
Optimization of 4-Anilinoquinolines as Dengue Virus Inhibitors


Abstract
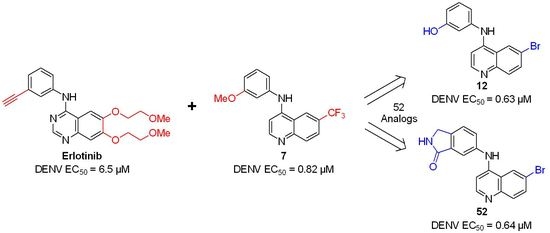
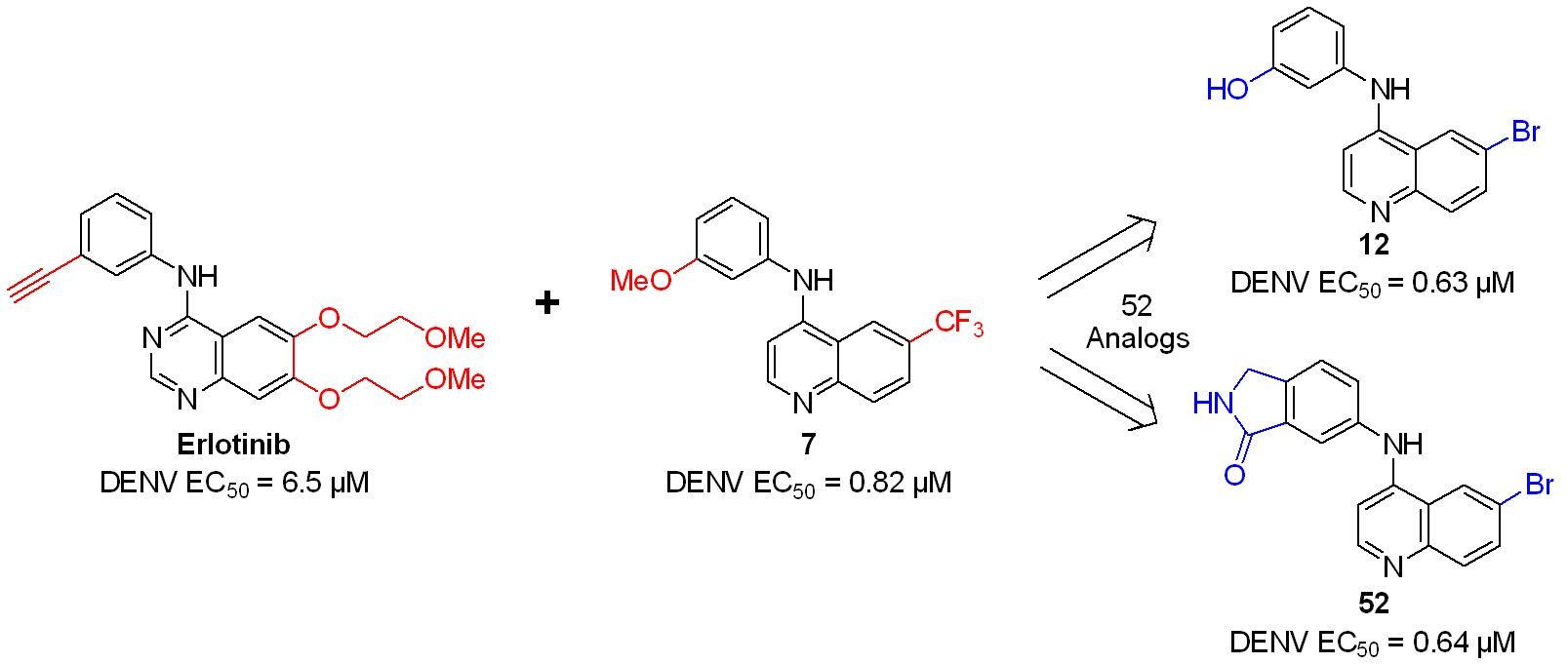
:
1. Introduction
2. Results
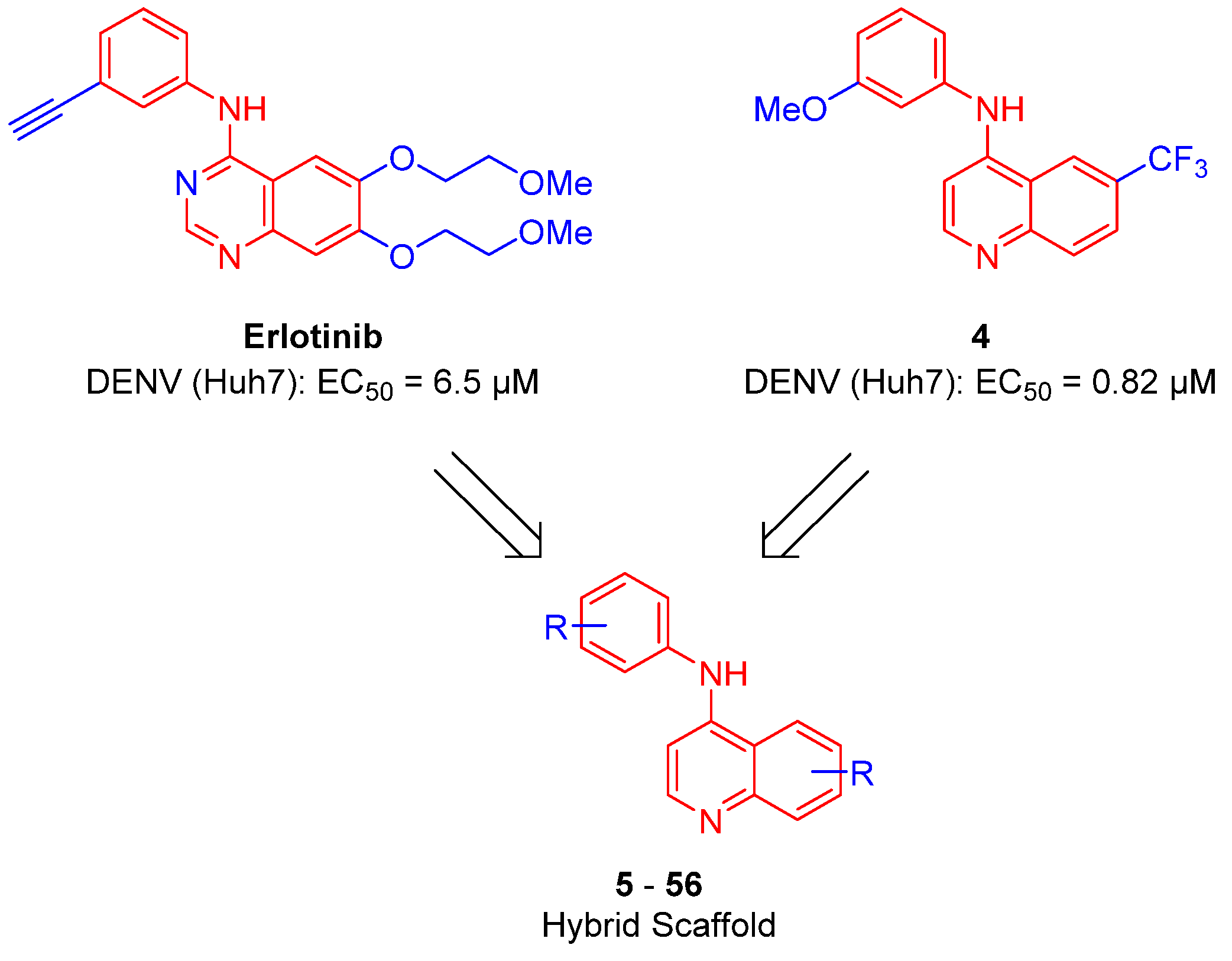
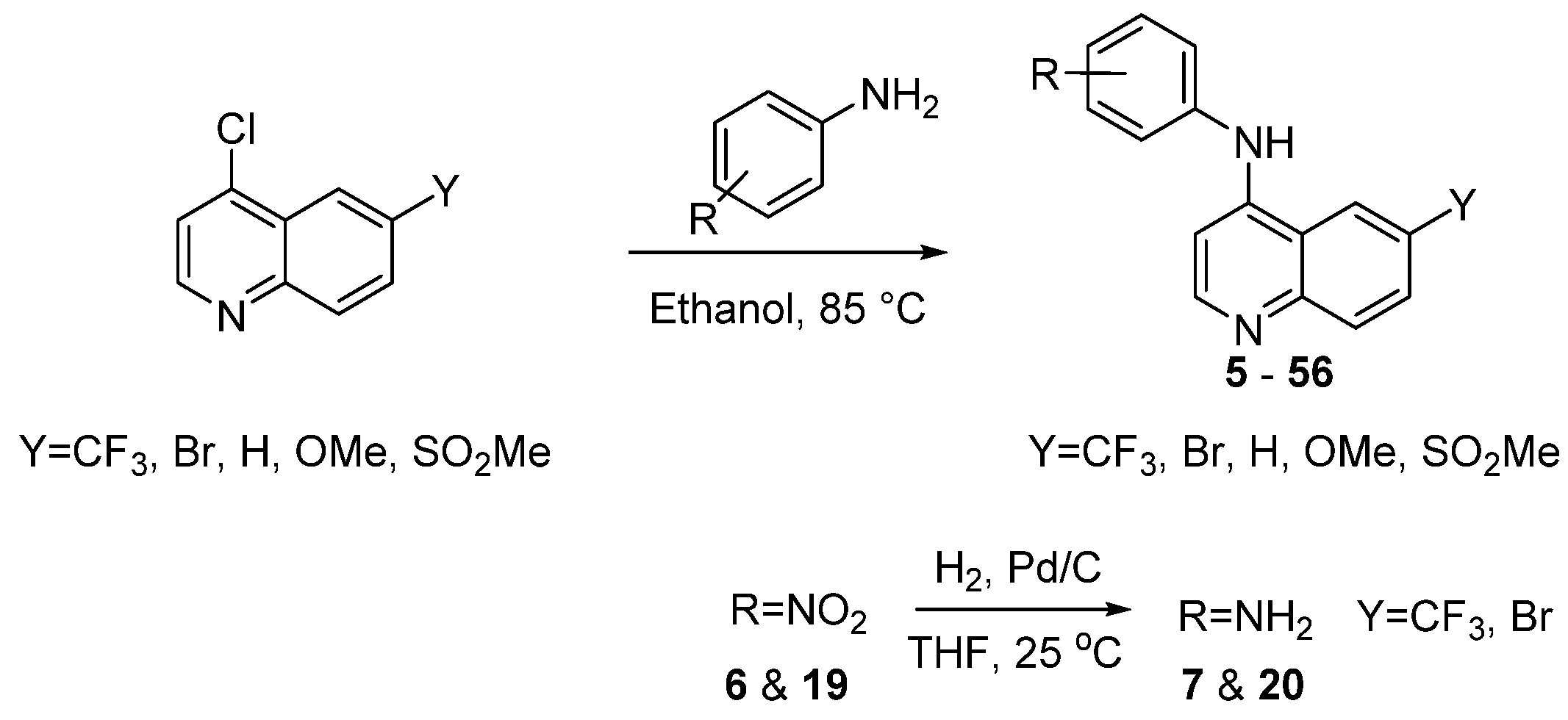
2.1. Synthesis
2.2. Antiviral Screening
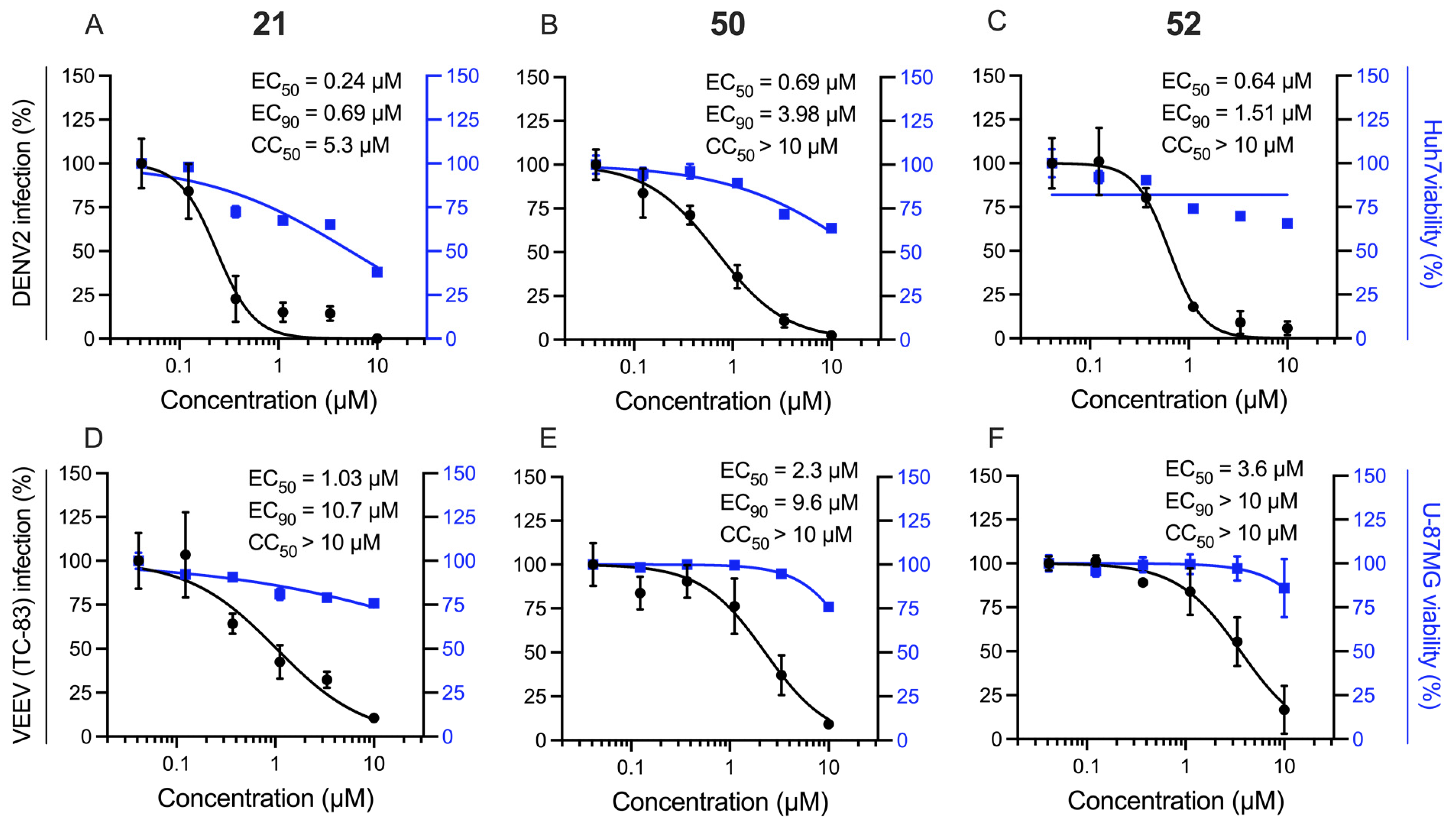
2.3. Extension Screening of Lead Compounds
3. Discussion
4. Materials and Methods
4.1. Chemistry Method
General Procedure for the Synthesis of 4-Anilinoquin(az)olines
4.2. Antiviral Screening
4.2.1. Virus Construct
4.2.2. Cells
4.2.3. Virus Production
4.2.4. Infection Assays
4.2.5. Viability Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Paixão, E.S.; Teixeira, M.G.; Rodrigues, L.C. Zika, chikungunya and dengue: The causes and threats of new and re-emerging arboviral diseases. BMJ Glob. Health 2017, 3, e000530. [Google Scholar] [CrossRef] [Green Version]
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Braack, L.; de Almeida, A.P.G.; Cornel, A.J.; Swanepoel, R.; de Jager, C. Mosquito-borne arboviruses of African origin: Review of key viruses and vectors. Parasit Vectors 2018, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.M.; Santos, N.C.; Martins, I.C. Dengue and Zika Viruses: Epidemiological History, Potential Therapies, and Promising Vaccines. Trop. Med. Infect. Dis. 2020, 5, 150. [Google Scholar] [CrossRef]
- Campbell, L.P.; Luther, C.; Moo-Llanes, D.; Ramsey, J.M.; Danis-Lozano, R.; Peterson, A.T. Climate change influences on global distributions of dengue and chikungunya virus vectors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140135. [Google Scholar] [CrossRef]
- Murray, N.E.; Quam, M.B.; Wilder-Smith, A. Epidemiology of dengue: Past, present and future prospects. Clin. Epidemiol. 2013, 5, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, M.G.; Gubler, D.J.; Izquierdo, A.; Martinez, E.; Halstead, S.B. Dengue infection. Nat. Rev. Dis. Primers 2016, 2, 16055. [Google Scholar] [CrossRef] [PubMed]
- Flipse, J.; Smit, J.M. The Complexity of a Dengue Vaccine: A Review of the Human Antibody Response. PLoS Negl. Trop. Dis. 2015, 9, e0003749. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Meng, Y.; Halloran, M.E.; Longini, I.M., Jr. Dependency of Vaccine Efficacy on Preexposure and Age: A Closer Look at a Tetravalent Dengue Vaccine. Clin. Infect. Dis. 2018, 66, 178–184. [Google Scholar] [CrossRef]
- Sharma, A.; Knollmann-Ritschel, B. Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development. Viruses 2019, 11, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán-Terán, C.; Calderón-Rangel, A.; Rodriguez-Morales, A.; Mattar, S. Venezuelan equine encephalitis virus: The problem is not over for tropical America. Ann. Clin. Microbiol Antimicrob. 2020, 19, 19. [Google Scholar] [CrossRef]
- Hawley, R.J.; Eitzen, E.M., Jr. Biological weapons—A primer for microbiologists. Annu. Rev. Microbiol. 2001, 55, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Stevens, A.J.; Gahan, M.E.; Mahalingam, S.; Keller, P.A. The medicinal chemistry of dengue fever. J. Med. Chem. 2009, 52, 7911–7926. [Google Scholar] [CrossRef] [PubMed]
- Bekerman, E.; Neveu, G.; Shulla, A.; Brannan, J.; Pu, S.Y.; Wang, S.; Xiao, F.; Barouch-Bentov, R.; Bakken, R.R.; Mateo, R.; et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J. Clin. Investig. 2017, 127, 1338. [Google Scholar] [CrossRef] [PubMed]
- Pu, S.Y.; Wouters, R.; Schor, S.; Rozenski, J.; Barouch-Bentov, R.; Prugar, L.I.; O’Brien, C.M.; Brannan, J.M.; Dye, J.M.; Herdewijn, P.; et al. Optimization of Isothiazolo[4,3-b]pyridine-Based Inhibitors of Cyclin G Associated Kinase (GAK) with Broad-Spectrum Antiviral Activity. J. Med. Chem. 2018, 61, 6178. [Google Scholar] [CrossRef]
- Verdonck, S.; Pu, S.Y.; Sorrell, F.J.; Elkins, J.M.; Froeyen, M.; Gao, L.J.; Prugar, L.I.; Dorosky, D.E.; Brannan, J.M.; Barouch-Bentov, R.; et al. Synthesis and Structure-Activity Relationships of 3,5-Disubstituted-pyrrolo[2,3-b]pyridines as Inhibitors of Adaptor-Associated Kinase 1 with Antiviral Activity. J. Med. Chem. 2019, 62, 5810. [Google Scholar] [CrossRef]
- Nitsche, C.; Steuer, C.; Klein, C.D. Arylcyanoacrylamides as inhibitors of the Dengue and West Nile virus proteases. Bioorg. Med. Chem. 2011, 19, 7318. [Google Scholar] [CrossRef]
- Yang, C.C.; Hu, H.S.; Wu, R.H.; Wu, S.H.; Lee, S.J.; Jiaang, W.T.; Chern, J.H.; Huang, Z.S.; Wu, H.N.; Chang, C.M.; et al. A novel dengue virus inhibitor, BP13944, discovered by high-throughput screening with dengue virus replicon cells selects for resistance in the viral NS2B/NS3 protease. Antimicrob. Agents Chemother. 2014, 58, 110. [Google Scholar] [CrossRef] [Green Version]
- Saudi, M.; Zmurko, J.; Kaptein, S.; Rozenski, J.; Neyts, J.; Van Aerschot, A. Synthesis and evaluation of imidazole-4,5- and pyrazine-2,3-dicarboxamides targeting dengue and yellow fever virus. Eur. J. Med. Chem. 2014, 87, 529. [Google Scholar] [CrossRef] [Green Version]
- Behnam, M.A.M.; Graf, D.; Bartenschlager, R.; Zlotos, D.P.; Klein, C.D. Discovery of Nanomolar Dengue and West Nile Virus Protease Inhibitors Containing a 4-Benzyloxyphenylglycine Residue. J. Med. Chem. 2015, 58, 9354. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, F.; Nilar, S.; Noble, C.G.; Lim, S.P.; Rao, R.; Tania, S.; Wang, G.; Lee, G.; Hunziker, J.; Karuna, R.; et al. Discovery of Potent Non-Nucleoside Inhibitors of Dengue Viral RNA-Dependent RNA Polymerase from a Fragment Hit Using Structure-Based Drug Design. J. Med. Chem. 2016, 59, 3935. [Google Scholar] [CrossRef] [PubMed]
- Bardiot, D.; Koukni, M.; Smets, W.; Carlens, G.; McNaughton, M.; Kaptein, S.; Dallmeier, K.; Chaltin, P.; Neyts, J.; Marchand, A. Discovery of Indole Derivatives as Novel and Potent Dengue Virus Inhibitors. J. Med. Chem. 2018, 61, 8390. [Google Scholar] [CrossRef] [PubMed]
- Millies, B.; von Hammerstein, F.; Gellert, A.; Hammerschmidt, S.; Barthels, F.; Göppel, U.; Immerheiser, M.; Elgner, F.; Jung, N.; Basic, M.; et al. Proline-Based Allosteric Inhibitors of Zika and Dengue Virus NS2B/NS3 Proteases. J. Med. Chem. 2019, 62, 11359. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xie, X.; Chen, H.; Zou, J.; Xue, Y.; Ye, N.; Shi, P.Y.; Zhou, J. Design, synthesis and biological evaluation of spiropyrazolopyridone derivatives as potent dengue virus inhibitors. Bioorg. Med. Chem Lett. 2020, 30, 127162. [Google Scholar] [CrossRef]
- Chen, W.C.; Simanjuntak, Y.; Chu, L.W.; Ping, Y.H.; Lee, Y.L.; Lin, Y.L.; Li, W.S. Benzenesulfonamide Derivatives as Calcium/Calmodulin-Dependent Protein Kinase Inhibitors and Antiviral Agents against Dengue and Zika Virus Infections. J. Med. Chem. 2020, 63, 1313. [Google Scholar] [CrossRef]
- Venkatesham, A.; Saudi, M.; Kaptein, S.; Neyts, J.; Rozenski, J.; Froeyen, M.; Van Aerschot, A. Aminopurine and aminoquinazoline scaffolds for development of potential dengue virus inhibitors. Eur. J. Med. Chem. 2017, 126, 101. [Google Scholar] [CrossRef]
- Yang, Y.; Cao, L.; Gao, H.; Wu, Y.; Wang, Y.; Fang, F.; Lan, T.; Lou, Z.; Rao, Y. Discovery, Optimization, and Target Identification of Novel Potent Broad-Spectrum Antiviral Inhibitors. J. Med. Chem. 2019, 62, 4056. [Google Scholar] [CrossRef]
- Opsenica, I.; Burnett, J.C.; Gussio, R.; Opsenica, D.; Todorović, N.; Lanteri, C.A.; Sciotti, R.J.; Gettayacamin, M.; Basilico, N.; Taramelli, D.; et al. A chemotype that inhibits three unrelated pathogenic targets: The botulinum neurotoxin serotype A light chain, P. falciparum malaria, and the Ebola filovirus. J. Med. Chem. 2011, 54, 1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, B.; Tong, X.K.; Tang, W.; Li, D.W.; He, P.L.; Garcia, J.M.; Zeng, L.M.; Gao, A.H.; Yang, L.; Li, J.; et al. Discovery and optimization of 2,4-diaminoquinazoline derivatives as a new class of potent dengue virus inhibitors. J. Med. Chem. 2012, 55, 3135. [Google Scholar] [CrossRef] [PubMed]
- Vincetti, P.; Caporuscio, F.; Kaptein, S.; Gioiello, A.; Mancino, V.; Suzuki, Y.; Yamamoto, N.; Crespan, E.; Lossani, A.; Maga, G.; et al. Discovery of Multitarget Antivirals Acting on Both the Dengue Virus NS5-NS3 Interaction and the Host Src/Fyn Kinases. J. Med. Chem. 2015, 58, 4964. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Patel, S.J.; Vangrevelinghe, E.; Xu, H.Y.; Rao, R.; Jaber, D.; Schul, W.; Gu, F.; Heudi, O.; Ma, N.L.; et al. A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saul, S.; Pu, S.Y.; Zuercher, W.J.; Einav, S.; Asquith, C.R.M. Potent antiviral activity of novel multi-substituted 4-anilinoquin(az)olines. Bioorg. Med. Chem. Lett. 2020, 30, 127284. [Google Scholar] [CrossRef] [PubMed]
- Saul, S.; Huang, P.T.; Einav, S.; Asquith, C.R.M. Evaluation and identification of 4-anilinoquin(az)olines as potent inhibitors of both dengue virus (DENV) and Venezuelan equine encephalitis virus (VEEV). Bioorg. Med. Chem. Lett. 2021, 128407. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.A.; Biggs, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Laitinen, T.; Bennett, J.M.; Godoi, P.H.; East, M.P.; Tizzard, G.H.; Graves, L.M.; Johnson, G.L.; Dornsife, R.E.; Wells, C.I.; et al. Identification and optimization of 4-anilinoquinolines as inhibitors of cyclin G associated kinase. ChemMedChem 2018, 13, 48–66. [Google Scholar] [CrossRef] [Green Version]
- Asquith, C.R.M.; Berger, B.T.; Wan, J.; Bennett, J.M.; Capuzzi, S.J.; Crona, D.J.; Drewry, D.H.; East, M.P.; Elkins, J.M.; Fedorov, O.; et al. SGC-GAK-1: A Chemical Probe for Cyclin G Associated Kinase (GAK). J. Med. Chem. 2019, 62, 2830. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Naegeli, K.M.; East, M.P.; Laitinen, T.; Havener, T.M.; Wells, C.I.; Johnson, G.L.; Drewry, D.H.; Zuercher, W.J.; Morris, D.C. Design of a Cyclin G Associated Kinase (GAK)/Epidermal Growth Factor Receptor (EGFR) Inhibitor Set to Interrogate the Relationship of EGFR and GAK in Chordoma. J. Med. Chem. 2019, 62, 4772–4778. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Treiber, D.K.; Zuercher, W.J. Utilizing comprehensive and mini-kinome panels to optimize the selectivity of quinoline inhibitors for cyclin G associated kinase (GAK). Bioorg. Med. Chem. Lett. 2019, 29, 1727. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Bennett, J.M.; Su, L.; Laitinen, T.; Elkins, J.M.; Pickett, J.E.; Wells, C.I.; Li, Z.; Willson, T.M.; Zuercher, W.J. Towards the Development of an In vivo Chemical Probe for Cyclin G Associated Kinase (GAK). Molecules 2019, 24, 4016. [Google Scholar] [CrossRef] [Green Version]
- Asquith, C.R.M.; Fleck, N.; Torrice, C.D.; Crona, D.J.; Grundner, C.; Zuercher, W.J. Anti-tubercular activity of novel 4-anilinoquinolines and 4-anilinoquinazolines. Bioorg. Med. Chem. Lett. 2019, 29, 2695–2699. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Maffuid, K.A.; Laitinen, T.; Torrice, C.D.; Tizzard, G.J.; Crona, D.J.; Zuercher, W.J. Targeting an EGFR Water Network with 4-Anilinoquin(az)oline Inhibitors for Chordoma. ChemMedChem 2019, 14, 1693–1700. [Google Scholar] [CrossRef] [Green Version]
- Carabajal, M.A.; Asquith, C.R.M.; Laitinen, T.; Tizzard, G.J.; Yim, L.; Rial, A.; Chabalgoity, J.A.; Zuercher, W.J.; García Véscovi, E. Quinazoline-Based Antivirulence Compounds Selectively Target Salmonella PhoP/PhoQ Signal Transduction System. Antimicrob. Agents Chemother. 2019, 64, e01744-19. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Laitinen, T.; Wells, C.I.; Tizzard, G.J.; Zuercher, W.J. New Insights into 4-Anilinoquinazolines as Inhibitors of Cardiac Troponin I-Interacting Kinase (TNNi3K). Molecules 2020, 25, 1697. [Google Scholar] [CrossRef] [Green Version]
- Asquith, C.R.M.; Laitinen, T.; Bennett, J.M.; Wells, C.I.; Elkins, J.M.; Zuercher, W.J.; Tizzard, G.J.; Poso, A. Design and Analysis of the 4-Anilinoquin(az)oline Kinase Inhibition Profiles of GAK/SLK/STK10 Using Quantitative Structure-Activity Relationships. ChemMedChem 2020, 15, 26. [Google Scholar] [CrossRef] [Green Version]
- Asquith, C.R.M.; Tizzard, G.J.; Bennett, J.M.; Wells, C.I.; Elkins, J.M.; Willson, T.M.; Poso, A.; Laitinen, T. Targeting the Water Network in Cyclin G-Associated Kinase (GAK) with 4-Anilino-quin(az)oline Inhibitors. ChemMedChem 2020, 15, 1200. [Google Scholar] [CrossRef] [PubMed]
- Savoie, P.R.; Welch, J.T. Preparation and utility of organic pentafluorosulfanyl-containing compounds. Chem. Rev. 2015, 115, 1130–1190. [Google Scholar] [CrossRef] [PubMed]
- Ammerman, N.C.; Beier-Sexton, M.; Azad, A.F. Growth and maintenance of Vero cell lines. Curr. Protoc. Microbiol. 2008, 11, A-4E. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, L. Non-kinase targets of protein kinase inhibitors. Nat. Rev. Drug Discov. 2017, 16, 424–440. [Google Scholar] [CrossRef]
- Haese, N.N.; May, N.A.; Taft-Benz, S.; Moukha-Chafiq, O.; Madadi, N.; Zhang, S.; Karyakarte, S.D.; Rodzinak, K.J.; Nguyen, T.H.; Denton, M.; et al. Identification of Quinolinones as Antivirals against Venezuelan Equine Encephalitis Virus. Antimicrob Agents Chemother. 2021, 65, e0024421. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.H.; Jonsson, C.B.; Tower, N.A.; Chu, Y.K.; Sahin, E.; Golden, J.E.; Noah, J.W.; Schroeder, C.E.; Sotsky, J.B.; Sosa, M.I.; et al. Discovery of a novel compound with anti-venezuelan equine encephalitis virus activity that targets the nonstructural protein 2. PLoS Pathog. 2014, 10, e1004213. [Google Scholar] [CrossRef] [PubMed]
- Persoons, L.; Vanderlinden, E.; Vangeel, L.; Wang, X.; Do, N.D.T.; Foo, S.C.; Leyssen, P.; Neyts, J.; Jochmans, D.; Schols, D.; et al. Broad spectrum anti-coronavirus activity of a series of anti-malaria quinoline analogues. Antiviral Res. 2021, 193, 105127. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Gayen, S.; Kang, C.; Yuan, Z.; Shi, P.-Y. Membrane topology and function of dengue virus NS2A protein. J. Virol. 2013, 87, 4609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, G.; Xu, H.Y.; Qing, M.; Wang, Q.Y.; Shi, P.Y. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antiviral Res. 2011, 91, 11. [Google Scholar] [CrossRef]
- Sun, C.; Gardner, C.L.; Watson, A.M.; Ryman, K.D.; Klimstra, W.B. Stable, high-level expression of reporter proteins from improved alphavirus expression vectors to track replication and dissemination during encephalitic and arthritogenic disease. J. Virol. 2014, 88, 2035. [Google Scholar] [CrossRef] [Green Version]
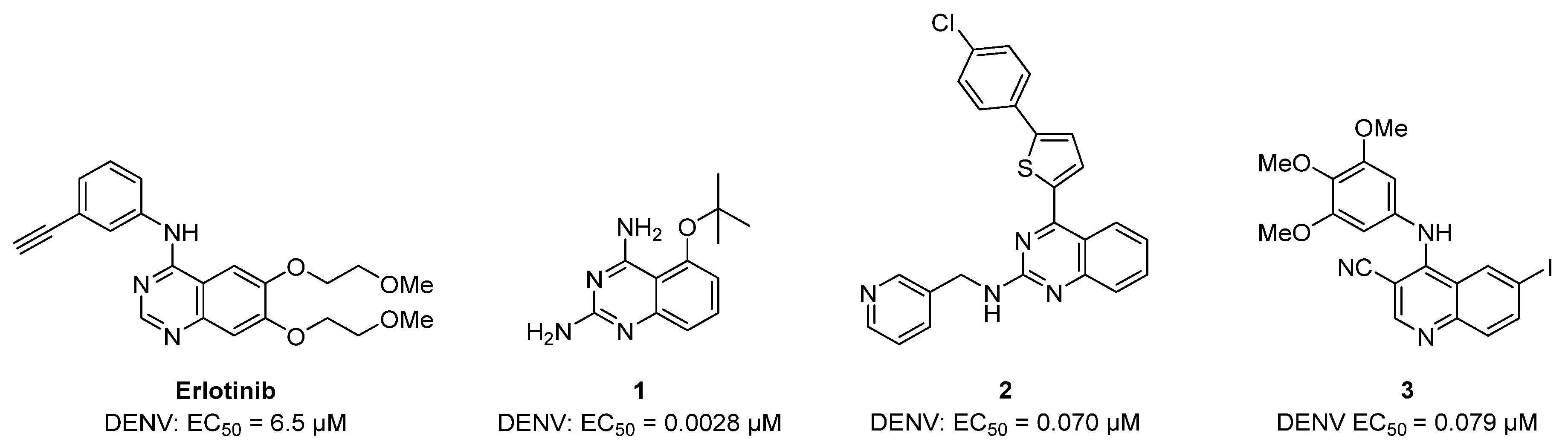




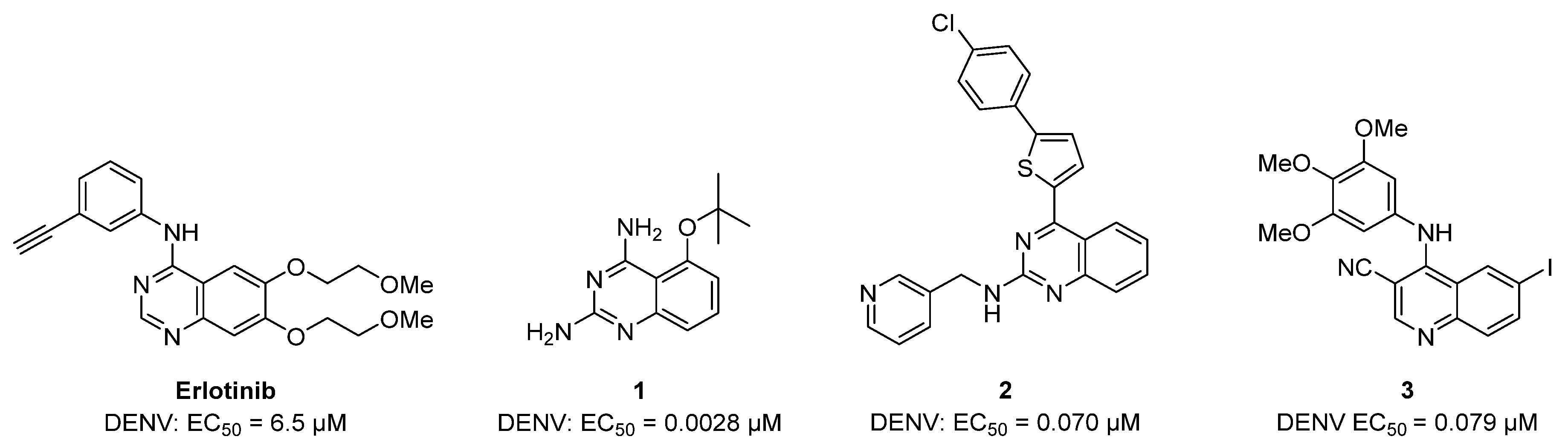
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cmpd | R | DENV Inhibition | Cell Viability |
|---|---|---|---|
| EC50 a | CC50 b | ||
| 4 | OMe | 0.82 | >10 |
| 5 | OH | 2.7 | >10 |
| 6 | NO2 | >10 | >10 |
| 7 | NH2 | 5.9 | >10 |
| 8 | NHMe | 8.9 | >10 |
| 9 | NMe2 | 2.3 | >10 |
| 10 | CH2OH | 6.6 | >10 |
| Cmpd | R1 | R2 | DENV Inhibition | Cell Viability |
|---|---|---|---|---|
| EC50 a | CC50 b | |||
| 4 | OMe | CF3 | 0.82 | >10 |
| 5 | OH | CF3 | 2.7 | >10 |
| 11 | OMe | Br | 0.84 | >10 |
| 12 | OH | Br | 0.63 | >10 |
| 13 | OMe | H | 2.5 | >10 |
| 14 | OH | H | >10 | >10 |
| 15 | OMe | OMe | 4.4 | >10 |
| 16 | OH | OMe | >10 | >10 |
| 17 | OMe | SO2Me | >10 | >10 |
| 18 | OH | SO2Me | >10 | >10 |
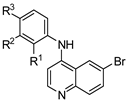
| Cmpd | R1 | R2 | R3 | DENV Inhibition | Cell Viability |
|---|---|---|---|---|---|
| EC50 a | CC50 b | ||||
| 12 | H | OH | H | 0.63 | >10 |
| 19 | H | NO2 | H | >10 | >10 |
| 20 | H | NH2 | H | >10 | >10 |
| 21 | H | NHMe | H | 0.24 | 5.3 |
| 22 | H | NMe2 | H | >10 | >10 |
| 23 | H | CH2OH | H | 3.7 | >10 |
| 24 | CH2OH | H | H | 7.1 | >10 |
| 25 | H | SF5 | H | 1.5 | >10 |
| 26 | H | OtBu | H | 2.3 | 9.2 |
| 27 | H | tBu | H | 0.94 | 6.0 |
| 28 | H | OCH2O | 1.7 | 8.6 | |
| 29 | H | CH2OCH2 | >10 | >10 | |
| 30 | OCH2O | H | >10 | >10 | |
| 31 | CH2OCH2 | H | >10 | >10 | |
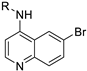 |  | ||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R | DENV Inhibition | Cell Viability | Cmpd | R | DENV Inhibition | Cell Viability |
| EC50 a | CC50 b | EC50 a | CC50 b | ||||
| 32 | 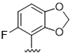 | 1.9 | >10 | 45 | 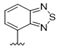 | 5.8 | >10 |
| 33 | 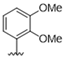 | 7.8 | >10 | 46 | 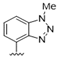 | 5.5 | >10 |
| 34 | 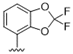 | 7.1 | >10 | 47 |  | 5.4 | >10 |
| 35 | 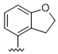 | 0.95 | >10 | 48 | 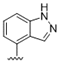 | 2.1 | >10 |
| 36 | 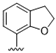 | 5.7 | >10 | 49 |  | 1.8 | >10 |
| 37 | 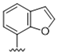 | 2.7 | >10 | 50 | 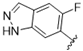 | 0.69 | >10 |
| 38 | 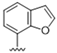 | 1.9 | >10 | 51 | 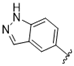 | 1.2 | >10 |
| 39 |  | >10 | >10 | 52 | 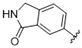 | 0.64 | >10 |
| 40 | 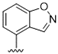 | >10 | >10 | 53 | 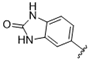 | >10 | >10 |
| 41 | 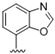 | 3.2 | >10 | 54 | 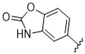 | >10 | >10 |
| 42 |  | >10 | >10 | 55 | 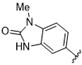 | 7.1 | >10 |
| 43 | 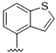 | 2.9 | >10 | 56 | 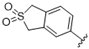 | 6.3 | >10 |
| 44 | 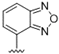 | 9.2 | >10 | - | - | - | - |
| Cmpd | DENV Inhibition a | VEEV Inhibition b | Vero Cells c | ||||
|---|---|---|---|---|---|---|---|
| EC50 | EC90 | CC50 | EC50 | EC90 | CC50 | CC50 | |
| 4 | 0.82 | 1.5 | >10 | >10 | >10 | >10 | >10 |
| 12 | 0.63 | 3.2 | >10 | 9.9 | >10 | >10 | >10 |
| 21 | 0.24 | 0.69 | 5.3 | 1.0 | >10 | >10 | >10 |
| 50 | 0.69 | 4.0 | >10 | 2.3 | 9.6 | >10 | >10 |
| 52 | 0.64 | 1.5 | >10 | 3.6 | >10 | >10 | >10 |
| Sunitinib | 1.1 | >10 | >10 | 4.4 | >10 | >10 | >10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, P.-T.; Saul, S.; Einav, S.; Asquith, C.R.M. Optimization of 4-Anilinoquinolines as Dengue Virus Inhibitors. Molecules 2021, 26, 7338. https://doi.org/10.3390/molecules26237338
Huang P-T, Saul S, Einav S, Asquith CRM. Optimization of 4-Anilinoquinolines as Dengue Virus Inhibitors. Molecules. 2021; 26(23):7338. https://doi.org/10.3390/molecules26237338
Chicago/Turabian StyleHuang, Pei-Tzu, Sirle Saul, Shirit Einav, and Christopher R. M. Asquith. 2021. "Optimization of 4-Anilinoquinolines as Dengue Virus Inhibitors" Molecules 26, no. 23: 7338. https://doi.org/10.3390/molecules26237338
APA StyleHuang, P.-T., Saul, S., Einav, S., & Asquith, C. R. M. (2021). Optimization of 4-Anilinoquinolines as Dengue Virus Inhibitors. Molecules, 26(23), 7338. https://doi.org/10.3390/molecules26237338