
Chromatin Accessibility Is Associated with Artemisinin Biosynthesis Regulation in Artemisia annua


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
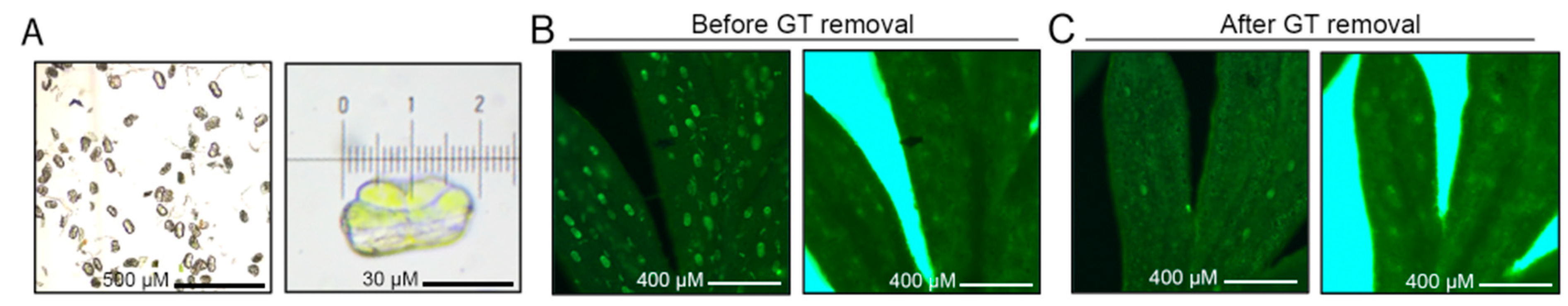
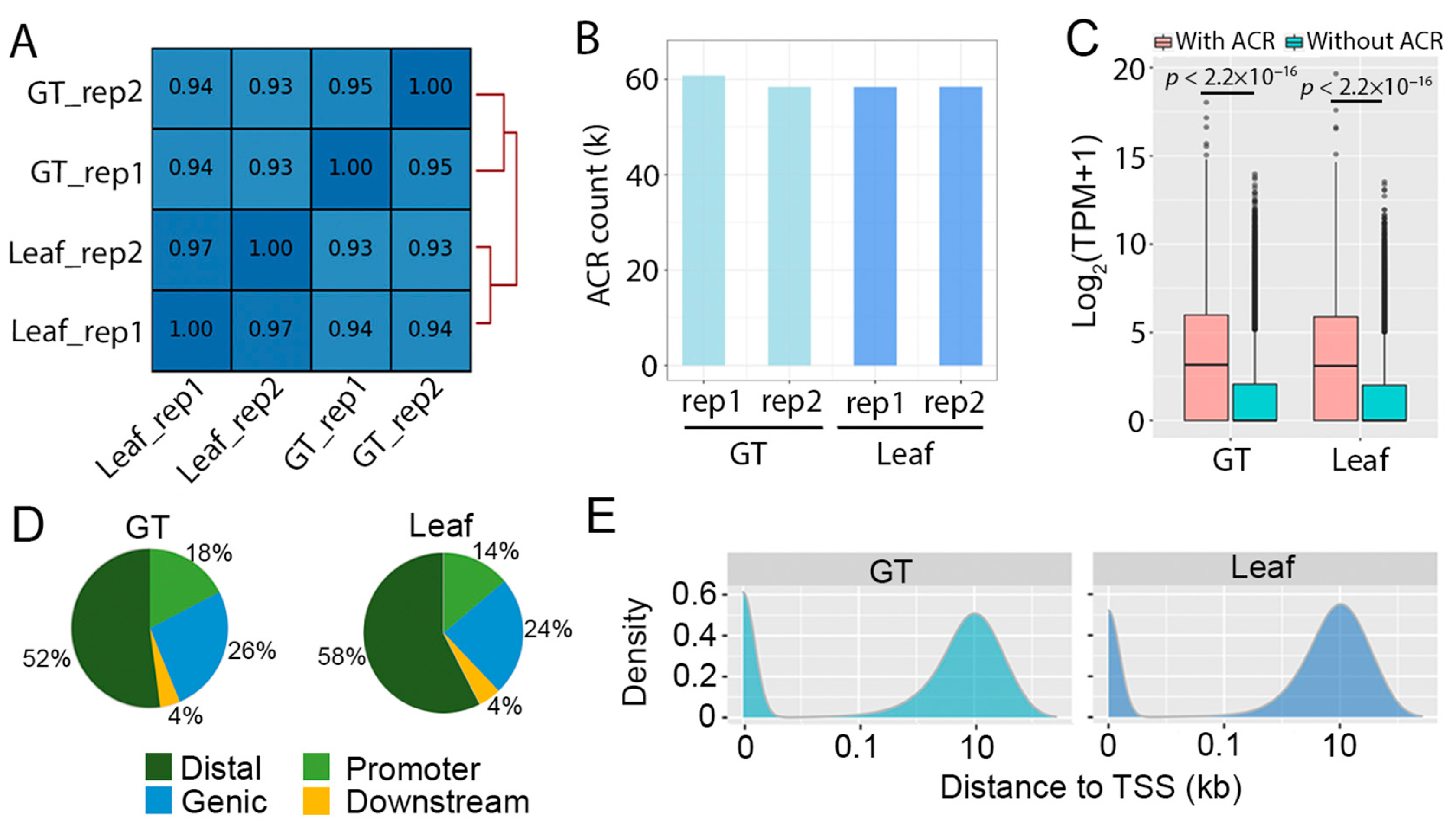
2.1. Mapping Accessible Chromatin in Artemisia annua
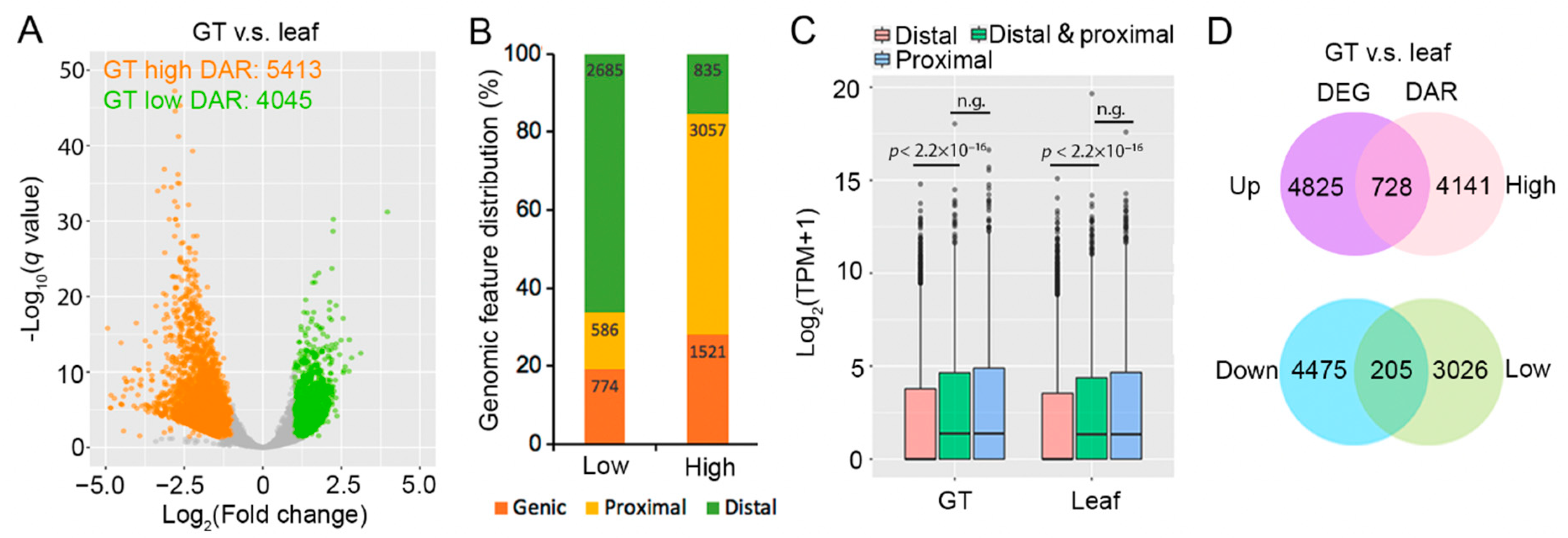
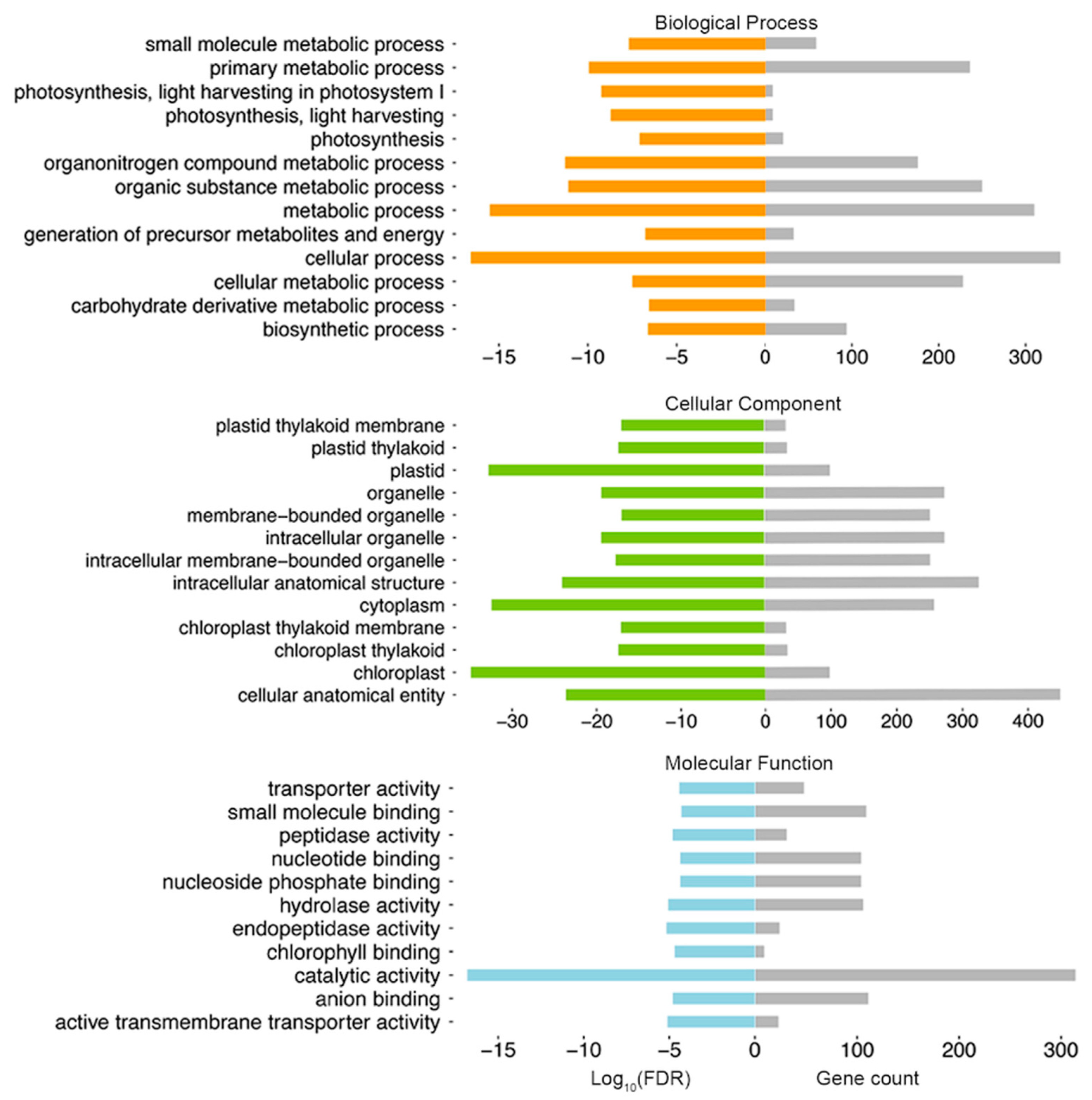
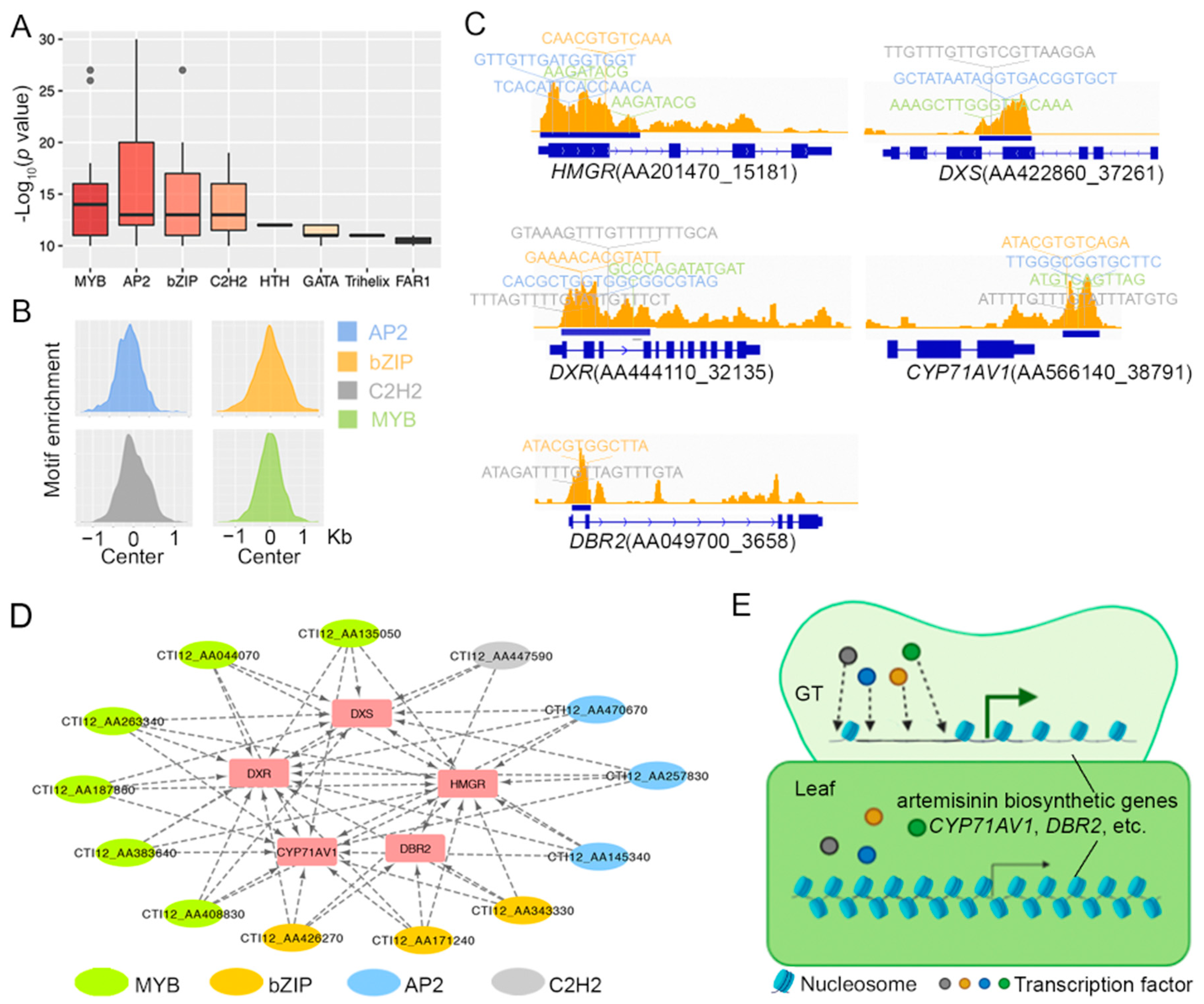
2.2. Glandular Trichome High-Accessible Differential Accessible Regions (DARs) Are Likely Involved in Regulating GT-Specific Cellular Function
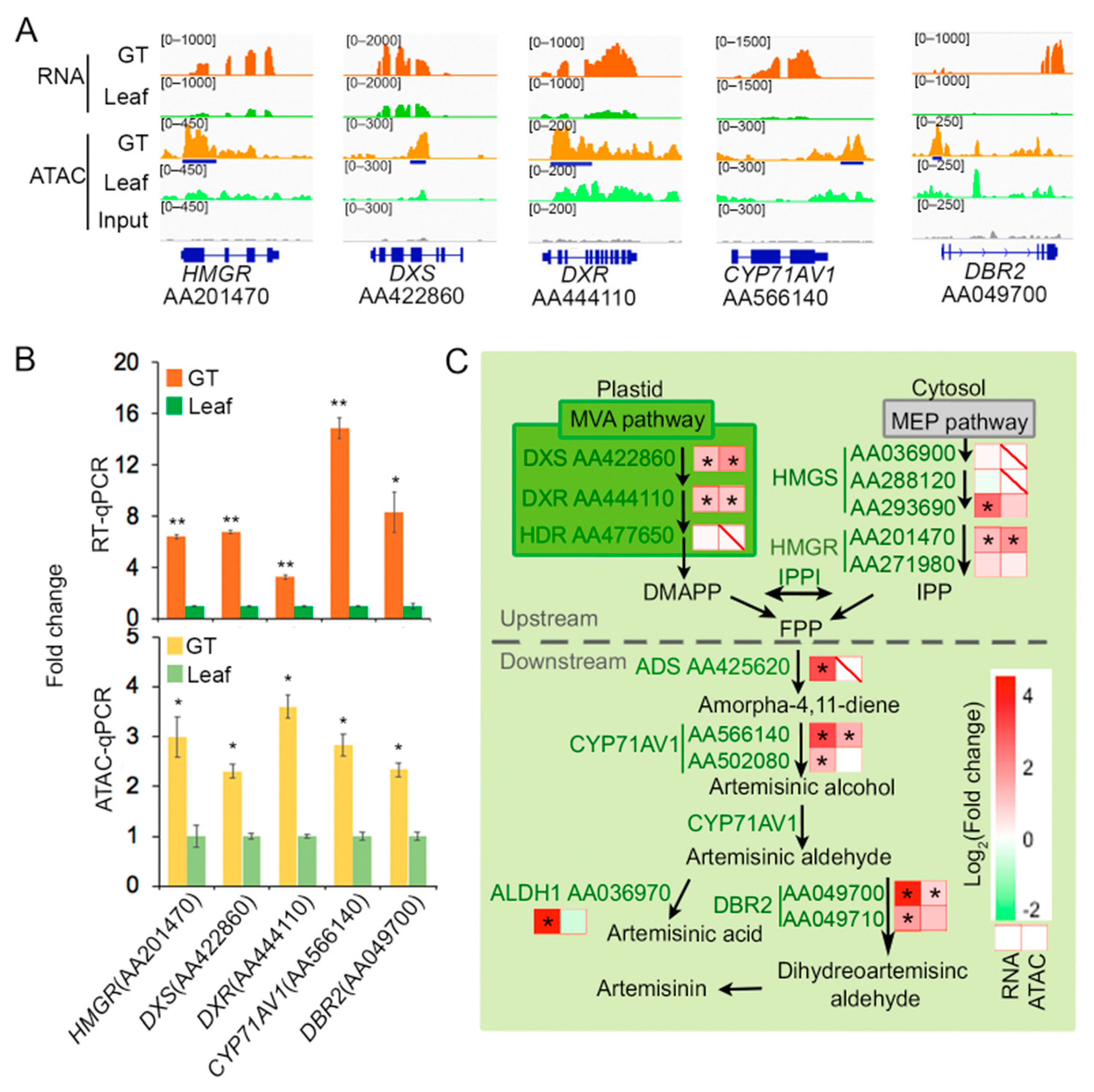
2.3. Some Artemisinin Pathway Gene Expressions Are Associated with Chromatin Accessibility
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sample Preparation
4.2. ATAC-seq Library Generation, Sequencing, and Mapping
4.3. Identification and Annotation of ACRs and Differential Accessible Regions (DARs)
4.4. GO Analysis
4.5. RNA-seq Library Generation, Sequencing, and Mapping
4.6. RT-qPCR and ATAC-qPCR
4.7. Network Construction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Qinghaosu, C.R.G.f. A new sesquiterpene lactone-qinghaosu. Kexue Tongbao 1977, 3, 142. (In Chinese) [Google Scholar]
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- German, P.I.; Aweeka, F.T. Clinical pharmacology of artemisinin-based combination therapies. Clin. Pharmacokinet. 2008, 47, 91–102. [Google Scholar] [PubMed]
- Haq, F.U.; Roman, M.; Ahmad, K.; Rahman, S.U.; Shah, S.M.A.; Suleman, N.; Ullah, S.; Ahmad, I.; Ullah, W. Artemisia annua: Trials are needed for COVID-19. Phytother. Res. 2020, 34, 2423–2424. [Google Scholar] [CrossRef]
- Law, S.; Leung, A.W.; Xu, C. Is the traditional Chinese herb “Artemisia annua” possible to fight against COVID-19? Integr. Med. Res. 2020, 9, 100474. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Zhang, L.; Liao, Z.; Wang, S.; Yan, T.; Shi, P.; Liu, M.; Fu, X.; Pan, Q.; Wang, Y.; et al. The Genome of Artemisia annua Provides Insight into the Evolution of Asteraceae Family and Artemisinin Biosynthesis. Mol. Plant 2018, 11, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Tan, H.; Zhang, L. Artemisia annua glandular secretory trichomes: The biofactory of antimalarial agent artemisinin. Sci. Bull. 2016, 61, 26–36. [Google Scholar] [CrossRef]
- Ro, D.K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Farhi, M.; Marhevka, E.; Ben-Ari, J.; Algamas-Dimantov, A.; Liang, Z.; Zeevi, V.; Edelbaum, O.; Spitzer-Rimon, B.; Abeliovich, H.; Schwartz, B.; et al. Generation of the potent anti-malarial drug artemisinin in tobacco. Nat. Biotechnol. 2011, 29, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Graham, I.A.; Besser, K.; Blumer, S.; Branigan, C.A.; Czechowski, T.; Elias, L.; Guterman, I.; Harvey, D.; Isaac, P.G.; Khan, A.M.; et al. The genetic map of Artemisia annua L. identifies loci affecting yield of the antimalarial drug artemisinin. Science 2010, 327, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Townsend, T.; Segura, V.; Chigeza, G.; Penfield, T.; Rae, A.; Harvey, D.; Bowles, D.; Graham, I.A. The use of combining ability analysis to identify elite parents for Artemisia annua F1 hybrid production. PLoS ONE 2013, 8, e61989. [Google Scholar] [CrossRef]
- Han, J.L.; Liu, B.Y.; Ye, H.C.; Wang, H.; Li, Z.Q.; Li, G.F. Effects of overexpression of the endogenous farnesyl diphosphate synthase on the artemisinin content in Artemisia annua L. J. Integr. Plant Biol. 2006, 48, 482–487. [Google Scholar] [CrossRef]
- Shi, P.; Fu, X.Q.; Liu, M.; Shen, Q.; Jiang, W.M.; Li, L.; Sun, X.F.; Tang, K.X. Promotion of artemisinin content in Artemisia annua by overexpression of multiple artemisinin biosynthetic pathway genes. Plant Cell Tissue Organ. Cult. 2017, 129, 251–259. [Google Scholar] [CrossRef]
- Van Nieuwerburgh, F.C.; Vande Casteele, S.R.; Maes, L.; Goossens, A.; Inze, D.; Van Bocxlaer, J.; Deforce, D.L. Quantitation of artemisinin and its biosynthetic precursors in Artemisia annua L. by high performance liquid chromatography-electrospray quadrupole time-of-flight tandem mass spectrometry. J. Chromatogr. A 2006, 1118, 180–187. [Google Scholar] [PubMed]
- Olsson, M.E.; Olofsson, L.M.; Lindahl, A.L.; Lundgren, A.; Brodelius, M.; Brodelius, P.E. Localization of enzymes of artemisinin biosynthesis to the apical cells of glandular secretory trichomes of Artemisia annua L. Phytochemistry 2009, 70, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Duke, M.V.; Paul, R.N.; Elsohly, H.N.; Sturtz, G.; Duke, S.O. Localization of artemisinin and artemisitene in foliar tissues of glanded and glandless biotypes of Artemisia annua L. Int. J. Plant Sci. 1994, 155, 365–372. [Google Scholar] [CrossRef]
- Judd, R.; Bagley, M.C.; Li, M.; Zhu, Y.; Lei, C.; Yuzuak, S.; Ekelof, M.; Pu, G.; Zhao, X.; Muddiman, D.C.; et al. Artemisinin Biosynthesis in Non-glandular Trichome Cells of Artemisia annua. Mol. Plant 2019, 12, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Duke, S.O.; Paul, R.N. Development and Fine-Structure of the Glandular Trichomes of Artemisia annua L. Int. J. Plant Sci. 1993, 154, 107–118. [Google Scholar] [CrossRef]
- Towler, M.J.; Weathers, P.J. Evidence of artemisinin production from IPP stemming from both the mevalonate and the nonmevalonate pathways. Plant Cell Rep. 2007, 26, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Mercke, P.; Bengtsson, M.; Bouwmeester, H.J.; Posthumus, M.A.; Brodelius, P.E. Molecular cloning, expression, and characterization of amorpha-4,11-diene synthase, a key enzyme of artemisinin biosynthesis in Artemisia annua L. Arch. Biochem. Biophys. 2000, 381, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Heo, K.; Chang, Y.J.; Park, S.H.; Rhee, S.K.; Kim, S.U. Cyclization mechanism of amorpha-4,11-diene synthase, a key enzyme in artemisinin biosynthesis. J. Nat. Prod. 2006, 69, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Teoh, K.H.; Polichuk, D.R.; Reed, D.W.; Nowak, G.; Covello, P.S. Artemisia annua L. (Asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome P450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. FEBS Lett. 2006, 580, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Teoh, K.H.; Reed, D.W.; Maes, L.; Goossens, A.; Olson, D.J.; Ross, A.R.; Covello, P.S. The molecular cloning of artemisinic aldehyde Delta11 reductase and its role in glandular trichome-dependent biosynthesis of artemisinin in Artemisia annua. J. Biol. Chem. 2008, 283, 21501–21508. [Google Scholar] [CrossRef]
- Teoh, K.H.; Polichuk, D.R.; Reed, D.W.; Covello, P.S. Molecular cloning of an aldehyde dehydrogenase implicated in artemisinin biosynthesis in Artemisia annua. Botany 2009, 87, 635–642. [Google Scholar] [CrossRef]
- Czechowski, T.; Larson, T.R.; Catania, T.M.; Harvey, D.; Brown, G.D.; Graham, I.A. Artemisia annua mutant impaired in artemisinin synthesis demonstrates importance of nonenzymatic conversion in terpenoid metabolism. Proc. Natl. Acad. Sci. USA 2016, 113, 15150–15155. [Google Scholar] [CrossRef] [PubMed]
- Matías-Hernández, L.; Jiang, W.; Yang, K.; Tang, K.; Brodelius, P.E.; Pelaz, S. AaMYB1 and its orthologue AtMYB61 affect terpene metabolism and trichome development in Artemisia annua and Arabidopsis thaliana. Plant J. 2017, 90, 520–534. [Google Scholar] [CrossRef]
- Han, J.; Wang, H.; Lundgren, A.; Brodelius, P.E. Effects of overexpression of AaWRKY1 on artemisinin biosynthesis in transgenic Artemisia annua plants. Phytochemistry 2014, 102, 89–96. [Google Scholar] [CrossRef]
- Chen, M.; Yan, T.; Shen, Q.; Lu, X.; Pan, Q.; Huang, Y.; Tang, Y.; Fu, X.; Liu, M.; Jiang, W.; et al. GLANDULAR TRICHOME-SPECIFIC WRKY 1 promotes artemisinin biosynthesis in Artemisia annua. New Phytol. 2017, 214, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, L.; Zhang, F.; Jiang, W.; Shen, Q.; Zhang, L.; Lv, Z.; Wang, G.; Tang, K. AaORA, a trichome-specific AP2/ERF transcription factor of Artemisia annua, is a positive regulator in the artemisinin biosynthetic pathway and in disease resistance to Botrytis cinerea. New Phytol. 2013, 198, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Xiao, L.; Gao, S.; Li, Q.; Chen, J.; Xiao, Y.; Ji, Q.; Chen, R.; Chen, W.; Zhang, L. TRICHOME AND ARTEMISININ REGULATOR 1 Is Required for Trichome Development and Artemisinin Biosynthesis in Artemisia annua. Mol. Plant 2015, 8, 1396–1411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fu, X.; Lv, Z.; Lu, X.; Shen, Q.; Zhang, L.; Zhu, M.; Wang, G.; Sun, X.; Liao, Z.; et al. A basic leucine zipper transcription factor, AabZIP1, connects abscisic acid signaling with artemisinin biosynthesis in Artemisia annua. Mol. Plant 2015, 8, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Xiao, J.; Shen, Y.; Ma, D.; Li, Z.; Pu, G.; Li, X.; Huang, L.; Liu, B.; Ye, H.; et al. Cloning and characterization of AabHLH1, a bHLH transcription factor that positively regulates artemisinin biosynthesis in Artemisia annua. Plant Cell Physiol. 2014, 55, 1592–1604. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.X.; Li, J.X.; Yang, C.Q.; Hu, W.L.; Wang, L.J.; Chen, X.Y. The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol. Plant 2012, 5, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Maher, K.A.; Bajic, M.; Kajala, K.; Reynoso, M.; Pauluzzi, G.; West, D.A.; Zumstein, K.; Woodhouse, M.; Bubb, K.; Dorrity, M.W.; et al. Profiling of Accessible Chromatin Regions across Multiple Plant Species and Cell Types Reveals Common Gene Regulatory Principles and New Control Modules. Plant Cell 2018, 30, 15–36. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.C.; Wang, J.; Schaller, G.E.; Kieber, J.J. Cytokinin modulates context-dependent chromatin accessibility through the type-B response regulators. Nat. Plants 2018, 4, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Marion, C. The structural organization of oligonucleosomes. J. Biomol. Struct. Dyn. 1984, 2, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.; Moore, I.K.; Fondufe-Mittendorf, Y.; Gossett, A.J.; Tillo, D.; Field, Y.; LeProust, E.M.; Hughes, T.R.; Lieb, J.D.; Widom, J.; et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 2009, 458, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.G.; Bussiek, M.; Langowski, J.; Widom, J. Spontaneous access to DNA target sites in folded chromatin fibers. J. Mol. Biol. 2008, 379, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Sabo, P.J.; Kuehn, M.S.; Thurman, R.; Johnson, B.E.; Johnson, E.M.; Cao, H.; Yu, M.; Rosenzweig, E.; Goldy, J.; Haydock, A.; et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat. Methods 2006, 3, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Mieczkowski, J.; Cook, A.; Bowman, S.K.; Mueller, B.; Alver, B.H.; Kundu, S.; Deaton, A.M.; Urban, J.A.; Larschan, E.; Park, P.J.; et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun. 2016, 7, 11485. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Marand, A.P.; Ricci, W.A.; Ethridge, C.L.; Zhang, X.; Schmitz, R.J. The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants 2019, 5, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Zhang, Q.; Qi, Y.; Guo, D. Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genom. 2009, 10, 465. [Google Scholar] [CrossRef] [PubMed]
- Schramek, N.; Wang, H.; Römisch-Margl, W.; Keil, B.; Radykewicz, T.; Winzenhörlein, B.; Beerhues, L.; Bacher, A.; Rohdich, F.; Gershenzon, J.; et al. Artemisinin biosynthesis in growing plants of Artemisia annua. A 13CO2 study. Phytochemistry 2010, 71, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Mudalige, C.; Gingras, A.C.; Pearlman, R.E.; Fillingham, J. The bromodomain-containing protein Ibd1 links multiple chromatin-related protein complexes to highly expressed genes in Tetrahymena thermophila. Epigenetics Chromatin 2018, 11, 10. [Google Scholar]
- Rawal, Y.; Chereji, R.V.; Qiu, H.; Ananthakrishnan, S.; Govind, C.K.; Clark, D.J.; Hinnebusch, A.G. SWI/SNF and RSC cooperate to reposition and evict promoter nucleosomes at highly expressed genes in yeast. Genes Dev. 2018, 32, 695–710. [Google Scholar] [CrossRef]
- Qiu, H.; Biernat, E.; Govind, C.K.; Rawal, Y.; Chereji, R.V.; Clark, D.J.; Hinnebusch, A.G. Chromatin remodeler Ino80C acts independently of H2A.Z to evict promoter nucleosomes and stimulate transcription of highly expressed genes in yeast. Nucleic Acids Res. 2020, 48, 8408–8430. [Google Scholar] [CrossRef] [PubMed]
- Bertea, C.M.; Voster, A.; Verstappen, F.W.; Maffei, M.; Beekwilder, J.; Bouwmeester, H.J. Isoprenoid biosynthesis in Artemisia annua: Cloning and heterologous expression of a germacrene A synthase from a glandular trichome cDNA library. Arch. Biochem. Biophys. 2006, 448, 3–12. [Google Scholar] [CrossRef]
- Laterre, R.; Pottier, M.; Remacle, C.; Boutry, M. Photosynthetic Trichomes Contain a Specific Rubisco with a Modified pH-Dependent Activity. Plant Physiol. 2017, 173, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Pajoro, A.; Madrigal, P.; Muiño, J.M.; Matus, J.T.; Jin, J.; Mecchia, M.A.; Debernardi, J.M.; Palatnik, J.F.; Balazadeh, S.; Arif, M.; et al. Dynamics of chromatin accessibility and gene regulation by MADS-domain transcription factors in flower development. Genome Biol. 2014, 15, R41. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Yang, H.; Zhang, T.; Naruse, K.; Tu, Q. Dynamic transcriptional and chromatin accessibility landscape of medaka embryogenesis. Genome Res. 2020, 30, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Ricci, W.A.; Lu, Z.; Ji, L.; Marand, A.P.; Ethridge, C.L.; Murphy, N.G.; Noshay, J.M.; Galli, M.; Mejía-Guerra, M.K.; Colomé-Tatché, M.; et al. Widespread long-range cis-regulatory elements in the maize genome. Nat. Plants 2019, 5, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Danko, C.G.; Hyland, S.L.; Core, L.J.; Martins, A.L.; Waters, C.T.; Lee, H.W.; Cheung, V.G.; Kraus, W.L.; Lis, J.T.; Siepel, A. Identification of active transcriptional regulatory elements from GRO-seq data. Nat. Methods 2015, 12, 433–438. [Google Scholar] [CrossRef]
- Crawford, G.E.; Davis, S.; Scacheri, P.C.; Renaud, G.; Halawi, M.J.; Erdos, M.R.; Green, R.; Meltzer, P.S.; Wolfsberg, T.G.; Collins, F.S. DNase-chip: A high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat. Methods 2006, 3, 503–509. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Huang, Y.; Wang, Q.; Guo, D. Chromatin Accessibility Is Associated with Artemisinin Biosynthesis Regulation in Artemisia annua. Molecules 2021, 26, 1194. https://doi.org/10.3390/molecules26041194
Zhou L, Huang Y, Wang Q, Guo D. Chromatin Accessibility Is Associated with Artemisinin Biosynthesis Regulation in Artemisia annua. Molecules. 2021; 26(4):1194. https://doi.org/10.3390/molecules26041194
Chicago/Turabian StyleZhou, Limeng, Yingzhang Huang, Qi Wang, and Dianjing Guo. 2021. "Chromatin Accessibility Is Associated with Artemisinin Biosynthesis Regulation in Artemisia annua" Molecules 26, no. 4: 1194. https://doi.org/10.3390/molecules26041194
APA StyleZhou, L., Huang, Y., Wang, Q., & Guo, D. (2021). Chromatin Accessibility Is Associated with Artemisinin Biosynthesis Regulation in Artemisia annua. Molecules, 26(4), 1194. https://doi.org/10.3390/molecules26041194