
Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy



Abstract
1. Introduction
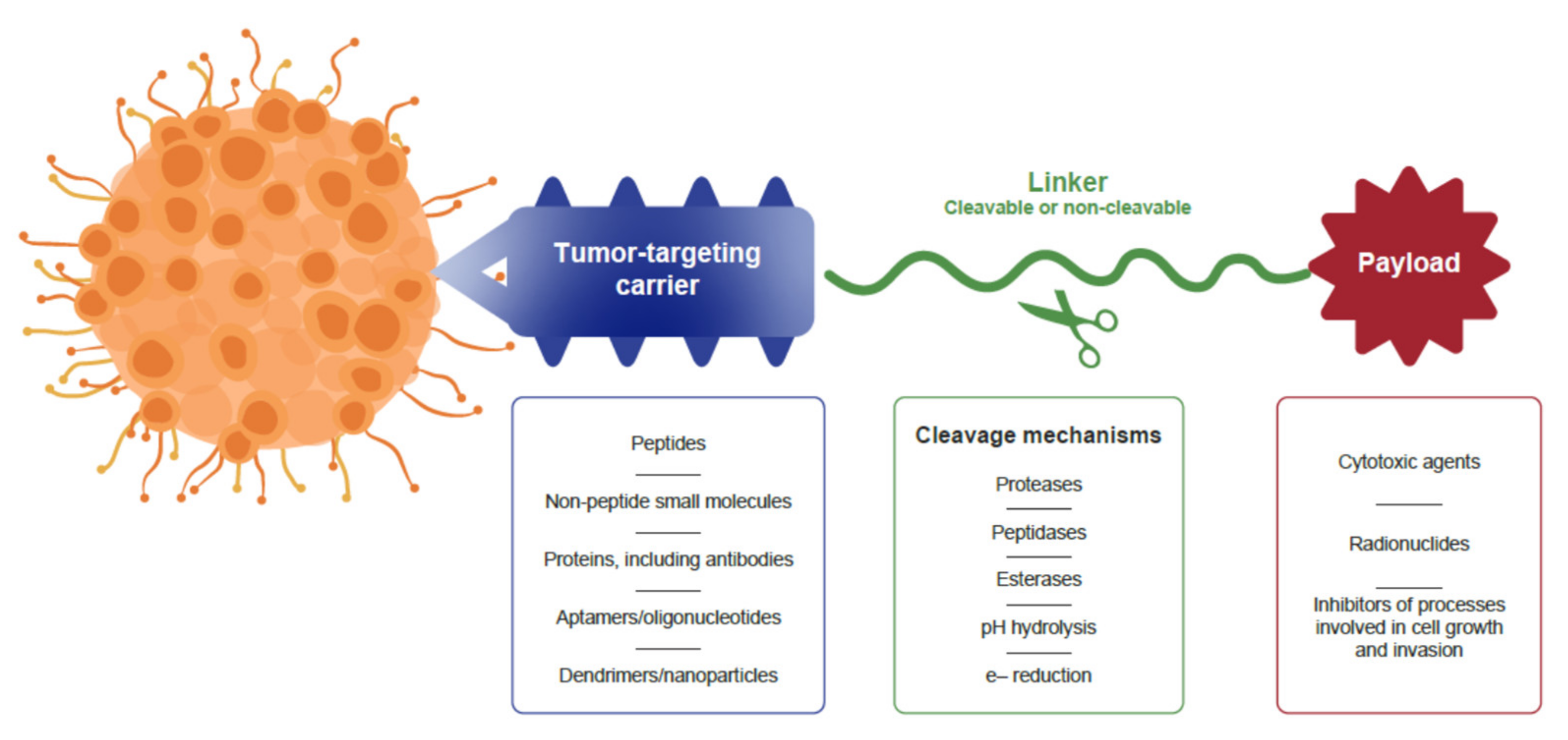
2. Structure of Anticancer Drug Conjugates
3. Limitations with Antibody-Drug Conjugates as Cancer Treatments
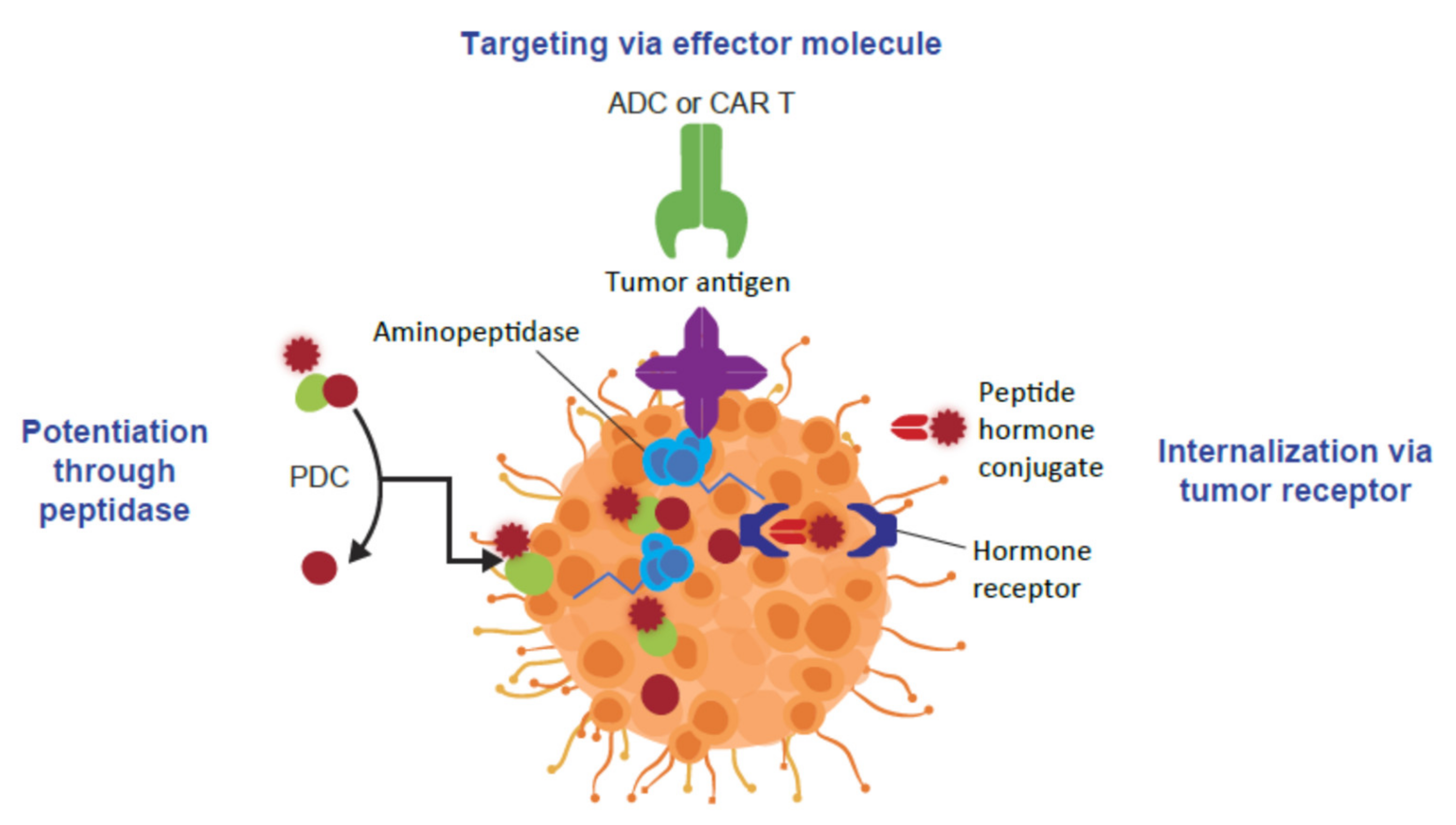
4. Peptide-Drug Conjugates as Targeted Cancer Treatments
5. The Clinical Application of Peptide-Drug Conjugates in Cancer
6. Future Directions with Peptide-Drug Conjugates
7. Conclusions and Perspectives on Progress with Drug Conjugates as Cancer Therapeutics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-drug conjugates and their targets in advanced cancer therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.M.; Iegre, J.; O’Donovan, D.H.; Ölwegård Halvarsson, M.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide-drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
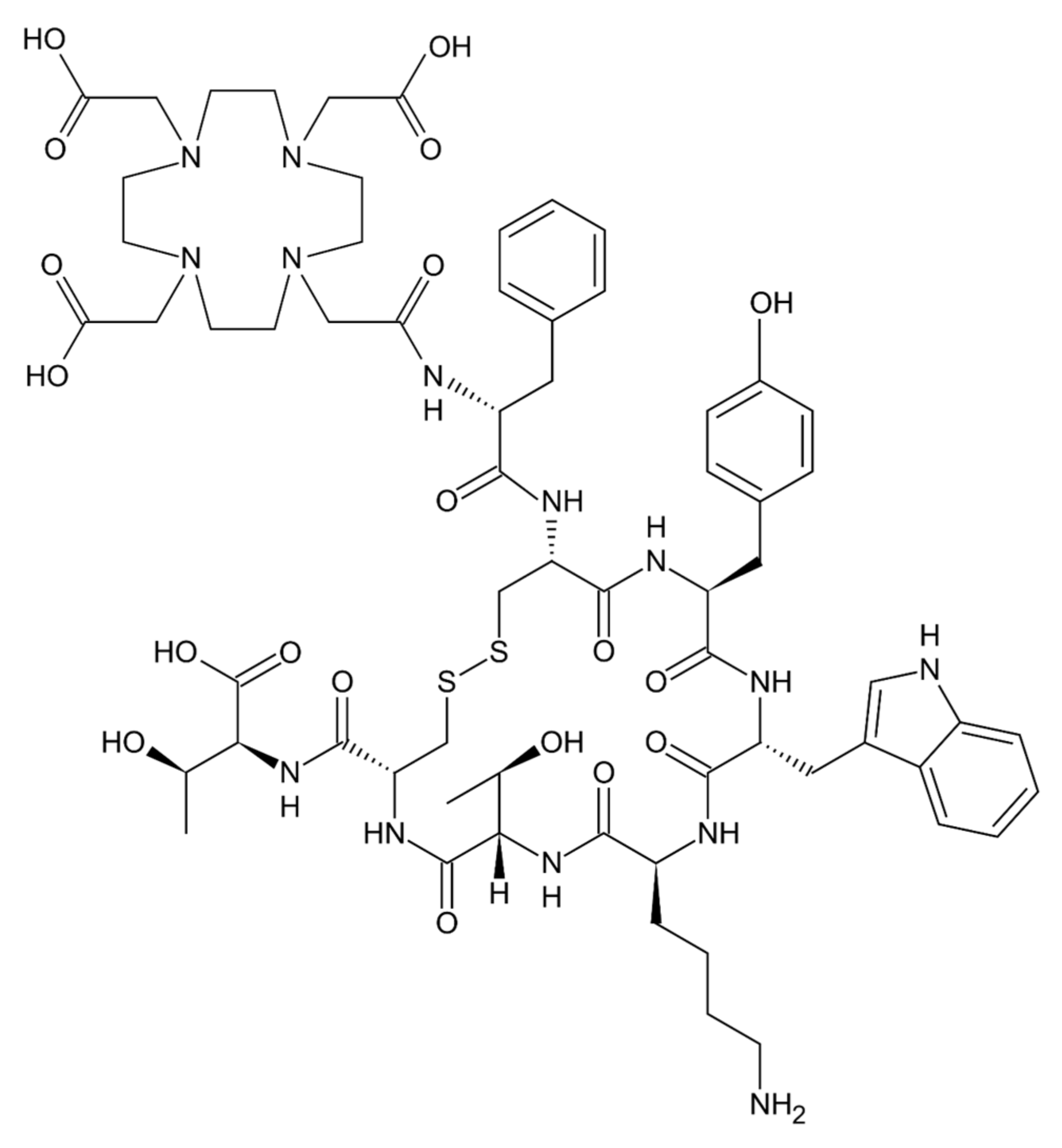
- Das, S.; Al-Toubah, T.; El-Haddad, G.; Strosberg, J. 177Lu-DOTATATE for the treatment of gastroenteropancreatic neuroendocrine tumors. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 1023–1031. [Google Scholar] [CrossRef]
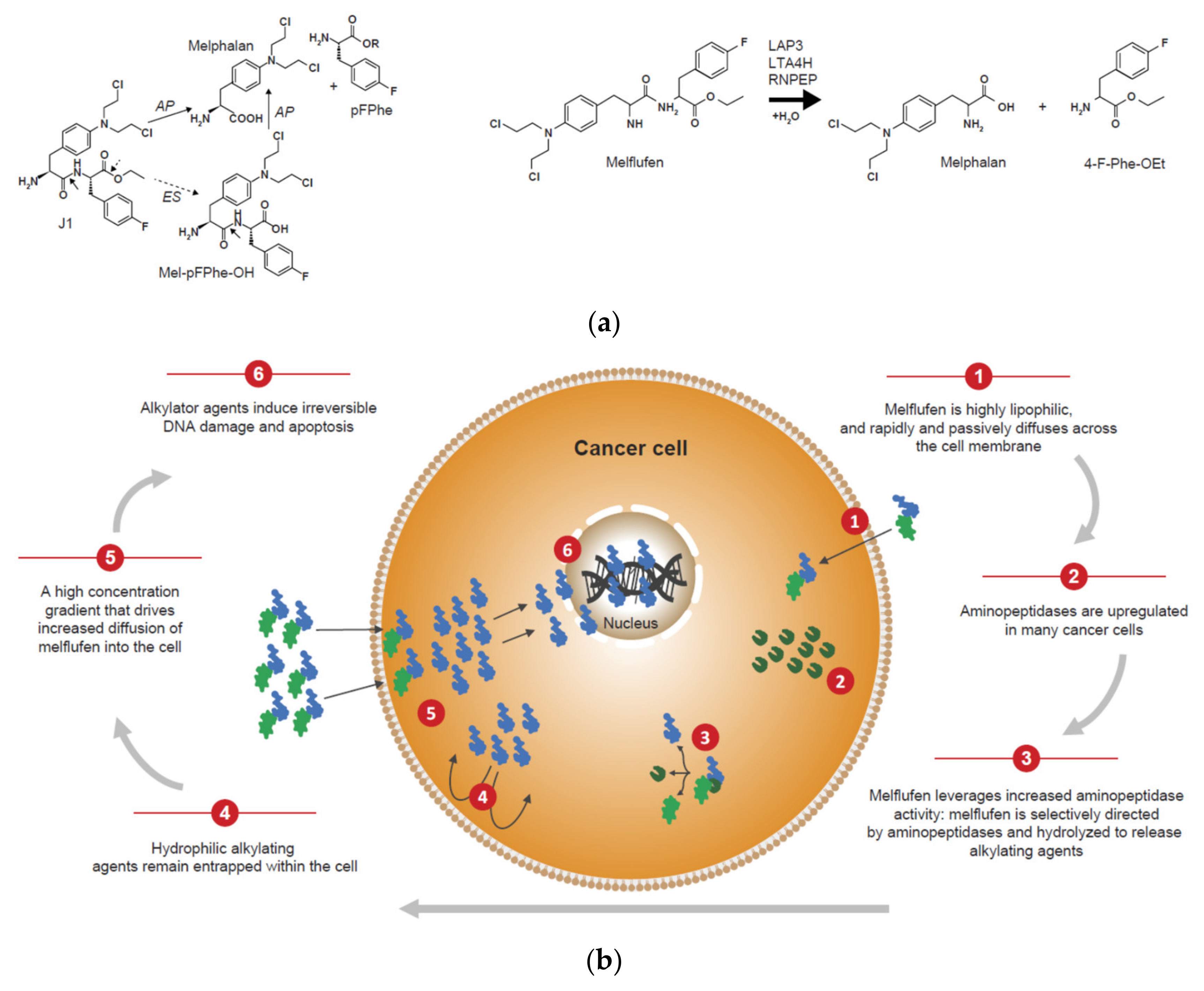
- Wickström, M.; Viktorsson, K.; Lundholm, L.; Aesoy, R.; Nygren, H.; Sooman, L.; Fryknäs, M.; Vogel, L.K.; Lewensohn, R.; Larsson, R.; et al. The alkylating prodrug J1 can be activated by aminopeptidase N, leading to a possible target directed release of melphalan. Biochem. Pharmacol. 2010, 79, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Ray, A.; Viktorsson, K.; Spira, J.; Paba-Prada, C.; Munshi, N.; Richardson, P.; Lewensohn, R.; Anderson, K.C. In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells. Clin. Cancer Res. 2013, 19, 3019–3031. [Google Scholar] [CrossRef] [PubMed]
- Wickström, M.; Nygren, P.; Larsson, R.; Harmenberg, J.; Lindberg, J.; Sjöberg, P.; Jerling, M.; Lehmann, F.; Richardson, P.; Anderson, K.; et al. Melflufen—A peptidase-potentiated alkylating agent in clinical trials. Oncotarget 2017, 8, 66641–66655. [Google Scholar] [CrossRef]
- Gullbo, J.; Wickström, M.; Tullberg, M.; Ehrsson, H.; Lewensohn, R.; Nygren, P.; Luthman, K.; Larsson, R. Activity of hydrolytic enzymes in tumour cells is a determinant for anti-tumour efficacy of the melphalan containing prodrug J1. J. Drug Target. 2003, 11, 355–363. [Google Scholar] [CrossRef]
- Ray, A.; Ravillah, D.; Das, D.S.; Song, Y.; Nordström, E.; Gullbo, J.; Richardson, P.G.; Chauhan, D.; Anderson, K.C. A novel alkylating agent melflufen induces irreversible DNA damage and cytotoxicity in multiple myeloma cells. Br. J. Haematol. 2016, 174, 397–409. [Google Scholar] [CrossRef]
- Yoo, J.; Park, C.; Yi, G.; Lee, D.; Koo, H. Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems. Cancers 2019, 11, 640. [Google Scholar] [CrossRef]
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.Å.; Löfblom, J. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef]
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef]
- Altai, M.; Liu, H.; Ding, H.; Mitran, B.; Edqvist, P.H.; Tolmachev, V.; Orlova, A.; Gräslund, T. Affibody-derived drug conjugates: Potent cytotoxic molecules for treatment of HER2 over-expressing tumors. J. Control. Release 2018, 288, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Brandl, F.; Busslinger, S.; Zangemeister-Wittke, U.; Plückthun, A. Optimizing the anti-tumor efficacy of protein-drug conjugates by engineering the molecular size and half-life. J. Control. Release 2020, 327, 186–197. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.B.; Gonzalez-Martin, A.; LoRusso, P.M.; Ferrero, J.M.; Smitt, M.; Yu, R.; Leung, A.C.; Wildiers, H.; TH3RESA study collaborators. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 689–699. [Google Scholar] [CrossRef]
- PEPAXTO Prescribing Information. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214383s000lbl.pdf (accessed on 1 August 2021).
- LUTATHERA Prescribing Information. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208700s000lbl.pdf (accessed on 1 August 2021).
- ZEVALIN Prescribing Information. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/125019s0156.pdf (accessed on 1 August 2021).
- BEXXAR Prescribing Information. 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125011s102lbl.pdf (accessed on 1 August 2021).
- KADCYLA Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf (accessed on 1 August 2021).
- ENHERTU Prescribing Information. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761139s000lbl.pdf (accessed on 1 August 2021).
- ADCETRIS US Prescribing Information. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125388_s056s078lbl.pdf (accessed on 1 August 2021).
- POLIVY Prescribing Information. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 1 August 2021).
- BLENREP Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf (accessed on 1 August 2021).
- MYLOTARG Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761060lbl.pdf (accessed on 1 August 2021).
- BESPONSA Prescribing Information. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf (accessed on 1 August 2021).
- PADCEV Prescribing Information. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf (accessed on 1 August 2021).
- TRODELVY Prescribing Information. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.pdf (accessed on 1 August 2021).
- ZYNLONTA Prescribing Information. 2021. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf (accessed on 1 August 2021).
- LUMOXITI Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761104s000lbl.pdf (accessed on 1 August 2021).
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Wagner-Rousset, E.; Janin-Bussat, M.C.; Colas, O.; Excoffier, M.; Ayoub, D.; Haeuw, J.F.; Rilatt, I.; Perez, M.; Corvaïa, N.; Beck, A. Antibody-drug conjugate model fast characterization by LC-MS following IdeS proteolytic digestion. mAbs 2014, 6, 273–285. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Impact Story: Developing the Tools to Evaluate Complex Drug Products: Peptides. 2019. Available online: https://www.fda.gov/drugs/regulatory-science-action/impact-story-developing-tools-evaluate-complex-drug-products-peptides (accessed on 1 August 2021).
- Ahrens, V.M.; Bellmann-Sickert, K.; Beck-Sickinger, A.G. Peptides and peptide conjugates: Therapeutics on the upward path. Future Med. Chem. 2012, 4, 1567–1586. [Google Scholar] [CrossRef]
- Miettinen, J.J.; Kumari, R.; Traustadottir, G.A.; Huppunen, M.E.; Sergeev, P.; Majumder, M.M.; Schepsky, A.; Gudjonsson, T.; Lievonen, J.; Bazou, D.; et al. Aminopeptidase expression in multiple myeloma associates with disease progression and sensitivity to melflufen. Cancers 2021, 13, 1527. [Google Scholar] [CrossRef]
- Lehmann, F.; Wennerberg, J. Evolution of nitrogen-based alkylating anticancer agents. Processes 2021, 9, 377. [Google Scholar] [CrossRef]
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journey of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433. [Google Scholar] [CrossRef] [PubMed]
- Wickström, M.; Larsson, R.; Nygren, P.; Gullbo, J. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 2011, 102, 501–508. [Google Scholar] [CrossRef]
- Hitzerd, S.M.; Verbrugge, S.E.; Ossenkoppele, G.; Jansen, G.; Peters, G.J. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids 2014, 46, 793–808. [Google Scholar] [CrossRef]
- Lee, J.; Vinh, N.B.; Drinkwater, N.; Yang, W.; Kannan Sivaraman, K.; Schembri, L.S.; Gazdik, M.; Grin, P.M.; Butler, G.S.; Overall, C.M.; et al. Novel human aminopeptidase N inhibitors: Discovery and optimization of subsite binding interactions. J. Med. Chem. 2019, 62, 7185–7209. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Adhikari, N.; Jha, T. Design of aminopeptidase N inhibitors as anti-cancer agents. J. Med. Chem. 2018, 61, 6468–6490. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide conjugates with small molecules designed to enhance efficacy and safety. Molecules 2019, 24, 1855. [Google Scholar] [CrossRef]
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. Melflufen and dexamethasone in heavily pretreated relapsed and refractory multiple myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Efebera, Y.A.; Hájek, R.; Granell, M.; Maisnar, V.; Straub, J.; Eveillard, J.R.; Karlin, L.; Ribrag, V.; Mateos, M.V.; et al. ANCHOR (OP-104): Melflufen plus dexamethasone (dex) and daratumumab (dara) or bortezomib (BTZ) in relapsed/refractory multiple myeloma (RRMM) refractory to an IMiD and/or a proteasome inhibitor (PI)-updated efficacy and safety. Blood 2019, 134 (Suppl. S1), 3124. [Google Scholar] [CrossRef]
- Oncopeptides AB. Oncopeptides Presents Phase 3 OCEAN Study Results at the IMW Meeting. Oncopeptides AB. 11 September 2021. Available online: https://www.oncopeptides.com/en/media/press-releases/oncopeptides-presents-phase-3-ocean-study-results-at-the-imw-meeting (accessed on 21 September 2021).
- Schepsky, A.; Traustadottir, G.A.; Joelsson, J.P.; Ingthorsson, S.; Kricker, J.; Bergthorsson, J.T.; Asbjarnarson, A.; Gudjonsson, T.; Nupponen, N.; Slipicevic, A.; et al. Melflufen, a peptide-conjugated alkylator, is an efficient anti-neo-plastic drug in breast cancer cell lines. Cancer Med. 2020, 9, 6726–6738. [Google Scholar] [CrossRef]
- Strese, S.; Wickström, M.; Fuchs, P.F.; Fryknäs, M.; Gerwins, P.; Dale, T.; Larsson, R.; Gullbo, J. The novel alkylating prodrug melflufen (J1) inhibits angiogenesis in vitro and in vivo. Biochem. Pharmacol. 2013, 86, 888–895. [Google Scholar] [CrossRef]
- Slipicevic, A.; Munawar, U.; Aschan, J.; Lehmann, F.; Miettinen, J.J.; Huppunen, M.-E.; Bargou, R.C.; Nupponen, N.N.; Rodriguez, P.; Richardson, P.; et al. Melflufen efficacy in multiple myeloma with TP53 aberrations. e-Poster presented at European Hematology Association. In Proceedings of the AACR Annual Meeting, Philadelphia, PA, USA, 22–24 June 2020. (Poster EP903). [Google Scholar]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 trial of 177Lu-dotatate for midgut neuroendocrine tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Sachpekidis, C.; Alberts, I.; Rominger, A.; Afshar-Oromieh, A. PSMA radioligand therapy in prostate cancer: Overview, latest advances and remaining challenges. Immunotherapy 2019, 11, 1267–1271. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Babich, J.W.; Kratochwil, C.; Giesel, F.L.; Eisenhut, M.; Kopka, K.; Haberkorn, U. The rise of PSMA ligands for diagnosis and therapy of prostate cancer. J. Nucl. Med. 2016, 57 (Suppl. S3), 79S–89S. [Google Scholar] [CrossRef]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [177Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Morris, M.J.; De Bono, J.S.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Phase III study of lutetium-177-PSMA-617 in patients with metastatic castration-resistant prostate cancer (VISION). J. Clin. Oncol. 2021, 39 (Suppl. S18), LBA4. [Google Scholar] [CrossRef]
- Theratechnologies, Inc. Theratechnologies Announces Publication of TH1902 Preclinical Data in Peer-Reviewed Journal, Cancer Science. News Release. Theratechnologies, Inc, 13 August 2021. Available online: https://www.theratech.com/news-releases/news-release-details/theratechnologies-announces-publication-th1902-preclinical-data (accessed on 21 September 2021).
- Miller, D.S.; Scambia, G.; Bondarenko, I.; Westermann, A.M.; Oaknin, A.; Oza, A.M.; Lisyanskaya, A.S.; Vergote, I.; Wenham, R.M.; Madhu Temkin, S.; et al. ZoptEC: Phase III randomized controlled study comparing zoptarelin with doxorubicin as second line therapy for locally advanced, recurrent, or metastatic endometrial cancer (NCT01767155). J. Clin. Oncol. 2018, 36 (Suppl. S15), 5503. [Google Scholar] [CrossRef]
- Drappatz, J.; Brenner, A.; Wong, E.T.; Eichler, A.; Schiff, D.; Groves, M.D.; Mikkelsen, T.; Rosenfeld, S.; Sarantopoulos, J.; Meyers, C.A.; et al. Phase I study of GRN1005 in recurrent malignant glioma. Clin. Cancer Res. 2013, 19, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Kumthekar, P.; Tang, S.; Brenner, A.J.; Kesari, S.; Anders, C.K.; Carrillo, J.A.; Chalasani, P.; Kabos, P.; Ahluwalia, M.S.; Ibrahim, N.K. OS7.2 A Phase II study of ANG1005, a novel BBB/BCB penetratant taxane in patients with recurrent brain metastases and leptomeningeal carcinomatosis from breast cancer. Neuro-Oncology 2016, 18 (Suppl. S4), iv16. [Google Scholar] [CrossRef][Green Version]
- Theratechnologies Inc. 2019. Available online: https://www.theratech.com/products/th-1902/ (accessed on 1 August 2021).
- Bragina, O.; von Witting, E.; Garousi, J.; Zelchan, R.; Sandström, M.; Orlova, A.; Medvedeva, A.; Doroshenko, A.; Vorobyeva, A.; Lindbo, S.; et al. Phase I study of 99mTc-ADAPT6, a scaffold protein-based probe for visualization of her2 expression in breast cancer. J. Nucl. Med. 2021, 62, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Xu, T.; Zhang, J.; Tolmachev, V.; Oroujeni, M.; Orlova, A.; Gräslund, T.; Vorobyeva, A. Affibody-derived drug conjugates targeting HER2: Effect of drug load on cytotoxicity and biodistribution. Pharmaceutics 2021, 13, 430. [Google Scholar] [CrossRef]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; Van Rietschoten, K.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE delivery using the bicycle toxin conjugate BT5528. Mol. Cancer Ther. 2020, 19, 1385–1394. [Google Scholar] [CrossRef]
- Bendell, J.C.; Wang, J.S.-Z.; Bashir, B.; Richardson, D.L.; Bennett, G.; Campbell, C.; Hennessy, M.G.; Jeffrey, P.; Kirui, J.; Mahnke, L.; et al. BT5528-100 phase I/II study of the safety, pharmacokinetics, and preliminary clinical activity of BT5528 in patients with advanced malignancies associated with EphA2 expression. J. Clin. Oncol. 2020, 38 (Suppl. S15), TPS3655. [Google Scholar] [CrossRef]
- Carvalho, M.R.; Reis, R.L.; Oliveira, J.M. Dendrimer nanoparticles for colorectal cancer applications. J. Mater. Chem. B 2020, 8, 1128–1138. [Google Scholar] [CrossRef]
- Carvalho, M.R.; Carvalho, C.R.; Maia, F.R.; Caballero, D.; Kundu, S.C.; Reis, R.L.; Oliveira, J.M. Peptide-modified dendrimer nanoparticles for targeted therapy of colorectal cancer. Adv. Ther. 2019, 2, 1900132. [Google Scholar] [CrossRef]
- Fan, Q.; Ji, Y.; Wang, J.; Wu, L.; Li, W.; Chen, R.; Chen, Z. Self-assembly behaviours of peptide-drug conjugates: Influence of multiple factors on aggregate morphology and potential self-assembly mechanism. R. Soc. Open Sci. 2018, 5, 172040. [Google Scholar] [CrossRef]
- Zhong, T.; Yao, X.; Zhang, S.; Guo, Y.; Duan, X.C.; Ren, W.; Huang, D.; Yin, Y.F.; Zhang, X. A self-assembling nanomedicine of conjugated linoleic acid-paclitaxel conjugate (CLA-PTX) with higher drug loading and carrier-free characteristic. Sci. Rep. 2016, 6, 36614. [Google Scholar] [CrossRef]
- Su, H.; Zhang, P.; Cheetham, A.G.; Koo, J.M.; Lin, R.; Masood, A.; Schiapparelli, P.; Quiñones-Hinojosa, A.; Cui, H. Supramolecular Crafting of Self-Assembling Camptothecin Prodrugs with Enhanced Efficacy against Primary Cancer Cells. Theranostics 2016, 6, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Little, N.; Lu, J. Self-assembling prodrug nanotherapeutics for synergistic tumor targeted drug delivery. Acta Biomater. 2020, 111, 20–28. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approved for: Tumor Type | Carrier Molecule | Linker | Payload | Molecular Weight | |
|---|---|---|---|---|---|
| Peptide-Drug Conjugates | |||||
| Melflufen [18] | Multiple myeloma | Di-amino acid | Enzymatically cleaved after cellular uptake | Nitrogen mustard; 2-bischloroethyl amino (alkylating agent) | ~0.5 kDa |
| 177Lu-dotatate [19] | Gastroenteropancreatic neuroendocrine tumors | Somatostatin peptide analog | Covalently-bound chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid | Radionuclide 177Lutetium | 1.6 kDa |
| Antibody-Drug Conjugates | |||||
| Ibritumomab tuixetan [20] | Non-Hodgkin lymphoma | Murine IgGI kappa anti-CD20 (ibritumomab) | Tiuxetan [N-[2-bis(carboxymethyl)amino]-3-(p-isothiocyanatophenyl)-propyl]-[N-[2-bis(carboxymethyl)amino]-2-( methyl) -ethyl]glycine | Radionuclide 111Indium-Ill or 90Yttrium-90 | 148 kDa |
| 131I-tositumomab [21] | Non-Hodgkin lymphoma | Murine IgG2a lambda anti-CD20 (tositumomab) | Covalent bond | Radionuclide 131Iodine | 150 kDa |
| Trastuzumab emtansine [22] | HER2-positive breast cancer | Humanized anti-HER2 IgG1 (trastuzumab) | Stable thioether (4-[N-maleimidomethyl] cyclohexane-1-carboxylate) | DM1 (microtubule inhibitor) | ~148.5 kDa |
| Trastuzumab deruxtecan [23] | HER2-positive breast cancer and gastric or gastroesophageal junction adenocarcinoma | Humanized anti-HER2 IgG1 (trastuzumab) | Protease-cleavable maleimide tetrapeptide linker | DXd (topoisomerase inhibitor) | NA |
| Brentuximab vedotin [24] | Lymphoma | Chimeric anti-CD30 IgG1 antibody | Protease cleavable linker | Monomethyl auristatin E (microtubule disrupting agent) | 153 kDa |
| Polatuzumab vedotin [25] | Diffuse large B-cell lymphoma | Humanized anti-CD79b IgG1 | Protease-cleavable maleimidocaproyl-valine-citrulline-p-aminobenzyloxycarbonyl | Monomethyl auristatin E (microtubule disrupting agent) | 150 kDa |
| Belantamab mafadotin [26] | Multiple myeloma | Afucosylated, humanized anti-B-cell maturation antigen IgG1 | Protease-resistant maleimidocaproyl linker | Monomethyl auristatin F (microtubule disrupting agent) | 152 kDa |
| Gemtuzumab ozogamicin [27] | Acute myeloid leukemia | Humanized anti-CD33 IgG4 kappa antibody | Covalent link | N-acetyl gamma calicheamicin (causes double-stranded DNA breaks) | NA |
| Inotuzumab ozogamicin [28] | Acute lymphoblastic leukemia | Humanized anti-CD22 IgG4 kappa antibody | Acid-cleavable linker | N-acetyl-gamma-calicheamicin (causes double-stranded DNA breaks) | 160 kDa |
| Enfortumab vedotin [29] | Urothelial cancer | Human anti-nectin-4 IgG1 kappa antibody | Protease-cleavable maleimidocaproyl valine-citrulline linker | Monomethyl auristatin E (microtubule disrupting agent) | 152 kDa |
| Sacituzumab govitecan [30] | Triple-negative breast cancer and urothelial cancer | Humanized anti-Trop-2 IgG1 kappa antibody | Hydrolysable linker (CL2A) | SN-38 (topoisomerase inhibitor) | 160 kDa |
| Loncastuximab tesirine-lpyl [31] | B-cell lymphoma | Human anti-CD19 IgG1 kappa antibody | Protease-cleavable valine-alanine linker | SG3199 (pyrrolobenzodiazepine dimer cytotoxic alkylating agent) | 151 kDa |
| Moxetumomab pasudotox-tdfk [32] | Hairy cell leukemia | Murine anti-CD22 Ig variable domain | Genetically fused | Truncated Pseudomonas exotoxin, PE38 (protein synthesis inhibitor) | 63 kDa |
| PDC | Structure | Clinical Proof of Principle Studies |
|---|---|---|
| Zoptarelin doxorubicin [59] | Doxorubicin linked to a small peptide agonist to the LHRH receptor | Zoptarelin failed to improve efficacy or safety versus doxorubicin alone in Phase III, randomized study in patients with pre-treated advanced/metastatic recurrent endometrial cancer (NCT01767155) |
| GRN1005 [60,61] | Paclitaxel linked to angiopep-2, a peptide ligand that crosses the BBB through an LRP-1-mediated mechanism | GRN1005 delivers paclitaxel across the BBB and achieves therapeutic concentrations in tumor tissue Similar toxicity to paclitaxel Clinical activity in recurrent glioma Intracranial partial responses in 8% of patients with breast cancer and brain metastases (NCT01480583) |
| TH1902 [62] | Docetaxel linked to a peptide targeting sortilin (SORT1) receptors | Awarded FDA fast-track designation March 2021: first patient entered into Phase I study of TH1902 in patients with advanced solid tumors (NCT04706962) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindberg, J.; Nilvebrant, J.; Nygren, P.-Å.; Lehmann, F. Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy. Molecules 2021, 26, 6042. https://doi.org/10.3390/molecules26196042
Lindberg J, Nilvebrant J, Nygren P-Å, Lehmann F. Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy. Molecules. 2021; 26(19):6042. https://doi.org/10.3390/molecules26196042
Chicago/Turabian StyleLindberg, Jakob, Johan Nilvebrant, Per-Åke Nygren, and Fredrik Lehmann. 2021. "Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy" Molecules 26, no. 19: 6042. https://doi.org/10.3390/molecules26196042
APA StyleLindberg, J., Nilvebrant, J., Nygren, P.-Å., & Lehmann, F. (2021). Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy. Molecules, 26(19), 6042. https://doi.org/10.3390/molecules26196042