
Utility of Bioluminescent Homogeneous Nucleotide Detection Assays in Measuring Activities of Nucleotide-Sugar Dependent Glycosyltransferases and Studying Their Inhibitors


Abstract
1. Introduction
2. Results and Discussion
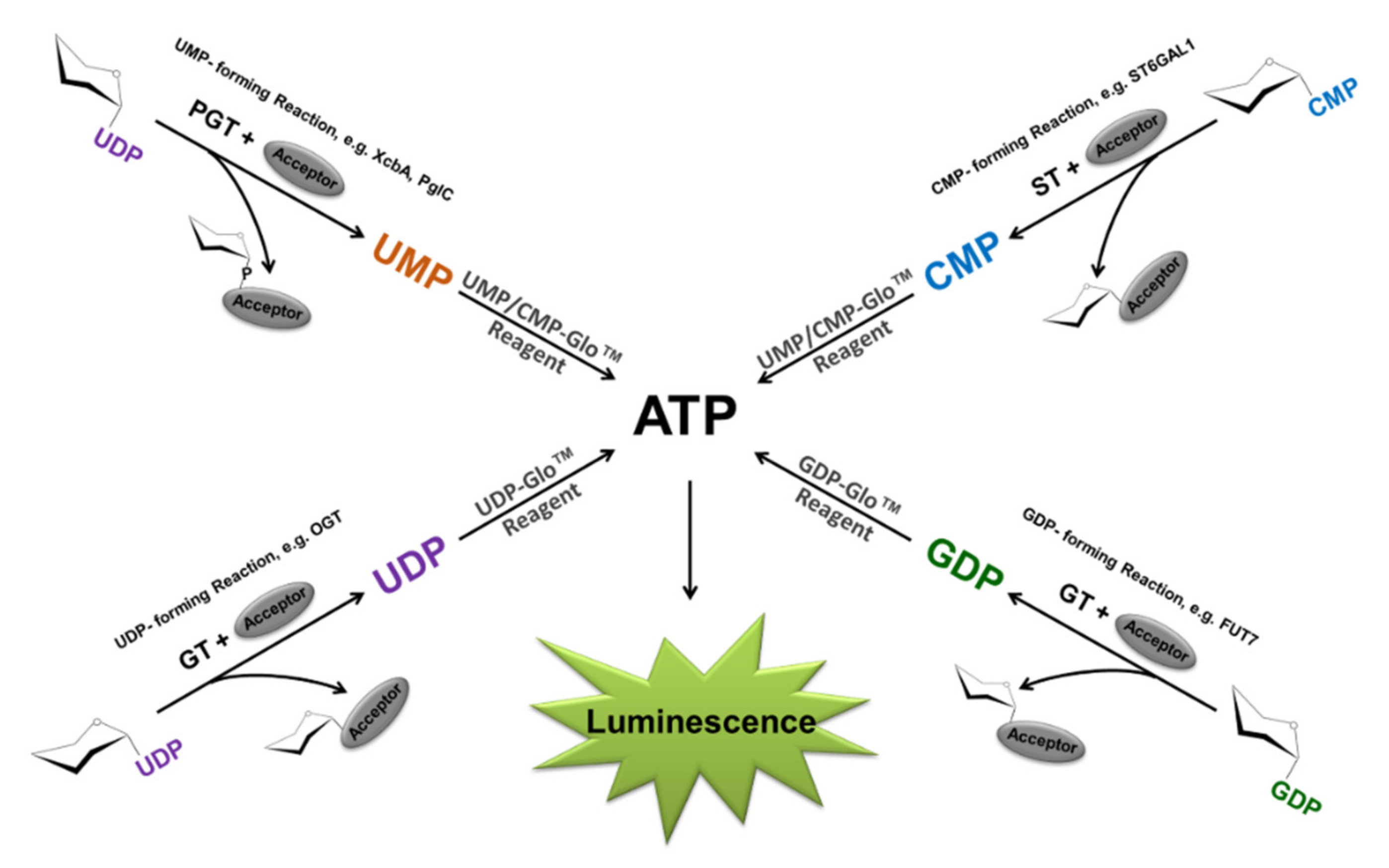
2.1. Bioluminescent Glycosyltransferase Assay Principle and Formats
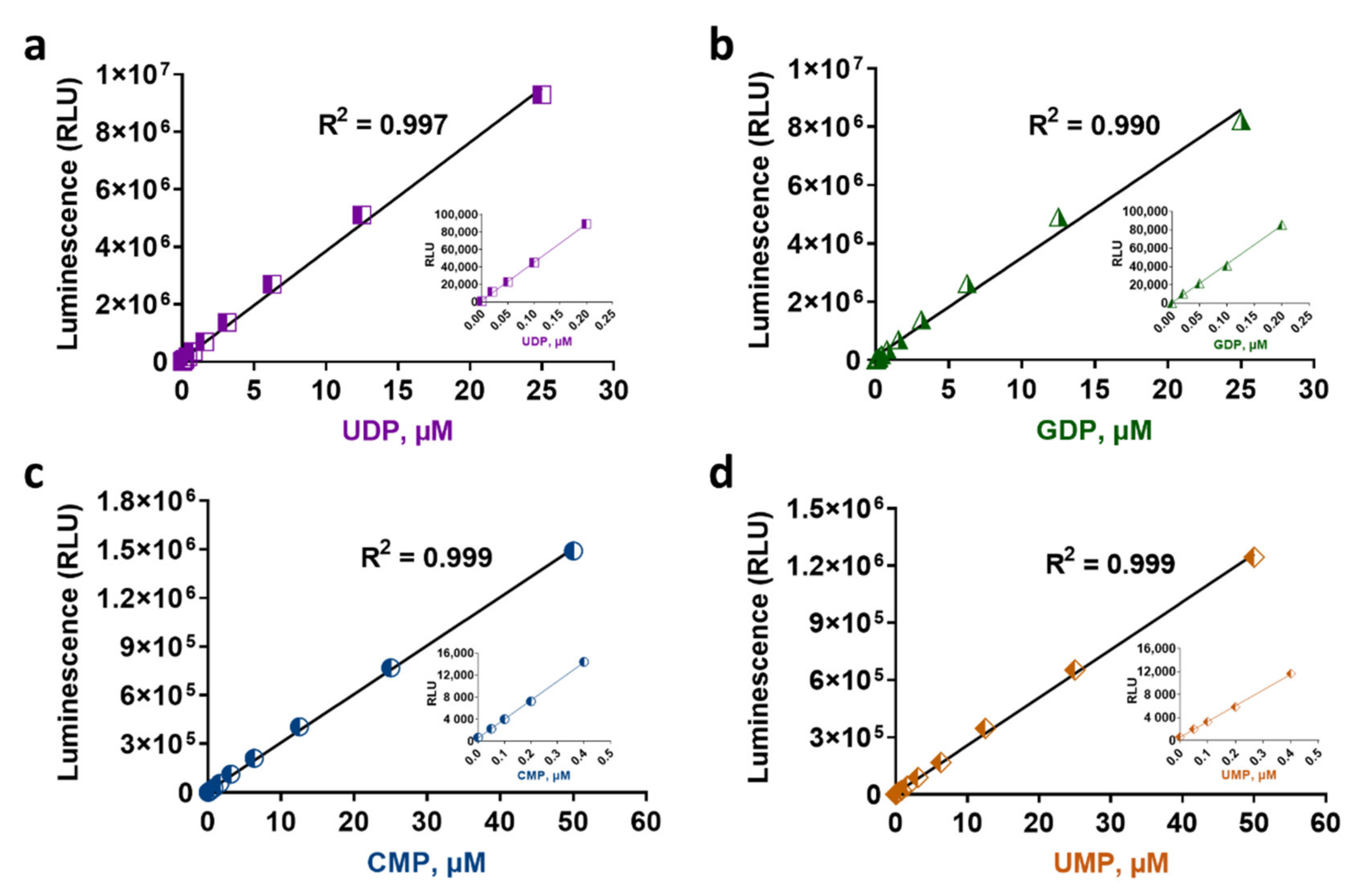
2.2. Glycosyltransferase Assays Sensitivity and Linearity
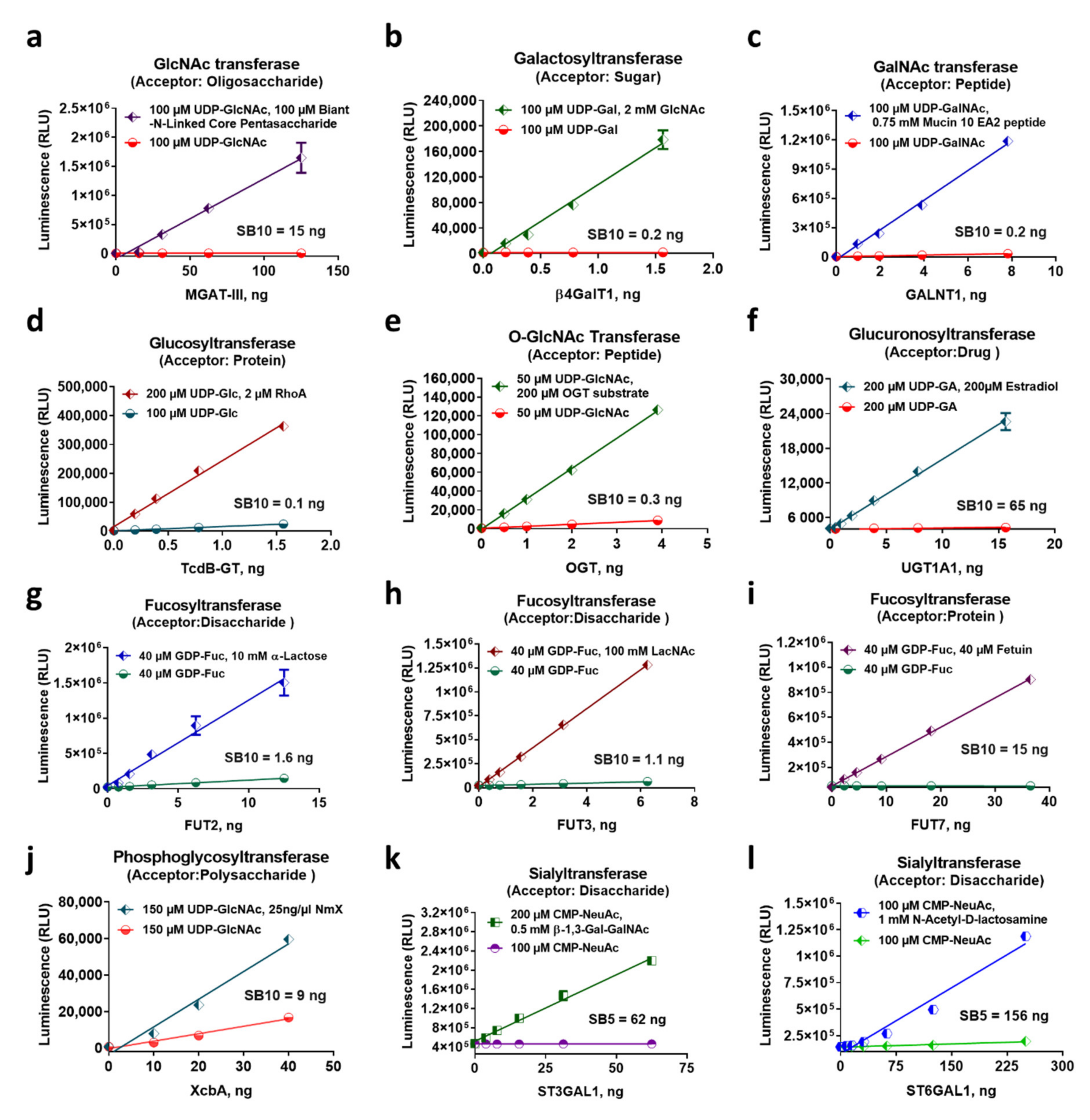
2.3. Characterization of Diverse Glycosyltransferase Activities
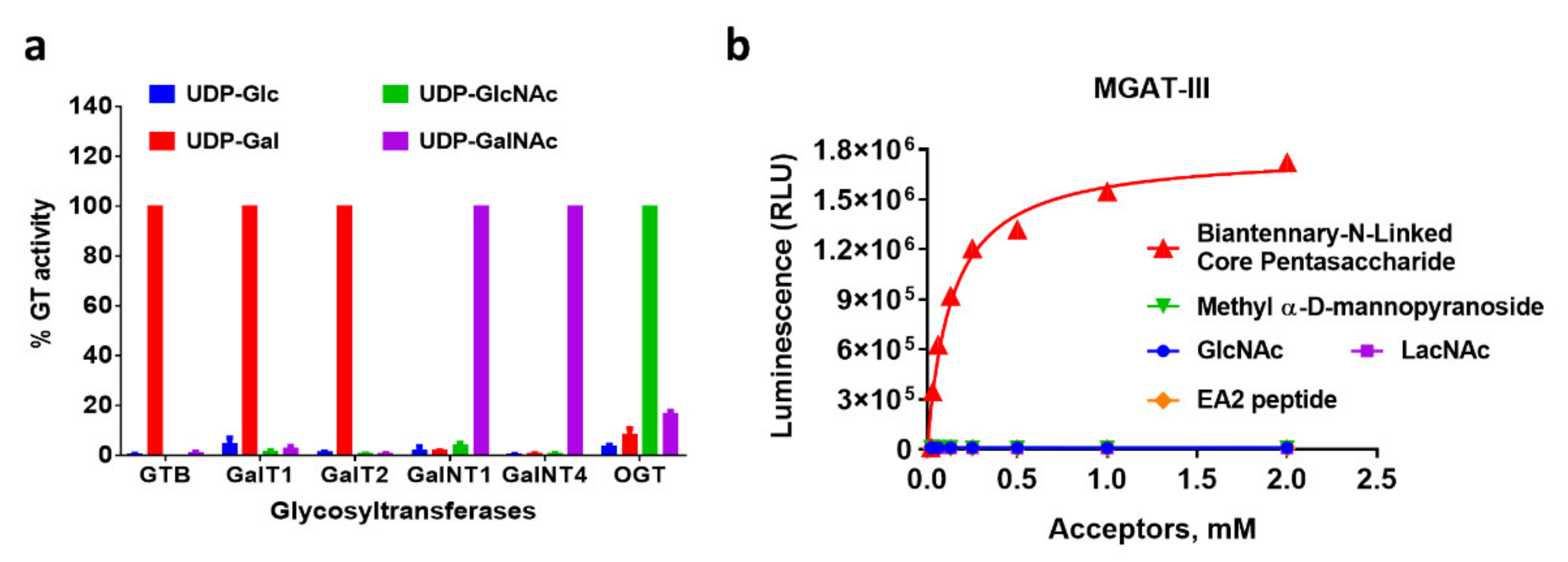
2.4. Profiling GT Substrate Selectivity with Nucleotide Detection
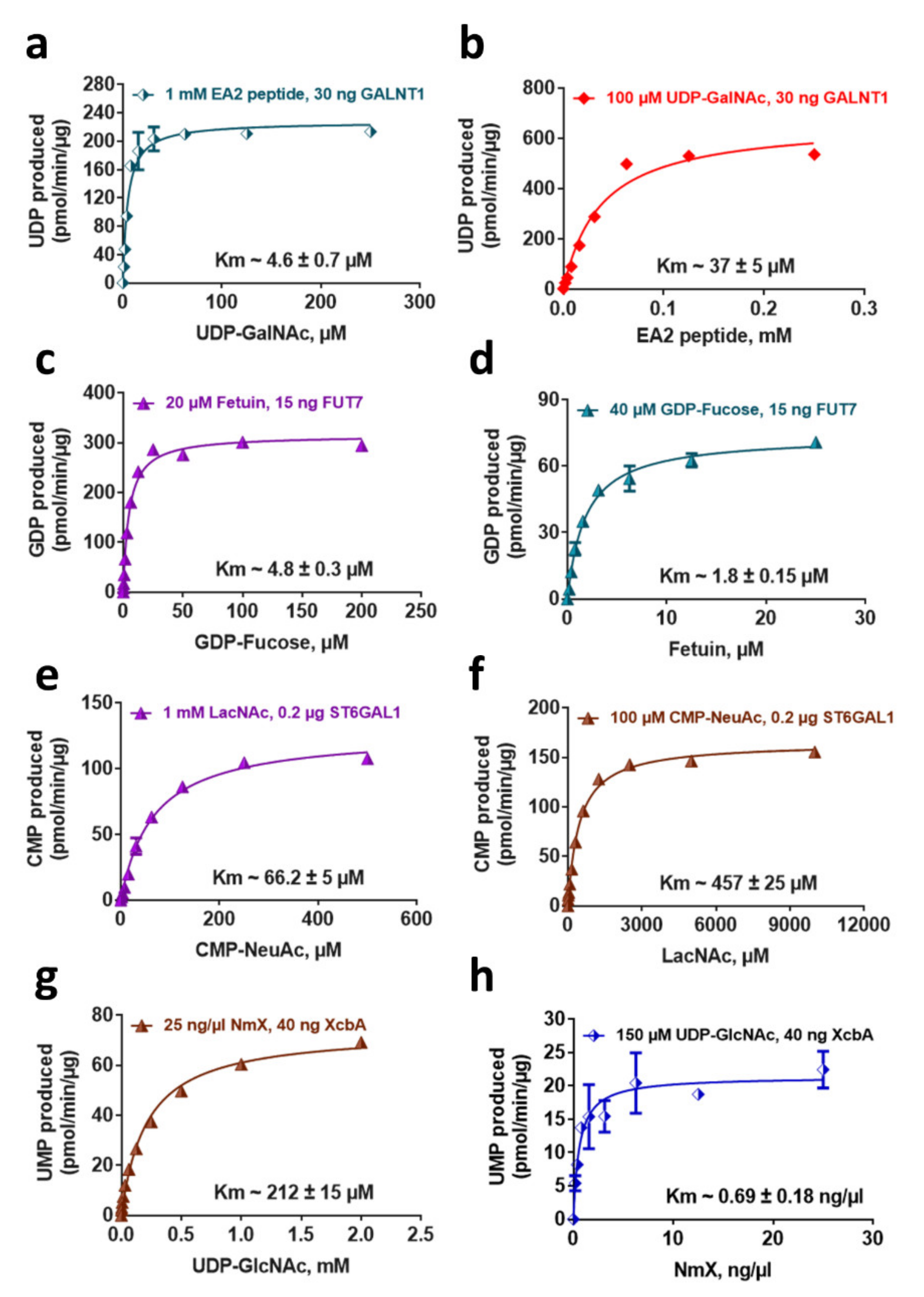
2.5. Determination of Enzyme Kinetic Parameters
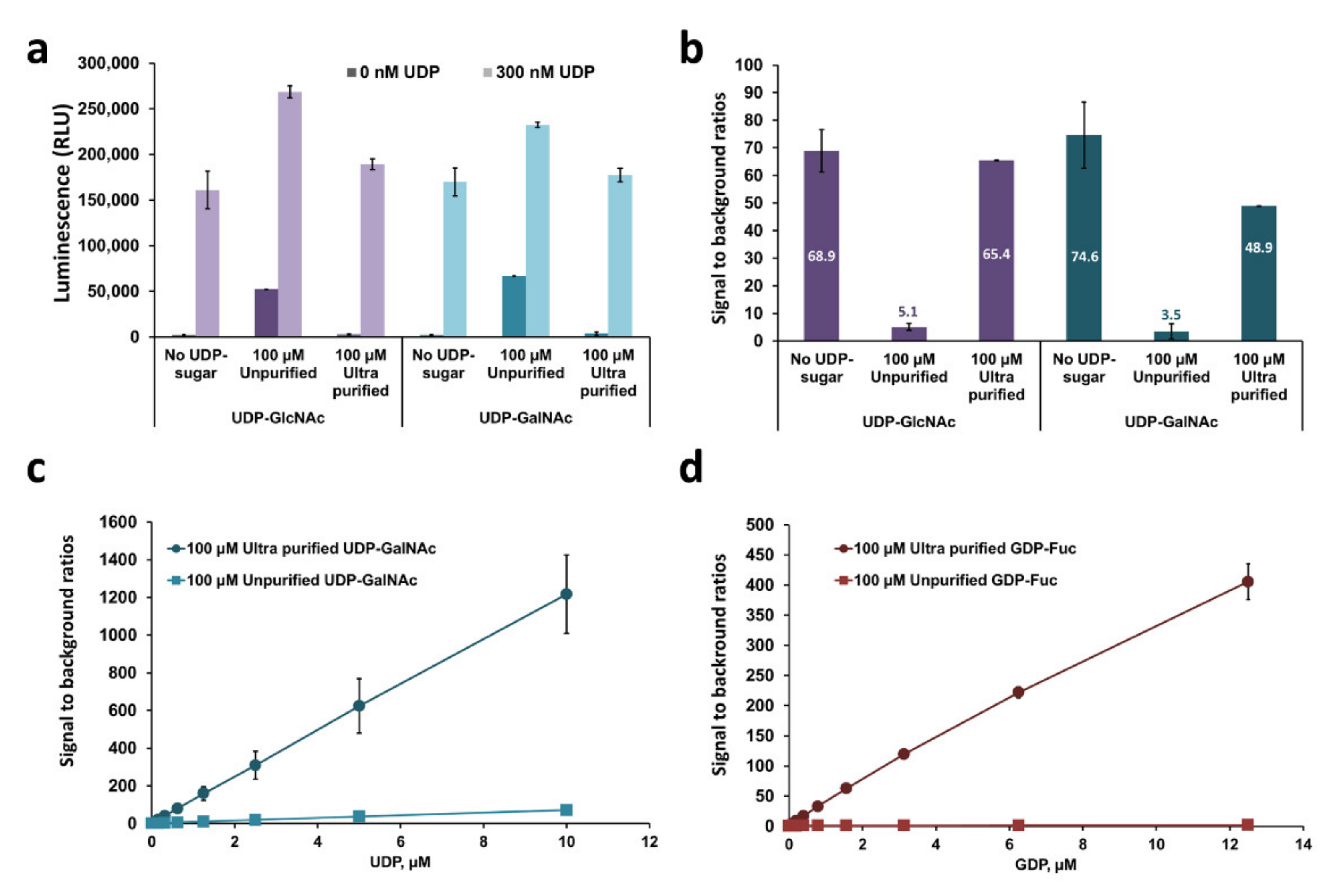
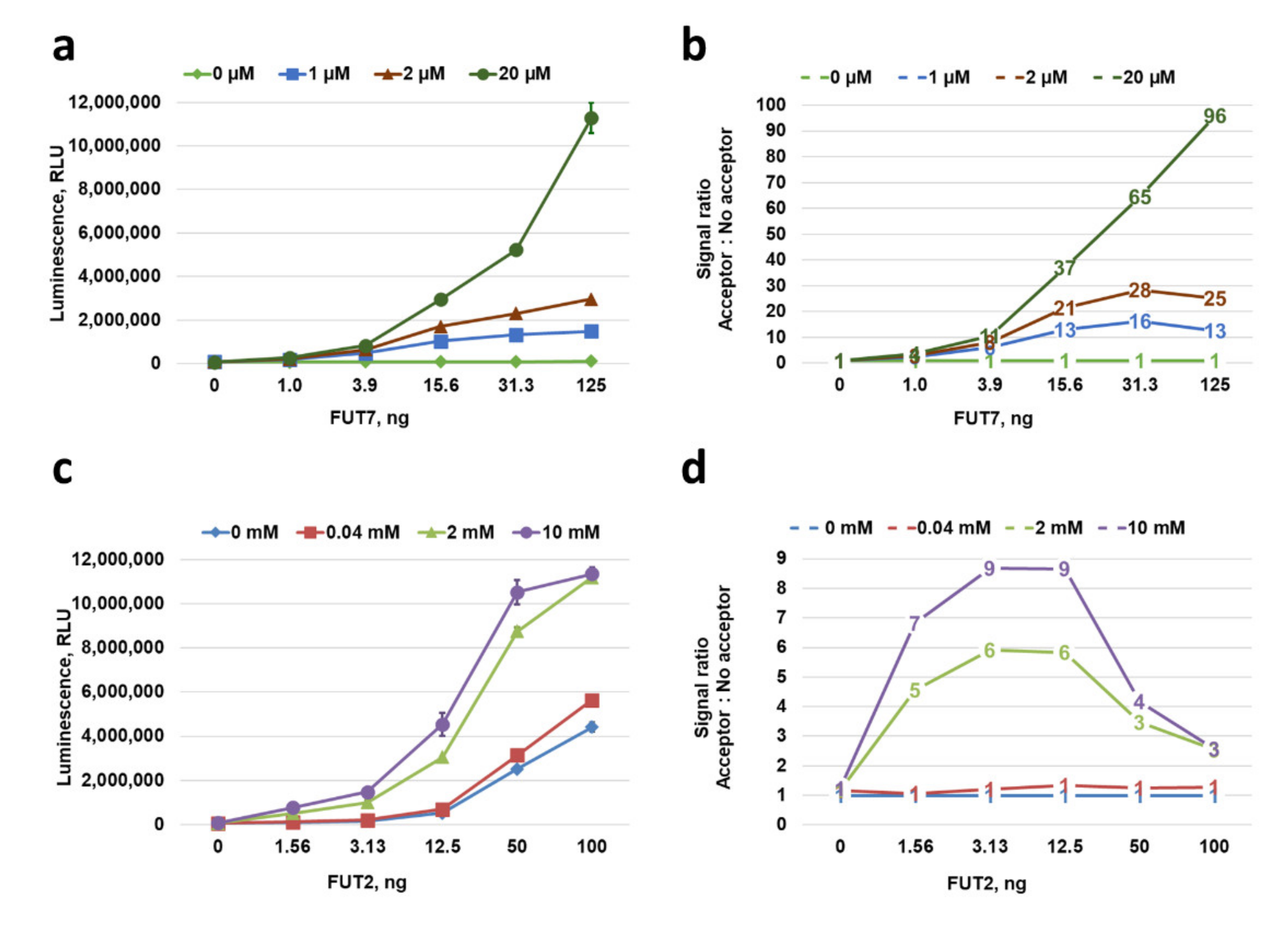
2.6. Characterization of GT Acceptor-Dependent and -Independent Nucleotide-Sugar Hydrolysis
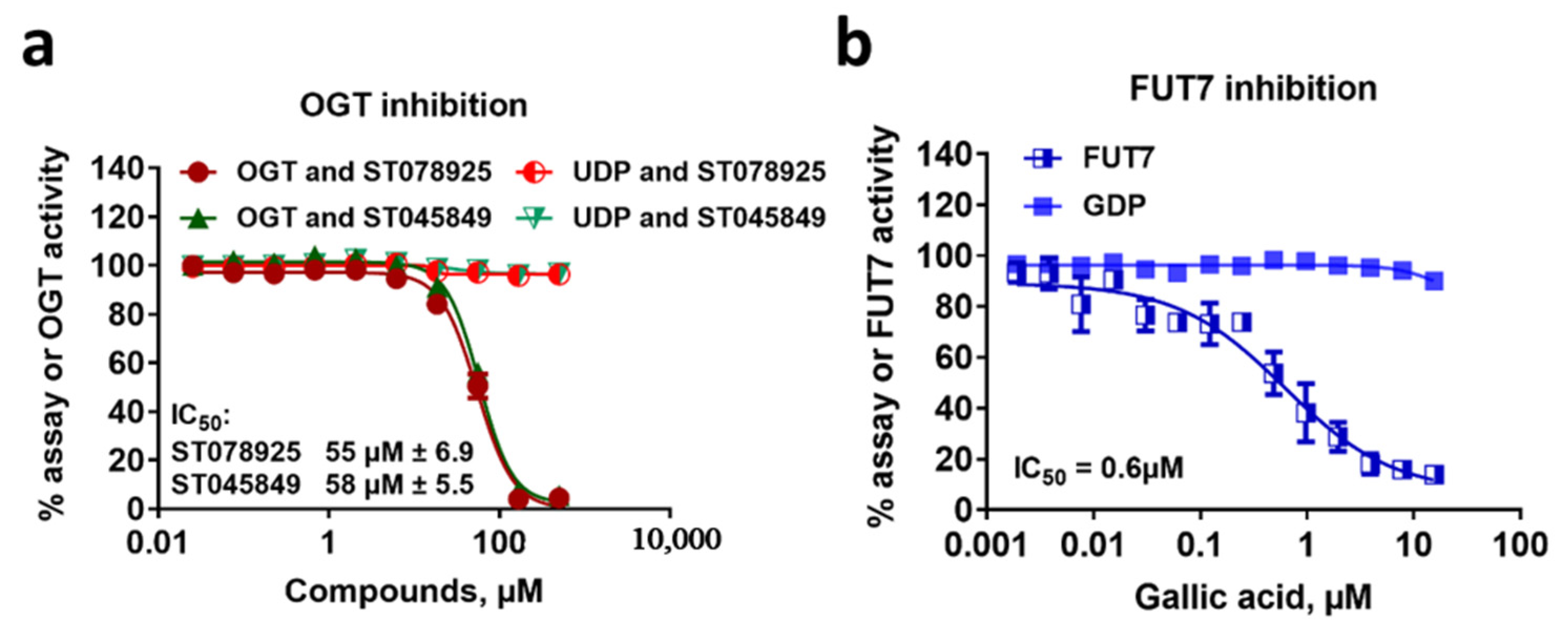
2.7. Glycosyltransferase Inhibition Assays
3. Materials and Methods
3.1. Glycosyltransferases and Substrates
3.2. Chemicals and Assay Components
3.3. Bioluminescent Nucleotide Detection Protocol
3.4. Nucleotide Standard Curves
3.5. Comparing Nucleotide-Sugars Purity
3.6. Glycosyltransferase Assay Conditions
3.7. Donor and Acceptor Substrate Specificity Studies
3.8. Substrate Km Determinations
3.9. Detection of Acceptor Substrate Dependent and Independent Enzyme Activity
3.10. OGT and FUT7 Inhibition with Chemical Compounds
3.11. Signal Detection and Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef]
- Weijers, C.A.G.M.; Franssen, M.C.R.; Visser, G.M. Glycosyltransferase-catalyzed synthesis of bioactive oligosaccharides. Biotechnol. Adv. 2008, 26, 436–456. [Google Scholar] [CrossRef]
- Kresge, N.; Simoni, R.D.; Hill, R.L.; Luis, F. Leloir and the Biosynthesis of Saccharides. J. Biol. Chem. 2005, 280, e16. [Google Scholar]
- Brown, G.D.; Gordon, S. Fungal β-Glucans and Mammalian Immunity. Immunity 2003, 19, 311–315. [Google Scholar] [CrossRef]
- Crocker, P.R.; Feizi, T. Carbohydrate recognition systems: Functional triads in cell—cell interactions. Curr. Opin. Struct. Biol. 1996, 6, 679–691. [Google Scholar] [CrossRef]
- Lowe, J.B. Glycan-dependent leukocyte adhesion and recruitment in inflammation. Curr. Opin. Cell Biol. 2003, 15, 531–538. [Google Scholar] [CrossRef]
- Bard, F.; Chia, J. Cracking the Glycome Encoder: Signaling, Trafficking, and Glycosylation. Trends Cell Biol. 2016, 26, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef]
- Sandhu, A.P.S.; Randhawa, G.S.; Dhugga, K.S. Plant Cell Wall Matrix Polysaccharide Biosynthesis. Mol. Plant. 2009, 2, 840–850. [Google Scholar] [CrossRef]
- Tytgat, H.L.P.; Lebeer, S. The Sweet Tooth of Bacteria: Common Themes in Bacterial Glycoconjugates. Microbiol. Mol. Biol. Rev. 2014, 78, 372–417. [Google Scholar] [CrossRef]
- Dube, D.H.; Bertozzi, C.R. Glycans in cancer and inflammation [mdash] potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 2005, 4, 477–488. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Marth, J.D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef]
- Jeyakumar, M.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Glycosphingolipid lysosomal storage diseases: Therapy and pathogenesis. Neuropathol. Appl. Neurobiol. 2002, 28, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Larson, C.J.; Koltun, E.S.; Hanel, A.; Chan, V.; Nachtigall, J.; Harrison, A.; Aay, N.; Du, H.; Arcalas, A.; et al. Discovery and Characterization of an Inhibitor of Glucosylceramide Synthase. J. Med. Chem. 2012, 55, 4322–4335. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, P.; Alfonso, P.; Atutxa, K.; Fernández-Galán, M.A.; Barez, A.; Franco, R.; Alonso, D.; Martin, A.; Latre, P.; Pocovi, M. Real-world clinical experience with long-term miglustat maintenance therapy in type 1 Gaucher disease: The ZAGAL project. Haematologica 2009, 94, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Palcic, M.M.; Sujino, K. Assays for glycosyltransferases. Trends Glycosci. Glycotechnol. 2001, 13, 361–370. [Google Scholar] [CrossRef]
- Wagner, G.K.; Pesnot, T. Glycosyltransferases and their Assays. ChemBioChem 2010, 11, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Ojida, A.; Takashima, I.; Kohira, T.; Nonaka, H.; Hamachi, I. Turn-On Fluorescence Sensing of Nucleoside Polyphosphates Using a Xanthene-Based Zn(II) Complex Chemosensor. J. Am. Chem. Soc. 2008, 130, 12095–12101. [Google Scholar] [CrossRef]
- Ryu, J.; Eom, M.S.; Ko, W.; Han, M.S.; Lee, H.S. A fluorescence-based glycosyltransferase assay for high-throughput screening. Bioorg. Med. Chem. 2014, 22, 2571–2575. [Google Scholar] [CrossRef]
- Zielinski, T.; Reichman, M.; Donover, P.S.; Lowery, R.G. Development and Validation of a Universal High-Throughput UDP-Glycosyltransferase Assay with a Time-Resolved FRET Signal. ASSAY Drug Dev. Technol. 2016, 14, 240–251. [Google Scholar] [CrossRef]
- Kumagai, K.; Kojima, H.; Okabe, T.; Nagano, T. Development of a highly sensitive, high-throughput assay for glycosyltransferases using enzyme-coupled fluorescence detection. Anal. Biochem. 2014, 447, 146–155. [Google Scholar] [CrossRef]
- Wu, Z.L.; Ethen, C.M.; Prather, B.; Machacek, M.; Jiang, W. Universal phosphatase-coupled glycosyltransferase assay. Glycobiology 2011, 21, 727–733. [Google Scholar] [CrossRef]
- Antczak, C.; Shum, D.; Radu, C.; Seshan, V.E.; Djaballah, H. Development and validation of a high-density fluorescence polarization-based assay for the trypanosoma RNA triphosphatase TbCet1. Comb. Chem. High Throughput Screen 2009, 12, 258–268. [Google Scholar] [CrossRef][Green Version]
- Li, J.-J.; Bugg, T.D.H. A fluorescent analogue of UDP-N-acetylglucosamine: Application for FRET assay of peptidoglycan translocase II (MurG). Chem. Commun. 2004, 2, 182–183. [Google Scholar] [CrossRef]
- Maeda, T.; Nishimura, S.-I. FRET-Based Direct and Continuous Monitoring of Human Fucosyltransferases Activity: An Efficient synthesis of Versatile GDP-L-Fucose Derivatives from Abundant d-Galactose. Chem. A Eur. J. 2008, 14, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.J.; Kraybill, B.C.; Walker, S. Discovery of O-GlcNAc Transferase Inhibitors. J. Am. Chem. Soc. 2005, 127, 14588–14589. [Google Scholar] [CrossRef]
- Helm, J.S.; Hu, Y.; Chen, L.; Gross, B.; Walker, S. Identification of Active-Site Inhibitors of MurG Using a Generalizable, High-Throughput Glycosyltransferase Screen. J. Am. Chem. Soc. 2003, 125, 11168–11169. [Google Scholar] [CrossRef] [PubMed]
- Glover, K.J.; Weerapana, E.; Chen, M.M.; Imperiali, B. Direct Biochemical Evidence for the Utilization of UDP-bacillosamine by PglC, an Essential Glycosyl-1-phosphate Transferase in the Campylobacter jejuni N-Linked Glycosylation Pathway. Biochemistry 2006, 45, 5343–5350. [Google Scholar] [CrossRef]
- Koresawa, M.; Okabe, T. High-Throughput Screening with Quantitation of ATP Consumption: A Universal Non-Radioisotope, Homogeneous Assay for Protein Kinase. ASSAY Drug Dev. Technol. 2004, 2, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ramteke, M.P.; Shelke, P.; Ramamoorthy, V.; Somavarapu, A.K.; Gautam, A.K.S.; Nanaware, P.P.; Karanam, S.; Mukhopadhyay, S.; Venkatraman, P. Identification of a novel ATPase activity in 14-3-3 proteins—Evidence from enzyme kinetics, structure guided modeling and mutagenesis studies. FEBS Lett. 2014, 588, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.; Li, Y.; Fan, B.; Ranjith-Kumar, C.T.; Kao, C.C. RNA binding by the NS3 protease of the Hepatitis C virus. Virus Res. 2012, 169, 80–90. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and Homogeneous ADP Monitoring Assay for Kinases. ASSAY Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Hsiao, K.; Goueli, S.A. A bioluminescent assay for monitoring conjugation of ubiquitin and ubiquitin-like proteins. Anal. Biochem. 2016, 510, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Bledsoe, R.; Chai, J.; Daka, P.; Deng, H.; Ding, Y.; Harris-Gurley, S.; Kryn, L.H.; Nartey, E.; Nichols, J.; et al. The Role of Phosphodiesterase 12 (PDE12) as a Negative Regulator of the Innate Immune Response and the Discovery of Antiviral Inhibitors. J. Biol. Chem. 2015, 290, 19681–19696. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.G.; Mela, I.; Wang, L.; Henderson, R.M.; Chapman, E.R.; Edwardson, J.M.; Audhya, A. Sar1 GTPase Activity Is Regulated by Membrane Curvature. J. Biol. Chem. 2016, 291, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Hsiao, K.; Goueli, S.A. A Homogenous Bioluminescent System for Measuring GTPase, GTPase Activating Protein, and Guanine Nucleotide Exchange Factor Activities. ASSAY Drug Dev. Technol. 2015, 13, 444–455. [Google Scholar] [CrossRef]
- Auld, D.S.; Zhang, Y.-Q.; Southall, N.T.; Rai, G.; Landsman, M.; MacLure, J.; Langevin, D.; Thomas, C.J.; Austin, C.P.; Inglese, J. A Basis for Reduced Chemical Library Inhibition of Firefly Luciferase Obtained from Directed Evolution. J. Med. Chem. 2009, 52, 1450–1458. [Google Scholar] [CrossRef]
- Fan, F.; Wood, K.V. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 2007, 5, 127–136. [Google Scholar] [CrossRef]
- Meisenheimer, P.L.; Uyeda, H.T.; Ma, D.; Sobol, M.; McDougall, M.G.; Corona, C.; Simpson, D.; Klaubert, D.H.; Cali, J.J. Proluciferin Acetals as Bioluminogenic Substrates for Cytochrome P450 Activity and Probes for CYP3A Inhibition. Drug Metab. Dispos. 2011, 39, 2403–2410. [Google Scholar] [CrossRef]
- Sheikh, M.O.; Halmo, S.M.; Patel, S.; Middleton, D.; Takeuchi, H.; Schafer, C.M.; West, C.M.; Haltiwanger, R.S.; Avci, F.Y.; Moremen, K.W.; et al. Rapid screening of sugar-nucleotide donor specificities of putative glycosyltransferases. Glycobiology 2017, 27, 206–212. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Jiang, J.; Gloster, T.M.; Zandberg, W.F.; Whitworth, G.E.; Vocadlo, D.J.; Walker, S. Structural snapshots of the reaction coordinate for O-GlcNAc transferase. Nat. Chem. Biol. 2012, 8, 966–968. [Google Scholar] [CrossRef]
- Halmo, S.M.; Singh, D.; Patel, S.; Wang, S.; Edlin, M.; Boons, G.-J.; Moremen, K.W.; Live, D.; Wells, L. Protein O-Linked Mannose β-1,4-N-Acetylglucosaminyl-transferase 2 (POMGNT2) Is a Gatekeeper Enzyme for Functional Glycosylation of α-Dystroglycan. J. Biol. Chem. 2017, 292, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Naegeli, A.; Michaud, G.; Schubert, M.; Lin, C.-W.; Lizak, C.; Darbre, T.; Reymond, J.-L.; Aebi, M. Substrate Specificity of Cytoplasmic N-Glycosyltransferase. J. Biol. Chem. 2014, 289, 24521–24532. [Google Scholar] [CrossRef] [PubMed]
- Praissman, J.L.; Willer, T.; Sheikh, M.O.; Toi, A.; Chitayat, D.; Lin, Y.-Y.; Lee, H.; Stalnaker, S.H.; Wang, S.; Prabhakar, P.K.; et al. The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition. eLife 2016, 5, e14473. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zhang, H.; Wu, H. A Conserved Domain Is Crucial for Acceptor Substrate Binding in a Family of Glucosyltransferases. J. Bacteriol. 2015, 197, 510–517. [Google Scholar] [CrossRef]
- Das, D.; Walvoort, M.T.C.; Lukose, V.; Imperiali, B. A Rapid and Efficient Luminescence-based Method for Assaying Phosphoglycosyltransferase Enzymes. Sci. Rep. 2016, 6, 33412. [Google Scholar] [CrossRef]
- McGraphery, K.; Schwab, W. Comparative Analysis of High-Throughput Assays of Family-1 Plant Glycosyltransferases. Int. J. Mol. Sci. 2020, 21, 2208. [Google Scholar] [CrossRef]
- Smyth, L.A.; Collins, I. Measuring and interpreting the selectivity of protein kinase inhibitors. J. Chem. Biol. 2009, 2, 131–151. [Google Scholar] [CrossRef]
- Alves, J.; Vidugiris, G.; Goueli, S.A.; Zegzouti, H. Bioluminescent High-Throughput Succinate Detection Method for Monitoring the Activity of JMJC Histone Demethylases and Fe(II)/2-Oxoglutarate-Dependent Dioxygenases. SLAS Discov. Adv. Sci. Drug Discov. 2017, 23, 242–254. [Google Scholar] [CrossRef]
- Leemhuis, H.; Dijkhuizen, L. Engineering of Hydrolysis Reaction Specificity in the Transglycosylase Cyclodextrin Glycosyltransferase. Biocatal. Biotransform. 2003, 21, 261–270. [Google Scholar] [CrossRef][Green Version]
- Ward, N.E.; O’Brian, C.A. The intrinsic ATPase activity of protein kinase C is catalyzed at the active site of the enzyme. Biochemistry 1992, 31, 5905–5911. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Hughes, M.A.; Johnson, G.L.; Scott, J.E. Development and Validation of a High-Throughput Intrinsic ATPase Activity Assay for the Discovery of MEKK2 Inhibitors. J. Biomol. Screen. 2012, 18, 388–399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kashem, M.A.; Nelson, R.M.; Yingling, J.D.; Pullen, S.S.; Prokopowicz, A.S., 3rd; Jones, J.W.; Wolak, J.P.; Rogers, G.R.; Morelock, M.M.; Snow, R.J.; et al. Three mechanistically distinct kinase assays compared: Measurement of intrinsic ATPase activity identified the most comprehensive set of ITK inhibitors. J. Biomol. Screen 2007, 12, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Fan, X.; Sun, J.; Ting, P.; Narula, S.; Lundell, D. Inhibition of fucosyltransferase VII by gallic acid and its derivatives. Arch. Biochem. Biophys. 2004, 425, 51–57. [Google Scholar] [CrossRef]
- Hsiao, K.; Zegzouti, H.; Goueli, S.A. Methyltransferase-Glo: A universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics 2016, 8, 321–339. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UDP-Glo Assay | Signal to Background Ratios (Fold) at Each Nucleotide Concentration (µM) 1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 25 | 12.5 | 6.25 | 3.13 | 1.56 | 0.78 | 0.39 | 0.20 | 0.10 | 0.05 | 0.02 | 0 | |
| UDP | 12,368 | 6803 | 3588 | 1828 | 917 | 459 | 227 | 119 | 60 | 30 | 16 | 1 |
| 441.7 | 284.6 | 153.4 | 76.2 | 38.7 | 17.1 | 10.6 | 5.3 | 3.0 | 1.0 | 0.6 | 0 | |
| CDP | 12,378 | 7086 | 3921 | 2012 | 1040 | 507 | 255 | 124 | 61 | 31 | 16 | 1 |
| 44.6 | 51.5 | 103.0 | 50.2 | 32.0 | 22.3 | 8.7 | 5.8 | 2.0 | 1.6 | 0.5 | 0 | |
| GDP-Glo assay | Signal to background ratios (fold) at each nucleotide concentration (µM) 1 | |||||||||||
| 25 | 12.5 | 6.25 | 3.13 | 1.56 | 0.78 | 0.39 | 0.20 | 0.10 | 0.05 | 0.02 | 0 | |
| GDP | 41,700 | 24,917 | 13,317 | 7028 | 3533 | 1788 | 898 | 436 | 208 | 110 | 54 | 1 |
| 2139.8 | 1848.1 | 338.0 | 446.7 | 53.0 | 77.6 | 11.6 | 32.0 | 15.5 | 7.2 | 4.2 | 0 | |
| UMP/CMP-Glo assay | Signal to background ratios (fold) at each nucleotide concentration (µM) 1 | |||||||||||
| 50 | 25 | 12.5 | 6.25 | 3.13 | 1.56 | 0.78 | 0.39 | 0.20 | 0.10 | 0.05 | 0 | |
| UMP | 1922 | 1009 | 535 | 259 | 139 | 68 | 34 | 18 | 9 | 5 | 3 | 1 |
| 32.33 | 1.70 | 2.53 | 4.40 | 1.40 | 0.53 | 0.48 | 0.21 | 0.09 | 0.07 | 0.05 | 0 | |
| CMP | 2186 | 1128 | 595 | 308 | 166 | 83 | 40 | 21 | 11 | 6 | 3 | 1 |
| 41.40 | 22.03 | 9.75 | 10.62 | 2.74 | 2.35 | 1.17 | 0.87 | 0.40 | 0.06 | 0.16 | 0 | |
| Glycosyltransferase | Buffer | Donor | Acceptor | Temp. | Time (min) |
|---|---|---|---|---|---|
| MGATIII N-Acetylglucosaminyl-transferase III | 50 mM Hepes 6.8, 5 mM MnCl2 | UDP-GlcNAc | Biantennary-N-linked core pentasaccharide | 23 °C | 60 |
| β4GalT1 β-1, 4-Galactosyl-transferase 1 | 50 mM Tris 7.5, 5 mM MnCl2, 1 mM DTT | UDP-Gal | GlcNAc | 23 °C | 60 |
| β4GalT2 β-1, 4-Galactosyl-transferase 1 | 50 mM Tris 7.5, 5 mM MnCl2, 2 mM CaCl2 | UDP-Gal | Glucose | 23 °C | 60 |
| GALNT1 Polypeptide GalNAc Transferase 1 | 50 mM Tris 8.0, 2.5 mM MnCl2, 1 mM CaCl2, 1 mM DTT | UDP-GalNAc | Mucin EA2 peptide | 37 °C | 60 |
| GALNT4 Polypeptide GalNAc Transferase 4 | 25 mM Tris 7.5, 5 mM MnCl2, 2.5 mM CaCl2 | UDP-GalNAc | Mucin EA2 peptide | 37 °C | 60 |
| TcdB C. difficile Toxin B Protein | 50 mM Hepes 7.5, 100 uM KCl, 2 mM MgCl2, 2 mM MnCl2, 1 mM DTT | UDP-Glc | RhoA protein | 23 °C | 60 |
| OGT O-GlcNAc Transferase | 25 mM Tris 7.5, 12.5 mM MgCl2, 0.062 mg/mL BSA, 1 mM DTT | UDP-GlcNAc | OGT-peptide substrate | 23 °C | 60 |
| UGT1A1 Glucuronosyltransferase 1A1 | 50 mM TES, 8 mM MgCl2, 25 mg/mL Alamethicin, 15 mM NaF pH 7.5 | UDP-GA | Estradiol | 37 °C | 60 |
| FUT2 Fucosyltransferase 2 | 5 mM Tris 7.5, 30 mM NaCl2, 2 mM MnCl2, 2 mM CaCl2 | GDP-Fucose | α-lactose | 37 °C | 30 |
| FUT3 Fucosyltransferase 3 | 5 mM Tris 7.5, 1 mM MnCl2 | GDP-Fucose | LAcNAc | 23 °C | 60 |
| FUT7 Fucosyltransferase 7 | 20 mM Tris 7.5, 2 mM MnCl2, 2 mM CaCl2 | GDP-Fucose | Fetuin | 37 °C | 30 |
| Xcb A Meningococcal X capsule N-acetylglucosamine-1-phosphotransferase | 50 mM Hepes 7.5, 25 mM MgCl2, 100 mM NaCl2, 2.4 mM imidazole | UDP-GlcNAc | NMX (α1→4)-linked GlcNAc-1-phosphate polymer | 23 °C | 60 |
| ST6Gal1 β-galactoside α-2,6-sialyltransferase 1 | 5 mM Tris 7.5, 150 mM NaCl2, 5 mM CaCl2, 5 mM MnCl2 | CMP-NANA | LAcNAc | 23 °C | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engel, L.; Alves, J.; Hennek, J.; Goueli, S.A.; Zegzouti, H. Utility of Bioluminescent Homogeneous Nucleotide Detection Assays in Measuring Activities of Nucleotide-Sugar Dependent Glycosyltransferases and Studying Their Inhibitors. Molecules 2021, 26, 6230. https://doi.org/10.3390/molecules26206230
Engel L, Alves J, Hennek J, Goueli SA, Zegzouti H. Utility of Bioluminescent Homogeneous Nucleotide Detection Assays in Measuring Activities of Nucleotide-Sugar Dependent Glycosyltransferases and Studying Their Inhibitors. Molecules. 2021; 26(20):6230. https://doi.org/10.3390/molecules26206230
Chicago/Turabian StyleEngel, Laurie, Juliano Alves, Jacquelyn Hennek, Said A. Goueli, and Hicham Zegzouti. 2021. "Utility of Bioluminescent Homogeneous Nucleotide Detection Assays in Measuring Activities of Nucleotide-Sugar Dependent Glycosyltransferases and Studying Their Inhibitors" Molecules 26, no. 20: 6230. https://doi.org/10.3390/molecules26206230
APA StyleEngel, L., Alves, J., Hennek, J., Goueli, S. A., & Zegzouti, H. (2021). Utility of Bioluminescent Homogeneous Nucleotide Detection Assays in Measuring Activities of Nucleotide-Sugar Dependent Glycosyltransferases and Studying Their Inhibitors. Molecules, 26(20), 6230. https://doi.org/10.3390/molecules26206230