
A Review of HER4 (ErbB4) Kinase, Its Impact on Cancer, and Its Inhibitors

 , ,
, ,
Abstract
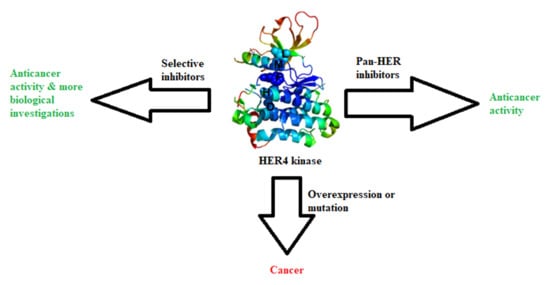
1. Introduction
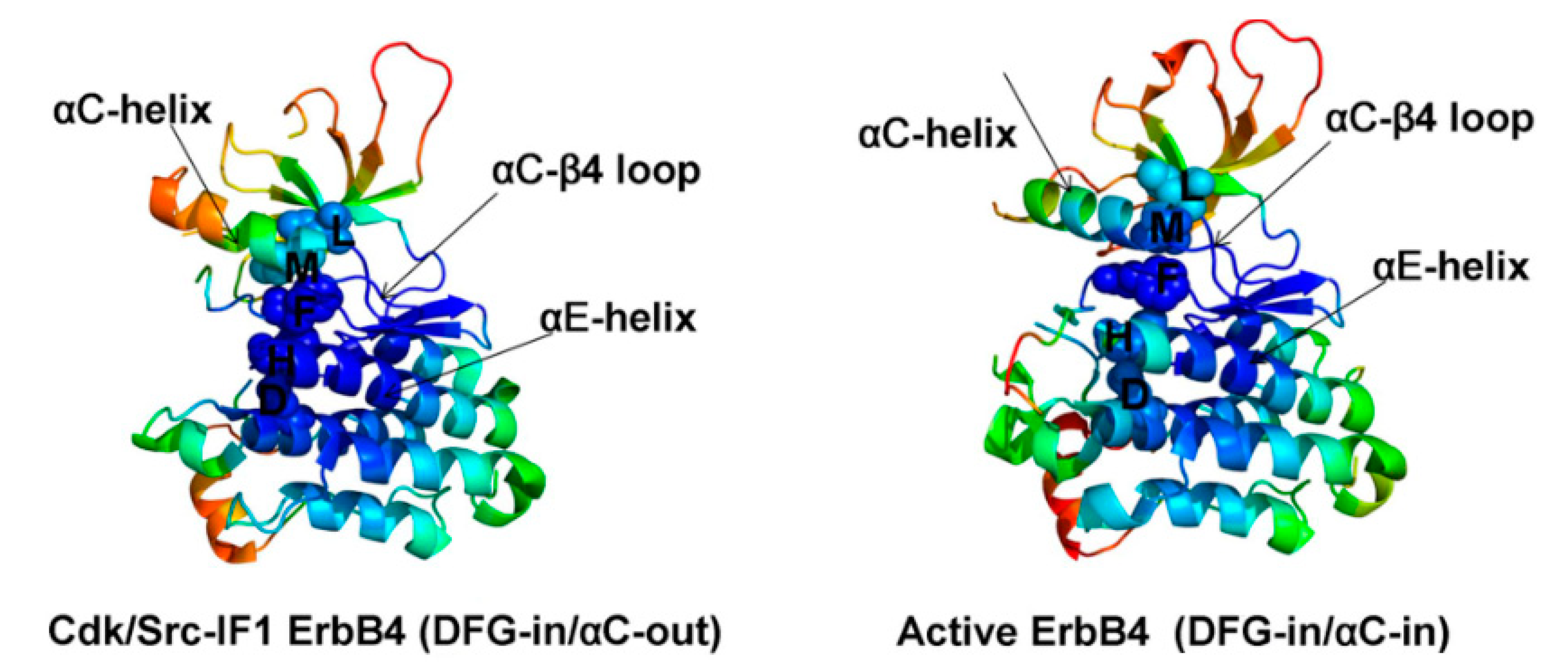
2. Structure of HER4 Kinase
3. Location of HER4 Kinase
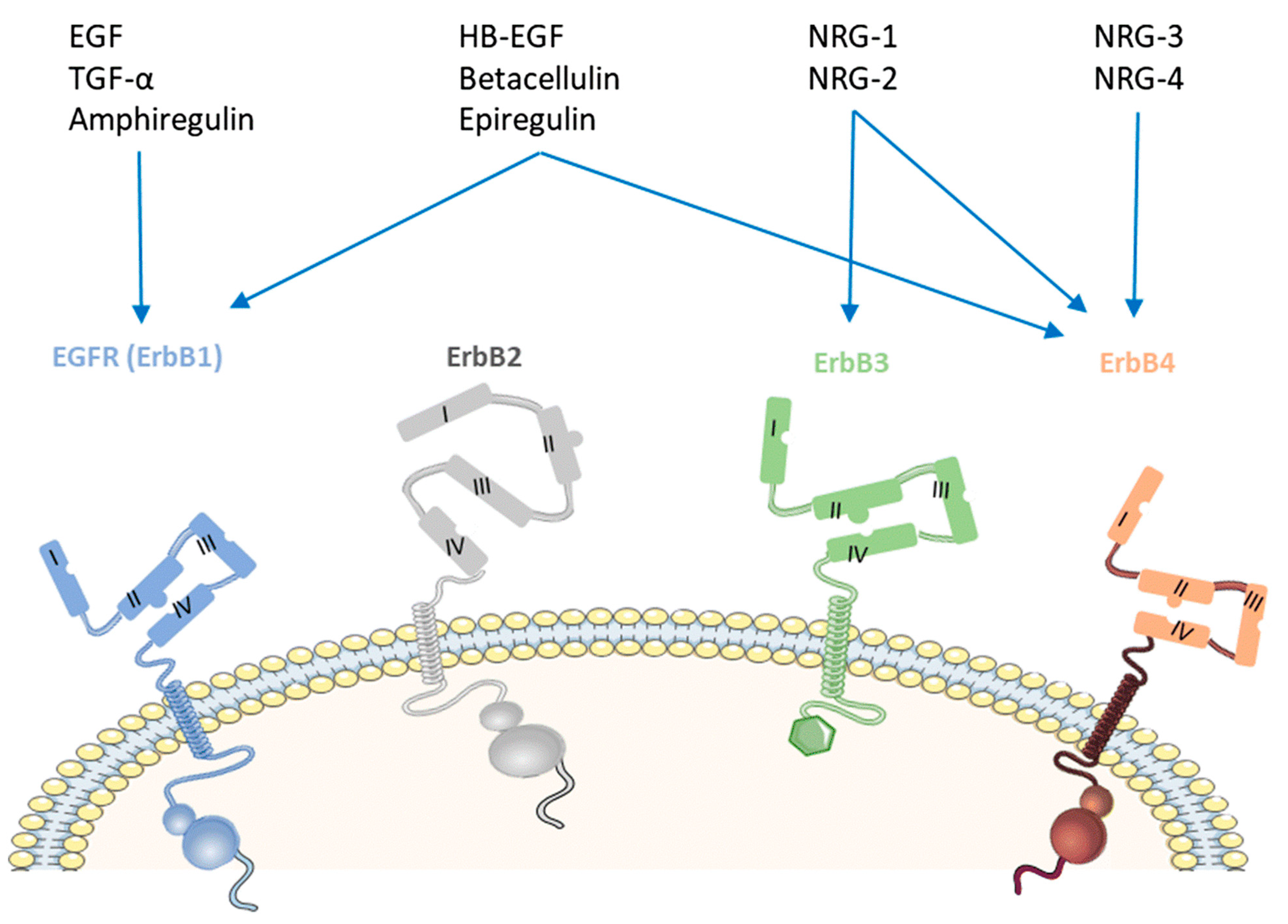
4. Ligands of HER4 Kinase
5. Physiological Roles and Functions of HER4
6. HER4 Relationship to Different Cancers
6.1. HER4 and Colorectal Cancer (CRC)
6.2. HER4 and Lung Cancer
6.3. HER4 and Gastric Cancer
6.4. HER4 and Hepatocellular Carcinoma
6.5. HER4 and Prostate Cancer
6.6. HER4 and Bladder Cancer
6.7. HER4 and Ovarian Cancer
6.8. HER4 and Breast Cancer
6.9. HER4 and Pancreatic Cancer
6.10. HER4 and Brain Cancer
6.11. HER4 and Melanoma
6.12. HER4 and Endometrial Cancer
6.13. HER4 and Osteosarcoma
7. HER4 Inhibitors
7.1. Quinazoline Inhibitors
7.1.1. Allitinib (AST-1306)
 |
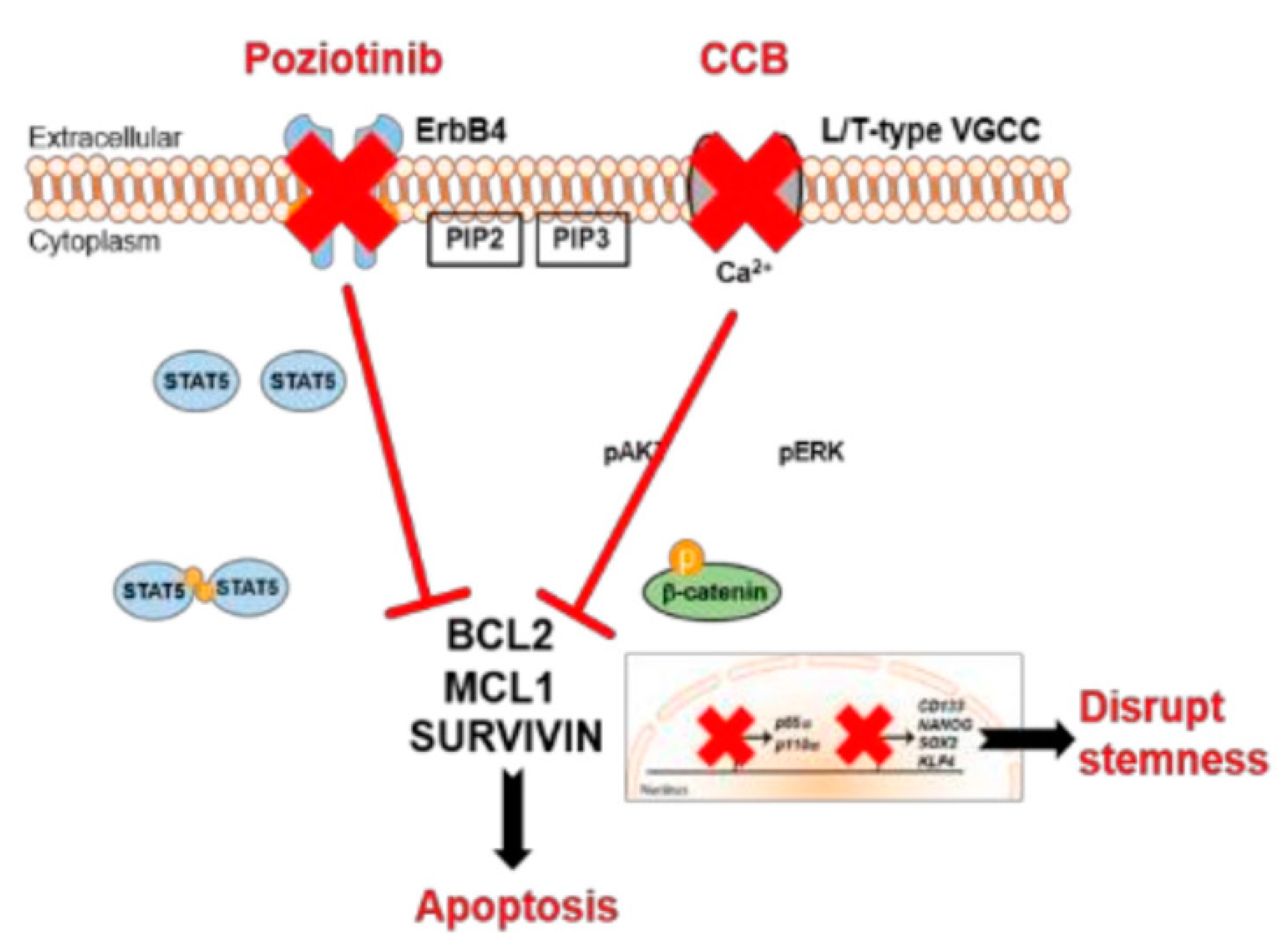
7.1.2. Poziotinib (HM781-36B)
 |
7.1.3. Dacomitinib (PF-00299804)
 |
7.1.4. Lapatinib
 |
7.1.5. Afatinib (BIBW2992)
 |
7.1.6. Canertinib (CI-1033)
 |
7.2. Quinoline Inhibitors
7.2.1. Neratinib (HKI-272)
 |
7.2.2. Pyrotinib (SHR1258)
 |
7.3. Other Inhibitors
7.3.1. Ibrutinib (PCI-32765)
 |
7.3.2. AC-480 (BMS-599626)
 |
7.3.3. Compounds I and II
 |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| BTC | betacellulin |
| BTK | Bruton’s tyrosine kinase |
| CLL | chronic lymphocytic leukemia |
| COX-2 | cyclooxygenase-2 |
| CRC | colorectal cancer |
| DEN | diethyl nitrosamine |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EPR | epiregulin |
| GBM | glioblastoma multiforme |
| GVHD | Graft-versus-host disease |
| HCC | hepatocellular carcinoma |
| HER | human epidermal growth factor receptor |
| MAPK | mitogen-activated protein kinase |
| MCL | mantle cell lymphoma |
| MZL | marginal zone lymphoma |
| NAPPA | nucleic acid programmable protein assays |
| NEU | neurogulin |
| NSCLC | non-small cell lung cancer |
| PI3K | phosphoinositide 3-kinase |
| RTK | receptor tyrosine kinase |
| RT-PCR | reverse transcription mRNA-PCRRA-level |
| TNF | tumor necrosis factor |
| WHO | World Health Organization |
| WM | Waldenstrom macroglobulinemia |
| WNT | wingless related integration site |
References
- Qiu, C.; Tarrant, M.K.; Choi, S.H.; Sathyamurthy, A.; Bose, R.; Banjade, S.; Pal, A.; Bornmann, W.G.; Lemmon, M.A.; Cole, P.A.; et al. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure 2008, 16, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Segers, V.F.M.; Dugaucquier, L.; Feyen, E.; Shakeri, H.; De Keulenaer, G.W. The role of ErbB4 in cancer. Cell. Oncol. 2020, 43, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Muraoka-Cook, R.S.; Feng, S.-M.; Strunk, K.E.; Earp, H.S. ErbB4/HER4: Role in Mammary Gland Development, Differentiation and Growth Inhibition. J. Mammary Gland. Biol. Neoplasia 2008, 13, 235–246. [Google Scholar] [CrossRef]
- Xu, J.; Gong, L.; Qian, Z.; Song, G.; Liu, J. ERBB4 promotes the proliferation of gastric cancer cells via the PI3K/Akt signaling pathway. Oncol. Rep. 2018, 39, 2892–2898. [Google Scholar] [CrossRef] [PubMed]
- Vickers, E. Treatments That Block Proteins Involved in Cell Communication. In A Beginner’s Guide to Targeted Cancer Treatments; John Wiley & Sons, Ltd.: Cambridge, MA, USA, 2018; pp. 65–109. [Google Scholar]
- Haryuni, R.D.; Watabe, S.; Yamaguchi, A.; Fukushi, Y.; Tanaka, T.; Kawasaki, Y.; Zhou, Y.; Yokoyama, S.; Sakurai, H. Negative feedback regulation of ErbB4 tyrosine kinase activity by ERK-mediated non-canonical phosphorylation. Biochem. Biophys. Res. Commun. 2019, 514, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J.; et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Tvorogov, D.; Sundvall, M.; Kurppa, K.; Hollmén, M.; Repo, S.; Johnson, M.S.; Elenius, K. Somatic Mutations of ErbB4: Selective loss-of-function phenotype affecting signal transduction pathways in cancer. J. Biol. Chem. 2009, 284, 5582–5591. [Google Scholar] [CrossRef]
- Walker, R.A. The erbB/HER type 1 tyrosine kinase receptor family. J. Pathol. 1998, 185, 234–235. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed]
- Plowman, G.D.; Culouscou, J.M.; Whitney, G.S.; Green, J.M.; Carlton, G.W.; Foy, L.; Neubauer, M.G.; Shoyab, M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc. Natl. Acad. Sci. USA 1993, 90, 1746–1750. [Google Scholar] [CrossRef]
- Carpenter, G. ErbB-4: Mechanism of action and biology. Exp. Cell Res. 2003, 284, 66–77. [Google Scholar] [CrossRef]
- Roskoski, R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacol. Res. 2014, 87, 42–59. [Google Scholar] [CrossRef]
- Bae, J.H.; Boggon, T.J.; Tomé, F.; Mandiyan, V.; Lax, I.; Schlessinger, J. Asymmetric receptor contact is required for tyrosine autophosphorylation of fibroblast growth factor receptor in living cells. Proc. Natl. Acad. Sci. USA 2010, 107, 2866–2871. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.P.; Bu, Z.; Lemmon, M.A. Ligand-Induced Structural Transitions in ErbB Receptor Extracellular Domains. Structure 2007, 15, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Red Brewer, M.; Choi, S.H.; Alvarado, D.; Moravcevic, K.; Pozzi, A.; Lemmon, M.A.; Carpenter, G. The Juxtamembrane Region of the EGF Receptor Functions as an Activation Domain. Mol. Cell 2009, 34, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-W.; Cheng, C.K.; Gustafson, W.C.; Charron, E.; Zipper, P.; Wong, R.A.; Chen, J.; Lau, J.; Knobbe-Thomsen, C.; Weller, M.; et al. EGFR Phosphorylates Tumor-Derived EGFRvIII Driving STAT3/5 and Progression in Glioblastoma. Cancer Cell 2013, 24, 438–449. [Google Scholar] [CrossRef] [PubMed]
- James, K.A.; Verkhivker, G.M. Structure-Based Network Analysis of Activation Mechanisms in the ErbB Family of Receptor Tyrosine Kinases: The Regulatory Spine Residues Are Global Mediators of Structural Stability and Allosteric Interactions. PLoS ONE 2014, 9, e113488. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Zhao, J.W. Analysis of regulatory mechanism after ErbB4 gene mutation based on local modeling methodology. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Carraway, K.L.; Sweeney, C. Localization and modulation of ErbB receptor tyrosine kinases. Curr. Opin. Cell Biol. 2001, 13, 125–130. [Google Scholar] [CrossRef]
- Karamouzis, M.V.; Badra, F.A.; Papavassiliou, A.G. Breast cancer: The upgraded role of HER-3 and HER-4. Int. J. Biochem. Cell Biol. 2007, 39, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Hoesl, C.; Röhrl, J.M.; Schneider, M.R.; Dahlhoff, M. The receptor tyrosine kinase ERBB4 is expressed in skin keratinocytes and influences epidermal proliferation. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2018, 1862, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Yamamoto-Ibusuki, M.; Yamamoto, Y.; Yamamoto, S.; Tomiguchi, M.; Takeshita, T.; Hayashi, M.; Sueta, A.; Iwase, H. The localization of HER4 intracellular domain and expression of its alternately-spliced isoforms have prognostic significance in ER+ HER2- breast cancer. Oncotarget 2014, 5, 3919. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Icli, B.; Bharti, A.; Pentassuglia, L.; Peng, X.; Sawyer, D.B. ErbB4 localization to cardiac myocyte nuclei, and its role in myocyte DNA damage response. Biochem. Biophys. Res. Commun. 2012, 418, 116–121. [Google Scholar] [CrossRef]
- Culouscou, J.M.; Plowman, G.D.; Carlton, G.W.; Green, J.M.; Shoyab, M. Characterization of a breast cancer cell differentiation factor that specifically activates the HER4/p180erbB4 receptor. J. Biol. Chem. 1993, 268, 18407–18410. [Google Scholar] [CrossRef]
- Tzahar, E.; Levkowitz, G.; Karunagaran, D.; Yi, L.; Peles, E.; Lavi, S.; Chang, D.; Liu, N.; Yayon, A.; Wen, D. ErbB-3 and ErbB-4 function as the respective low and high affinity receptors of all Neu differentiation factor/heregulin isoforms. J. Biol. Chem. 1994, 269, 25226–25233. [Google Scholar] [CrossRef]
- Plowman, G.D.; Green, J.M.; Culouscou, J.-M.; Carlton, G.W.; Rothwell, V.M.; Buckley, S. Heregulin induces tyrosine phosphorylation of HER4/p180erbB4. Nature 1993, 366, 473–475. [Google Scholar] [CrossRef]
- Zhang, D.; Sliwkowski, M.X.; Mark, M.; Frantz, G.; Akita, R.; Sun, Y.; Hillan, K.; Crowley, C.; Brush, J.; Godowski, P.J. Neuregulin-3 (NRG3): A novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA 1997, 94, 9562–9567. [Google Scholar] [CrossRef]
- Ebner, R.; Derynck, R. Epidermal growth factor and transforming growth factor-alpha: Differential intracellular routing and processing of ligand-receptor complexes. Cell Regul. 1991, 2, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Lai, C.; Riese, D.J.; Diamonti, A.J.; Cantley, L.C.; Carraway, K.L. Ligand Discrimination in Signaling through an ErbB4 Receptor Homodimer. J. Biol. Chem. 2000, 275, 19803–19807. [Google Scholar] [CrossRef]
- Riese, D.J.; Kim, E.D.; Elenius, K.; Buckley, S.; Klagsbrun, M.; Plowman, G.D.; Stern, D.F. The Epidermal Growth Factor Receptor Couples Transforming Growth Factor-α, Heparin-binding Epidermal Growth Factor-like Factor, and Amphiregulin to Neu, ErbB-3, and ErbB-4. J. Biol. Chem. 1996, 271, 20047–20052. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Bu, Z.; Ladbury, J.E.; Zhou, M.; Pinchasi, D.; Lax, I.; Engelman, D.M.; Schlessinger, J. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997, 16, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Riese, D.J.; van Raaij, T.M.; Plowman, G.D.; Andrews, G.C.; Stern, D.F. The cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol. Cell. Biol. 1995, 15, 5770–5776. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Peles, E.; Cupples, R.; Suggs, S.V.; Bacus, S.S.; Luo, Y.; Trail, G.; Hu, S.; Silbiger, S.M.; Levy, R.B.; et al. Neu differentiation factor: A transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell 1992, 69, 559–572. [Google Scholar] [CrossRef]
- Tidcombe, H.; Jackson-Fisher, A.; Mathers, K.; Stern, D.F.; Gassmann, M.; Golding, J.P. Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proc. Natl. Acad. Sci. USA 2003, 100, 8281–8286. [Google Scholar] [CrossRef] [PubMed]
- Chuu, C.-P.; Chen, R.-Y.; Barkinge, J.L.; Ciaccio, M.F.; Jones, R.B. Systems-Level Analysis of ErbB4 Signaling in Breast Cancer: A Laboratory to Clinical Perspective. Mol. Cancer Res. 2008, 6, 885. [Google Scholar] [CrossRef] [PubMed]
- Hollmén, M.; Määttä, J.A.; Bald, L.; Sliwkowski, M.X.; Elenius, K. Suppression of breast cancer cell growth by a monoclonal antibody targeting cleavable ErbB4 isoforms. Oncogene 2009, 28, 1309–1319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kountourakis, P.; Pavlakis, K.; Psyrri, A.; Rontogianni, D.; Xiros, N.; Patsouris, E.; Pectasides, D.; Economopoulos, T. Prognostic significance of HER3 and HER4 protein expression in colorectal adenocarcinomas. BMC Cancer 2006, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Kishore, C.; Bhadra, P. Current advancements and future perspectives of immunotherapy in colorectal cancer research. Eur. J. Pharmacol. 2021, 893, 173819. [Google Scholar] [CrossRef] [PubMed]
- de Wit, M.; Fijneman, R.J.A.; Verheul, H.M.W.; Meijer, G.A.; Jimenez, C.R. Proteomics in colorectal cancer translational research: Biomarker discovery for clinical applications. Clin. Biochem. 2013, 46, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Favreau-Lessard, A.J.; deKay, J.T.; Herrmann, Y.R.; Robich, M.P.; Koza, R.A.; Prudovsky, I.; Sawyer, D.B.; Ryzhov, S. Protective role of ErbB3 signaling in myeloid cells during adaptation to cardiac pressure overload. J. Mol. Cell. Cardiol. 2021, 152, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.R.; Edelblum, K.L.; Mullane, M.T.; Liang, D.; Polk, D.B. The ErbB4 Growth Factor Receptor Is Required for Colon Epithelial Cell Survival in the Presence of TNF. Gastroenterology 2009, 136, 217–226. [Google Scholar] [CrossRef]
- Keates, S.; Sougioultzis, S.; Keates, A.C.; Zhao, D.; Peek, R.M.; Shaw, L.M.; Kelly, C.P. cag+ Helicobacter pylori Induce Transactivation of the Epidermal Growth Factor Receptor in AGS Gastric Epithelial Cells. J. Biol. Chem. 2001, 276, 48127–48134. [Google Scholar] [CrossRef]
- Williams, C.S.; Bernard, J.K.; Demory Beckler, M.; Almohazey, D.; Washington, M.K.; Smith, J.J.; Frey, M.R. ERBB4 is over-expressed in human colon cancer and enhances cellular transformation. Carcinogenesis 2015, 36, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sullivan, L.L.; Nair, S.S.; Williams, C.C.; Pandey, A.K.; Marrero, L.; Vadlamudi, R.K.; Jones, F.E. Coregulation of Estrogen Receptor by ERBB4/HER4 Establishes a Growth-Promoting Autocrine Signal in Breast Tumor Cells. Cancer Res. 2006, 66, 7991–7998. [Google Scholar] [CrossRef]
- Thien, C.B.F.; Langdon, W.Y. Tyrosine kinase activity of the EGF receptor is enhanced by the expression of oncogenic 70Z-Cbl. Oncogene 1997, 15, 2909–2919. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.B.; Sengupta, D. Resolving the conformational dynamics of ErbB growth factor receptor dimers. J. Struct. Biol. 2019, 207, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, K.; Suda, K.; Onozato, R.; Kuwano, H.; Yatabe, Y.; Mitsudomi, T. Analysis of ERBB4 Mutations and Expression in Japanese Patients with Lung Cancer. J. Thorac. Oncol. 2010, 5, 1859–1861. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Denessiouk, K.; Johnson, M.S.; Elenius, K. Activating ERBB4 mutations in non-small cell lung cancer. Oncogene 2016, 35, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Li, R.; Chang, D.W.; Ye, Y.; Minna, J.D.; Roth, J.A.; Han, B.; Wu, X. Genetic variations in cancer-related significantly mutated genes and lung cancer susceptibility. Ann. Oncol. 2017, 28, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Wada, K.; Akamatsu, T.; Yonezawa, M.; Noguchi, H.; Mizoguchi, A.; Kasuga, M.; Sakamoto, C. A Novel Epidermal Growth Factor-like Molecule Containing Two Follistatin Modules Stimulates Tyrosine Phosphorylation of erbB-4 in MKN28 Gastric Cancer Cells. Biochem. Biophys. Res. Commun. 1999, 266, 593–602. [Google Scholar] [CrossRef]
- Song, G.; Zhang, H.; Chen, C.; Gong, L.; Chen, B.; Zhao, S.; Shi, J.; Xu, J.; Ye, Z. miR-551b regulates epithelial-mesenchymal transition and metastasis of gastric cancer by inhibiting ERBB4 expression. Oncotarget 2017, 8, 45725. [Google Scholar] [CrossRef]
- Chen, K.; Yang, D.; Li, X.; Sun, B.; Song, F.; Cao, W.; Brat, D.J.; Gao, Z.; Li, H.; Liang, H.; et al. Mutational landscape of gastric adenocarcinoma in Chinese: Implications for prognosis and therapy. Proc. Natl. Acad. Sci. USA 2015, 112, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, L.; Ni, H.; Sun, L.; Jiao, W.; Chen, L.; Zhou, Q.; Shen, T.; Cui, H.; Gao, T.; et al. ERBB4 acts as a suppressor in the development of hepatocellular carcinoma. Carcinogenesis 2017, 38, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Park, S.T.; Jang, J.W.; Kim, G.D.; Park, J.A.; Hur, W.; Woo, H.Y.; Kim, J.D.; Kwon, J.H.; Yoo, C.R.; Bae, S.H.; et al. Beneficial effect of metronomic chemotherapy on tumor suppression and survival in a rat model of hepatocellular carcinoma with liver cirrhosis. Cancer Chemother. Pharmacol. 2010, 65, 1029–1037. [Google Scholar] [CrossRef]
- Edwards, J.; Traynor, P.; Munro, A.F.; Pirret, C.F.; Dunne, B.; Bartlett, J.M.S. The Role of HER1-HER4 and EGFRvIII in Hormone-Refractory Prostate Cancer. Clin. Cancer Res. 2006, 12, 123–130. [Google Scholar] [CrossRef]
- Hernes, E.; Fosså, S.D.; Berner, A.; Otnes, B.; Nesland, J.M. Expression of the epidermal growth factor receptor family in prostate carcinoma before and during androgen-independence. Br. J. Cancer 2004, 90, 449–454. [Google Scholar] [CrossRef]
- Ping, P.; Zhang, J.; Zheng, Y.-T.; Li, R.C.X.; Dawn, B.; Tang, X.-L.; Takano, H.; Balafanova, Z.; Bolli, R. Demonstration of Selective Protein Kinase C–Dependent Activation of Src and Lck Tyrosine Kinases During Ischemic Preconditioning in Conscious Rabbits. Circ. Res. 1999, 85, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.M.; Bryant, I.; Fry, R.; Williams, E.E.; Riese, D.J. Phosphorylation of ErbB4 on Tyr1056 is critical for inhibition of colony formation by prostate tumor cell lines. Biochem. Biophys. Res. Commun. 2006, 349, 372–382. [Google Scholar] [CrossRef][Green Version]
- Tsai, Y.-S.; Cheng, H.-L.; Tzai, T.-S.; Chow, N.-H. Clinical Significance of ErbB Receptor Family in Urothelial Carcinoma of the Bladder: A Systematic Review and Meta-Analysis. Adv. Urol. 2012, 2012, 181964. [Google Scholar] [CrossRef]
- Memon, A.A.; Sorensen, B.S.; Meldgaard, P.; Fokdal, L.; Thykjaer, T.; Nexo, E. The relation between survival and expression of HER1 and HER2 depends on the expression of HER3 and HER4: A study in bladder cancer patients. Br. J. Cancer 2006, 94, 1703–1709. [Google Scholar] [CrossRef]
- El Badadawy, N.; Youssef, N.; Ismail, A.; Ragheb, F.; Hakim, S. Evaluation of the role of immunohistochemical expression of EGFR (ERB B1) and HER4 (ERB B4) in urinary bladder urothelial carcinoma. Pathology 2016, 48, S123. [Google Scholar] [CrossRef]
- Saglam, O.; Xiong, Y.; Marchion, D.C.; Strosberg, C.; Wenham, R.M.; Johnson, J.J.; Saeed-Vafa, D.; Cubitt, C.; Hakam, A.; Magliocco, A.M. ERBB4 Expression in Ovarian Serous Carcinoma Resistant to Platinum-Based Therapy. Cancer Control. 2017, 24, 89–95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sundvall, M.; Veikkolainen, V.; Kurppa, K.; Salah, Z.; Tvorogov, D.; Zoelen, E.J.V.; Aqeilan, R.; Elenius, K. Cell Death or Survival Promoted by Alternative Isoforms of ErbB4. Mol. Biol. Cell 2010, 21, 4275–4286. [Google Scholar] [CrossRef]
- Muraoka-Cook, R.S.; Sandahl, M.; Husted, C.; Hunter, D.; Miraglia, L.; Feng, S.-M.; Elenius, K.; Earp, S.I.H. The Intracellular Domain of ErbB4 Induces Differentiation of Mammary Epithelial Cells. Mol. Biol. Cell 2006, 17, 4118–4129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Junttila, T.T.; Sundvall, M.; Määttä, J.A.; Elenius, K. ErbB4 and Its Isoforms: Selective Regulation of Growth Factor Responses by Naturally Occurring Receptor Variants. Trends Cardiovasc. Med. 2000, 10, 304–310. [Google Scholar] [CrossRef]
- Gullick, W.J. c-erbB-4/HER4: Friend or foe? J. Pathol. 2003, 200, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Koutras, A.K.; Kalogeras, K.T.; Dimopoulos, M.A.; Wirtz, R.M.; Dafni, U.; Briasoulis, E.; Pectasides, D.; Gogas, H.; Christodoulou, C.; Aravantinos, G.; et al. Evaluation of the prognostic and predictive value of HER family mRNA expression in high-risk early breast cancer: A Hellenic Cooperative Oncology Group (HeCOG) study. Br. J. Cancer 2008, 99, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Sassen, A.; Rochon, J.; Wild, P.; Hartmann, A.; Hofstaedter, F.; Schwarz, S.; Brockhoff, G. Cytogenetic analysis of HER1/EGFR, HER2, HER3 and HER4 in 278 breast cancer patients. Breast Cancer Res. 2008, 10, R2. [Google Scholar] [CrossRef] [PubMed]
- Bacus, S.S.; Chin, D.; Yarden, Y.; Zelnick, C.R.; Stern, D.F. Type 1 receptor tyrosine kinases are differentially phosphorylated in mammary carcinoma and differentially associated with steroid receptors. Am. J. Pathol. 1996, 148, 549–558. [Google Scholar]
- Kew, T.Y.; Bell, J.A.; Pinder, S.E.; Denley, H.; Srinivasan, R.; Gullick, W.J.; Nicholson, R.I.; Blamey, R.W.; Ellis, I.O. c-erbB-4 protein expression in human breast cancer. Br. J. Cancer 2000, 82, 1163–1170. [Google Scholar] [CrossRef]
- Bièche, I.; Onody, P.; Tozlu, S.; Driouch, K.; Vidaud, M.; Lidereau, R. Prognostic value of ERBB family mRNA expression in breast carcinomas. Int. J. Cancer 2003, 106, 758–765. [Google Scholar] [CrossRef]
- Lodge, A.J.; Anderson, J.J.; Gullick, W.J.; Haugk, B.; Leonard, R.C.F.; Angus, B. Type 1 growth factor receptor expression in node positive breast cancer: Adverse prognostic significance of c-erbB-4. J. Clin. Pathol. 2003, 56, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Määttä, J.A.; Sundvall, M.; Junttila, T.T.; Peri, L.; Laine, V.J.O.; Isola, J.; Egeblad, M.; Elenius, K. Proteolytic Cleavage and Phosphorylation of a Tumor-associated ErbB4 Isoform Promote Ligand-independent Survival and Cancer Cell Growth. Mol. Biol. Cell 2006, 17, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Sundvall, M.; Lundin, M.; Lundin, J.; Tanner, M.; Härkönen, P.; Joensuu, H.; Isola, J.; Elenius, K. Cleavable ErbB4 Isoform in Estrogen Receptor–Regulated Growth of Breast Cancer Cells. Cancer Res. 2005, 65, 1384–1393. [Google Scholar] [CrossRef]
- Lynch, C.C.; Vargo-Gogola, T.; Martin, M.D.; Fingleton, B.; Crawford, H.C.; Matrisian, L.M. Matrix Metalloproteinase 7 Mediates Mammary Epithelial Cell Tumorigenesis through the ErbB4 Receptor. Cancer Res. 2007, 67, 6760–6767. [Google Scholar] [CrossRef]
- Muraoka-Cook, R.S.; Caskey, L.S.; Sandahl, M.A.; Hunter, D.M.; Husted, C.; Strunk, K.E.; Sartor, C.I.; Rearick, W.A.; McCall, W.; Sgagias, M.K.; et al. Heregulin-Dependent Delay in Mitotic Progression Requires HER4 and BRCA1. Mol. Cell. Biol. 2006, 26, 6412–6424. [Google Scholar] [CrossRef] [PubMed]
- Naresh, A.; Long, W.; Vidal, G.A.; Wimley, W.C.; Marrero, L.; Sartor, C.I.; Tovey, S.; Cooke, T.G.; Bartlett, J.M.S.; Jones, F.E. The ERBB4/HER4 Intracellular Domain 4ICD Is a BH3-Only Protein Promoting Apoptosis of Breast Cancer Cells. Cancer Res. 2006, 66, 6412–6420. [Google Scholar] [CrossRef] [PubMed]
- Sundvall, M.; Iljin, K.; Kilpinen, S.; Sara, H.; Kallioniemi, O.-P.; Elenius, K. Role of ErbB4 in Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2008, 13, 259–268. [Google Scholar] [CrossRef]
- Hollmén, M.; Liu, P.; Kurppa, K.; Wildiers, H.; Reinvall, I.; Vandorpe, T.; Smeets, A.; Deraedt, K.; Vahlberg, T.; Joensuu, H.; et al. Proteolytic Processing of ErbB4 in Breast Cancer. PLoS ONE 2012, 7, e39413. [Google Scholar] [CrossRef] [PubMed]
- te Velde, E.A.; Franke, A.C.; van Hillegersberg, R.; Elshof, S.M.; de Weger, R.W.; Borel Rinkes, I.H.M.; van Diest, P.J. HER-family gene amplification and expression in resected pancreatic cancer. Eur. J. Surg. Oncol. 2009, 35, 1098–1104. [Google Scholar] [CrossRef][Green Version]
- Mill, C.P.; Gettinger, K.L.; Riese, D.J. Ligand stimulation of ErbB4 and a constitutively-active ErbB4 mutant result in different biological responses in human pancreatic tumor cell lines. Exp. Cell Res. 2011, 317, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Thybusch-Bernhardt, A.; Beckmann, S.; Juhl, H. Comparative analysis of the EGF-receptor family in pancreatic cancer: Expression of HER-4 correlates with a favourable tumor stage. Int. J. Surg. Investig. 2001, 2, 393–400. [Google Scholar] [PubMed]
- Graber, H.U.; Friess, H.; Kaufmann, B.; Willi, D.; Zimmermann, A.; Korc, M.; Büchler, M.W. ErbB-4 mRNA expression is decreased in non-metastatic pancreatic cancer. Int. J. Cancer 1999, 84, 24–27. [Google Scholar] [CrossRef]
- Jones, D.C.; Scanteianu, A.; DiStefano, M.; Bouhaddou, M.; Birtwistle, M.R. Analysis of copy number loss of the ErbB4 receptor tyrosine kinase in glioblastoma. PLoS ONE 2018, 13, e0190664. [Google Scholar] [CrossRef]
- Andersson, U.; Guo, D.; Malmer, B.; Bergenheim, A.T.; Brännström, T.; Hedman, H.; Henriksson, R. Epidermal growth factor receptor family (EGFR, ErbB2–4) in gliomas and meningiomas. Acta Neuropathol. 2004, 108, 135–142. [Google Scholar] [CrossRef]
- Donoghue, J.F.; Kerr, L.T.; Alexander, N.W.; Greenall, S.A.; Longano, A.B.; Gottardo, N.G.; Wang, R.; Tabar, V.; Adams, T.E.; Mischel, P.S.; et al. Activation of ERBB4 in Glioblastoma Can Contribute to Increased Tumorigenicity and Influence Therapeutic Response. Cancers 2018, 10, 243. [Google Scholar] [CrossRef]
- Prickett, T.D.; Agrawal, N.S.; Wei, X.; Yates, K.E.; Lin, J.C.; Wunderlich, J.R.; Cronin, J.C.; Cruz, P.; Rosenberg, S.A.; Samuels, Y.; et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 2009, 41, 1127–1132. [Google Scholar] [CrossRef]
- Nielsen, T.O.; Poulsen, S.S.; Journe, F.; Ghanem, G.; Sorensen, B.S. HER4 and its cytoplasmic isoforms are associated with progression-free survival of malignant melanoma. Melanoma Res. 2014, 24, 88–91. [Google Scholar] [CrossRef]
- Kurppa, K.; Elenius, K. Mutated ERBB4: A novel drug target in metastatic melanoma? Pigment. Cell Melanoma Res. 2009, 22, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Settleman, J. A Therapeutic Opportunity in Melanoma: ErbB4 Makes a Mark on Skin. Cancer Cell 2009, 16, 278–279. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Ejskjær, K.; Sørensen, B.S.; Poulsen, S.S.; Forman, A.; Nexø, E.; Mogensen, O. Expression of the epidermal growth factor system in endometrioid endometrial cancer. Gynecol. Oncol. 2007, 104, 158–167. [Google Scholar] [CrossRef]
- Jaffe, N. Osteosarcoma: Review of the Past, Impact on the Future. The American Experience. In Pediatric and Adolescent Osteosarcoma; Jaffe, N., Bruland, O.S., Bielack, S., Eds.; Springer: Boston, MA, USA, 2010; pp. 239–262. [Google Scholar]
- Dai, X.; Ma, W.; He, X.; Jha, R.K. Review of therapeutic strategies for osteosarcoma, chondrosarcoma, and Ewing’s sarcoma. Med. Sci. Monit. 2011, 17, RA177–RA190. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, Q.; Wang, S.; Huang, Z.; Yu, F.; Lin, J. HER4 promotes the growth and metastasis of osteosarcoma via the PI3K/AKT pathway. Acta Biochim. Biophys. Sin. 2020, 52, 345–362. [Google Scholar] [CrossRef]
- Xie, H.; Lin, L.; Tong, L.; Jiang, Y.; Zheng, M.; Chen, Z.; Jiang, X.; Zhang, X.; Ren, X.; Qu, W.; et al. AST1306, A Novel Irreversible Inhibitor of the Epidermal Growth Factor Receptor 1 and 2, Exhibits Antitumor Activity Both In Vitro and In Vivo. PLoS ONE 2011, 6, e21487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cao, J.; Li, J.; Zhang, Y.; Chen, Z.; Peng, W.; Sun, S.; Zhao, N.; Wang, J.; Zhong, D.; et al. A phase I study of AST1306, a novel irreversible EGFR and HER2 kinase inhibitor, in patients with advanced solid tumors. J. Hematol. Oncol. 2014, 7, 22. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Brattain, M.G.; Zhong, H. Dual Inhibitors of PI3K/mTOR or mTOR-Selective Inhibitors: Which Way Shall We Go? Curr. Med. Chem. 2011, 18, 5528–5544. [Google Scholar] [CrossRef]
- Ahammad, I.; Sarker, M.R.I.; Khan, A.M.; Islam, S.; Hossain, M. Virtual Screening to Identify Novel Inhibitors of Pan ERBB Family of Proteins from Natural Products with Known Anti-tumorigenic Properties. Int. J. Pept. Res. Ther. 2020, 26, 1923–1938. [Google Scholar] [CrossRef]
- To Evaluate the Efficacy and Safety of Anlotinib Combined with Allitinib in Lung Cancer. Available online: https://ClinicalTrials.gov/show/NCT04671303 (accessed on 30 November 2021).
- Lee, H.; Kim, J.W.; Choi, D.K.; Yu, J.H.; Kim, J.H.; Lee, D.-S.; Min, S.-H. Poziotinib suppresses ovarian cancer stem cell growth via inhibition of HER4-mediated STAT5 pathway. Biochem. Biophys. Res. Commun. 2020, 526, 158–164. [Google Scholar] [CrossRef]
- Subramanian, J.; Katta, A.; Masood, A.; Vudem, D.R.; Kancha, R.K. Emergence of ERBB2 Mutation as a Biomarker and an Actionable Target in Solid Cancers. Oncologist 2019, 24, e1303–e1314. [Google Scholar] [CrossRef]
- Cha, M.Y.; Lee, K.-O.; Kim, M.; Song, J.Y.; Lee, K.H.; Park, J.; Chae, Y.J.; Kim, Y.H.; Suh, K.H.; Lee, G.S.; et al. Antitumor activity of HM781-36B, a highly effective pan-HER inhibitor in erlotinib-resistant NSCLC and other EGFR-dependent cancer models. Int. J. Cancer 2012, 130, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- A Study of Poziotinib in Combination with T-DM1 in HER2-Positive Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT03429101 (accessed on 30 November 2021).
- Poziotinib in Patients with NSCLC Having EGFR or HER2 Exon 20 Insertion Mutation. Available online: https://ClinicalTrials.gov/show/NCT04044170 (accessed on 30 November 2021).
- NOV120101 (Poziotinib) for 1st Line Monotherapy in Patients with Lung Adenocarcinoma. Available online: https://ClinicalTrials.gov/show/NCT01819428 (accessed on 30 November 2021).
- Study of Poziotinib in Patients with HER2-Positive Metastatic Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT02659514 (accessed on 30 November 2021).
- A Mass Balance and Pharmacokinetics Study of 14C-Labeled Poziotinib in Cancer Patients Suitable for Treatment with Poziotinib. Available online: https://ClinicalTrials.gov/show/NCT03804515 (accessed on 30 November 2021).
- Poziotinib in Patients with HER2+ Recurrent Stage IV BC Who Have Received at Least 2 Prior HER2-Directed Regimens. Available online: https://ClinicalTrials.gov/show/NCT02418689 (accessed on 30 November 2021).
- Search Result of Poziotinib in clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/results?recrs=&cond=&term=poziotinib&cntry=&state=&city=&dist=%20 (accessed on 30 November 2021).
- Lee, H.; Kim, J.W.; Lee, D.-S.; Min, S.-H. Combined Poziotinib with Manidipine Treatment Suppresses Ovarian Cancer Stem-Cell Proliferation and Stemness. Int. J. Mol. Sci. 2020, 21, 7379. [Google Scholar] [CrossRef] [PubMed]
- Motohara, T.; Katabuchi, H. Ovarian Cancer Stemness: Biological and Clinical Implications for Metastasis and Chemotherapy Resistance. Cancers 2019, 11, 907. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Girard, N. Optimizing outcomes in EGFR mutation-positive NSCLC: Which tyrosine kinase inhibitor and when? Future Oncol. 2018, 14, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; de Marinis, F. Dacomitinib in EGFR-positive non-small cell lung cancer: An attractive but broken option. Transl. Lung Cancer Res. 2018, 7, S100–S102. [Google Scholar] [CrossRef] [PubMed]
- Bilancia, D.; Rosati, G.; Dinota, A.; Germano, D.; Romano, R.; Manzione, L. Lapatinib in breast cancer. Ann. Oncol. 2007, 18, vi26–vi30. [Google Scholar] [CrossRef]
- Tsang, R.Y.; Sadeghi, S.; Finn, R.S. Lapatinib, a Dual-Targeted Small Molecule Inhibitor of EGFR and HER2, in HER2-Amplified Breast Cancer: From Bench to Bedside. Clin. Med. Insights Ther. 2011, 3, CMT.S3783. [Google Scholar] [CrossRef]
- Wind, S.; Schnell, D.; Ebner, T.; Freiwald, M.; Stopfer, P. Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. 2017, 56, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, N.; Kumbla, R.A.; Mita, M. Chapter 1—Current Trends in Cancer Therapy. In Cardio-Oncology; Gottlieb, R.A., Mehta, P.K., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 1–24. [Google Scholar]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; et al. Novel D761Y and Common Secondary T790M Mutations in Epidermal Growth Factor Receptor–Mutant Lung Adenocarcinomas with Acquired Resistance to Kinase Inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.-H.; Wu, Y.-L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Manzo, A.; Montanino, A.; Costanzo, R.; Sandomenico, C.; Palumbo, G.; Schettino, C.; Daniele, G.; Morabito, A.; Perrone, F.; Piccirillo, M.C. Chapter 33—EGFR Mutations: Best Results from Second- and Third-Generation Tyrosine Kinase Inhibitors. In Oncogenomics; Dammacco, F., Silvestris, F., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 477–486. [Google Scholar]
- Keating, G.M. Afatinib: A Review of Its Use in the Treatment of Advanced Non-Small Cell Lung Cancer. Drugs 2014, 74, 207–221. [Google Scholar] [CrossRef]
- Sachdev, J.C.; Jahanzeb, M. Blockade of the HER Family of Receptors in the Treatment of HER2-Positive Metastatic Breast Cancer. Clin. Breast Cancer 2012, 12, 19–29. [Google Scholar] [CrossRef]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- A Phase II Study of CI-1033 in Treating Patients with Metastatic (Stage IV) Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT00051051 (accessed on 30 November 2021).
- PH 1 Evaluation Of Oral CI-1033 In Combination With Paclitaxel/Carboplatin As 1st Line Chemotherapy In NSCLC Patients. Available online: https://ClinicalTrials.gov/show/NCT00174356 (accessed on 30 November 2021).
- A Phase 2, Randomized, Open-Label Study Of Single Agent CI-1033 in Patients with Advanced Non-Small Cell Lung Cancer. Available online: https://ClinicalTrials.gov/show/NCT00050830 (accessed on 30 November 2021).
- Singh, P.K.; Singh, H.; Silakari, O. Kinases inhibitors in lung cancer: From benchside to bedside. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2016, 1866, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; Conlon, N.T.; Kannan, S.; Verma, C.S.; Eli, L.D.; Lalani, A.S.; Crown, J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Prové, A.; Dirix, L. Neratinib for the treatment of breast cancer. Expert Opin. Pharmacother. 2016, 17, 2243–2248. [Google Scholar] [CrossRef]
- Xu, B.; Yan, M.; Ma, F.; Hu, X.; Feng, J.; Ouyang, Q.; Tong, Z.; Li, H.; Zhang, Q.; Sun, T.; et al. Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2-positive metastatic breast cancer (PHOEBE): A multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 351–360. [Google Scholar] [CrossRef]
- Gao, Z.; Song, C.; Li, G.; Lin, H.; Lian, X.; Zhang, N.; Cao, B. Pyrotinib treatment on HER2-positive gastric cancer cells promotes the released exosomes to enhance endothelial cell progression, which can be counteracted by apatinib. OncoTargets Ther. 2019, 12, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Pyrotinib: First Global Approval. Drugs 2018, 78, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- A Study of Pyrotinib Plus Vinorelbine in Patients with Brain Metastases from HER2-Positive Metastatic Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT03933982 (accessed on 30 November 2021).
- Inetetamab Combined with Pyrotinib and Chemotherapy in the Treatment of HER2 Positive Metastatic Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT04681911 (accessed on 30 November 2021).
- Berglöf, A.; Hamasy, A.; Meinke, S.; Palma, M.; Krstic, A.; Månsson, R.; Kimby, E.; Österborg, A.; Smith, C.I.E. Targets for Ibrutinib Beyond B Cell Malignancies. Scand. J. Immunol. 2015, 82, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Demetri, G.D.; Mehren, M.V.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.M.; Roberts, P.J.; Heinz, D.; et al. Long-Term Results From a Randomized Phase II Trial of Standard- Versus Higher-Dose Imatinib Mesylate for Patients With Unresectable or Metastatic Gastrointestinal Stromal Tumors Expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.M.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Bowles, K.M.; Barrera, L.N.; Murray, M.Y.; Zaitseva, L.; MacEwan, D.J. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-κB. Cell. Signal. 2013, 25, 106–112. [Google Scholar] [CrossRef]
- Wang, X.; Wong, J.; Sevinsky, C.J.; Kokabee, L.; Khan, F.; Sun, Y.; Conklin, D.S. Bruton’s Tyrosine Kinase Inhibitors Prevent Therapeutic Escape in Breast Cancer Cells. Mol. Cancer Ther. 2016, 15, 2198–2208. [Google Scholar] [CrossRef]
- Smith, M.R. Ibrutinib in B lymphoid malignancies. Expert Opin. Pharmacother. 2015, 16, 1879–1887. [Google Scholar] [CrossRef] [PubMed]
- Rauf, F.; Festa, F.; Park, J.G.; Magee, M.; Eaton, S.; Rinaldi, C.; Betanzos, C.M.; Gonzalez-Malerva, L.; LaBaer, J. Ibrutinib inhibition of ERBB4 reduces cell growth in a WNT5A-dependent manner. Oncogene 2018, 37, 2237–2250. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.W.; Lee, F.Y.; Yu, C.; Luo, F.R.; Oppenheimer, S.; Zhang, H.; Smykla, R.A.; Mastalerz, H.; Fink, B.E.; Hunt, J.T.; et al. Preclinical Antitumor Activity of BMS-599626, a pan-HER Kinase Inhibitor That Inhibits HER1/HER2 Homodimer and Heterodimer Signaling. Clin. Cancer Res. 2006, 12, 6186–6193. [Google Scholar] [CrossRef] [PubMed]
- Gill, L.A.; Verdonk, M.; Boyle, G.R.; Taylor, R. A Comparison of Physicochemical Property Profiles of Marketed Oral Drugs and Orally Bioavailable Anti-Cancer Protein Kinase Inhibitors in Clinical Development. Curr. Top. Med. Chem. 2007, 7, 1408–1422. [Google Scholar] [PubMed]
- Ashar, Y.V.; Zhou, J.; Gupta, P.; Teng, Q.-X.; Lei, Z.-N.; Reznik, S.E.; Lusvarghi, S.; Wurpel, J.; Ambudkar, S.V.; Chen, Z.-S. BMS-599626, a Highly Selective Pan-HER Kinase Inhibitor, Antagonizes ABCG2-Mediated Drug Resistance. Cancers 2020, 12, 2502. [Google Scholar] [CrossRef] [PubMed]
- Pharmacokinetics (PK) Study of AC480 for Recurrent Glioma. Available online: https://ClinicalTrials.gov/show/NCT00979173 (accessed on 30 November 2021).
- BMS-599626 in Treating Patients with Metastatic Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT00093730 (accessed on 30 November 2021).
- BMS-599626 in Patients with Advanced Solid Malignancies. Available online: https://ClinicalTrials.gov/show/NCT00095537 (accessed on 30 November 2021).
- MAD Refractory: Solid Tumor QD w/o Break. Available online: https://ClinicalTrials.gov/show/NCT00207012 (accessed on 30 November 2021).
- Safety Study for Intravenous (IV) AC480 (AC480IV) to Treat Advanced Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT01245543 (accessed on 30 November 2021).
- Haluska, P.; Carboni, J.M.; TenEyck, C.; Attar, R.M.; Hou, X.; Yu, C.; Sagar, M.; Wong, T.W.; Gottardis, M.M.; Erlichman, C. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol. Cancer Ther. 2008, 7, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
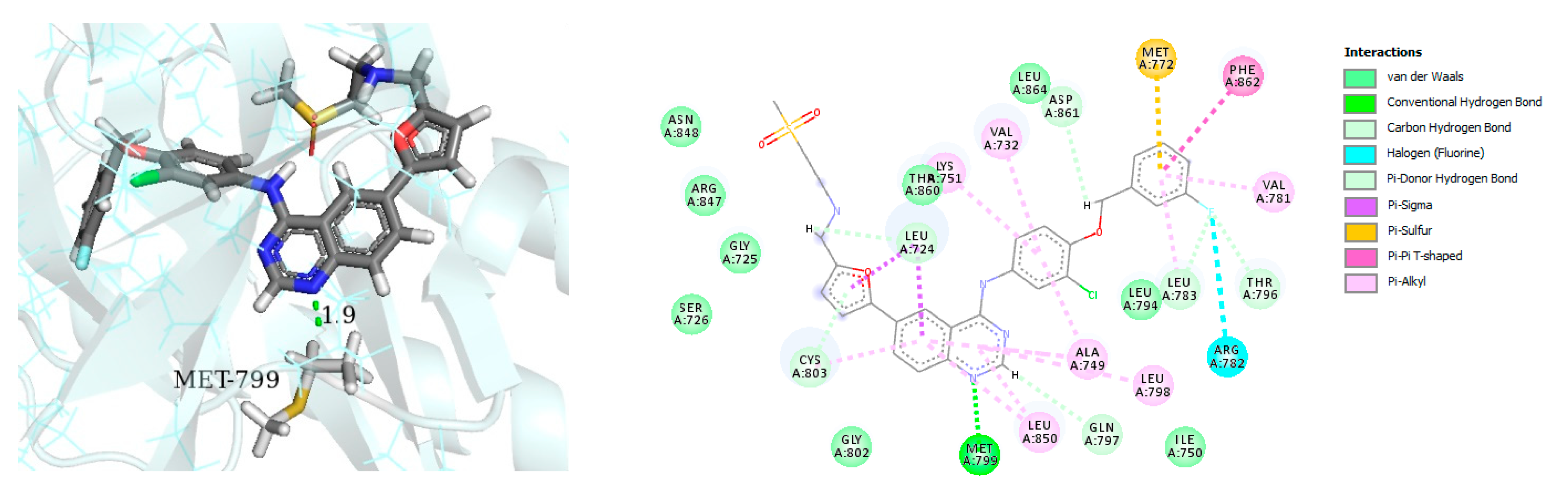
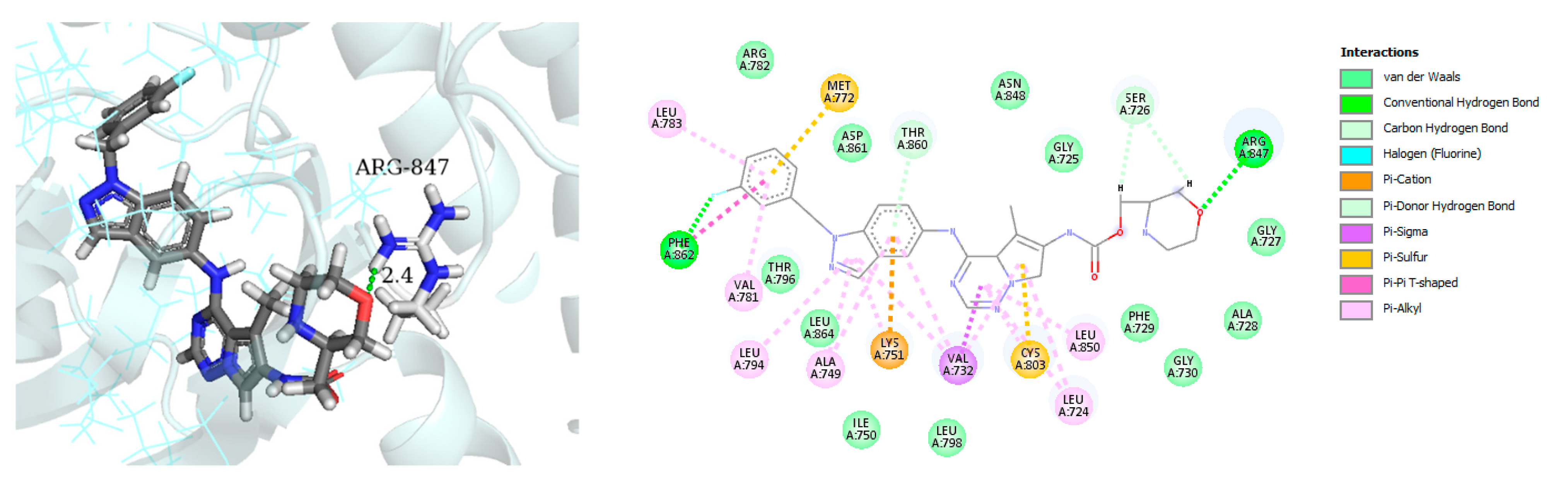
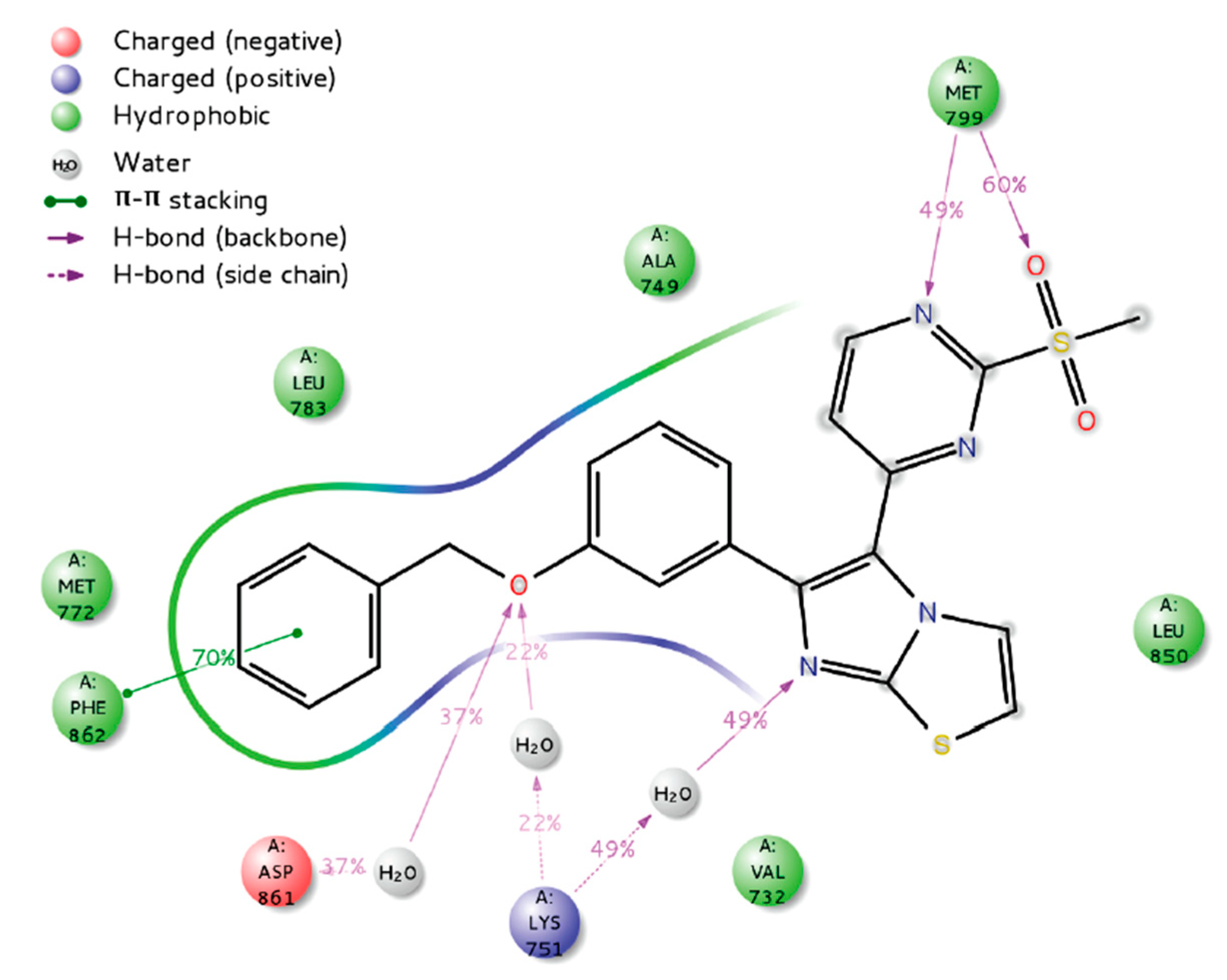
- Zaraei, S.-O.; Sbenati, R.M.; Alach, N.N.; Anbar, H.S.; El-Gamal, R.; Tarazi, H.; Shehata, M.K.; Abdel-Maksoud, M.S.; Oh, C.-H.; El-Gamal, M.I. Discovery of first-in-class imidazothiazole-based potent and selective ErbB4 (HER4) kinase inhibitors. Eur. J. Med. Chem. 2021, 224, 113674. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Type of Inhibitor | IC50 against HER4 Kinase | Other Biological Activity | Status of Clinical Trials | Company |
|---|---|---|---|---|---|---|
| Allitinib |  | Irreversible | 0.8 nM | In vitro:
| Active but not recruiting: One clinical study to analyze the efficacy and safety of anlotinib in combination with allitinib in the treatment of lung cancer. | Investigational |
| Poziotinib |  | Irreversible | 23.5 nM | In vitro:
| Active but not recruiting:
3 studies | Hanmi Pharmaceutical, South Korea |
| Dacomitinib |  | Irreversible | 73.7 nM | In vitro:
| Completed:
| Pfizer |
| Lapatinib |  | Reversible | 3.67 nM | In vitro:
The growth of BT474 and HN5 xenografts is significantly inhibited by oral administration of Lapatinib (100 mg/kg) twice daily in a dose-dependent manner. | Terminated Study as protocol would not be able to approach stated accrual. | GlaxoSmithKline |
| Afatinib |  | Irreversible selective inhibitor | 1 nM | In vitro:
| Completed:
| Boehringer Ingelheim |
| Canertinib |  | Irreversible | 7 nM | In vitro: Canertinib alone suppresses constitutively activated Akt and MAP kinase. In vivo: At 5 mg/kg body weight, canertinib displays remarkable activity against A431 xenografts in nude mice. | Unknown Canertinib has had poor clinical outcomes, and the presence of side effects such as diarrhea and rash in advanced NSCLC patients has restricted its clinical applications. | Pfizer, development discontinued |
| Neratinib |  | Irreversible | 19 nM | In vitro:
| Recruited Phase I trial that focuses on the side effects and best dose of Neratinib in combination with everolimus, palbociclib, or trametinib in patients who have solid tumors with EGFR mutations/amplification, HER2 mutations/amplification, HER3/4 mutations, or KRAS mutations that have spread to other parts of the body and are refractory to treatment (advanced or metastatic). Neratinib, palbociclib, and trametinib can inhibit tumor cell growth by inhibiting certain enzymes required for cell growth. | Wyeth & Pfizer |
| Pyrotinib |  | Irreversible | unknown | In vitro: Pyrotinib, a dual tyrosine kinase inhibitor for EGFR and HER2, has excellent in vitro potency, selectivity, and PK profiles. In vivo: Pyrotinib has shown to have potent anti-tumor effects in HER2-overexpressing xenograft models, as well as adequate safety windows in animals and beneficial pharmacokinetic properties in humans. | Recruited: Clinical study of Pyrotinib plus Vinorelbine as the therapy of brain metastases from HER2-positive metastatic breast cancer. Running: Pyrotinib, on the other hand, is being examined in another study, which is divided into two sections. Investigators will assess the protection and tolerability of pyrotinib Plus Capecitabine combined with brain radiotherapy in phase Ib trial. | Shanghai Hengrui Pharmaceutical |
| Ibrutinib |  | Irreversible | Unknown | In vitro:
Tumor volumes in ibrutinib-responsive mouse xenograft tumors were reduced with ibrutinib therapy. | Active, not recruiting: A phase I/II study of ibrutinib in previously treated EGFR mutant NSCLC. | Janssen |
| AC-480 |  | Reversible | 190 nM | In vitro:
| Completed:
| Bristol Myers Squibb |
| Compound I |  | Reversible | 15.24 nM |
| - | Investigational |
| Compound II |  | Reversible | 17.70 nM |
| - | Investigational |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Gamal, M.I.; Mewafi, N.H.; Abdelmotteleb, N.E.; Emara, M.A.; Tarazi, H.; Sbenati, R.M.; Madkour, M.M.; Zaraei, S.-O.; Shahin, A.I.; Anbar, H.S. A Review of HER4 (ErbB4) Kinase, Its Impact on Cancer, and Its Inhibitors. Molecules 2021, 26, 7376. https://doi.org/10.3390/molecules26237376
El-Gamal MI, Mewafi NH, Abdelmotteleb NE, Emara MA, Tarazi H, Sbenati RM, Madkour MM, Zaraei S-O, Shahin AI, Anbar HS. A Review of HER4 (ErbB4) Kinase, Its Impact on Cancer, and Its Inhibitors. Molecules. 2021; 26(23):7376. https://doi.org/10.3390/molecules26237376
Chicago/Turabian StyleEl-Gamal, Mohammed I., Nada H. Mewafi, Nada E. Abdelmotteleb, Minnatullah A. Emara, Hamadeh Tarazi, Rawan M. Sbenati, Moustafa M. Madkour, Seyed-Omar Zaraei, Afnan I. Shahin, and Hanan S. Anbar. 2021. "A Review of HER4 (ErbB4) Kinase, Its Impact on Cancer, and Its Inhibitors" Molecules 26, no. 23: 7376. https://doi.org/10.3390/molecules26237376
APA StyleEl-Gamal, M. I., Mewafi, N. H., Abdelmotteleb, N. E., Emara, M. A., Tarazi, H., Sbenati, R. M., Madkour, M. M., Zaraei, S.-O., Shahin, A. I., & Anbar, H. S. (2021). A Review of HER4 (ErbB4) Kinase, Its Impact on Cancer, and Its Inhibitors. Molecules, 26(23), 7376. https://doi.org/10.3390/molecules26237376