
Anti-Inflammatory, Antidiabetic Properties and In Silico Modeling of Cucurbitane-Type Triterpene Glycosides from Fruits of an Indian Cultivar of Momordica charantia L.

 ,
, 
 ,
,  and
and 
Abstract
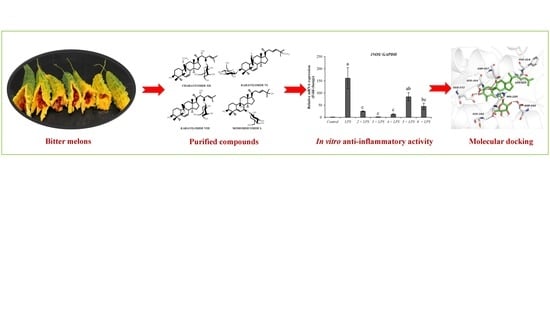
1. Introduction
2. Results and Discussion
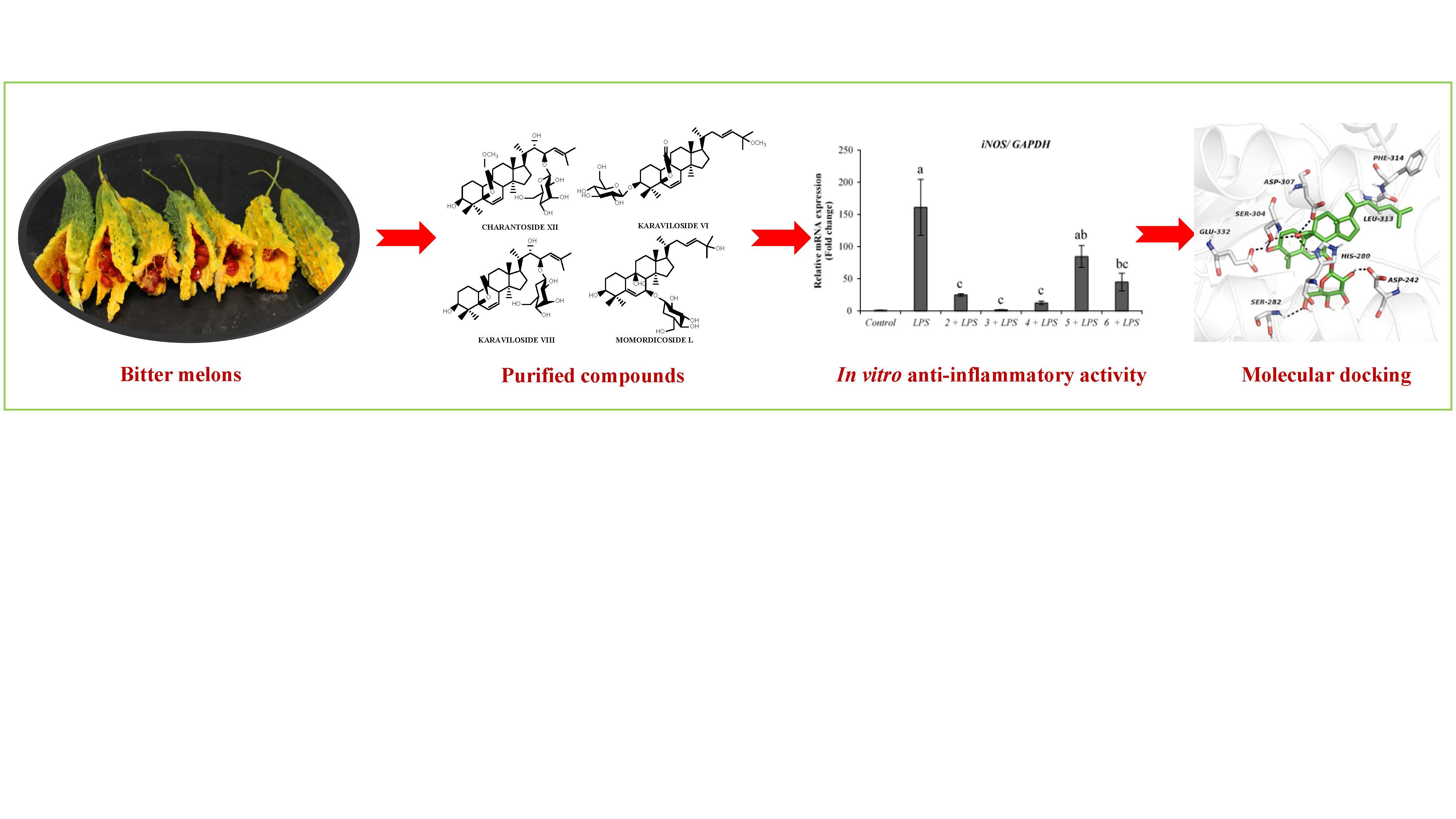
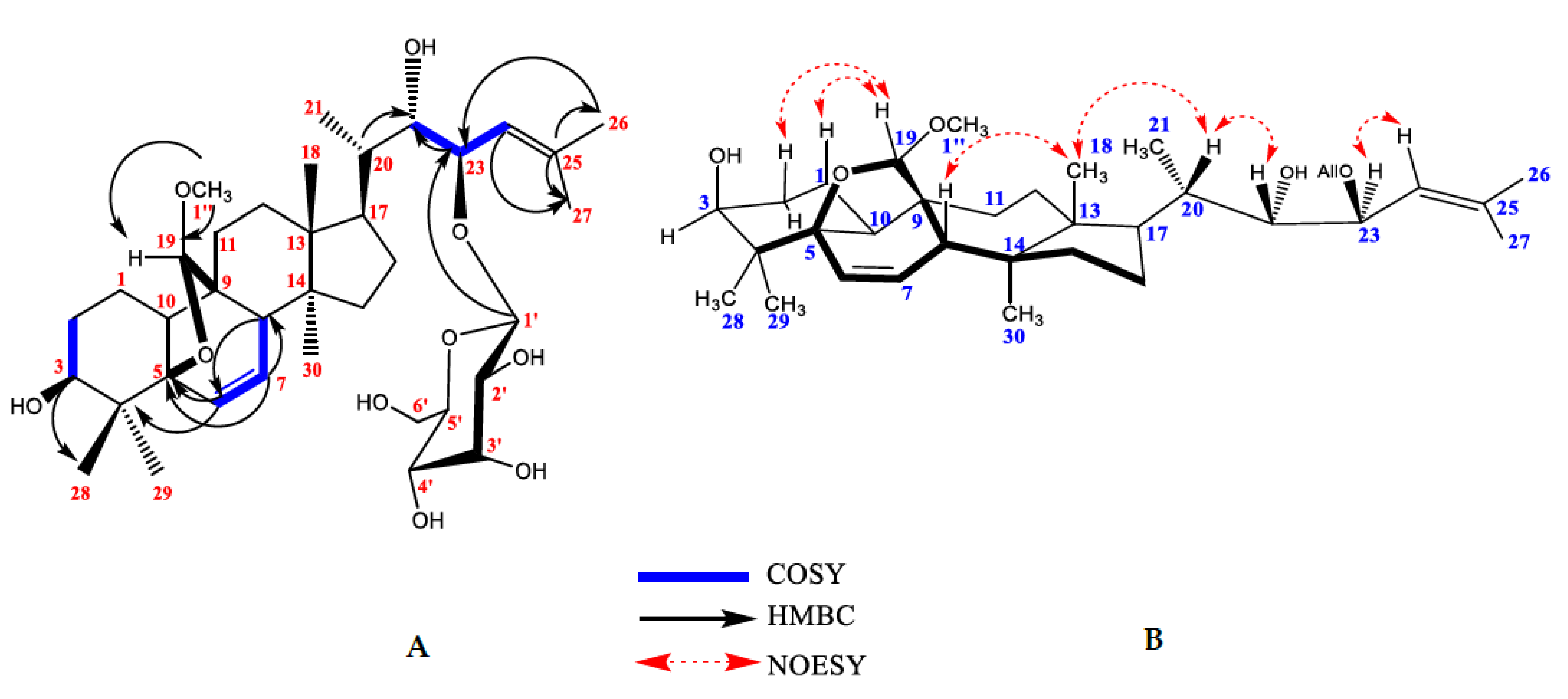
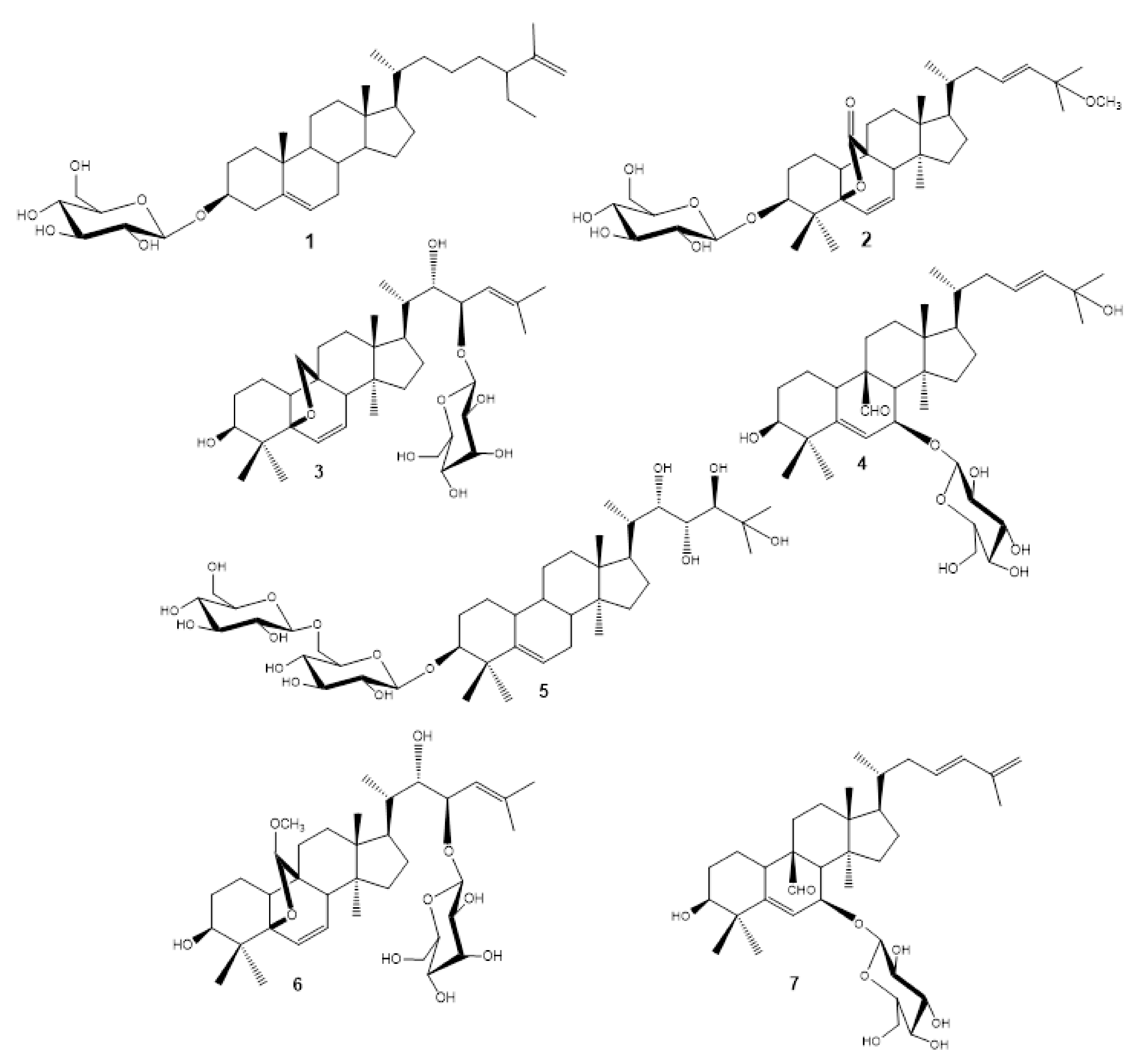
2.1. Isolation of Compounds
2.2. Bioassays
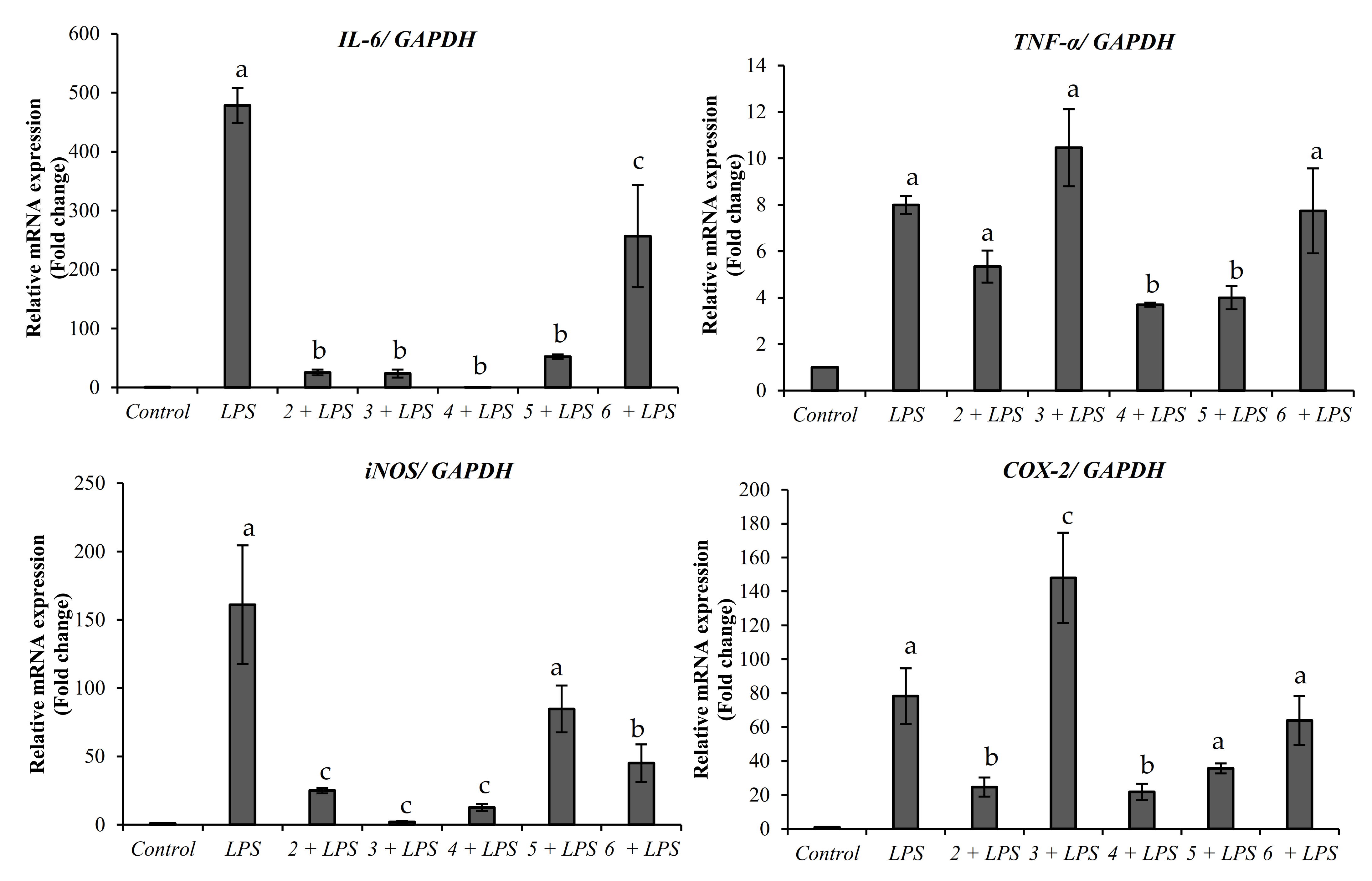
2.2.1. Anti-Inflammatory Activity
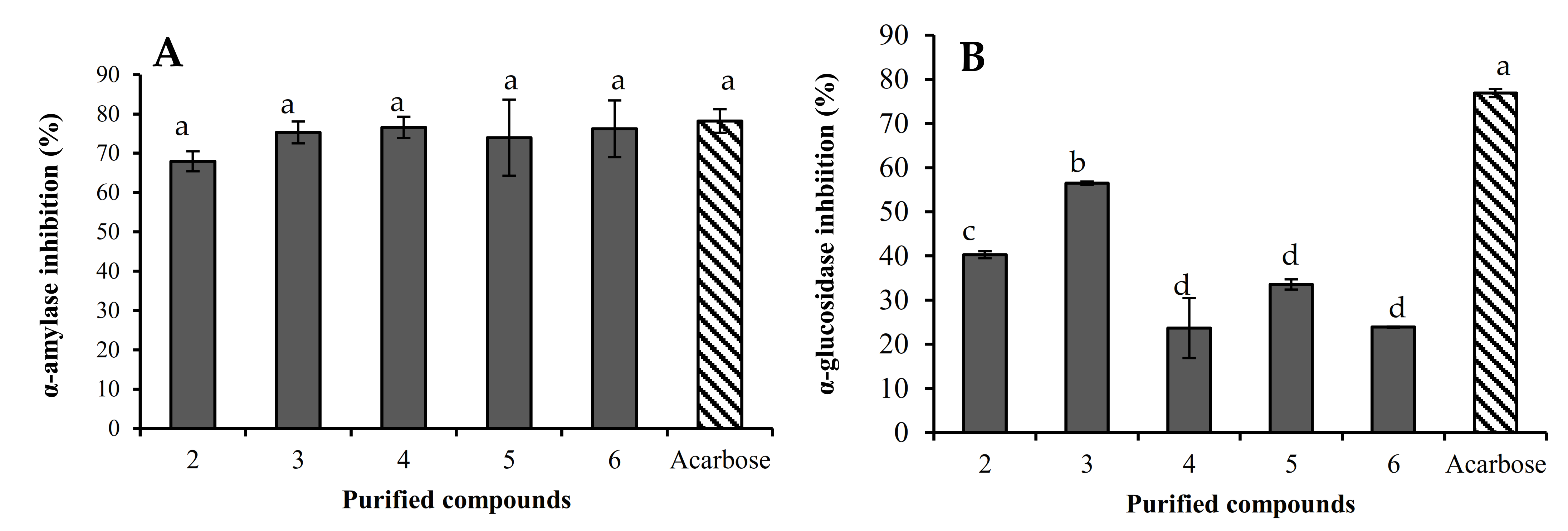
2.2.2. α-Amylase and α-Glucosidase
2.3. Molecular Docking
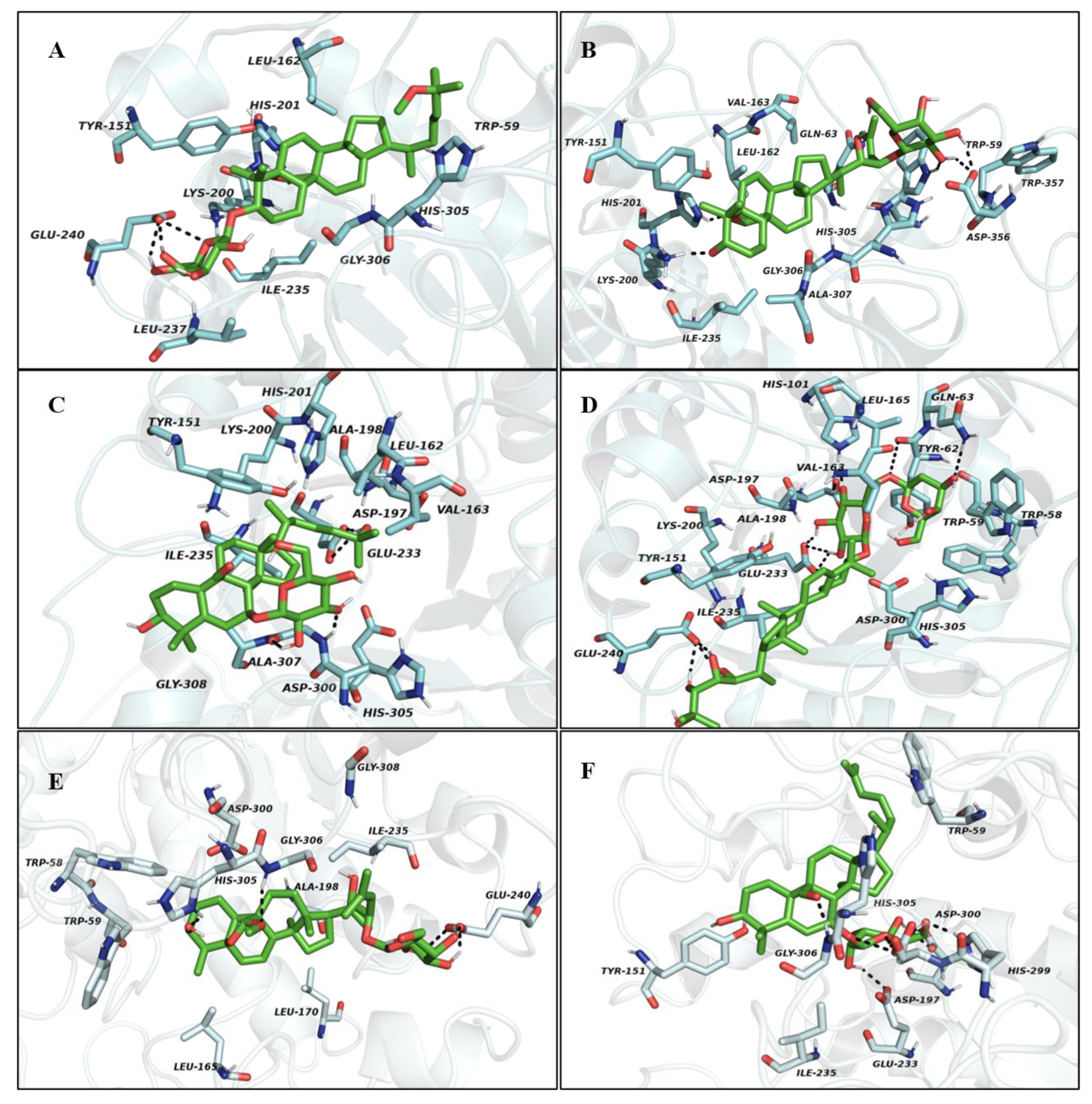
2.3.1. Molecular Docking Study with α-Amylase
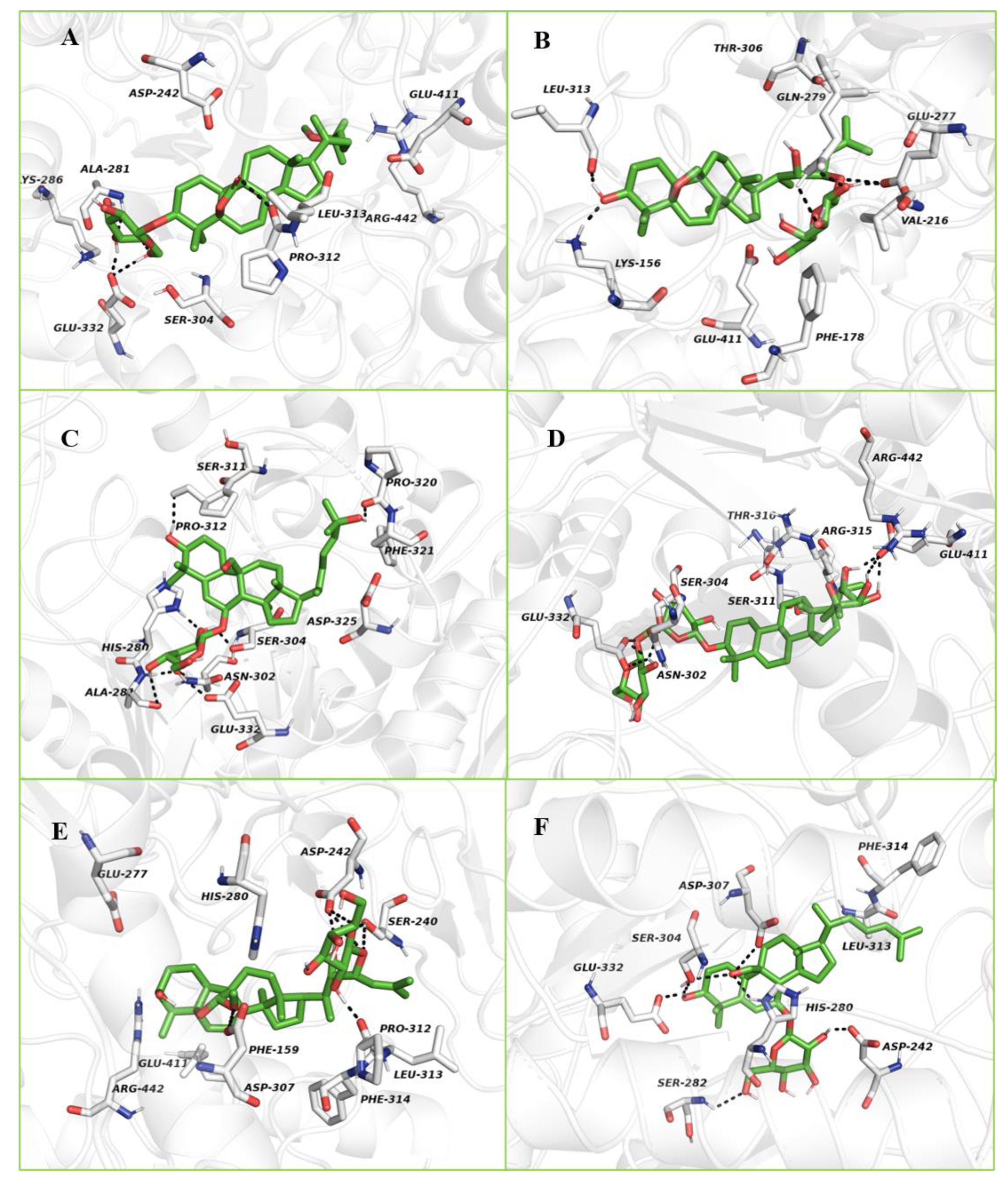
2.3.2. Molecular Docking Study with α-Glucosidase
3. Materials and Methods
3.1. Chemicals
3.2. General Experimental Procedure
3.3. Plant Material
3.4. Soxhlet Extraction
3.5. Purification Procedure
3.6. LC-HR-MS Analysis
3.7. Determination of the Absolute Configuration of the Sugar Unit
3.8. Physicochemical Parameters of Charantoside XV (6)
3.9. Bioassays
3.9.1. Cell Culture and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
3.9.2. In Vitro α-Amylase Assay
3.9.3. In Vitro α-Glucosidase Assay
3.10. In Silico Docking Study
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bharathi, L.K.; John, K.J. Momordica Genus in Asia—An Overview; Springer: New Delhi, India, 2013. [Google Scholar]
- Snee, L.S.; Nerurkar, V.R.; Dooley, D.A.; Efird, J.T.; Shovic, A.C.; Nerurkar, P.V. Strategies to improve palatability and increase consumption intentions for Momordica charantia (bitter melon): A vegetable commonly used for diabetes management. Nutr. J. 2011, 10, 78–88. [Google Scholar] [CrossRef]
- Deshaware, S.; Gupta, S.; Singhal, R.S.; Joshi, M.; Variyar, P.S. Debittering of bitter gourd juice using β-cyclodextrin: Mechanism and effect on antidiabetic potential. Food Chem. 2018, 262, 78–85. [Google Scholar] [CrossRef]
- Tan, M.-J.; Ye, J.-M.; Turner, N.; Hohnen-Behrens, C.; Ke, C.-Q.; Tang, C.-P.; Chen, T.; Weiss, H.-C.; Gesing, E.-R.; Rowland, A. Antidiabetic activities of triterpenoids isolated from bitter melon associated with activation of the AMPK pathway. Chem. Biol. 2008, 15, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Nazaruk, J.; Borzym-Kluczyk, M. The role of triterpenes in the management of diabetes mellitus and its complications. Phytochem. Rev. 2015, 14, 675–690. [Google Scholar] [CrossRef]
- Chang, C.-I.; Chen, C.-R.; Liao, Y.-W.; Shih, W.-L.; Cheng, H.-L.; Tzeng, C.-Y.; Li, J.-W.; Kung, M.-T. Octanorcucurbitane triterpenoids protect against tert-butyl hydroperoxide-induced hepatotoxicity from the stems of Momordica charantia. Chem. Pharm. Bull. 2010, 58, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Nakamura, S.; Murakami, T. Structures of New Cucurbitane-Type Triterpenes and Glycosides, Karavilagenins D and E, and Karavilosides 6, 7, 8, 9, 10, and 11, from the Fruit of Momordica charantia. Heterocycles 2007, 71, 331–341. [Google Scholar]
- Tuan, N.Q.; Lee, D.-H.; Oh, J.; Kim, C.S.; Heo, K.-S.; Myung, C.-S.; Na, M. Inhibition of proliferation of vascular smooth muscle cells by Cucurbitanes from Momordica charantia. J. Nat. Prod. 2017, 80, 2018–2025. [Google Scholar] [CrossRef]
- Yue, J.; Xu, J.; Cao, J.; Zhang, X.; Zhao, Y. Cucurbitane triterpenoids from Momordica charantia L. and their inhibitory activity against α-glucosidase, α-amylase and protein tyrosine phosphatase 1B (PTP1B). J. Funct. Foods 2017, 37, 624–631. [Google Scholar] [CrossRef]
- Akihisa, T.; Higo, N.; Tokuda, H.; Ukiya, M.; Akazawa, H.; Tochigi, Y.; Kimura, Y.; Suzuki, T.; Nishino, H. Cucurbitane-type triterpenoids from the fruits of Momordica charantia and their cancer chemopreventive effects. J. Nat. Prod. 2007, 70, 1233–1239. [Google Scholar] [PubMed]
- Hasani-Ranjbar, S.; Nayebi, N.; Larijani, B.; Abdollahi, M. A systematic review of the efficacy and safety of herbal medicines used in the treatment of obesity. World J. Gastroenterol. 2009, 15, 3073–3085. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.; Birtwhistle, R.; Kotecha, J.; Hannah, S.; Cuthbertson, S. Anti-diabetic and hypoglycaemic effects of Momordica charantia (bitter melon): A mini review. Br. J. Nutr. 2009, 102, 1703–1708. [Google Scholar] [CrossRef]
- Chen, J.-C.; Bik-San Lau, C.; Chan, J.Y.-W.; Fung, K.-P.; Leung, P.-C.; Liu, J.-Q.; Zhou, L.; Xie, M.-J.; Qiu, M.-H. The antigluconeogenic activity of cucurbitacins from Momordica charantia. Planta Med. 2015, 81, 327–332. [Google Scholar] [CrossRef]
- Bao, B.; Chen, Y.-G.; Zhang, L.; Xu, Y.L.N.; Wang, X.; Liu, J.; Qu, W. Momordica charantia (Bitter Melon) reduces obesity-associated macrophage and mast cell infiltration as well as inflammatory cytokine expression in adipose tissues. PLoS ONE 2013, 8, e84075. [Google Scholar] [CrossRef]
- Wang, X.; Bao, W.; Liu, J.; OuYang, Y.-Y.; Wang, D.; Rong, S.; Xiao, X.; Shan, Z.-L.; Zhang, Y.; Yao, P. Inflammatory markers and risk of type 2 diabetes: A systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar]
- Persaud, S.J.; Burns, C.J.; Belin, V.D.; Jones, P.M. Glucose-induced regulation of COX-2 expression in human islets of Langerhans. Diabetes 2004, 53, S190–S192. [Google Scholar] [CrossRef] [PubMed]
- Abascal, K.; Yarnell, E. Using bitter melon to treat diabetes. Altern. Complement. Ther. 2005, 11, 179–184. [Google Scholar] [CrossRef]
- Shivanagoudra, S.R.; Perera, W.H.; Perez, J.L.; Athrey, G.; Sun, Y.; Jayaprakasha, G.; Patil, B.S. Cucurbitane-type compounds from Momordica charantia: Isolation, in vitro antidiabetic, anti-inflammatory activities and in silico modeling approaches. Bioorg. Chem. 2019, 87, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Shivanagoudra, S.R.; Perera, W.H.; Perez, J.L.; Athrey, G.; Sun, Y.; Wu, C.S.; Jayaprakasha, G.; Patil, B.S. In vitro and in silico elucidation of antidiabetic and anti-inflammatory activities of bioactive compounds from Momordica charantia L. Bioorg. Med. Chem. 2019, 27, 3097–3109. [Google Scholar] [CrossRef]
- Perez, J.L.; Jayaprakasha, G.; Patil, B.S. Separation and Identification of Cucurbitane-Type Triterpenoids from Bitter Melon. In Instrumental Methods for the Analysis and Identification of Bioactive Molecules; ACS Symposium Series 1185; American Chemical Society: Washington, DC, USA, 2014; pp. 51–78. [Google Scholar]
- Liaw, C.-C.; Huang, H.-C.; Hsiao, P.-C.; Zhang, L.-J.; Lin, Z.-H.; Hwang, S.-Y.; Hsu, F.-L.; Kuo, Y.-H. 5β,19-epoxycucurbitane triterpenoids from Momordica charantia and their anti-inflammatory and cytotoxic activity. Planta Med. 2015, 81, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Murakami, T.; Nakamura, J.; Kobayashi, H.; Matsuda, H.; Yoshikawa, M. Structures of new cucurbitane-type triterpenes and glycosides, karavilagenins and karavilosides, from the dried fruit of Momordica charantia L. in Sri Lanka. Chem. Pharm. Bull. 2006, 54, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Y.; Chen, H.B.; Liu, Z.M.; Wang, B.; Zhao, Y.Y. Cucurbitane triterpenoids from Momordica charantia. Magn. Reson. Chem. 2007, 45, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Q.; Chen, J.-C.; Wang, C.-F.; Qiu, M.-H. New cucurbitane triterpenoids and steroidal glycoside from Momordica charantia. Molecules 2009, 14, 4804–4813. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Jian-Feng, L.; Li-Ping, K.; He-Shui, Y.; Zhang, L.-J.; Xin-Bo, S.; Bai-Ping, M. A new C30 sterol glycoside from the fresh fruits of Momordica charantia. Chin. J. Nat. Med. 2012, 2, 88–91. [Google Scholar]
- Nhiem, N.X.; Kiem, P.V.; Minh, C.V.; Ban, N.K.; Cuong, N.X.; Ha, L.M.; Tai, B.H.; Quang, T.H.; Tung, N.H.; Kim, Y.H. Cucurbitane—type triterpene glycosides from the fruits of Momordica charantia. Magn. Reson. Chem. 2010, 48, 392–396. [Google Scholar]
- Okabe, H.; Miyahara, Y.; Yamauchi, T.; Miyahara, K.; Kawasaki, T. Studies on the constituents of Momordica charantia LI Isolation and characterization of momordicosides A and B, glycosides of a pentahydroxy-cucurbitane triterpene. Chem. Pharm. Bull. 1980, 28, 2753–2762. [Google Scholar] [CrossRef]
- Chen, J.C.; Lu, L.; Zhang, X.M.; Zhou, L.; Li, Z.R.; Qiu, M.H. Eight New Cucurbitane Glycosides, Kuguaglycosides A–H, from the Root of Momordica charantia L. Helv. Chim. Acta 2008, 91, 920–929. [Google Scholar] [CrossRef]
- Tabata, K.; Hamano, A.; Akihisa, T.; Suzuki, T. Kuguaglycoside C, a constituent of Momordica charantia, induces caspase—independent cell death of neuroblastoma cells. Cancer Sci. 2012, 103, 2153–2158. [Google Scholar]
- Hsiao, P.-C.; Liaw, C.-C.; Hwang, S.-Y.; Cheng, H.-L.; Zhang, L.-J.; Shen, C.-C.; Hsu, F.-L.; Kuo, Y.-H. Antiproliferative and hypoglycemic cucurbitane-type glycosides from the fruits of Momordica charantia. J. Agric. Food Chem. 2013, 61, 2979–2986. [Google Scholar] [CrossRef]
- Kobori, M.; Nakayama, H.; Fukushima, K.; Ohnishi-Kameyama, M.; Ono, H.; Fukushima, T.; Akimoto, Y.; Masumoto, S.; Yukizaki, C.; Hoshi, Y. Bitter gourd suppresses lipopolysaccharide-induced inflammatory responses. J. Agric. Food Chem. 2008, 56, 4004–4011. [Google Scholar] [CrossRef]
- Lii, C.-K.; Chen, H.-W.; Yun, W.-T.; Liu, K.-L. Suppressive effects of wild bitter gourd (Momordica charantia Linn. var. abbreviata ser.) fruit extracts on inflammatory responses in RAW 264.7 macrophages. J. Ethnopharmacol. 2009, 122, 227–233. [Google Scholar] [CrossRef]
- Svobodova, B.; Barros, L.; Calhelha, R.C.; Heleno, S.; Alves, M.J.; Walcott, S.; Bittova, M.; Kuban, V.; Ferreira, I.C. Bioactive properties and phenolic profile of Momordica charantia L. medicinal plant growing wild in Trinidad and Tobago. Ind. Crops Prod. 2017, 95, 365–373. [Google Scholar] [CrossRef]
- Hsu, C.-L.; Fang, S.-C.; Liu, C.-W.; Chen, Y.-F. Inhibitory effects of new varieties of bitter melon on lipopolysaccharide-stimulated inflammatory response in RAW 264.7 cells. J. Funct. Foods 2013, 5, 1829–1837. [Google Scholar]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef]
- Alhadramy, M.S. Diabetes and oral therapies: A review of oral therapies for diabetes mellitus. J. Taibah Univ. Med. Sci. 2016, 11, 317–329. [Google Scholar]
- Ceriello, A.; Davidson, J.; Hanefeld, M.; Leiter, L.; Monnier, L.; Owens, D.; Tajima, N.; Tuomilehto, J. Postprandial hyperglycaemia and cardiovascular complications of diabetes: An update. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, Y.; Zhou, W. In vitro and in silico studies of the inhibition activity of anthocyanins against porcine pancreatic α-amylase. J. Funct. Foods 2016, 21, 50–57. [Google Scholar] [CrossRef]
- Qian, M.; Spinelli, S.; Payan, F.; Driguez, H. Structure of a pancreatic α-amylase bound to a substrate analogue at 2.03 Å resolution. Protein Sci. 1997, 6, 2285–2296. [Google Scholar]
- Lo Piparo, E.; Scheib, H.; Frei, N.; Williamson, G.; Grigorov, M.; Chou, C.J. Flavonoids for controlling starch digestion: Structural requirements for inhibiting human α-amylase. J. Med. Chem. 2008, 51, 3555–3561. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, G.; Yan, J.; Gong, D. Inhibitory effect of morin on tyrosinase: Insights from spectroscopic and molecular docking studies. Food Chem. 2014, 163, 226–233. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Oliveira, E.F.; Sousa, J.L.; Tomé, S.M.; Ramos, M.J.; Silva, A.M.; Fernandes, P.A.; Fernandes, E. α-Glucosidase inhibition by flavonoids: An in vitro and in silico structure–activity relationship study. J. Enzyme Inhib. Med. Chem. 2017, 32, 1216–1228. [Google Scholar] [CrossRef] [PubMed]
- Perera, W.H.; Ghiviriga, I.; Rodenburg, D.L.; Alves, K.; Bowling, J.J.; Avula, B.; Khan, I.A.; McChesney, J.D. Rebaudiosides T and U, minor C-19 xylopyranosyl and arabinopyranosyl steviol glycoside derivatives from Stevia rebaudiana (Bertoni) Bertoni. Phytochem. 2017, 135, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Dirsch, V.M.; Kiemer, A.K.; Wagner, H.; Vollmar, A.M. Effect of allicin and ajoene, two compounds of garlic, on inducible nitric oxide synthase. Atherosclerosis 1998, 139, 333–339. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Compound 6 (MeOH-d4) | |
|---|---|---|
| δH | δC | |
| 1 | 1.15; 1.50 m, 2H | 18.5 |
| 2 | 0.82; 1.84 m, 2H | 28.3 |
| 3 | 3.36 m, 1H | 77.8 |
| 4 | − | 38.5 |
| 5 | − | 88.1 |
| 6 | 5.98 dd (J = 9.8, 2.1 Hz, 1H) | 132.2 |
| 7 | 5.58 dd (J = 9.8, 3.7 Hz, 1H) | 133.8 |
| 8 | 2.86 m, 1H | 43.2 |
| 9 | − | 49.7 |
| 10 | 2.44 m, 1H | 42.1 |
| 11 | 1.54; 1.75 m, 2H | 24.3 |
| 12 | 1.54; 1.54 m, 2H | 32.0 |
| 13 | − | 46.5 |
| 14 | − | 49.0 |
| 15 | 1.32; 1.32 m, 2H | 35.0 |
| 16 | 1.34; 1.34 m, 2H | 29.0 |
| 17 | 1.97 m, 1H | 47.7 |
| 18 | 0.84 s, 3H | 14.8 |
| 19 | 4.70 s, 1H | 113.3 |
| 20 | 1.77 dd (J = 11.3, 1.0 Hz, 1H) | 42.1 |
| 21 | 0.96 d (J = 6 Hz, 3H) | 14.7 |
| 22 | 3.63 m, 1H | 77.3 |
| 23 | 4.29 m, 1H | 81.5 |
| 24 | 5.24 d (J = 10.2 Hz, 1H) | 124.6 |
| 25 | − | 137.4 |
| 26 | 1.74, 3H | 26.7 |
| 27 | 1.77, 3H | 19.1 |
| 28 | 1.18 s (3H) | 20.9 |
| 29 | 0.87 s (3H) | 24.6 |
| 30 | 0.87 s (3H) | 20.6 |
| 23-O-All | ||
| 1′ | 4.73 d (J = 8 Hz, 1H) | 102.6 |
| 2′ | 3.32, 1H | 73.1 |
| 3′ | 4.05, 1H | 73.0 |
| 4′ | 3.48, 1H | 68.8 |
| 5′ | 3.58, 1H | 75.7 |
| 6′ | 3.60; 3.76, 2H | 63.1 |
| 1″ | 3.41, 3H | 58.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, W.H.; Shivanagoudra, S.R.; Pérez, J.L.; Kim, D.M.; Sun, Y.; K. Jayaprakasha, G.; S. Patil, B. Anti-Inflammatory, Antidiabetic Properties and In Silico Modeling of Cucurbitane-Type Triterpene Glycosides from Fruits of an Indian Cultivar of Momordica charantia L. Molecules 2021, 26, 1038. https://doi.org/10.3390/molecules26041038
Perera WH, Shivanagoudra SR, Pérez JL, Kim DM, Sun Y, K. Jayaprakasha G, S. Patil B. Anti-Inflammatory, Antidiabetic Properties and In Silico Modeling of Cucurbitane-Type Triterpene Glycosides from Fruits of an Indian Cultivar of Momordica charantia L. Molecules. 2021; 26(4):1038. https://doi.org/10.3390/molecules26041038
Chicago/Turabian StylePerera, Wilmer H., Siddanagouda R. Shivanagoudra, Jose L. Pérez, Da Mi Kim, Yuxiang Sun, Guddadarangavvanahally K. Jayaprakasha, and Bhimanagouda S. Patil. 2021. "Anti-Inflammatory, Antidiabetic Properties and In Silico Modeling of Cucurbitane-Type Triterpene Glycosides from Fruits of an Indian Cultivar of Momordica charantia L." Molecules 26, no. 4: 1038. https://doi.org/10.3390/molecules26041038
APA StylePerera, W. H., Shivanagoudra, S. R., Pérez, J. L., Kim, D. M., Sun, Y., K. Jayaprakasha, G., & S. Patil, B. (2021). Anti-Inflammatory, Antidiabetic Properties and In Silico Modeling of Cucurbitane-Type Triterpene Glycosides from Fruits of an Indian Cultivar of Momordica charantia L. Molecules, 26(4), 1038. https://doi.org/10.3390/molecules26041038