.9, 10.2 Hz, 1H), 2.29 (ddd, J = 5.8, 11.5, 13.2, Hz, 1H), 2.15 (ddd, J = 6.9, 9.3, 16.3 Hz, 1H), (dddd, J = 6.1, 9.5, 13.6, 13.6 Hz, 1H), 1.92–1.83 (m, 1H), 1.45–1.29 (m, 2H), 1.07 (s, 3H), 1.05 (s, 12H), 0.82 (s, 3H).
To a solution of the so-formed aldehyde (1.73 g, 4.23 mmol) in DCM (42.3 mL, 0.1 M) at 0 °C under inert atmosphere (Ar), we added 7.8 g of PPh3, and when it was dissolved, 5.0 g of CBr4 was added in portions. The mixture was stirred overnight. Next, the solvent was removed under reduced pressure until one-quarter of the volume remained. Then, 50 mL of Et2O was added and the suspension was filtered through celite. Finally, the solvent from the filtrate was removed in vacuo. The crude was purified by flash chromatography (silica gel, n-hexane/DCM, 98:2) to obtain 2.17 g (91% yield) of the compound 4.
1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 7.69–7.65 (m, 4H), 7.45–7.36 (m, 6H). 6.61 (s, 1H), 3.69 (dd, J = 6.9, 10.1 Hz, 1H), 3.56 (dd, J = 7.0, 10.1 Hz, 1H), 2.09–1.81 (m, 4H), 1.31–1.23 (m, 1H), 1.06 (s, 3H), 1.05 (s, 9H), 0.94 (s, 3H), 0.70 (s, 3H).
13C-NMR (125 MHz, CDCl3, 298 K): δ ppm 144.1 (d), 135.6 (d), 133.9 (s), 133.9 (s), 129.6 (d), 129.5 (d), 127.6 (d), 85.3 (s), 65.6, (t), 53.4 (s), 47.4 (d), 45.8 (s), 33.8 (t), 26.9 (q), 24.9 (t), 22.2 (q), 20.9 (q), 19.4 (q), 19.2 (s).
HRMS (ESI): m/z: calculated for C27H3679Br81BrOSi [M + Na+]: 587.0779, found: 587.0780.
[α]25D = + 12.8 (c 2.95, CHCl3).
3.5.4. Compounds 5–9
General procedure: Compound 4 was dissolved in dry THF (6 mL, 0.15M), and under inert atmosphere (Ar), we added 3 equiv of n-BuLi solution (2.3 M in hexane) at 0 °C. The reaction mixture was stirred for 15 min and then the corresponding aldehyde (i.e., heptanal → compound 5; para-formaldehyde → compound 6; acetaldehyde → compound 7; benzaldehyde → compound 8; 4-pentanal → compound 9) was added at −78 °C. The mixture was allowed to reach RT while it was stirred overnight. The reaction was quenched by adding 20 mL of saturated NH4Cl solution, and then it was extracted with Et2O (3 × 20 mL). The organic layer was collected, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude was purified by flash chromatography (silica gel) yielding the corresponding propargyl alcohol.
1-((1R,3S)-3-(((tert-butyldiphenylsilyl)oxy)methyl)-1,2,2-trimethylcyclopentyl)non-1-yn-3-ol (5). 1H-NMR (600 MHz, CDCl3, 298 K): δ ppm 7.68–7.65 (m, 4H), 7.44–7.36 (m, 6H), 4.35 (ddd, J = 2.2, 6.5, 6.5 Hz, 1H), 3.71 (dd, J = 6.7, 10.0 Hz, 1H), 3.57 (dd, J = 7.5, 10.0 Hz, 1H), 2.08–2.02 (m, 1H), 1.97–1.91 (m, 1H), 1.88–1.81 (m, 1H), 1.70–1.59 (m, 2H), 1.34–1.26 (m, 8H), 1.13 (s, 3H), 1.05 (s, 9H), 0.96 (s, 3H), 0.90 (s, 3H), 0.89 (t, J = 6.7 Hz, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 135.6 (d), 134.0 (s), 133.9 (s), 129.5 (d), 129.5 (d), 127.6 (d), 90.9 (s), 83.0 (s), 66.0 (t), 62.8 (d), 48.1 (d), 45.7 (s), 45.3 (s), 45.3 (s), 38.3 (t), 37.5 (t), 37.5 (t), 31.7 (t), 28.9 (t), 26.9 (q), 25.3 (t), 25.2 (t), 24.1 (q), 23.4 (q), 22.5 (t), 20.6 (q), 19.2 (s), 14.0 (q).
Column chromatography: n-hexane/EtOAc (95:5).
3-((1R,3S)-3-(((tert-butyldiphenylsilyl)oxy)methyl)-1,2,2-trimethylcyclopentyl)prop-2-yn-1-ol (6). 1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 7.69–7.66 (m, 4H), 7.45–7.36 (m, 6H), 4.25 (s, 2H), 3.71 (dd, J = 6.7, 10.0 Hz, 1H), 3.57 (dd, J = 7.4, 10.0 Hz, 1H), 2.10–2.02 (m, 1H), 1.99–1.91 (m, 1H), 1.90–1.81 (m, 1H), 1.64–1.57 (m, 1H), 1.42 (br, 1H), 1.39–1.30 (m, 1H), 1.14 (s, 3H), 1.05 (s, 9H), 0.97 (s, 3H), 0.91 (s, 3H).
13C-NMR (125 MHz, CDCl3, 298 K): δ ppm 135.6 (d), 134.0 (s), 133.9 (s), 129.5 (d), 129.5 (d), 127.6 (d), 91.9 (s), 79.9 (s), 66.0 (t), 51.5 (t), 48.1 (d), 45.7 (s), 45.3 (s), 37.4 (t), 26.9 (q), 25.3 (t), 24.0 (q), 23.4 (q), 22.5 (t), 20.6 (q), 19.2 (s).
HRMS (ESI): m/z: calculated for C28H38O2Si [M + Na+]: 457.2539, found: 457.2535.
Column chromatography: n-hexane:EtOAc (80:20).
4-((1R,3S)-3-(((tert-butyldiphenylsilyl)oxy)methyl)-1,2,2-trimethylcyclopentyl)but-3-yn-2-ol (7). 1H-NMR (600 MHz, CDCl3, 298 K): δ ppm 7.67 (d, J = 7.8 Hz, 4H), 7.44–7.40 (m, 6H), 4.51 (dd, J = 6.0, 12.4 Hz, 1H), 3.71 (dd, J = 6.7, 10.1 Hz, 1H), 3.56 (dd, J = 7.7, 9.6 Hz, 1H), 2.08–2.02 (m, 1H), 1.96–1.90 (m, 1H), 1.89–1.81 (m, 1H), 1.63 (br, 1H), 1.59 (ddd, J = 4.7, 10.1, 13.7 Hz, 1H), 1.40 (d, J = 6.5 Hz, 3H), 1.37–1.30 (m, 1H), 1.13 (s, 3H), 1.05 (s, 9H), 0.96 (s, 3H), 0.90 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 135.6 (d), 134.0 (s), 133.9 (s), 129.5 (d), 129.5 (d), 127.6 (d), 90.1 (s), 83.9 (s), 66.0 (t), 58.6 (d), 48.1 (t), 45.5 (s), 45.3 (s), 45.3 (s), 37.5 (t), 37.4 (t), 26.9 (q), 25.3 (t), 24.9 (q), 24.0 (q), 23.4 (q), 20.6(q), 19.2 (s).
HRMS (ESI): m/z: calculated for C29H40O2Si [M + Na+]: 471.2695, found: 471.2696.
Column chromatography: n-hexane/EtOAc (gradient 95:5–80:20).
3-((1R,3S)-3-(((tert-butyldiphenylsilyl)oxy)methyl)-1,2,2-trimethylcyclopentyl)-1-phenylprop-2-yn-1-ol (8). 1H-NMR (600 MHz, CDCl3, 298 K): δ ppm 7.66 (d, J = 7.5 Hz, 8H), 7.54 (d, J = 7.5 Hz, 4H), 7.44–7.40 (m, 4H), 7.40–7.34 (m, 12H), 7.33–7.29 (m, 2H), 5.45 (s, 2H), 3.71 (dd, J = 6.9, 10.0 Hz, 2H), 3.57 (dd, J = 7.6, 9.6 Hz, 2H), 2.10–2.04 (m, 2H), 2.03–1.97 (m, 4H), 1.90–1.83 (m, 2H), 1.67–1.61 (br, 2H), 1.38–1.31 (m, 2H), 1.19 (s, 3H), 1.18 (s, 3H), 1.04, (s, 18H), 0.97 (s, 3H), 0.98 (s, 3H), 0.93 (s, 3H), 0.92 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 141.4 (s), 141.4 (s), 135.6 (d), 134.0 (s), 133.9 (s), 129.5 (d), 129.5 (d), 128.5 (d), 128.1 (d), 127.6 (d), 126.7 (d), 93.1 (s), 81.5 (s), 65.9 (t), 64.9 (d), 48.1 (d), 45.8 (s), 45.5 (s), 37.5 (t), 37.4 (t), 26.9 (q), 25.3 (t), 24.1 (q), 24.0 (q), 23.4 (q), 20.7 (q), 20.7 (q), 19.2 (s).
HRMS (ESI): m/z: calculated for C34H42O2Si [M + Na+]: 533.2852, found: 533.2857.
Column chromatography: n-hexane/EtOAc (gradient 95:5–90:10).
1-((1R,3S)-3-(((tert-butyldiphenylsilyl)oxy)methyl)-1,2,2-trimethylcyclopentyl)hept-6-en-1-yn-3-ol (9). 1H-NMR (600 MHz, CDCl3, 298 K): δ ppm 7.68–7.65 (m, 8H), 7.44–7.36 (m, 12H), 5.87–5.80, (m, 2H), 5.05, (dd, J = 1.6, 17.1 Hz, 2H), 4.98 (ddd, J = 0.9, 0.9, 10.2 Hz, 2H), 4.38 (ddd, J = 2.2, 6.5, 6.5 Hz, 2H), 3.71 (dd, J = 6.8, 10.0 Hz, 2H), 3.56 (dd, J = 7.4, 10.0 Hz, 2H), 2.24–2.19 (m, 4H), 2.08–2.02 (m, 2H), 1.97–1.91 (m, 2H), 1.89–1.81 (m, 2H), 1.80–1.70 (m, 4H), 1.62–1.57 (m, 4H), 1.36–1.3 (m, 2H), 1.14 (s, 6H), 1.05 (s, 18H), 0.96 (s, 6H), 0.91 (s, 3H), 0.90 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 137.9 (d), 135.6 (d), 134.0 (s), 133.9 (s), 129.5 (d), 129.5 (d), 127.6 (d), 115.1 (t), 91.3 (s), 82.6 (s), 66.0 (t), 62.3 (d), 48.1 (d), 45.7 (s), 45.3 (s), 37.5 (t), 37.4 (t), 29.6 (t), 26.9 (q), 25.3 (t), 24.1 (q), 23.9 (q), 20.6 (q), 19.2 (s).
HRMS (ESI): m/z: calculated for C32H44O2Si [M + Na+]: 511.3008, found: 511.3015.
Column chromatography: n-hexane/EtOAc (gradient 95:5–90:10).
3.5.5. Compounds 10–14
Compounds 5–9 were each dissolved in THF (0.1 M concentration), and 1.5 equiv of TBAF solution (1.0 M in THF) was added to the mixture, which was stirred until monitorization by TLC showed that reaction was finished. Next, 15 mL of water was added to the reaction mixture, and after that it was extracted with Et2O (3 × 20 mL). The organic layer was collected, dried over anhydrous MgSO4, then filtered and concentrated by rotavap. The crude was purified by flash chromatography (silica gel) to furnish products 10–14.
1-((1R,3S)-3-(hydroxymethyl)-1,2,2-trimethylcyclopentyl)non-1-yn-3-ol (10). H-NMR (500 MHz, CDCl3, 298 K): δ ppm 4.37 (t, J = 6.6 Hz, 1H), 3.74 (dd, J = 5.6, 10.2 Hz, 1H), 3.55 (dd, J = 8.0, 10.2 Hz, 1H), 2.04–1.96 (m, 2H), 1.95–1.88 (m, 1H), 1.73–1.60 (m, 4H), 1.47–1.38 (m, 3H), 1.36–1.24 (m, 7H), 1.15 (s, 3H), 0.99 (s, 3H), 0.94 (s, 3H), 0.88 (t, J = 6.9 Hz, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 90.6 (s), 83.2 (s), 65.4 (t), 62.8 (d), 48.6 (d), 45.6 (s), 45.3 (s), 45.3 (s), 38.3 (t), 37.5 (t), 37.5 (t), 31.7 (t), 28.9 (t), 25.6 (t), 25.2 (t), 23.9 (q), 23.9 (q), 23.4 (q), 22.5 (t), 20.7 (q), 14.0 (q).
HRMS (ESI): m/z: calculated for C18H32O2 [M + Na+]: 303.2300, found: 303.2303.
Column chromatography: n-hexane:EtOAc (50:50).
3-((1R,3S)-3-(hydroxymethyl)-1,2,2-trimethylcyclopentyl)prop-2-yn-1-ol (11). 1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 4.28 (d, J = 4.5 Hz, 2H), 3.74 (dd, J = 5.5, 10.2 Hz, 1H), 3.57 (dd, J = 8.2, 10.0 Hz, 1H), 2.04–1.89 (m, 3H), 1.69–1.63 (m, 1H), 1.54 (br, 1H), 1.47–1.39 (m, 1H), 1.27 (br, 1H), 1.16 (s, 3H), 0.99 (s, 3H), 0.95 (s, 3H).
13C-NMR (125 MHz, CDCl3, 298 K): δ ppm 91.6 (s), 80.2 (s), 65.3 (t), 51.5 (t), 48.5 (d), 45.6 (s), 45.3 (s), 37.5 (t), 25.5 (t), 23.9 (q), 23.3 (q), 20.7 (q).
HRMS (ESI): m/z: calculated for C12H20O2 [M + Na+]: 219.1361, found: 219.1369.
Column chromatography: n-hexane/EtOAc (40:60).
4-((1R,3S)-3-(hydroxymethyl)-1,2,2-trimethylcyclopentyl)but-3-yn-2-ol (12). 1H-NMR (600 MHz, CDCl3, 298 K): δ ppm 4.54 (dd, J = 6.5, 13.1 Hz, 1H), 3.74 (dd, J = 5.7, 10.4 Hz, 1H), 3.56 (dd, J = 8.3, 10.1 Hz, 1H), 2.03–1.89 (m, 3H), 1.68–1.62 (m, 1H), 1.43 (d, J = 6.5 Hz, 4H), 1.15 (s, 3H), 0.98 (s, 3H), 0.94 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 89.9 (s), 84.1 (s), 65.4 (t), 58.6 (d), 48.6 (d), 45.5 (s), 45.4 (s), 45.3 (s), 37.5 (t), 37.5 (t), 25.6 (t), 24.9 (q), 23.9 (q), 23.4 (q), 20.6 (q).
HRMS (ESI): m/z: calculated for C13H22O2 [M + Na+]: 233.1517, found: 233.1513.
Column chromatography: n-hexane/EtOAc (50:50).
3-((1R,3S)-3-(hydroxymethyl)-1,2,2-trimethylcyclopentyl)-1-phenylprop-2-yn-1-ol (13). 1H-NMR (600 MHz, CDCl3, 298 K): δ ppm 7.55 (d, J = 7.4 Hz, 4H), 7.38 (t, J = 7.5 Hz, 4H), 7.32 (t, J = 7.3 Hz, 2H), 5.48 (s, 2H), 3.73 (dd, J = 5.6, 10.3 Hz, 2H), 3.54 (dd, J = 8.1, 10.1 Hz, 1H), 2.12 (br, 2H), 2.09–1.91 (m, 6H), 1.73–1.67 (m, 2H), 1.58 (br, 2H), 1.48–1.41 (m, 2H), 1.21 (s, 3H), 1.21 (s, 3H), 1.01 (s, 3H), 1.01 (s, 3H), 0.97 (s, 3H), 0.96 (s, 3H).
13C-NMR R (150 MHz, CDCl3, 298 K): δ ppm 141.4 (s), 128.5 (d), 128.2 (d), 126.7 (d), 92.8 (s), 81.8 (s), 65.3 (t), 64.9 (d), 48.6 (d), 45.8 (s), 45.5 (s), 37.5 (t), 37.5 (t), 25.6 (t), 23.9 (q), 23.9 (q), 23.4 (q), 20.8 (q).
HRMS (ESI): m/z: calculated for C18H24O2 [M + Na+]: 295.1674, found: 295.1675.
Column chromatography: DCM/EtOAc (gradient 90:10–50:50).
1-((1R,3S)-3-(hydroxymethyl)-1,2,2-trimethylcyclopentyl)hept-6-en-1-yn-3-ol (14). 1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 5.89–5.79, (m, 1H), 5.07, (dd, J = 1.7, 17.1 Hz, 1H), 5.00–4.96 (m, 1H), 4.40 (t, J = 6.5 Hz, 1H), 3.73 (dd, J = 5.6, 10.2 Hz, 1H), 3.56 (dd, J = 8.0, 10.2 Hz, 1H), 2.28–2.16 (m, 2H), 2.05–1.88 (m, 3H), 1.84–1.71 (m, 3H), 1.69–1.62 (m, 1H), 1.46–1.38 (m, 1H), 1.16 (s, 3H), 0.99 (s, 3H), 0.94 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 137.9 (d), 115.1 (t), 91.0 (s), 82.9 (s), 65.3 (t), 62.2 (d), 48.5 (d), 45.6 (s), 45.3 (s), 45.3 (s), 37.5 (t), 37.5 (t), 37.3 (t), 29.6 (t), 25.6 (t), 23.9 (q), 23.4 (q), 20.7 (q).
HRMS (ESI): m/z: calculated for C16H26O2 [M + Na+]: 273.1831, found: 273.1834.
Column chromatography: n-hexane/EtOAc (70:30).
3.5.6. Compounds 1, 20–23
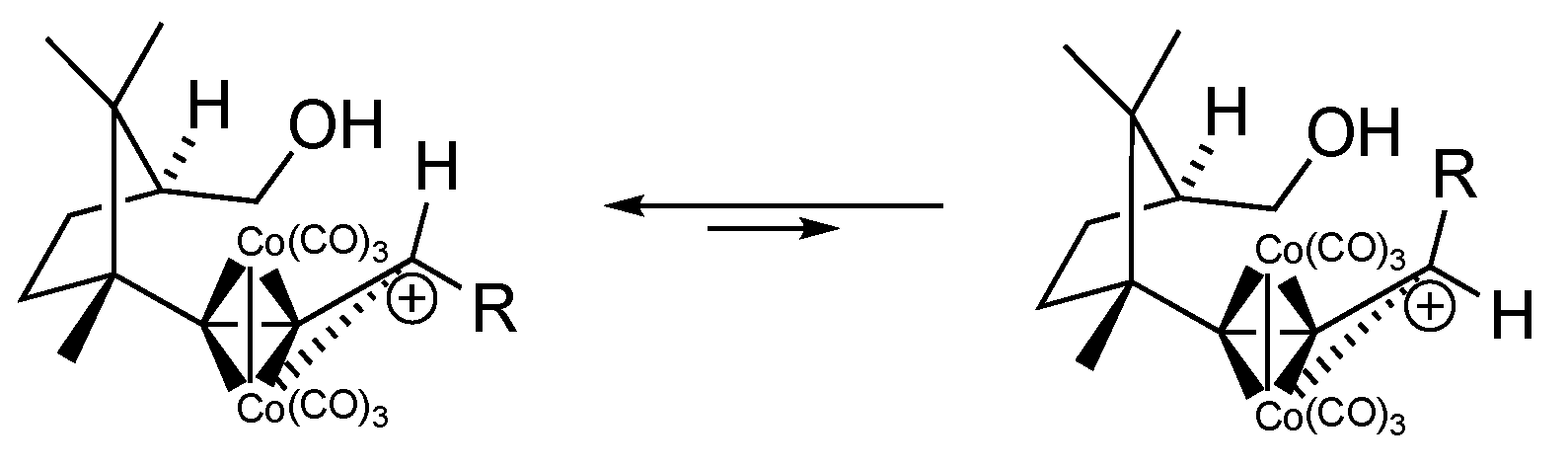
General procedure: To a solution of the corresponding diol (10–14) in DCM (0.1 M) under inert atmosphere (Ar), we added dicobalt octacarbonyl complex (1.2 equiv), and the mixture was stirred until the reaction was finished, which was monitored by TLC. Afterwards, the solvent was evaporated in vacuo, and the crude solid was adsorbed in silica gel and then separated by flash chromatography. The resulting compounds were again dissolved in DCM (0.05 M) under inert atmosphere (Ar), and the solution was cooled to −20 °C before dropwise addition of 1.3 equiv of BF3·Et2O. The resulting mixture was placed in a fridge to maintain the temperature at −20 °C during 36 h. Next, the reaction mixture was poured in a vigorously stirred solution of saturated NaHCO3 (30 mL). Then, the organic layer was collected, and the aqueous layer was extracted with DCM (3 × 20 mL). The organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The alkyne–Co2(CO)6 complexes from the crude were isolated by flash chromatography (silica gel). Finally, 6 equiv of 4-methylmorpholine N-oxide (NMO) were added to a DCM solution containing the resulting complexes under inert atmosphere (Ar), and the mixture was stirred for 36 h. After filtration of the NMO residues, the solvent was removed by rotavap and the crude was purified by flash chromatography (silica gel) obtaining compounds 1 and 21–23.
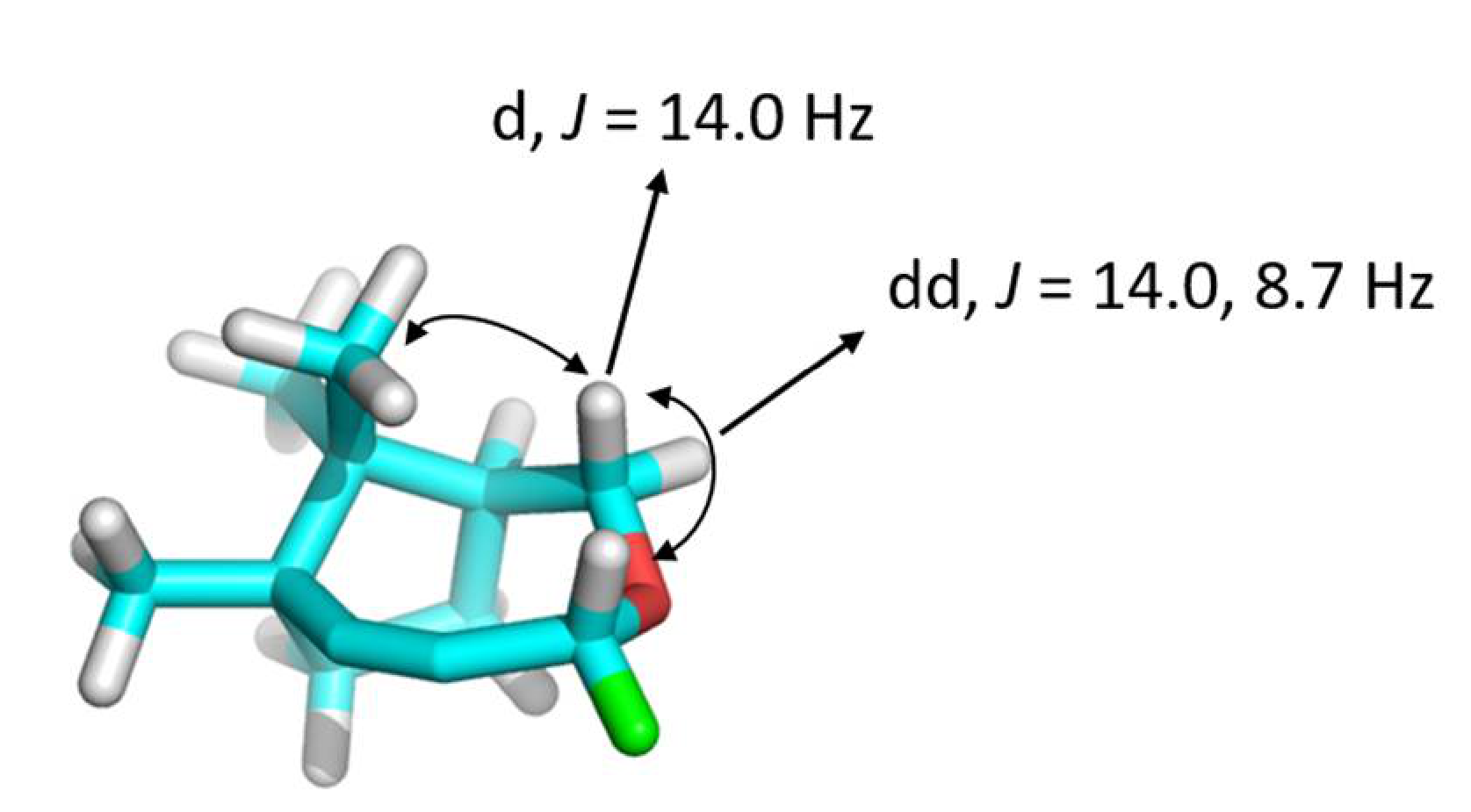
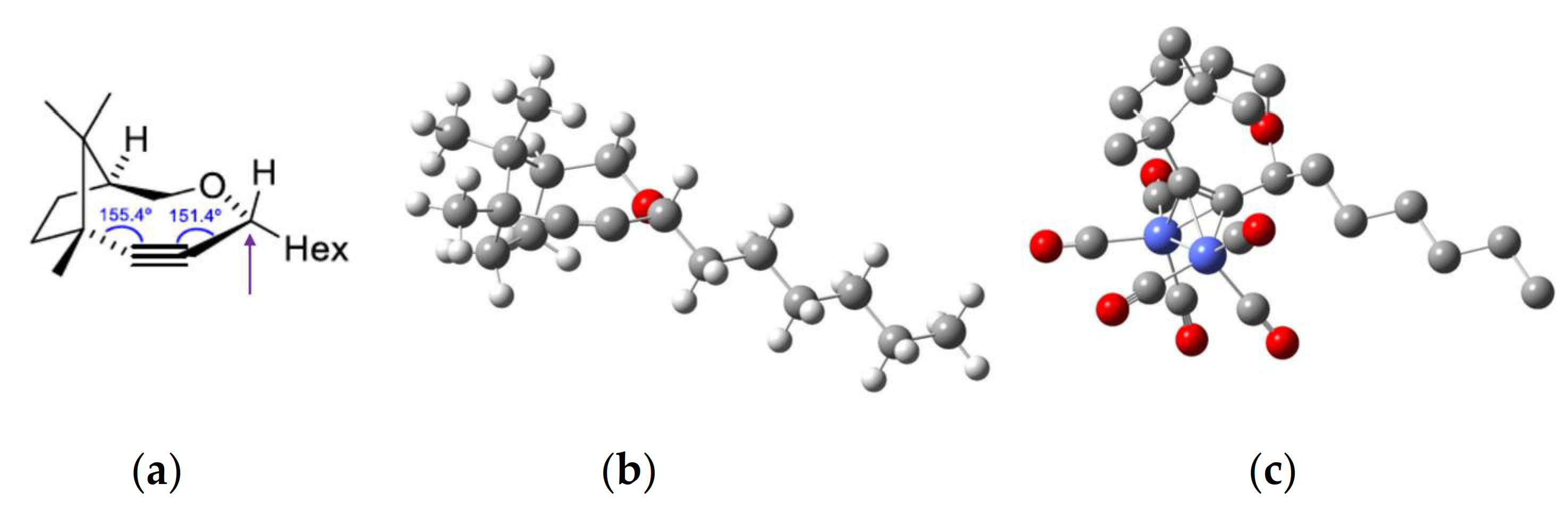
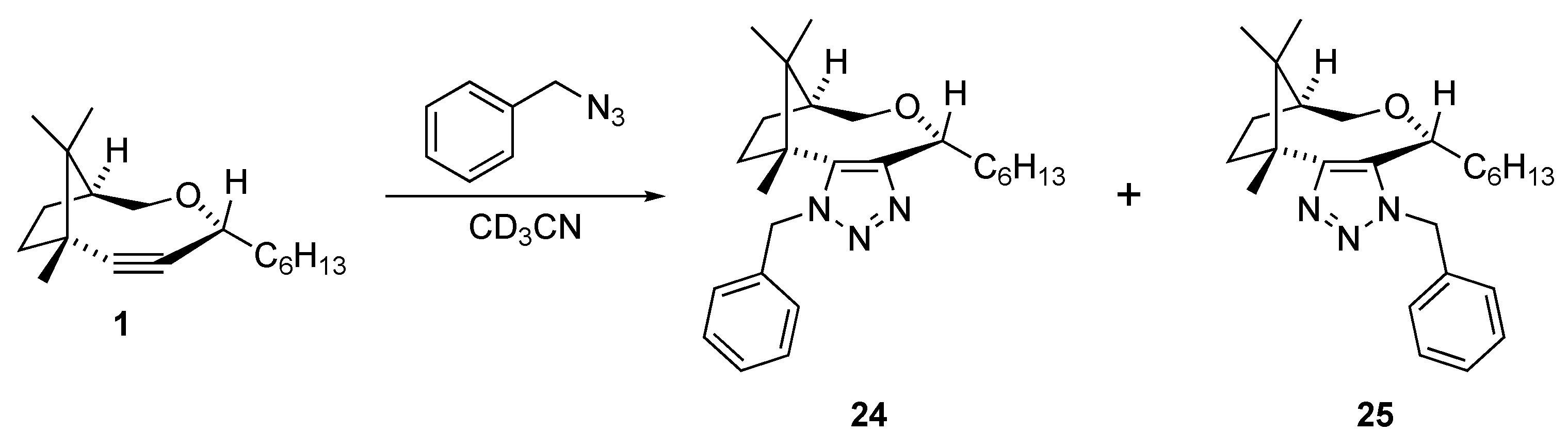
(1S,4R,7R)-4-hexyl-7,10,10-trimethyl-3-oxabicyclo[5.2.1]dec-5-yne (1). 1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 4.24 (dd, J = 5.9, 6.9 Hz, 1H), 3.85 (dd, J = 8.7, 14.0 Hz, 1H), 3.54 (d, J = 14.0 Hz, 1H), 2.26–2.16 (m, 1H), 1.88–1.71 (m, 4H), 1.65–1.52 (m, 2H), 1.41–1.22 (m, 8H), 1.15 (s, 3H), 0.97 (s, 3H), 0.87 (t, 6.9 Hz, 3H), 0.86 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 110.5 (s), 93.1 (s), 73.6 (d), 67.7 (t), 54.3 (s), 50.1 (d), 47.4 (s), 38.4 (t), 34.2 (t), 31.7 (t), 29.1 (t), 26.0 (q), 25.3 (t), 22.5 (t), 20.6 (q), 18.8 (t), 15.4 (q), 14.1 (q).
HRMS (ESI): m/z: calculated for C18H30O [M + H+]: 263.2375, found: 263.2377.
Column chromatography: n-hexane/EtOAc (gradient 85:15–70:30).
(1S,7R)-4,7,10,10-tetramethyl-3-oxabicyclo[5.2.1]dec-5-yne (21). 1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 4.33 (dd, J = 6.6, 13.2 Hz, 2H), 3.83 (dd, J = 8.6, 14.0 Hz, 1H), 3.55 (d, J = 14.0 Hz, 1H), 2.26–2.18 (m, 1H), 1.88–1.71 (m, 4H), 1.30 (d, J = 6.6 Hz, 1H), 1.14 (s, 3H), 0.97 (s, 3H), 0.86 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 109.6 (s), 93.9 (s), 68.9 (d), 67.8 (t), 54.4 (s), 50.3 (d), 47.4 (s), 38.5 (t), 26.1 (q), 20.6 (q), 20.6 (q), 18.9 (t), 15.4 (q).
HRMS (ESI): m/z: calculated for C13H20O [M + Na+]: 215.1412, found: 215.1414.
Column chromatography: n-hexane/DCM (gradient 90:10–80:20).
(1S,4R,7R)-7,10,10-trimethyl-4-phenyl-3-oxabicyclo[5.2.1]dec-5-yne (22). 1H-NMR (500 MHz, CDCl3, 298 K): δ ppm 7.46–7.46 (m, 2H), 7.37–7.28 (m, 3H), 5.30 (s, 1H), 3.99 (dd, J = 8.7, 13.9 Hz, 1H), 3.77 (d, J = 13.9 Hz, 1H), 2.35–2.28 (m, 1H), 1.98–1.91 (m, 1H), 1.88–1.78 (m, 1H), 1.24 (s, 3H), 1.03 (s, 3H), 0.90 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 138.1 (s), 128.4 (d), 128.4 (d), 127.5 (d), 112.4 (s), 91.3 (s), 75.5 (d), 68.2 (t), 54.5 (s), 50.2 (d), 47.5 (s), 38.5 (t), 26.0 (q), 20.7 (q), 19.0 (t), 15.3 (q).
HRMS (ESI): m/z: calculated for C18H22O [M + Na+]: 277.1568, found: 277.1570.
Column chromatography: n-hexane/DCM (gradient 85:15–70:30).
(1S,4R,7R)-4-(but-3-en-1-yl)-7,10,10-trimethyl-3-oxabicyclo[5.2.1]dec-5-yne (23).1H-NMR (500 MHz, CDCl3 298 K): δ ppm 5.82–5.74 (m, 1H), 5.01 (dd, J = 1.7, 17.2 Hz, 1H), 4.93 (d, J = 10.2 Hz, 1H), 4.25 (t, J = 6.2 Hz, 1H), 3.81 (dd, J = 8.7, 14.0 Hz, 1H), 3.51 (d, J = 14.0 Hz, 1H), 2.20–2.07 (m, 2H), 1.86–1.59 (m, 3H), 1.12 (s, 3H), 0.95 (s, 3H), 0.83 (s, 3H).
13C-NMR (150 MHz, CDCl3, 298 K): δ ppm 137.8 (d), 114.8 (t), 111.0 (s), 92.6 (s), 72.9 (d), 67.6 (t), 54.3 (s), 50.1 (d), 47.3 (s), 38.4 (t), 33.2 (t), 29.4 (t), 26.0 (q), 20.6 (q), 18.8 (t), 15.3 (q).
HRMS (ESI): m/z: calculated for C16H24O [M + Na+]: 255.1725, found: 255.1720.
Column chromatography: n-hexane/EtOAc (gradient 95:5–70:30).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





