
Tuning π-Acceptor/σ-Donor Ratio of the 2-Isocyanoazulene Ligand: Non-Fluorinated Rival of Pentafluorophenyl Isocyanide and Trifluorovinyl Isocyanide Discovered

 ,
, 
Abstract
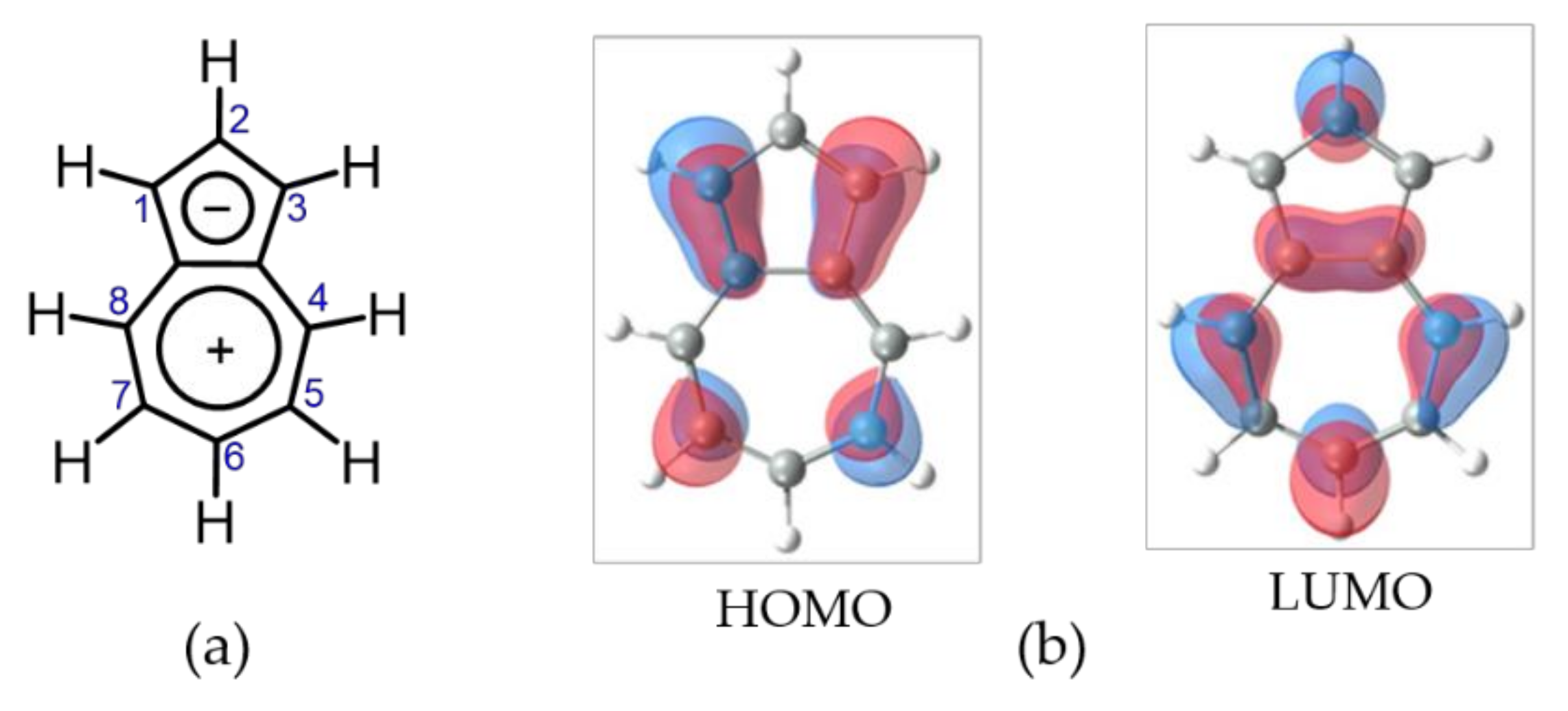
1. Introduction
2. Results
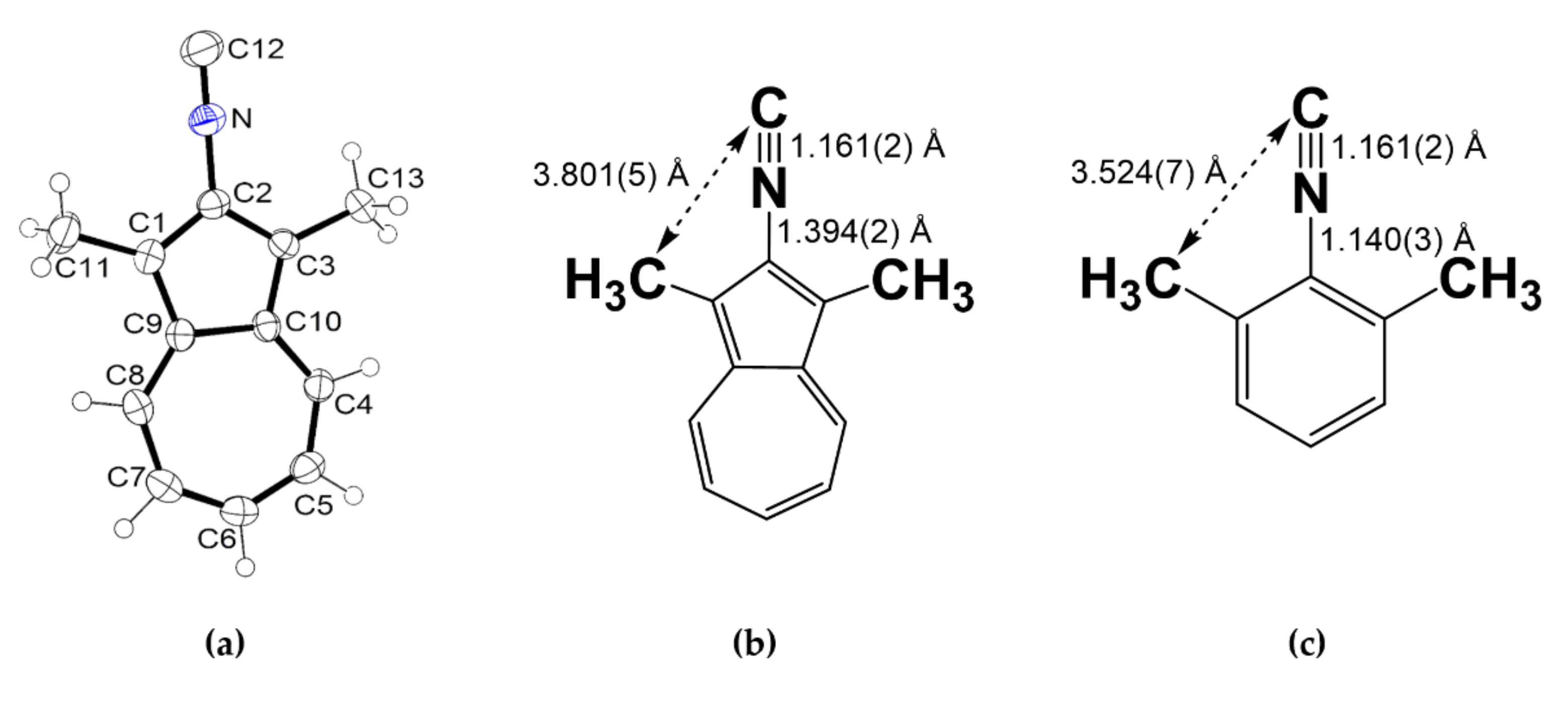
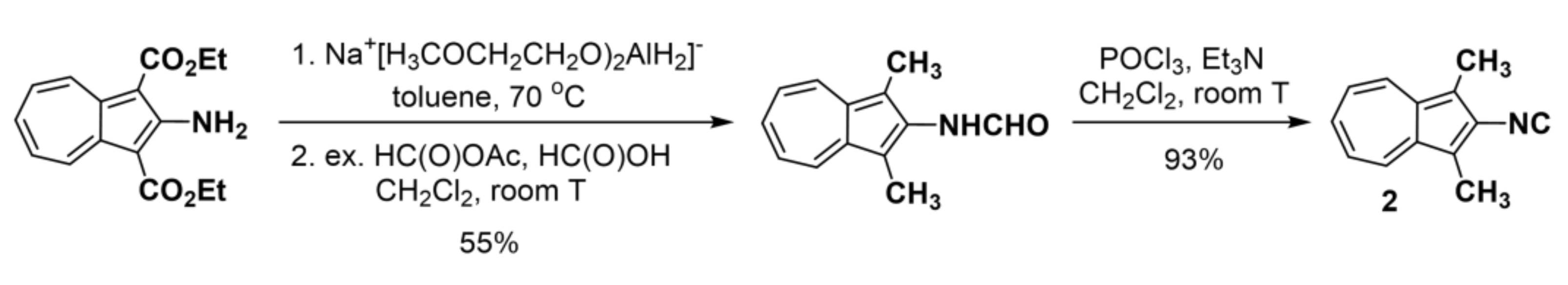
2.1. Synthesis and Properties of 2-Isocyano-1,3-Dimethylazulene (2)
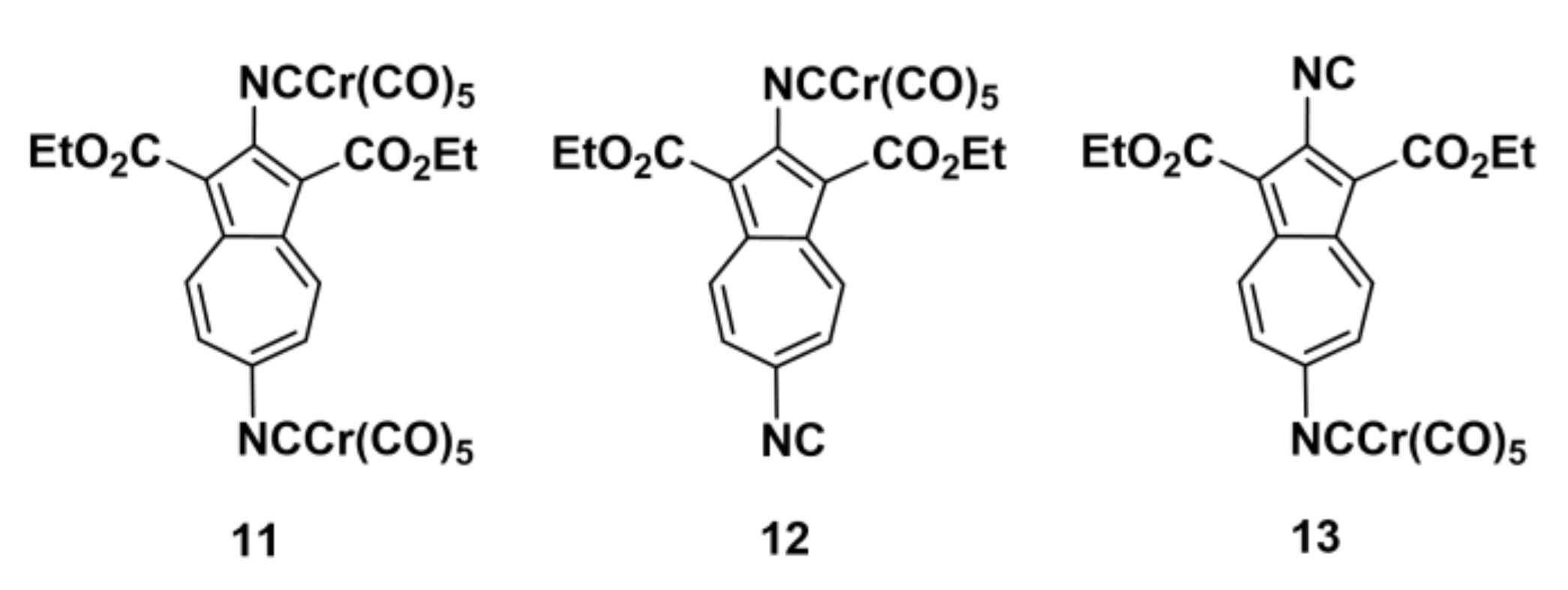
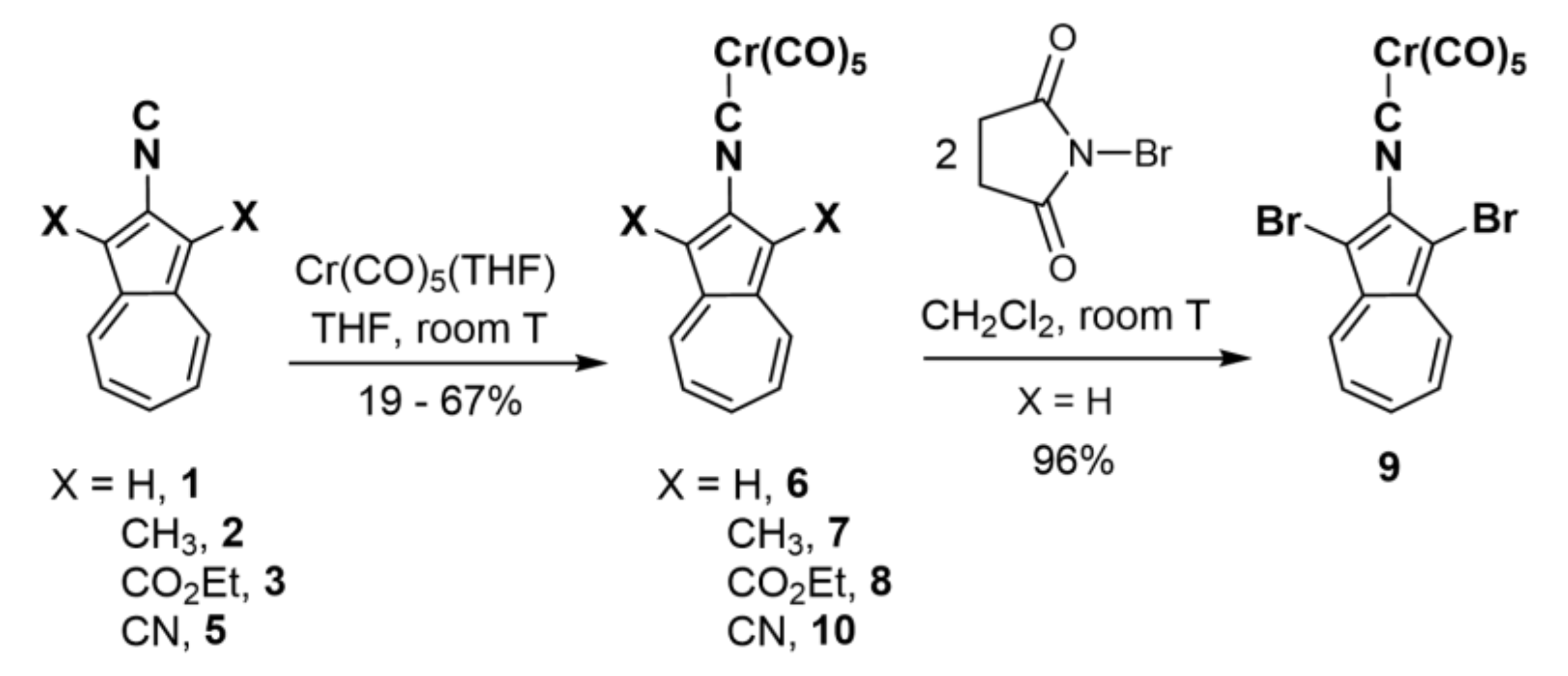
2.2. Preparation of Complexes [(OC)5Cr(2-Isocyano-1,3-X2-Azulene)], X = H, CH3, CO2Et, Br, and CN
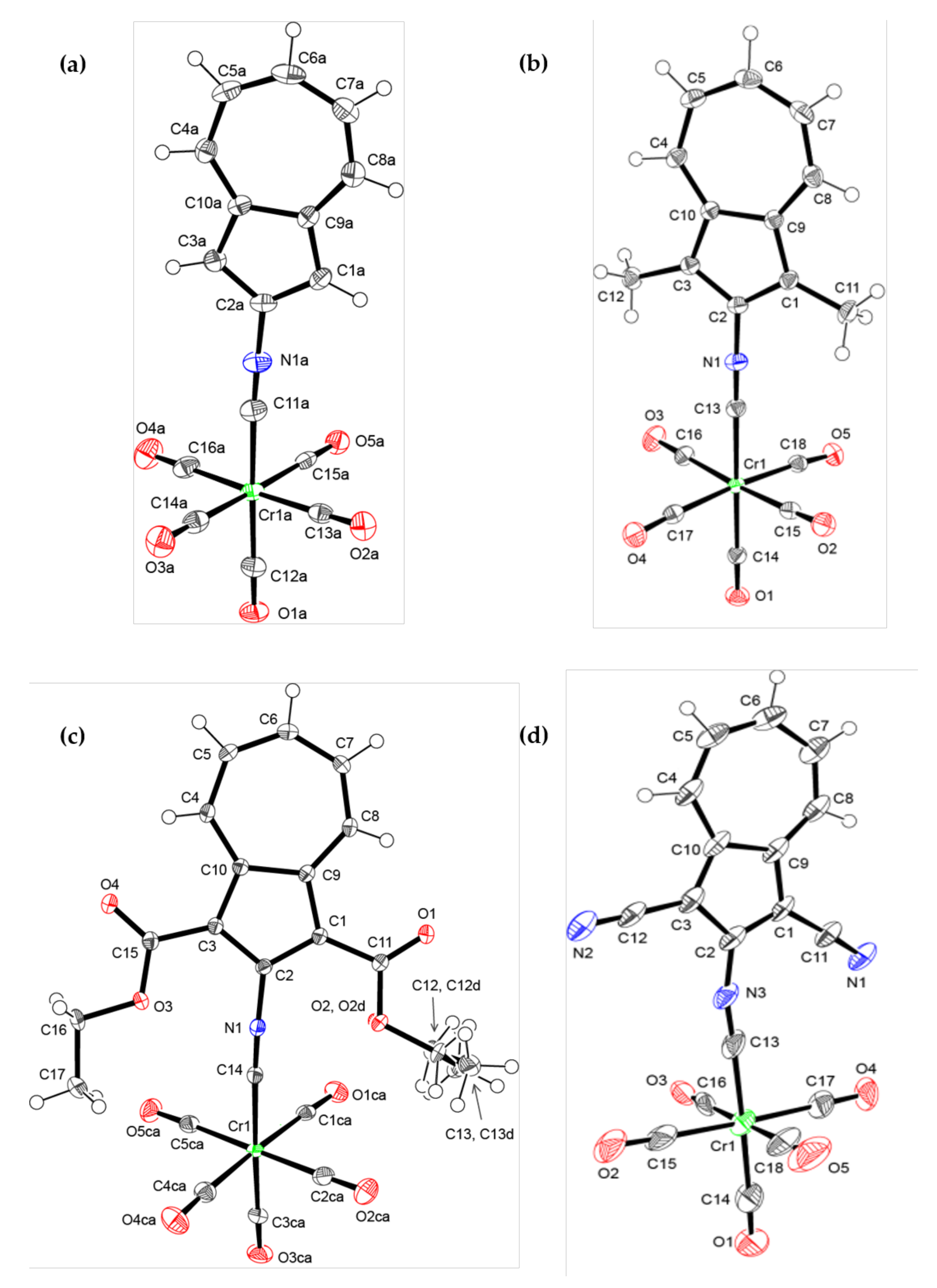
2.3. X-Ray Crystallographic Analysis of 6, 7, 8, and 10
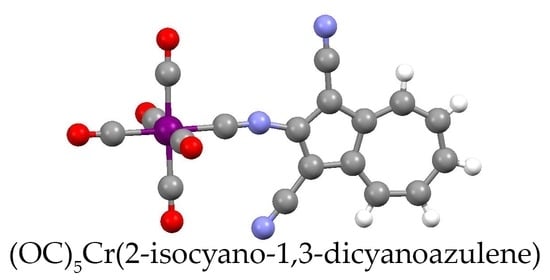
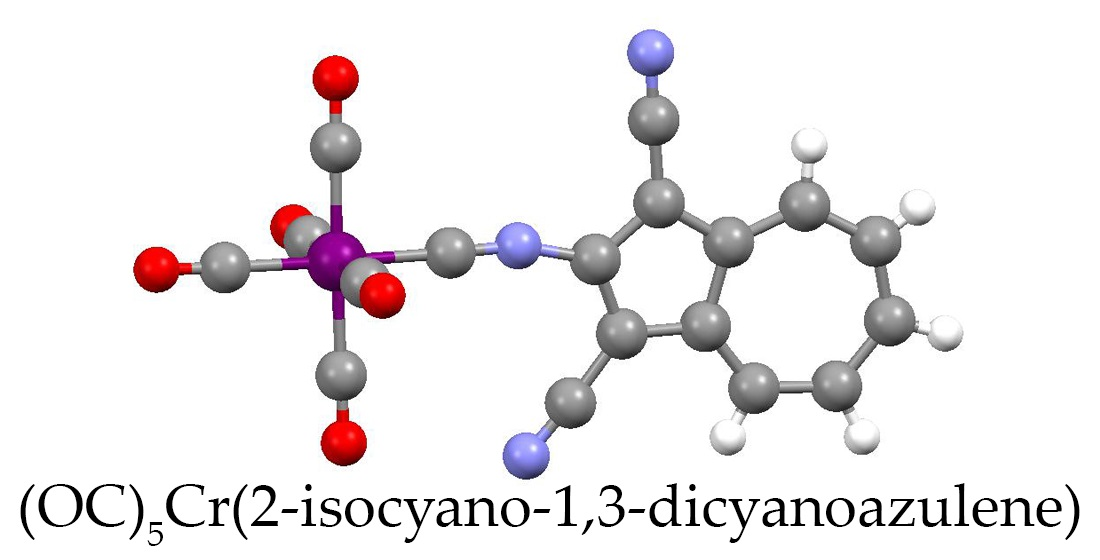
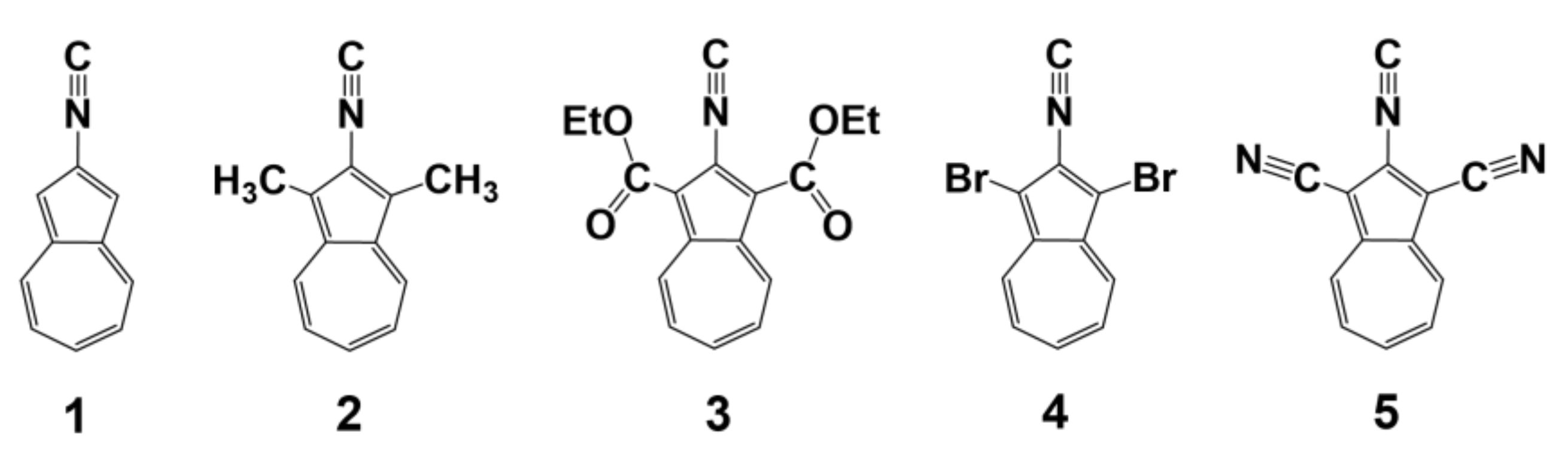
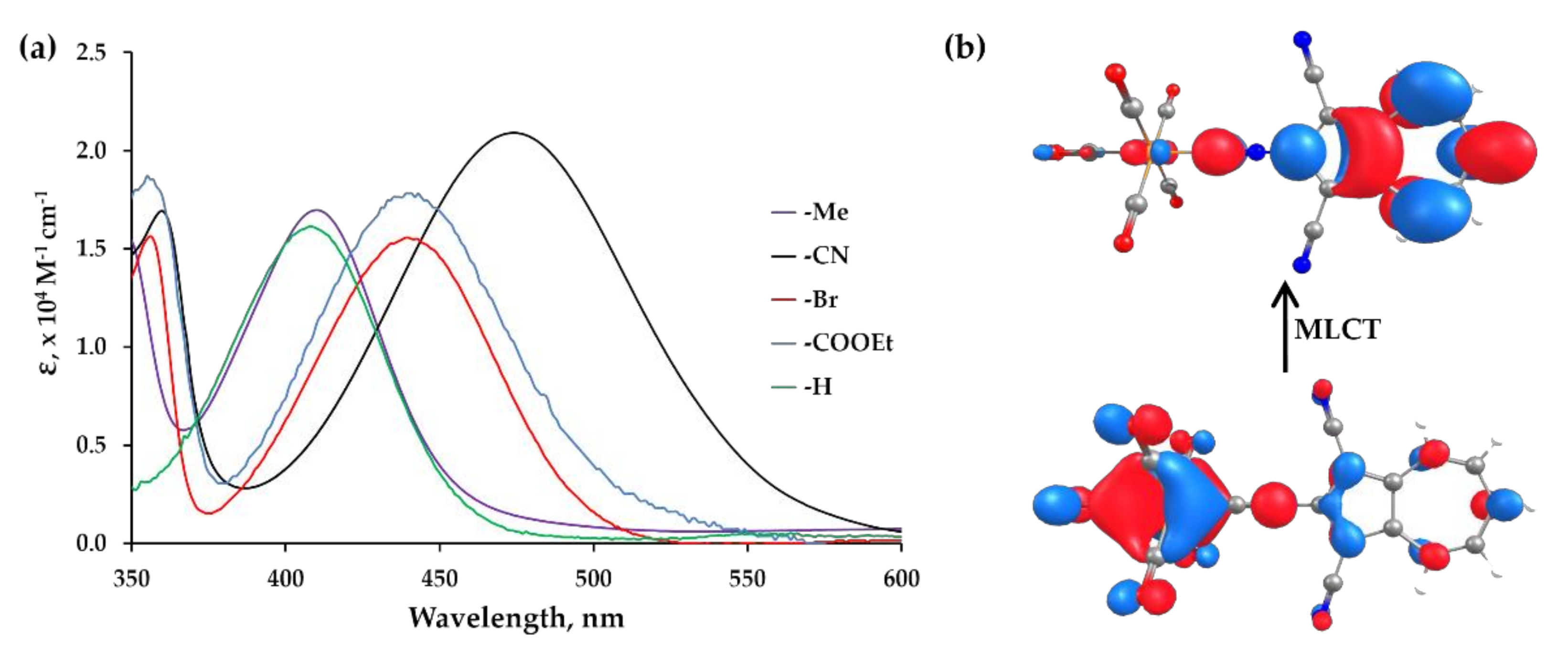
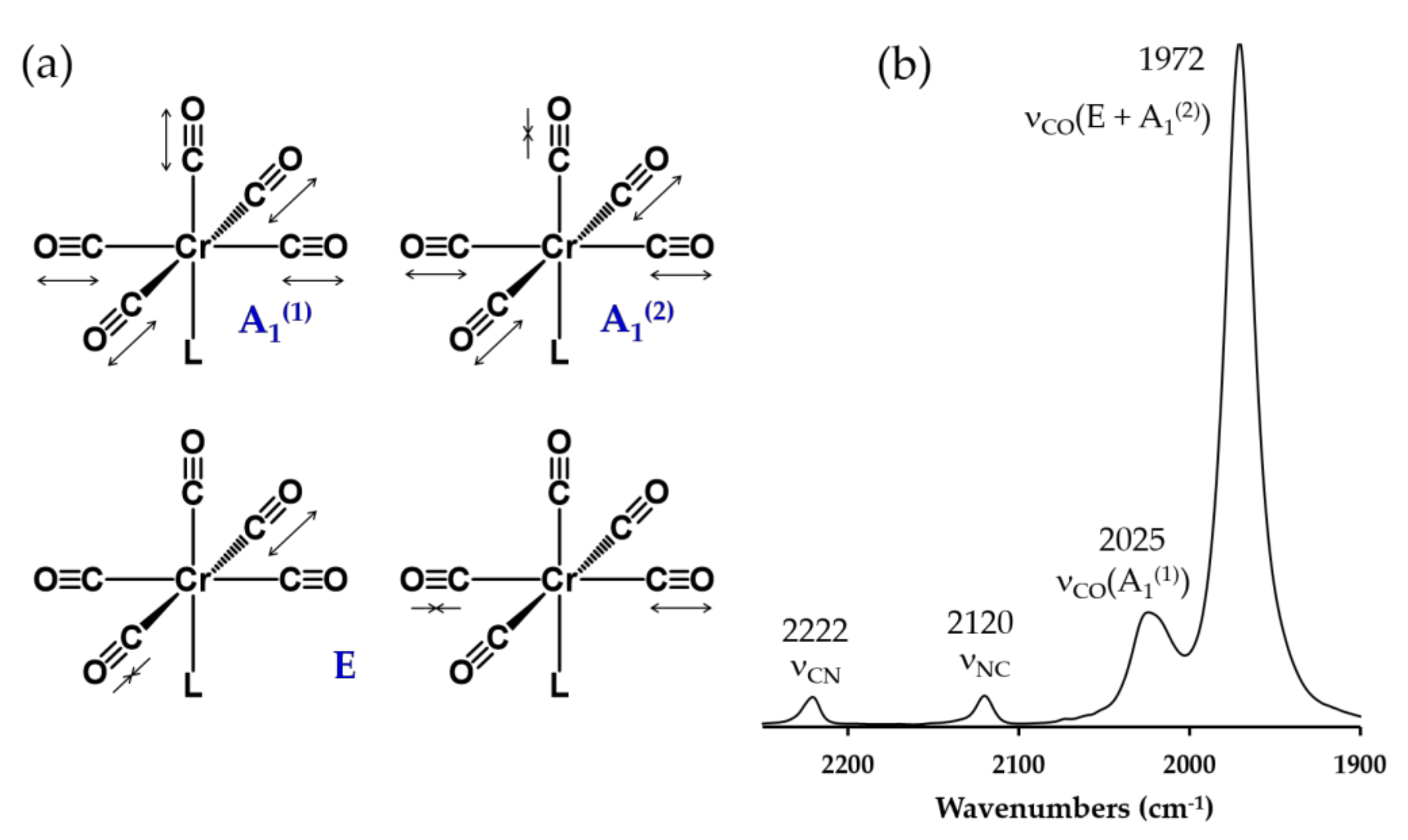
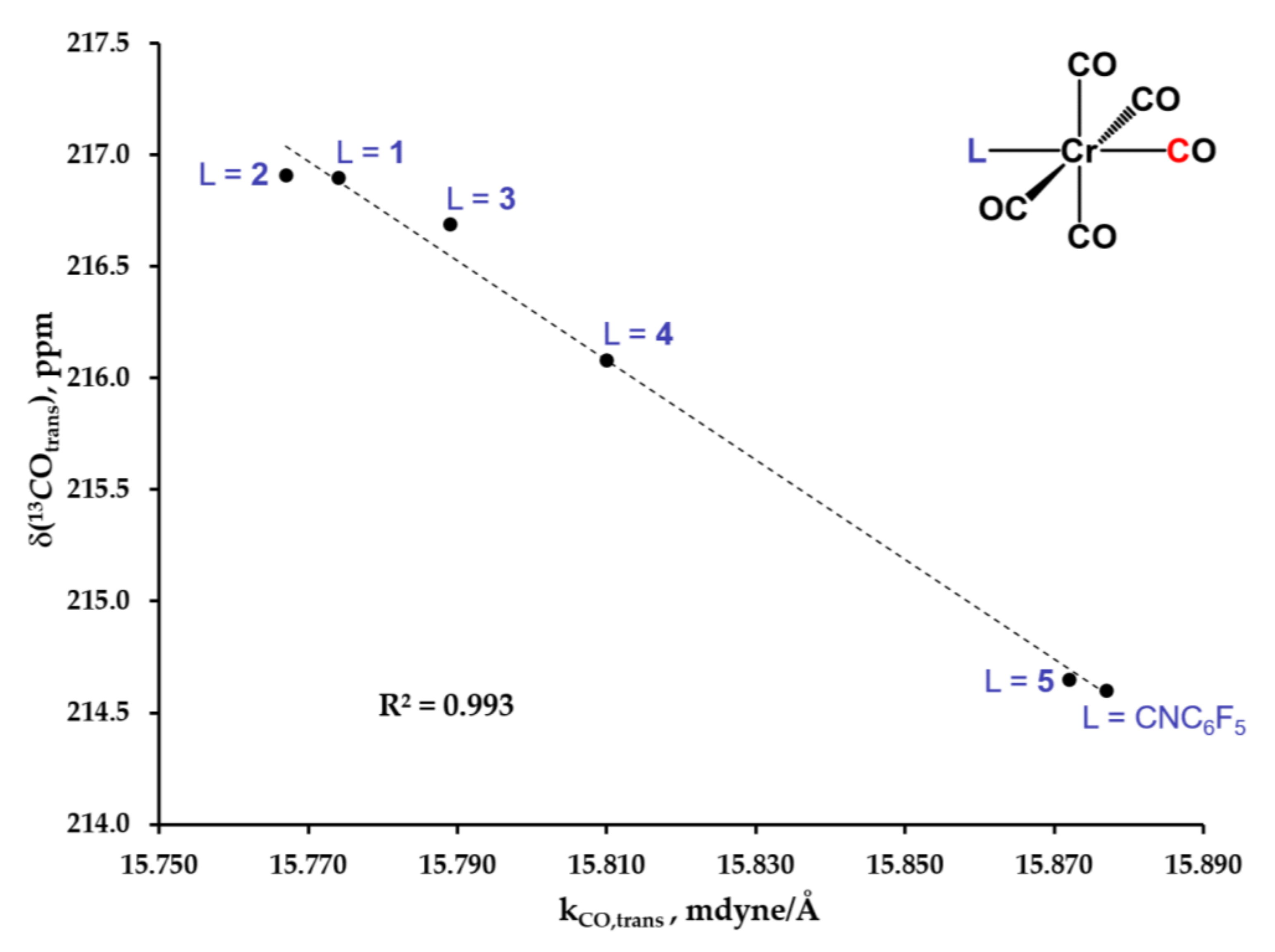
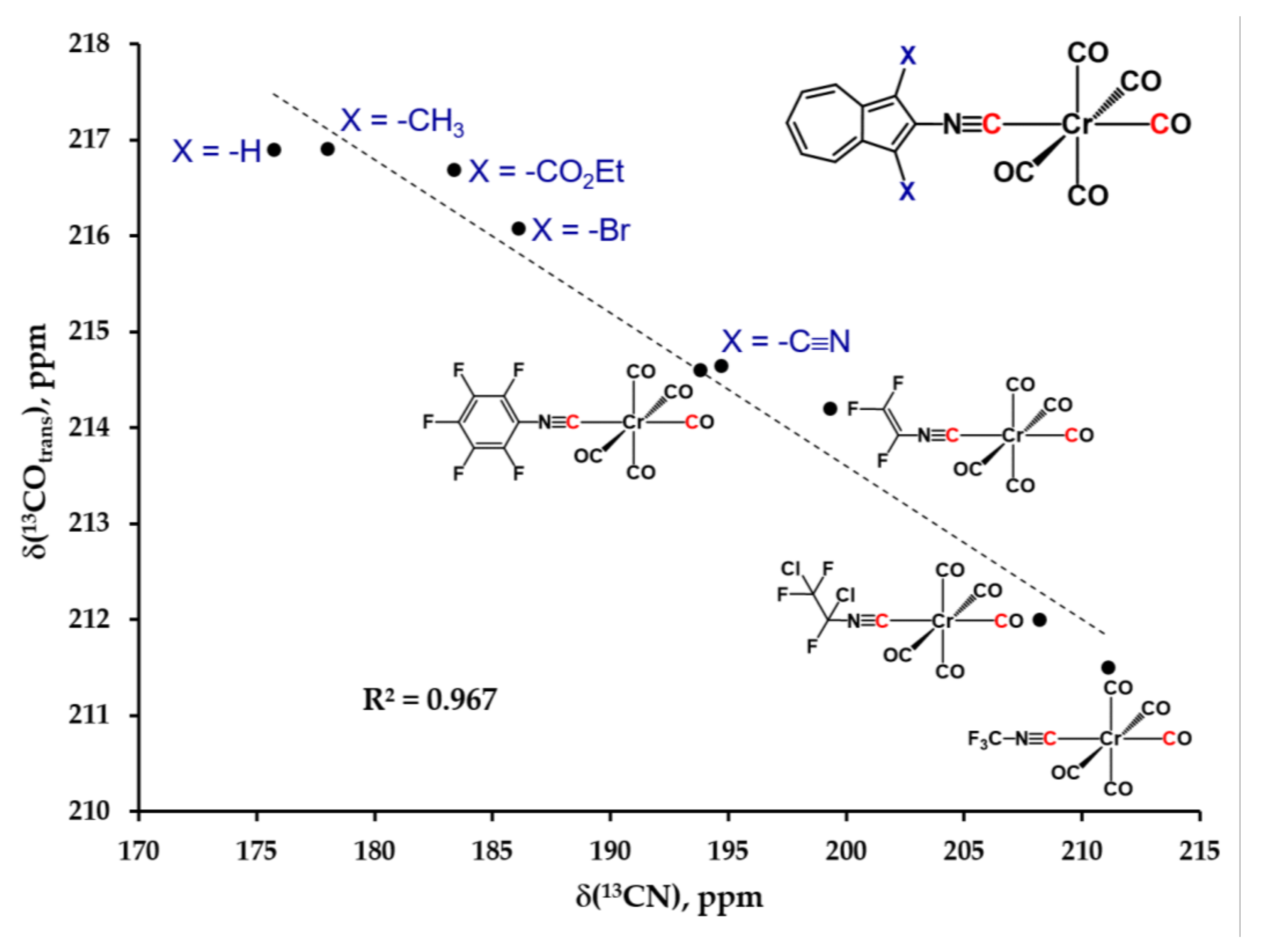
2.4. Electronic Absorption, Infrared, and 13C NMR Spectroscopic Studies of 6, 7, 8, 9 and 10
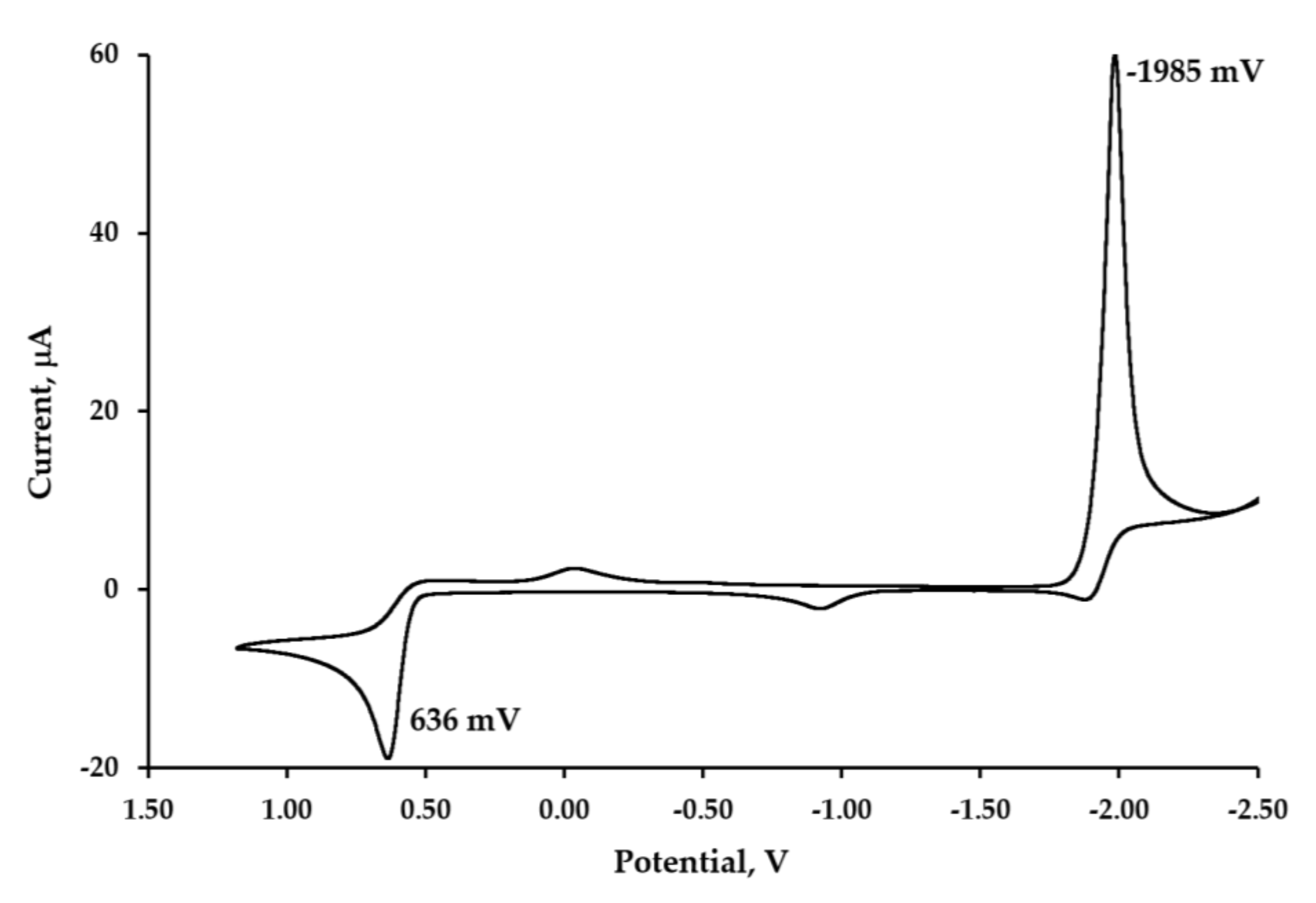
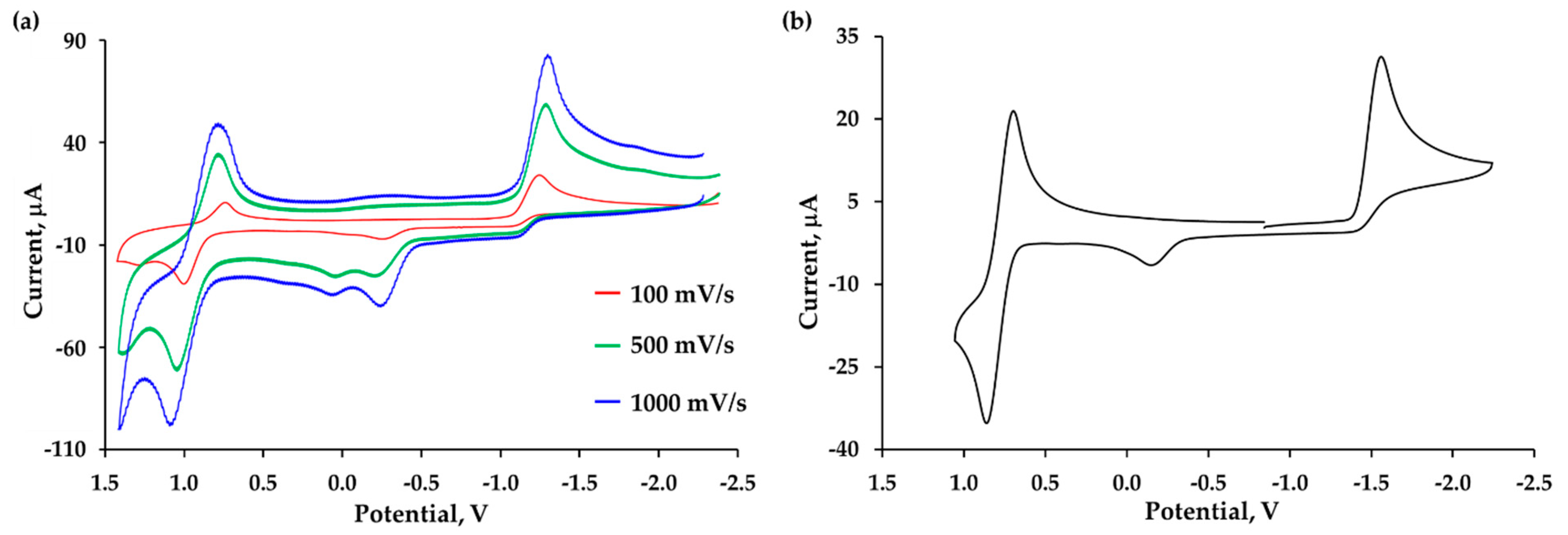
2.5. Electrochemical Studies
3. Conclusions
4. Materials and Methods
4.1. General Procedures, Starting Materials, and Equipment
4.2. Synthesis of 2-Formamido-1,3-Dimethylazulene
4.3. Synthesis of 2-Isocyano-1,3-Dimethylazulene (2)
4.4. Synthesis of [(OC)5Cr(2-Isocyanoazulene)] (6)
4.5. Synthesis of [(OC)5Cr(2-Isocyano-1,3-Dimethylazulene)] (7)
4.6. Synthesis of [(OC)5Cr(2-Isocyano-1,3-Dibromoazulene)] (9)
4.7. Synthesis of [(OC)5Cr(2-Isocyano-1,3-Dicyanoazulene)] (10)
4.8. X-ray Crystallographic Work
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoffmann, R. Building Bridges Between Inorganic and Organic Chemistry (Nobel Lecture). Angew. Chem. Int. Ed. 1982, 21, 711–724. [Google Scholar] [CrossRef]
- Giustiniano, M.; Basso, A.; Mercalli, V.; Massarotti, A.; Novellino, E.; Tron, G.C.; Zhu, J. To each his own: Isonitriles for all flavors. Functionalized isocyanides as valuable tools in organic synthesis. Chem. Soc. Rev. 2017, 46, 1295–1357. [Google Scholar] [CrossRef] [PubMed]
- Barybin, M.V.; Meyers, J.J.J.; Neal, B.M. ChemInform Abstract: Renaissance of Isocyanoarenes as Ligands in Low-Valent Organometallics. Chemin 2013, 44, 493–529. [Google Scholar] [CrossRef]
- Treichel, P.M. Transition Metal-Isocyanide Complexes. Adv. Organomet. Chem. 1973, 11, 21–86. [Google Scholar]
- Boyarskiy, V.P.; Bokach, N.A.; Luzyanin, K.V.; Kukushkin, V.Y. Metal-Mediated and Metal-Catalyzed Reactions of Iso-cyanides. Chem. Soc. Rev. 2015, 115, 2698–2779. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Mokhtarzadeh, C.C.; Ripatti, D.S.; Havrylyuk, I.; Kamezawa, R.; Moore, C.E.; Rheingold, A.L.; Figueroa, J.S. Comparative Measure of the Electronic Influence of Highly Substituted Aryl Isocyanides. Inorg. Chem. 2015, 54, 2936–2944. [Google Scholar] [CrossRef]
- Barnett, B.R.; Figueroa, J.S. Zero-valent isocyanides of nickel, palladium and platinum as transition metal σ-type Lewis bases. Chem. Commun. 2016, 52, 13829–13839. [Google Scholar] [CrossRef] [PubMed]
- Lentz, D. Trifluormethylisocyanid, ein starker π-Akzeptor-Ligand – Pentacarbonyl(thrifluormethylisocyanid)chrom und –wolfram. Chem. Ber. 1984, 117, 415–418. [Google Scholar] [CrossRef]
- Oberhammer, H.; Lentz, D. Gas-phase structure of pentacarbonyl(trifluoromethyl isocyanide)chromium, (CF3NC)Cr(CO)5. Inorg. Chem. 1985, 24, 1271–1273. [Google Scholar] [CrossRef]
- Lentz, D.; Preugschat, D. Preparation of the First Fluorinated Alkenyl lsocyanide F2C=CF-NC by Flash Vacuum Pyrolysis of [Cr(CO)5(CN-CF=CF2)]. Crystal and Molecular Structure of [Cr(CO)5(CN-CF=CF2)] at 125 K. Chem. Commun. 1992, 1523–1524. [Google Scholar] [CrossRef]
- Mujkic, M.; Lentz, D. Synthesis of highly unsaturated isocyanides via organometallic pathways. Dalton Trans. 2012, 41, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Lentz, D.; Anibarro, M.; Preugschat, D.; Bertrand, G. Transition metal complexes and cycloaddition products of pentafluor-ophenyl isocyanide. J. Fluorine Chem. 1998, 89, 73–81. [Google Scholar] [CrossRef]
- Robinson, R.E.; Holovics, T.C.; Deplazes, S.F.; Powell, D.R.; Lushington, G.H.; Thompson, W.H.; Barybin, M.V. Five Possible Isocyanoazulenes and Electron-Rich Complexes Thereof: A Quantitative Organometallic Approach for Probing Electronic Inhomogeneity of the Azulenic Framework. Organometallics 2005, 24, 2386–2397. [Google Scholar] [CrossRef]
- Anderson, A.G.; Steckler, B.M. Azulene. VIII. A Study of the Visible Absorption Spectra and Dipole Moments of Some 1- and 1,3-Substituted Azulenes1,2. J. Am. Chem. Soc. 1959, 81, 4941–4946. [Google Scholar] [CrossRef]
- Liu, R.S.H.; Asato, A.E. Tuning the color and excited state properties of the azulenic chromophore: NIR absorbing pigments and materials. J. Photochem. Photobiol. C Photochem. Rev. 2003, 4, 179–194. [Google Scholar] [CrossRef]
- Barybin, M.V. Nonbenzenoid aromatic isocyanides: New coordination building blocks for organometallic and surface chemistry. Co-ord. Chem. Rev. 2010, 254, 1240–1252. [Google Scholar] [CrossRef]
- Sun, S.; Zhuang, X.; Wand, L.; Zhang, B.; Ding, J.; Zhang, F.; Chen, Y. Azulene-Bridged Coordinated Framework Based Qua-si-Molecular Rectifier. J. Mater. Chem. C. 2017, 5, 2223–2229. [Google Scholar] [CrossRef]
- Yang, C.; Schellhammer, K.S.; Ortmann, F.; Sun, S.; Dong, R.; Karakus, M.; Mics, Z.; Löffler, M.; Zhang, F.; Zhuang, X.; et al. Coordination Polymer Framework Based On-Chip Micro-Supercapacitors with AC Line-Filtering Performance. Angew. Chem. Int. Ed. 2017, 56, 3920–3924. [Google Scholar] [CrossRef] [PubMed]
- Applegate, J.C.; Okeowo, M.K.; Erickson, N.R.; Neal, B.M.; Berrie, C.L.; Gerasimchuk, N.N.; Barybin, M.V. First π-linker featuring mercapto and isocyano anchoring groups within the same molecule: Synthesis, heterobimetallic complexation and self-assembly on Au(III). Chem. Sci. 2016, 7, 1422–1429. [Google Scholar] [CrossRef]
- DuBose, D.L.; Moody, D.; Robinson, R.E.; Holovics, T.C.; Weintrob, E.C.; Berrie, C.L.; Barybin, M.V. Interaction of Mono- and Diisocyanoazulene Derivatives with Gold Surface: First Examples of Self-Assembled Monolayer Films Involving Azulenic Scaffolds. Langmuir 2006, 22, 4599–4606. [Google Scholar] [CrossRef]
- Neal, B.M.; Vorushilov, A.S.; Delarosa, A.M.; Robinson, R.E.; Berrie, C.L.; Barybin, M.V. Ancillary nitrile substituents as convenient IR spectroscopic reporters for self-assembly of mercapto- and isocyanoazulenes on Au(III). Chem. Commun. 2011, 47, 10803–10805. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.R.; Spaeth, A.D.; Neal, B.M.; Berrie, C.L.; Thompson, W.H.; Day, V.W.; Barybin, M.V. Linear 6,6’-Biazulenyl Framework Featuring Isocyanide Termini: Synthesis, Structure, Redox Behavior, Complexation, and Self-assembly on Gold(III). J. Am. Chem. Soc. 2010, 132, 15924–15926. [Google Scholar] [CrossRef] [PubMed]
- Kühne, T.; Au-Yeung, K.H.; Eisenhut, F.; Aiboudi, O.; Ryndyk, D.A.; Cuniberti, G.; Lissel, F.; Moresco, F. STM induced ma-nipulation of azulene-based molecules and nanostructures: The role of the dipole moment. Nanoscale 2020, 12, 24471–24476. [Google Scholar] [CrossRef]
- Fathi-Rasekh, M.; Rohde, G.T.; Hart, M.D.; Nakakita, T.; Zatsikha, Y.V.; Valiev, R.R.; Barybin, M.V.; Nemykin, V.N. Posi-tional Isomers of Isocyanoazulenes as Axial Ligands Coordinated to Ruthenium(II) Tetraphenylporphyrin: Fine-Tuning Redox and Optical Profiles. Inorg. Chem. 2019, 58, 9316–9325. [Google Scholar] [CrossRef]
- De Domingo, E.; Barcenilla, M.; Martín-Alvarez, J.M.; Miguel, J.A.; Coco, S. The 2-isocyanoazulene-gold(I) fragment as a versatile element for organometallic dyes and liquid crystals. Dye. Pigment. 2020, 176, 108195. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Ghorai, S. Very few isocyanides with non-offensive smell are known: Versatile, Fragrant, Convertible Isonitriles. J. Am. Chem. Soc. 2006, 128, 11772–11773. [Google Scholar] [CrossRef]
- Drakenberg, T.; Dahlqvist, K.-I.; Forsén, S.; Ragnarsson, U.; Rasmussen, S.E.; Sunde, E.; Sørensen, N.A. The Barrier to Internal Rotation in Amides. II. N,N-Dimethytrichloroacetamide. Experimental Errors in the Evaluation of Rate Constants with the NMR Lineshape Method. Acta Chem. Scand. 1970, 24, 694–702. [Google Scholar] [CrossRef][Green Version]
- Patil, P.; Ahmadian-Moghaddam, M.; Dömling, A. Isocyanide 2.0. Green Chem. 2020, 22, 6902. [Google Scholar] [CrossRef]
- Minelli, M.; Maley, W.J. Multinuclear NMR studies of molybdenum and tungsten carbonyl isocyanide complexes. Inorg. Chem. 1989, 28, 2954–2958. [Google Scholar] [CrossRef]
- Mathieson, T.; Schier, A.; Schmidbaur, H. Supramolecular chemistry of gold(I) thiocyanate complexes with thiophene, phosphine and isocyanide ligands, and the structure of 2,6-dimethylphenyl isocyanide. J. Chem. Soc. Dalton Trans. 2001, 1196–1200. [Google Scholar] [CrossRef]
- Ree, N.; Andersen, C.L.; Kilde, M.D.; Hammerich, O.; Nielsen, M.B.; Mikkelsen, K.V. The quest for determining one-electron redox potentials of azulene-1-carbonitriles by calculation. Phys. Chem. Chem. Phys. 2018, 20, 7438–7446. [Google Scholar] [CrossRef] [PubMed]
- Sattler, W.; Parkin, G. Synthesis of transition metal isocyanide compounds from carbonyl complexes via reaction with Li[Me3SiNR]. Chem. Commun. 2009, 7566–7568. [Google Scholar] [CrossRef]
- Holovics, T.C.; Deplazes, S.F.; Toriyama, M.; Powell, D.R.; Lushington, G.H.; Barybin, M.V. Organometallic Isocyanocyclopentadienides: A Combined Synthetic, Spectroscopic, Structural, Electrochemical and Theoretical Investigation. Organo Met. 2004, 23, 2927–2938. [Google Scholar] [CrossRef]
- Holovics, T.C.; Robinson, R.E.; Weintrob, E.C.; Toriyama, M.; Lushington, A.G.H.; Barybin, M.V. The 2,6-Diisocyanoazulene Motif: Synthesis and Efficient Mono- and Heterobimetallic Complexation with Controlled Orientation of the Azulenic Dipole. J. Am. Chem. Soc. 2006, 128, 2300–2309. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A.; Kraihanzel, C.S. Vibrational Spectra and Bonding in Metal Carbonyls. I. Infrared Spectra of Phos-phine-substituted Group VI Carbonyls in the CO Stretching Region. J. Am. Chem. Soc. 1962, 84, 4432–4438. [Google Scholar] [CrossRef]
- Karakaş, D.; Kaya, C. Force constant calculations for octahedral complexes of the type M(CO)5L, based on the CO-factored force field. J. Organomet. Chem. 2001, 640, 37–40. [Google Scholar] [CrossRef]
- Johnston, R.F. Effects of Multiple Substituents on the Physical Properties of Pentacarbonylarylisocyanidechromium Com-pounds. J. Coord. Chem. 1994, 31, 57–66. [Google Scholar] [CrossRef]
- Ugi, I.; Fetzer, U.; Eholzer, U.; Knupfer, H.; Offermann, K. Isonitril-Synthesen. Angew. Chem. 1965, 77, 492–504. [Google Scholar] [CrossRef]
- Dewar, M.J.S. The Molecular Orbital Theory of Organic Chemistry; McGraw-Hill: New York, NY, USA, 1969. [Google Scholar]
- Nozoe, T.; Takase, K.; Nakazawa, T.; Fukuda, S. The formation of azulene derivatives from 2H-cyclohepta[b]furan-2-one derivatives. Tetrahedron 1971, 27, 3357–3368. [Google Scholar] [CrossRef]
- Krimen, K.I. Acetic Formic Anhydride. Org. Synth. 1970, 50, 1–3. [Google Scholar]
- Neese, F. ORCA—An ab initio, Density Functional and Semiempirical Program Package, version 3.0; University of Bonn: Bonn, Germany, 2012. [Google Scholar]
- Becke, A.D. Density functional calculations of molecular bond energies. J. Chem. Phys. 1986, 84, 4524–4529. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CNR | υN≡C, cm−1 | δ(13C≡N), ppm | δ(C≡14N), ppm |
|---|---|---|---|
| 1 | 2127 1 | 168.6 2,5 | 175.3 3,5 |
| 2 | 2115 1 | 170.7 2 | 172.3 3 |
| 2,6-CNXyl | 2117 1 | 167.7 2 | 175.6 4,6 |
| Complex | d(Cr–CN) | d(C≡N) | d(Cr-COtrans) | ∠C-N-C |
|---|---|---|---|---|
| [(OC)5Cr(CNtBu)] 1 | 2.016 | 1.150 | 1.872 | 177.9 |
| 62 | 1.974(4) 1.987(4) | 1.155(4) 1.153(4) | 1.879(4) 1.882(4) | 178.(4) 170.5(3) |
| 7 | 1.981(1) | 1.165(2) | 1.885(1) | 178.9(2) |
| 8 | 1.977(2) | 1.163(2) | 1.881(2) | 168.9(2) |
| [(OC)5Cr(CNp-FArDArF2)] 3 | 1.967(2) | 1.162(3) | 1.894(2) | 172.6(2) |
| [(OC)5Cr(CNC2F3)] 4 | 1.942(2) | 1.162(2) | 1.909(2) | 173.6(2) |
| 10 | 1.937(2) | 1.176(3) | 1.901(3) | 164.8(3) |
| (OC)5Cr(CNAz) Complex | λmax, nm | υmax, cm−1 |
|---|---|---|
| 6 | 409 | 24,450 |
| 7 | 410 | 24,390 |
| 8 | 441 1 | 22,676 |
| 9 | 440 | 22,727 |
| 10 | 474 | 21,097 |
| Complex | υN≡C(A1), cm−1 | υC≡O(A1(1)), cm−1 | νC≡O(A1(2)), cm−1 | νC≡O(E), cm−1 | kC≡O,trans, mdyne/Å |
|---|---|---|---|---|---|
| 72 | 2129 | 2050 | 1957 | 1957 | 15.767 |
| 62 | 2138 | 2052 | 1957 | 1957 | 15.774 |
| 82 | 2140 | 2049 | 1959 | 1959 | 15.789 |
| 92 | 2132 | 2042 | 1963 | 1963 | 15.816 |
| 102 | 2120 | 2025 | 1972 | 1972 | 15.875 |
| [(OC)5Cr(CNC6F5)] 3 | 2125 | 2041 | 1968 | 1968 | 15.877 |
| Complex | δ(13CN), ppm | δ(13COtrans), ppm | δ(13COcis), ppm |
|---|---|---|---|
| 6 | 175.64 | 216.83 | 214.58 |
| 7 | 177.99 | 216.91 | 214.68 |
| 81 | 183.36 | 216.69 | 214.60 |
| 9 | 186.07 | 216.08 | 214.10 |
| 10 | 194.69 | 214.65 | 213.09 |
| [(OC)5Cr(CNC6F5)] 2 | 193.8 | 214.6 | 213.3 |
| [(OC)5Cr(CNC2F3)] 3 | 199.3 | 214.2 | 213.0 |
| [(OC)5Cr(CNC(ClF)C(ClF2))] 3 | 208.2 | 212.0 | 212.0 |
| [(OC)5Cr(CNCF3)] 4 | 211.1 | 211.5 | 211.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hart, M.D.; Meyers, J.J., Jr.; Wood, Z.A.; Nakakita, T.; Applegate, J.C.; Erickson, N.R.; Gerasimchuk, N.N.; Barybin, M.V. Tuning π-Acceptor/σ-Donor Ratio of the 2-Isocyanoazulene Ligand: Non-Fluorinated Rival of Pentafluorophenyl Isocyanide and Trifluorovinyl Isocyanide Discovered. Molecules 2021, 26, 981. https://doi.org/10.3390/molecules26040981
Hart MD, Meyers JJ Jr., Wood ZA, Nakakita T, Applegate JC, Erickson NR, Gerasimchuk NN, Barybin MV. Tuning π-Acceptor/σ-Donor Ratio of the 2-Isocyanoazulene Ligand: Non-Fluorinated Rival of Pentafluorophenyl Isocyanide and Trifluorovinyl Isocyanide Discovered. Molecules. 2021; 26(4):981. https://doi.org/10.3390/molecules26040981
Chicago/Turabian StyleHart, Mason D., John J. Meyers, Jr., Zachary A. Wood, Toshinori Nakakita, Jason C. Applegate, Nathan R. Erickson, Nikolay N. Gerasimchuk, and Mikhail V. Barybin. 2021. "Tuning π-Acceptor/σ-Donor Ratio of the 2-Isocyanoazulene Ligand: Non-Fluorinated Rival of Pentafluorophenyl Isocyanide and Trifluorovinyl Isocyanide Discovered" Molecules 26, no. 4: 981. https://doi.org/10.3390/molecules26040981
APA StyleHart, M. D., Meyers, J. J., Jr., Wood, Z. A., Nakakita, T., Applegate, J. C., Erickson, N. R., Gerasimchuk, N. N., & Barybin, M. V. (2021). Tuning π-Acceptor/σ-Donor Ratio of the 2-Isocyanoazulene Ligand: Non-Fluorinated Rival of Pentafluorophenyl Isocyanide and Trifluorovinyl Isocyanide Discovered. Molecules, 26(4), 981. https://doi.org/10.3390/molecules26040981