
Synthesis of Indoloquinolines: An Intramolecular Cyclization Leading to Advanced Perophoramidine-Relevant Intermediates


 , and
, and 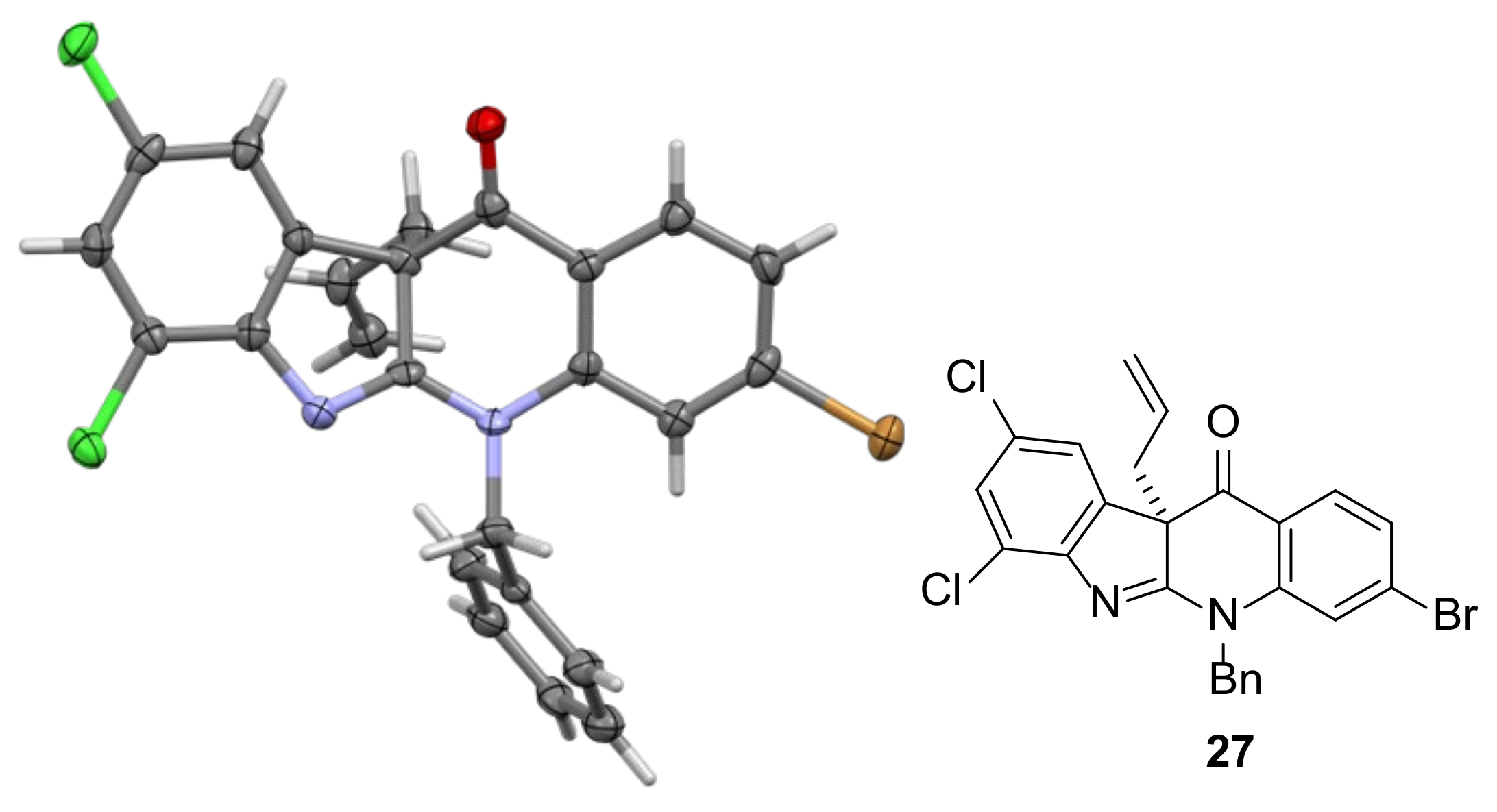{kind=link}
{kind=link}
{kind=link}
{kind=link}
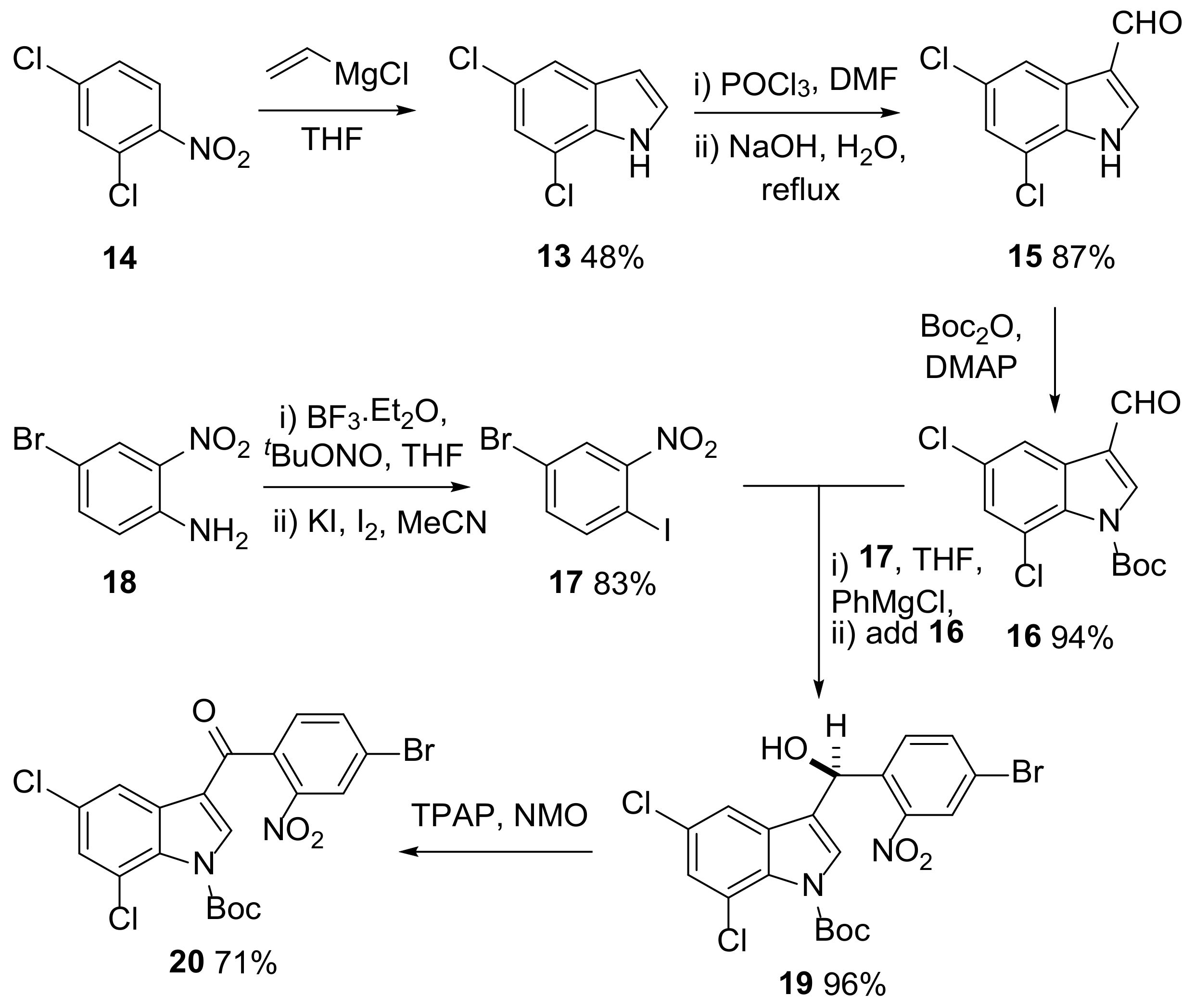{kind=link}
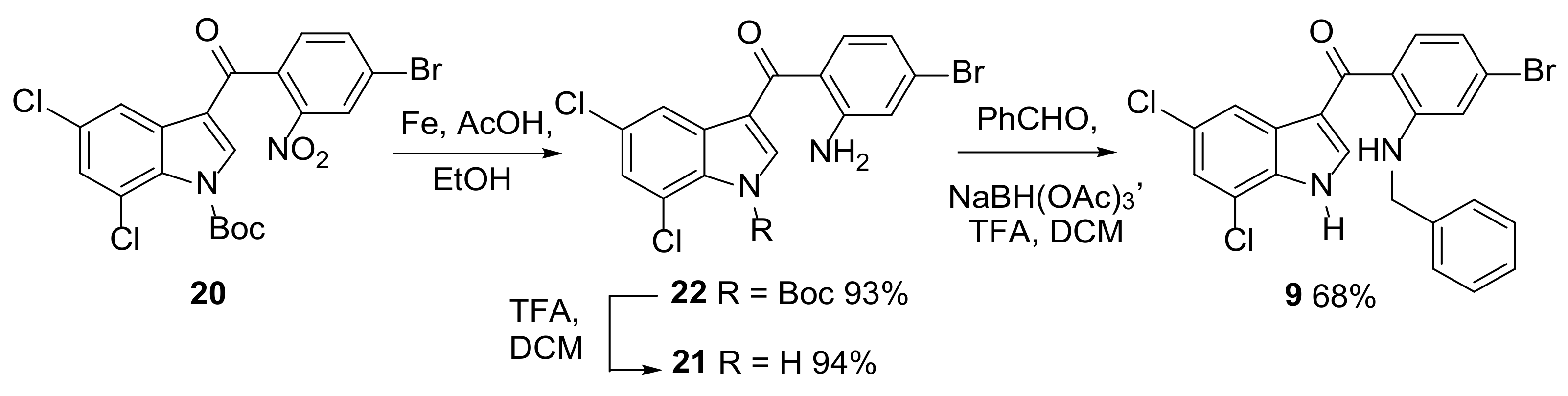{kind=link}
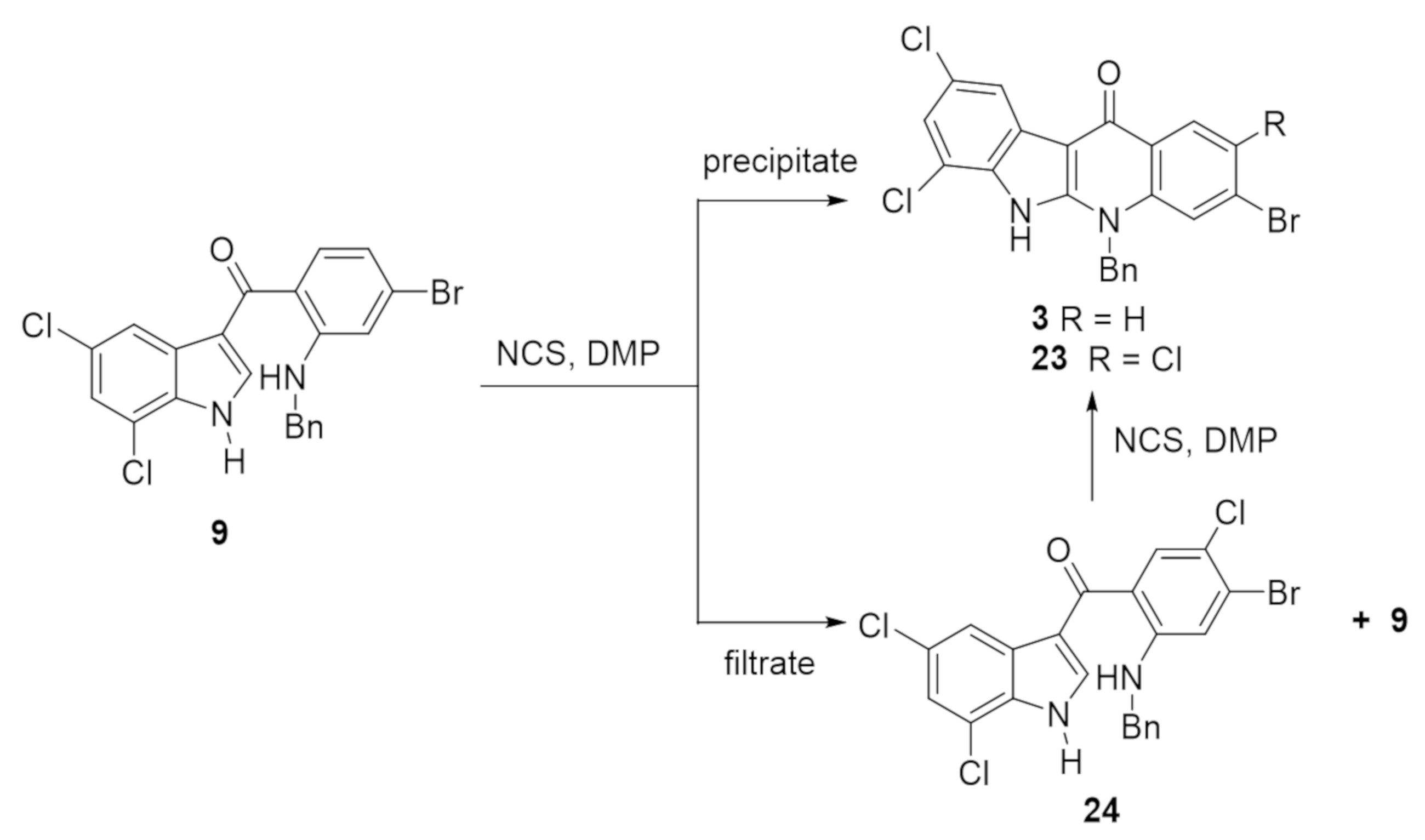{kind=link}
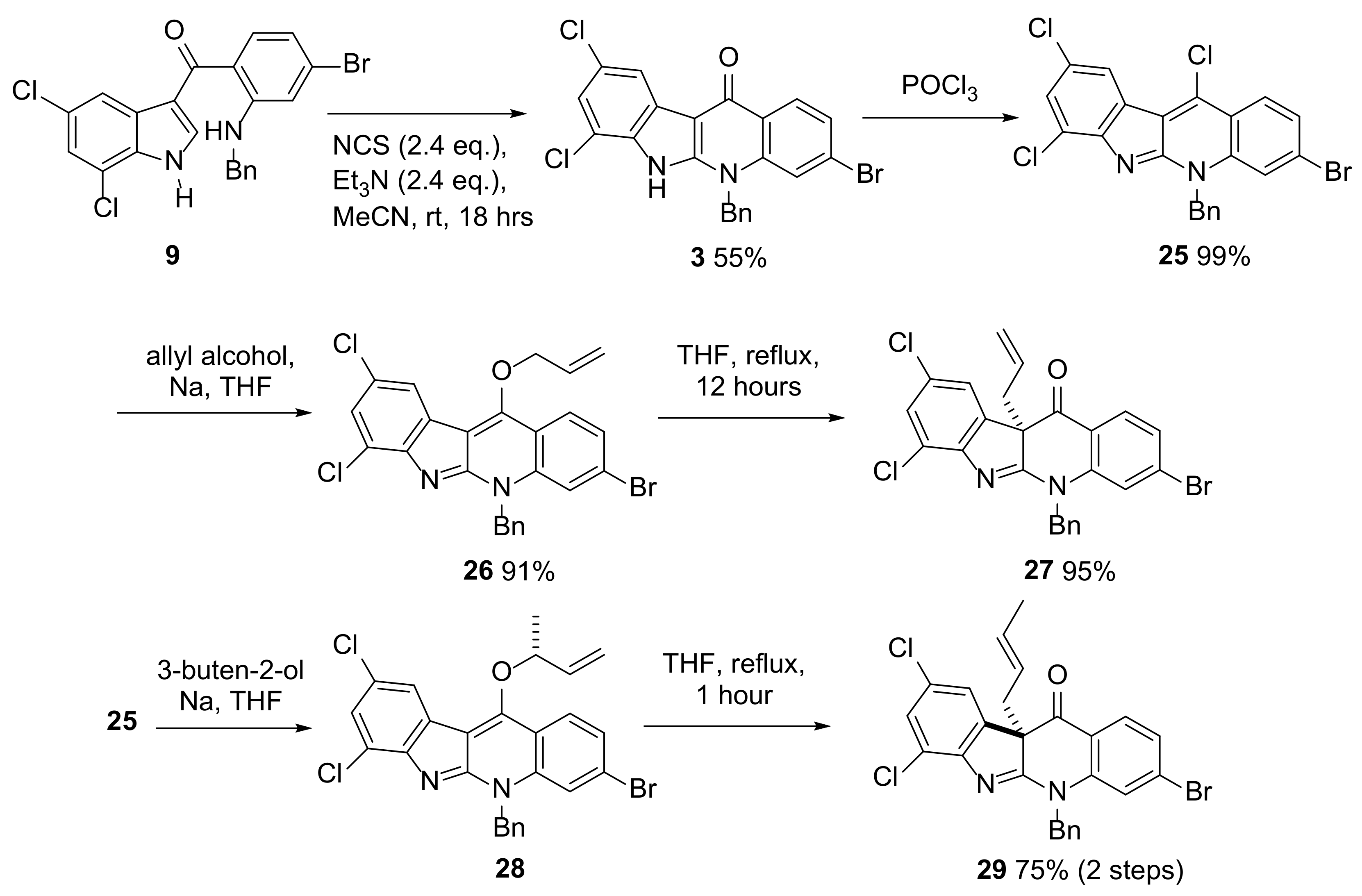{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
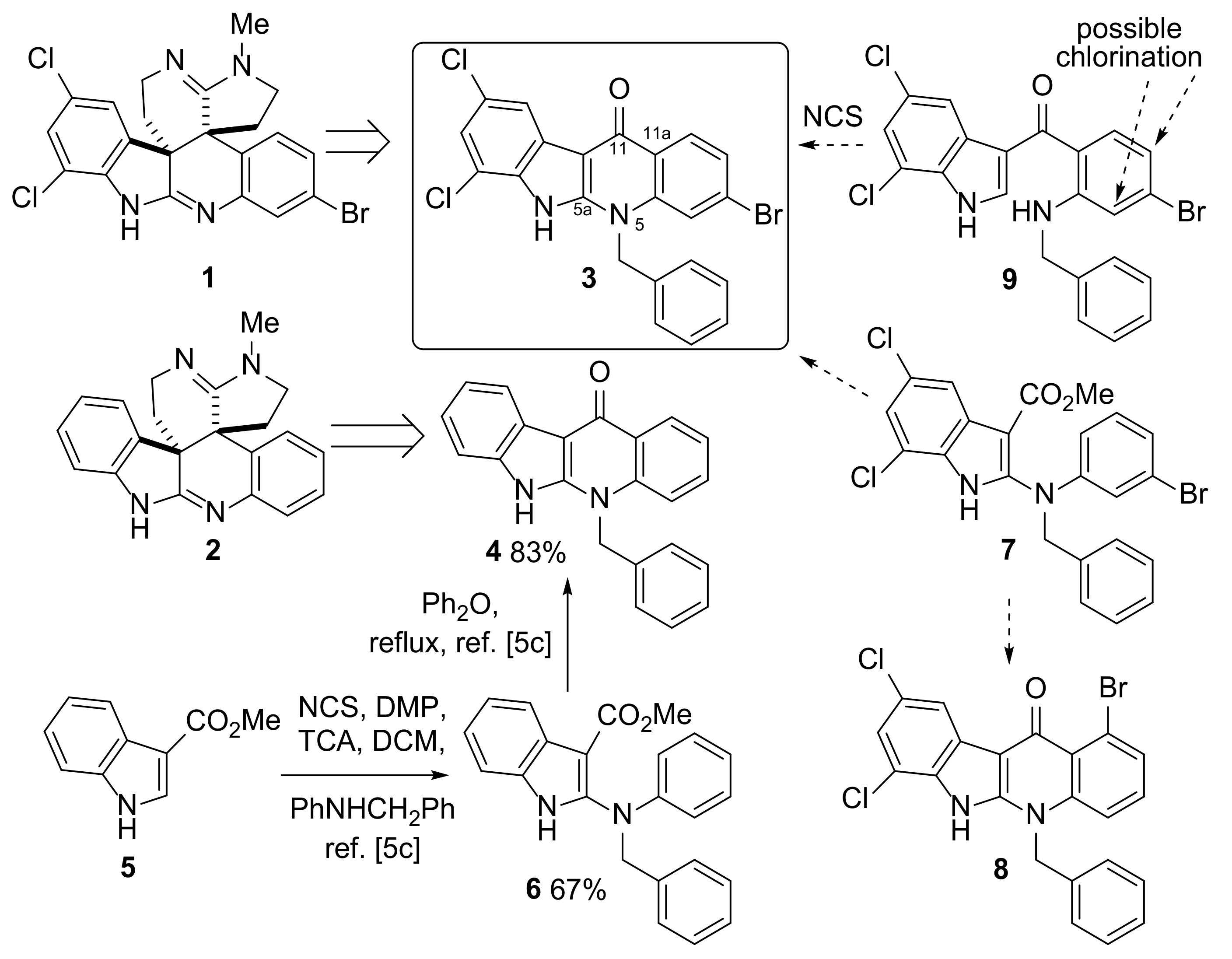
2.1. Proposed Route to Halogenated Intermediate 3
2.2. Synthesis of Halogenated Intermediate 3
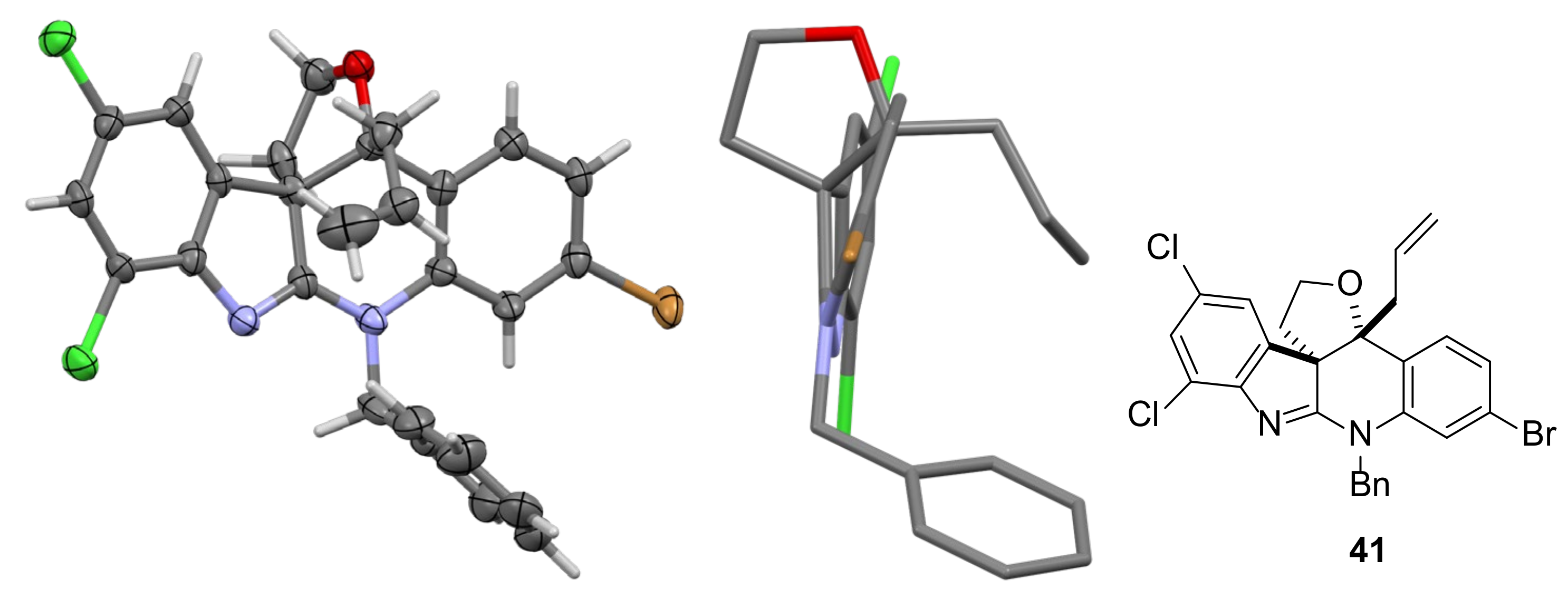
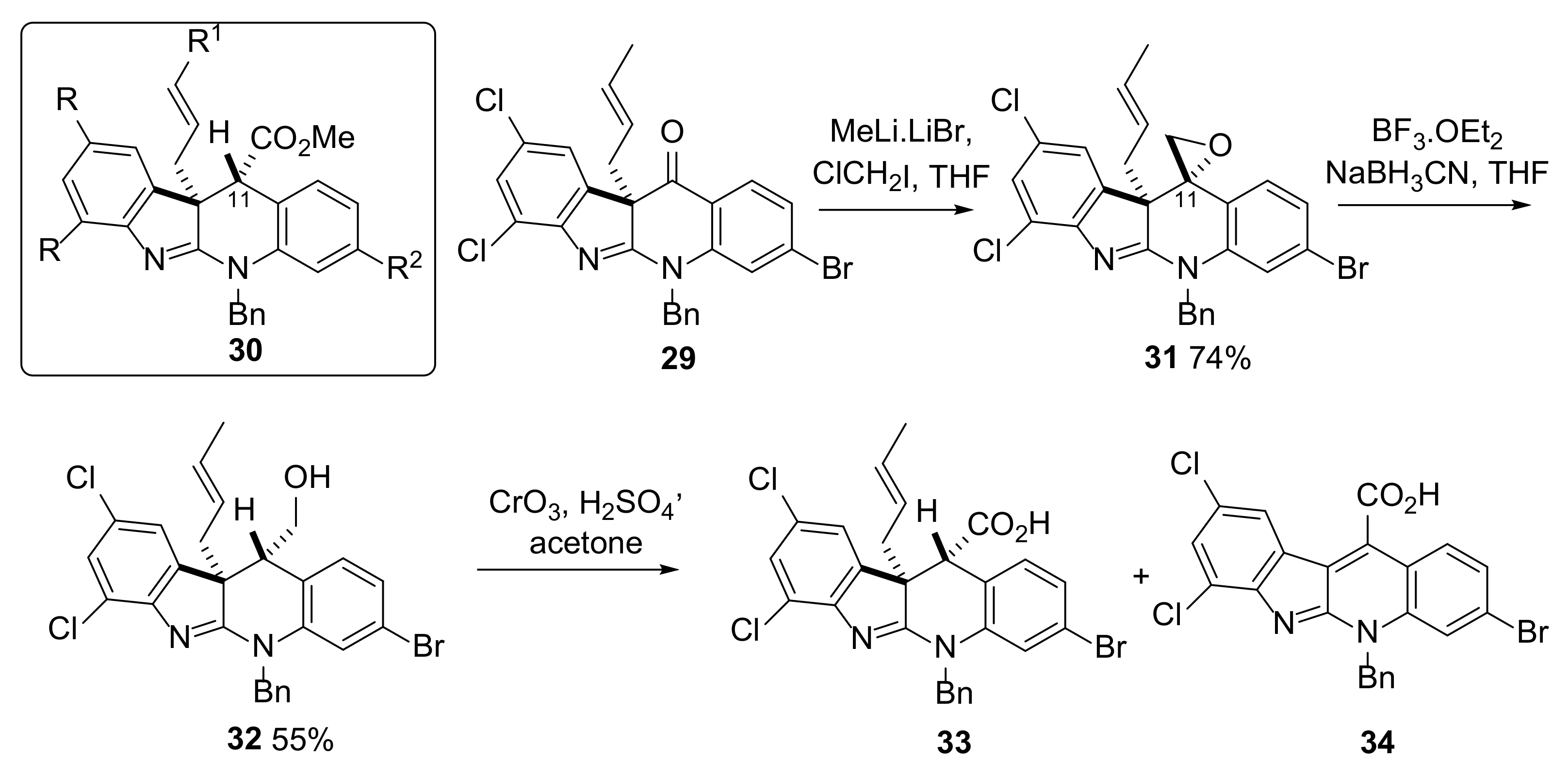
2.3. Installation of the First All-Carbon Quaternary Center and X-ray Structure Determination of Advanced Intermediate 27
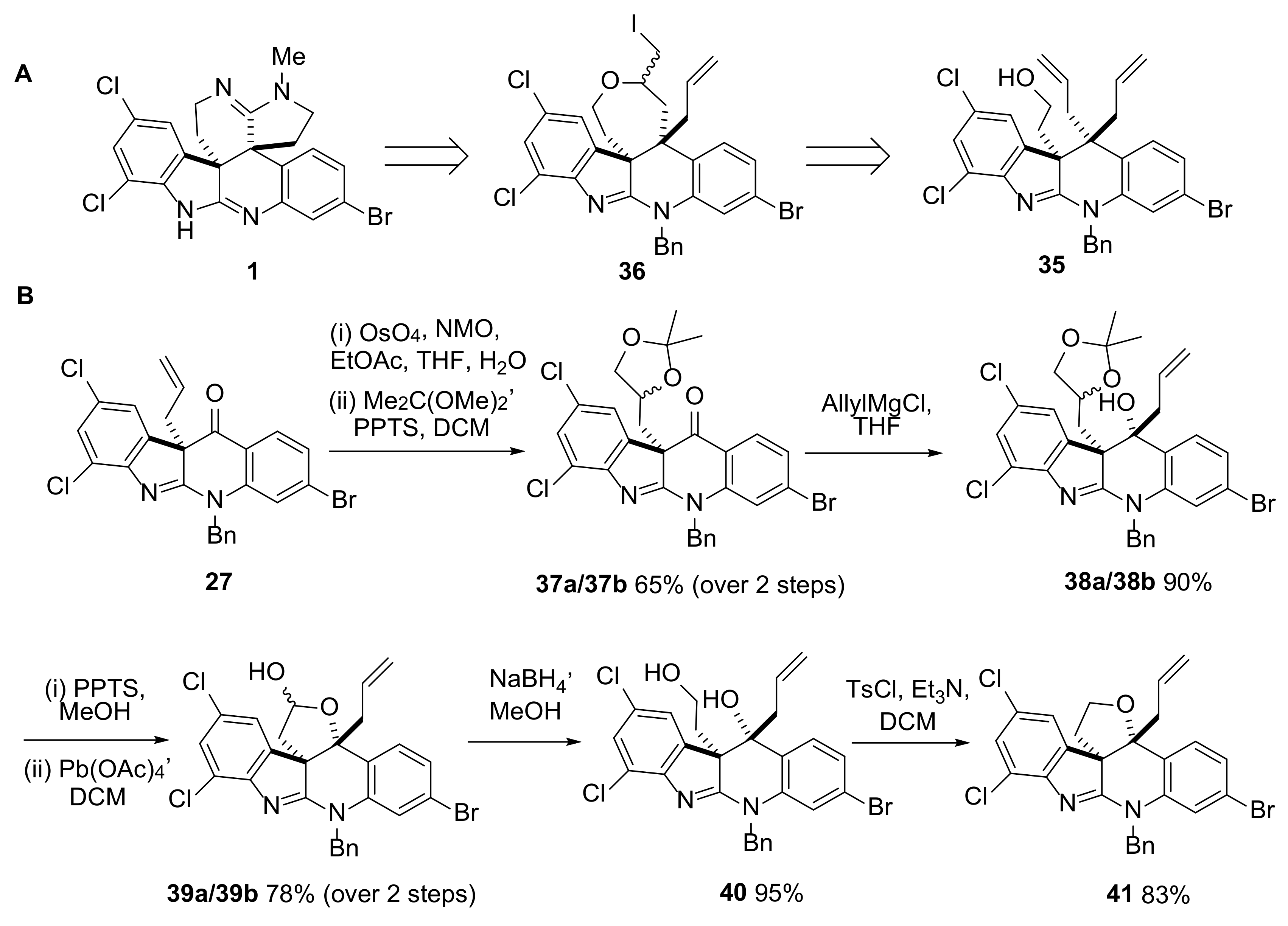
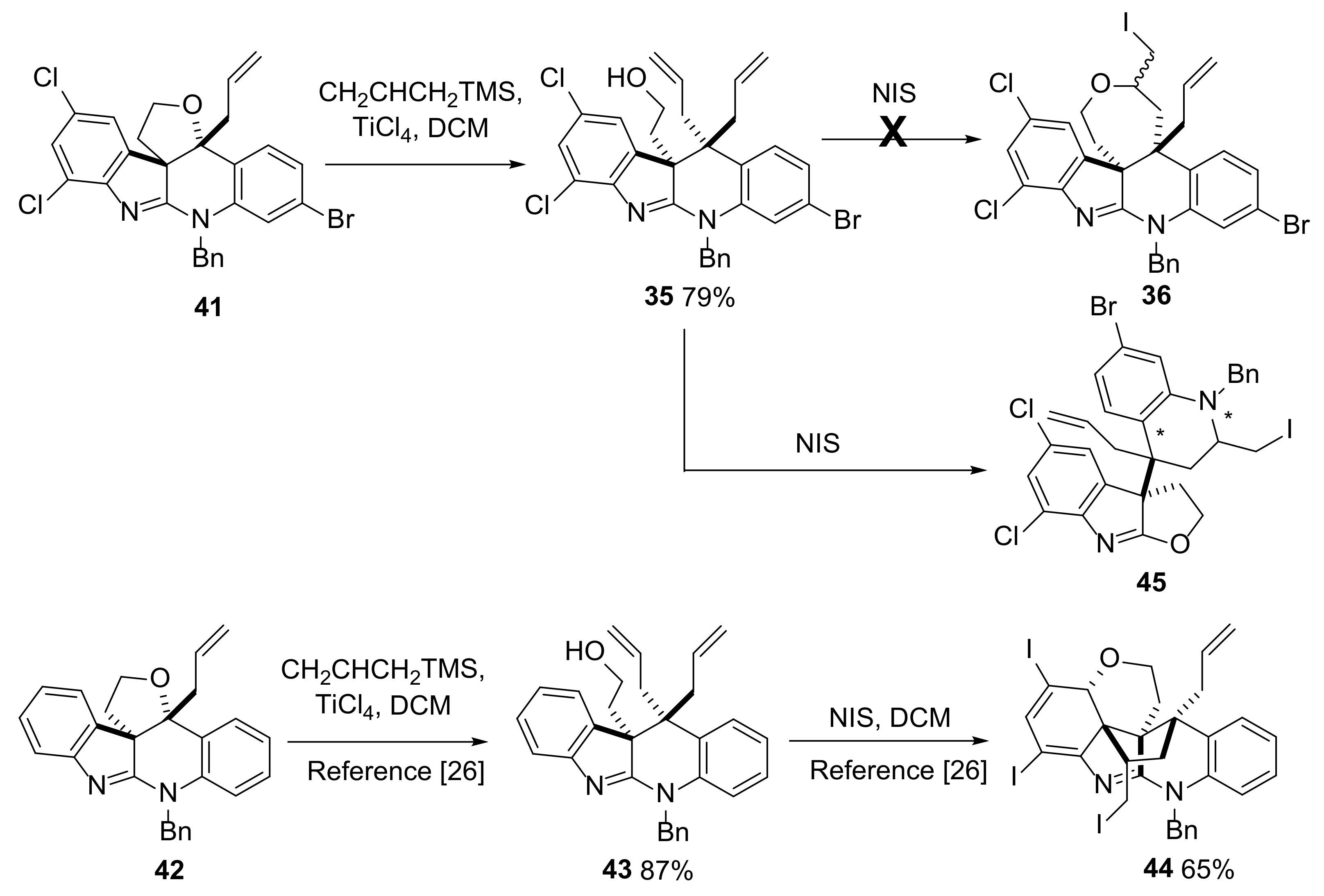
2.4. Attempted Progress towards Perophoramidine 1
3. Experimental
3.1. General Experimental Details
3.2. X-ray Structure Determination for 27 and 41
3.3. Preparation of Selected Compounds
3.3.1. N-Boc-5,7-dichloroindole-3-carbaldehyde 16
3.3.2. tert-Butyl 3-((4-bromo-2-nitrophenyl)(hydroxy)methyl)-5,7-dichloro-1H-indole-1-carboxylate 19
3.3.3. tert-Butyl 3-(4-bromo-2-nitrobenzoyl)-5,7-dichloro-1H-indole-1-carboxylate 20
3.3.4. (2-Amino-4-bromophenyl)(5,7-dichloro-1H-indol-3-yl)methanone 21
3.3.5. tert-Butyl 3-(2-amino-4-bromobenzoyl)-5,7-dichloro-1H-indole-1-carboxylate 22
3.3.6. (2-Benzylamino)-4-bromophenyl)(5,7-dichloro-1H-indol-3-yl)methanone 9
3.3.7. 5-Benzyl-3-bromo-7,9-dichloro-5H-indolo[2,3-b]quinolin-11(6H)-one 3
3.3.8. 5-Benzyl-3-bromo-7,9,11-trichloro-5H-indolo[2,3-b]quinoline 25
3.3.9. 11-(Allyloxy)-5-benzyl-3-bromo-7,9-dichloro-5H-indolo[2,3-b]quinoline 26
3.3.10. 10b-Allyl-5-benzyl-3-bromo-7,9-dichloro-5H-indolo[2,3-b]quinolin-11(10bH)-one 27
3.3.11. (E)-5-Benzyl-3-bromo-10b-(but-2-en-1-yl)-7,9-dichloro-5H-indolo[2,3-b]quinolin-11(10bH)-one 29
3.3.12. 5-Benzy-3-bromo-10b-((E)-but-2-en-1-yl)-7,9-dichloro-5,10b-dihydrospiro[indolo[2,3-b]quinoline-11,2′-oxirane] 31
3.3.13. (10bR,11R)-5-Benzyl-3-bromo-10b-((E)-but-2-en-1-yl)-7,9-dichloro-10b,11-dihydro-5H-indolo[2,3-b]quinolin-11-yl)methanol 32
3.3.14. (10bR,11R)-11-Allyl-5-benzyl-3-bromo-7,9-dichloro-10b-(2-hydroxyethyl)-10b,11-dihydro-5H-indolo[2,3-b]quinolin-11-ol 40
3.3.15. (3aR,13bR)-13b-Allyl-9-benzyl-11-bromo-5,7-dichloro-2,3,9,13b-tetrahydrofuro[3,2-c]indolo [2,3-b]quinoline 41
3.3.16. (R)-2-(11,11-Diallyl-5-benzyl-3-bromo-7,9-dichloro-10b,11-dihydro-5H-indolo[2,3-b]quinolin-10b-yl)ethanol 35
3.3.17. 3a-(4-Allyl-1-benzyl-7-bromo-2-(iodomethyl)-1,2,3,4-tetrahydroquinolin-4-yl)-5,7-dichloro-3,3a-dihydro-2H-furo[2,3-b]indole 45
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Verbitski, S.M.; Mayne, C.L.; Davis, R.A.; Concepcion, G.P.; Ireland, C.M. Isolation, structure determination, and biological activity of a novel alkaloid, perophoramidine, from the Philippine ascidian Perophora namei. J. Org. Chem. 2002, 67, 7124–7126. [Google Scholar] [CrossRef]
- Siengalewicz, P.; Gaich, T.; Mulzer, J. It all began with an error: The nomofungin/communesin story. Angew. Chem. Int. Ed. 2008, 47, 8170–8176. [Google Scholar] [CrossRef]
- Robinson, R.; Teuber, H.J. Reactions with nitrosodisulfonate. IV. Calycanthine and calycanthidine. Chem. Ind. 1954, 46, 783–784. [Google Scholar]
- Woodward, R.B.; Yang, N.C.; Katz, T.J.; Clark, V.M.; Harley-Mason, J.; Ingleby, R.F.; Shepard, N. Calycanthine: The structure of the alkaloid and its degradation product, calycanine. Proc. Chem. Soc. 1960, 76–78. [Google Scholar]
- Hendrickson, B.; Rees, R.; Goschke, R.R. Total synthesis of the calycanthaceous alkaloids. Chimonanthine. Proc. Chem. Soc. 1962, 383–384. [Google Scholar]
- Hamor, T.A.; Robertson, J.M.; Shrivastava, H.N.; Silverton, J.V. The structure of calycanthine. Proc. Chem. Soc. 1960, 78–80. [Google Scholar]
- Hamor, T.A.; Robertson, J.M. The structure of calycanthine. X-ray analysis of the dihydrobromide dehydrate. J. Chem. Soc. 1962, 194–205. [Google Scholar] [CrossRef]
- Grant, I.J.; Hamor, T.A.; Robertson, J.M.; Sim, G.A. Structure of chimonanthine. Proc. Chem. Soc. 1962, 148–149. [Google Scholar]
- Grant, I.J.; Hamor, T.A.; Robertson, J.M.; Sim, G.A. The structure of chimonanthine. X-ray analysis of chimonanthine dihydrobromide. J. Chem. Soc. 1965, 5678–5696. [Google Scholar] [CrossRef]
- Verotta, L.; Pilati, T.; Tato, M.; Elisabetsky, E.; Amador, T.A.; Nunes, D.S. Pyrrolidinoindoline Alkaloids from Psychotria colorata. J. Nat. Prod. 1998, 61, 392–396. [Google Scholar] [CrossRef]
- Fuchs, J.R.; Funk, R.L. Total Synthesis of (±)-Perophoramidine. J. Am. Chem. Soc. 2004, 126, 5068–5069. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xue, F.; Xiao, X.; Qin, Y. Total Synthesis of (+)-Perophoramidine and Determination of the Absolute Configuration. J. Am. Chem. Soc. 2010, 132, 14052–14053. [Google Scholar] [CrossRef]
- Zhang, H.; Hong, L.; Kang, H.; Wang, R. Construction of Vicinal All-Carbon Quaternary Stereocenters by Catalytic Asymmetric Alkylation Reaction of 3-Bromooxindoles with 3-Substituted Indoles: Total Synthesis of (+)-Perophoramidine. J. Am. Chem. Soc. 2013, 135, 14098–14101. [Google Scholar] [CrossRef]
- Han, S.-J.; Vogt, F.; May, J.A.; Krishnan, S.; Gatti, M.; Virgil, S.C.; Stoltz, B.M. Evolution of a Unified, Stereodivergent Approach to the Synthesis of Communesin F and Perophoramidine. J. Org. Chem. 2015, 80, 528–547. [Google Scholar] [CrossRef]
- Han, S.-J.; Vogt, F.; Krishnan, S.; May, J.A.; Gatti, M.; Virgil, S.C.; Stoltz, B.M. A Diastereodivergent Synthetic Strategy for the Syntheses of Communesin F and Perophoramidine. Org. Lett. 2014, 16, 3316–3319. [Google Scholar] [CrossRef]
- Trost, B.M.; Osipov, M.; Kruger, S.; Zhang, Y. A catalytic asymmetric total synthesis of (-)-perophoramidine. Chem. Sci. 2015, 6, 349–353. [Google Scholar] [CrossRef]
- Artman, G.D., III; Weinreb, S.M. An Approach to the Total Synthesis of the Marine Ascidian Metabolite Perophoramidine via a Halogen-Selective Tandem Heck/Carbonylation Strategy. Org. Lett. 2003, 5, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, Q.-R.; Huang, J.-R.; Li, Y.; Su, F.; Dong, L. The application of Morita-Baylis-Hillman reaction: Synthetic studies on perophoramidine. Tetrahedron 2017, 73, 3966–3972. [Google Scholar] [CrossRef]
- Sabahi, A.; Novikov, A.; Rainier, J.D. 2-Thioindoles as precursors to spiro-fused indolines: Synthesis of (±)-dehaloperophoramidine. Angew. Chem. Int. Ed. 2006, 45, 4317–4320. [Google Scholar] [CrossRef]
- Ishida, T.; Ikota, H.; Kurahashi, K.; Tsukano, C.; Takemoto, Y. Dearomatizing Conjugate Addition to Quinolinyl Amidines for the Synthesis of Dehaloperophoramidine through Tandem Arylation and Allylation. Angew. Chem. Int. Ed. 2013, 52, 10204–10207. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, R.P.; Neal, A.R.; Johnston, C.A.; Voute, N.; Lancefield, C.S.; Stell, M.D.; Medda, F.; Makiyi, E.F.; Turner, E.M.; Ojo, S.O.; et al. Total synthesis of dehaloperophoramidine using a highly diastereoselective Hosomi-Sakurai reaction. Chem. Commun. 2016, 52, 10747–10750. [Google Scholar] [CrossRef]
- Hoang, A.; Popov, K.; Somfai, P. An Efficient Synthesis of (±)-Dehaloperophoramidine. J. Org. Chem. 2017, 82, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Voute, N.; Philp, D.; Slawin, A.M.Z.; Westwood, N.J. Studies on the Claisen rearrangements in the indolo[2,3-b]quinoline system. Org. Biomol. Chem. 2010, 8, 442–450. [Google Scholar] [CrossRef] [PubMed]
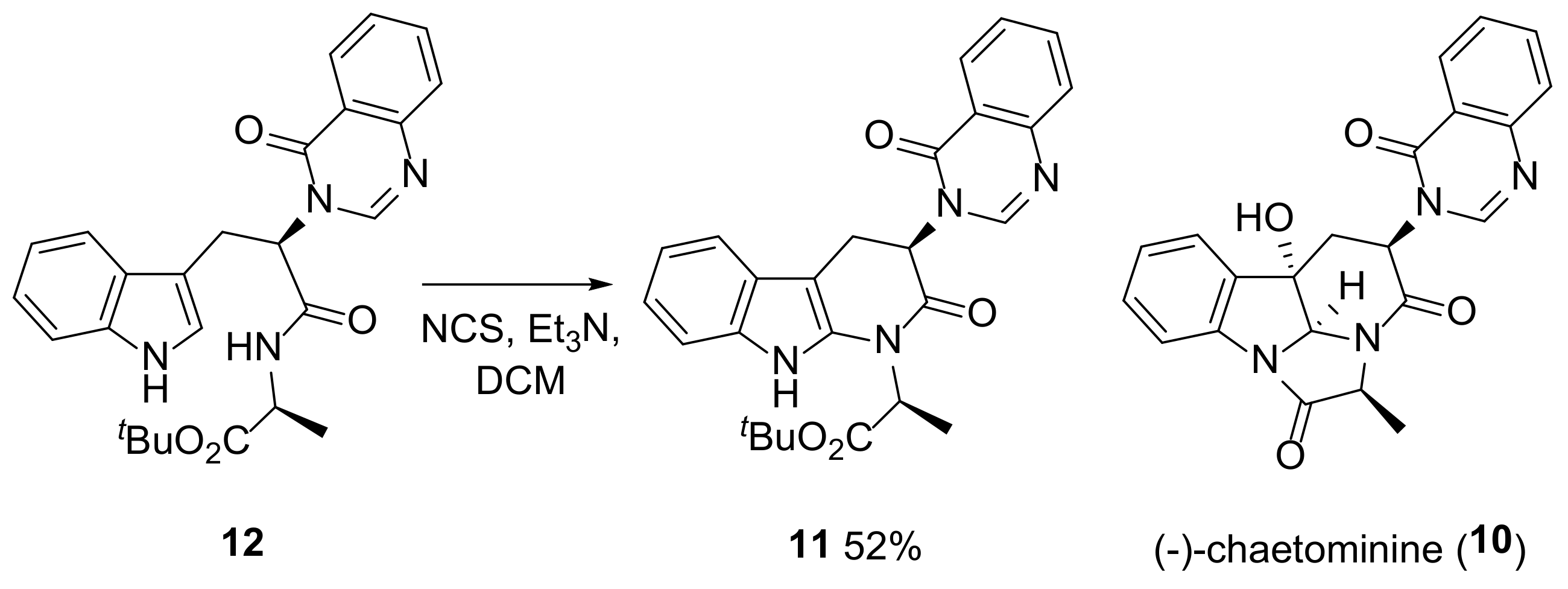
- Malgesini, B.; Forte, B.; Borghi, D.; Quartieri, F.; Gennari, C.; Papeo, G. A Straightforward Total Synthesis of (-)-Chaetominine. Chem. Eur. J. 2009, 15, 7922–7929. [Google Scholar] [CrossRef]
- Arcadi, A.; Bianchi, G.; Inesi, A.; Marinelli, F.; Rossi, L. Electrochemical-mediated cyclization of 2-alkynylanilines: A clean and safe synthesis of indole derivatives. Eur. J. Org. Chem. 2008, 5, 783–787. [Google Scholar] [CrossRef]
- Alfonsi, M.; Arcadi, A.; Aschi, M.; Bianchi, G.; Marinelli, F. Gold-Catalyzed Reactions of 2-Alkynyl-phenylamines with α,β-Enones. J. Org. Chem. 2005, 70, 2265–2273. [Google Scholar] [CrossRef]
- Bartoli, G.; Palmieri, G.; Bosco, M.; Dalpozzo, R. The reaction of vinyl Grignard reagents with 2-substituted nitroarenes: A new approach to the synthesis of 7-substituted indoles. Tetrahedron Lett. 1989, 30, 2129–2132. [Google Scholar] [CrossRef]
- Teng, X.; Degterev, A.; Jagtap, P.; Xing, X.; Choi, S.; Denu, R.; Yuan, J.; Cuny, G.D. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5039–5044. [Google Scholar] [CrossRef] [PubMed]
- Vilsmeier, A.; Haack, A. Über die einwirkung von halogenphosphor auf alkyl-formanilide. Eine neue methode zur darstellung sekundärer und tertiärer p-alkylamino-benzaldehyde. Eur. J. Org. Chem. 1927, 60, 119–122. [Google Scholar] [CrossRef]
- Flatt, A.K.; Yao, Y.; Maya, F.; Tour, J.M. Orthogonally Functionalized Oligomers for Controlled Self-Assembly. J. Org. Chem. 2004, 69, 1752–1755. [Google Scholar] [CrossRef]
- Sapountzis, I.; Knochel, P. General preparation of functionalized o-nitroarylmagnesium halides through an iodine-magnesium exchange. Angew. Chem. Int. Ed. 2002, 41, 1610–1611. [Google Scholar] [CrossRef]
- Sapountzis, I.; Dube, H.; Lewis, R.; Gommermann, N.; Knochel, P. Synthesis of Functionalized Nitroarylmagnesium Halides via an Iodine-Magnesium Exchange. J. Org. Chem. 2005, 70, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Griffith, W.P.; Ley, S.V.; Whitcombe, G.P.; White, A.D. Preparation and use of tetra-n-butylammonium per-ruthenate (TBAP reagent) and tetra-n-propylammonium per-ruthenate (TPAP reagent) as new catalytic oxidants for alcohols. Chem. Commun. 1987, 21, 1625–1627. [Google Scholar] [CrossRef]
- Boros, E.E.; Thompson, J.B.; Katamreddy, S.R.; Carpenter, A.J. Facile Reductive Amination of Aldehydes with Electron-Deficient Anilines by Acyloxyborohydrides in TFA: Application to a Diazaindoline Scale-Up. J. Org. Chem. 2009, 74, 3587–3590. [Google Scholar] [CrossRef]
- Mangette, J.E.; Chen, X.; Krishnamoorthy, R.; Samuel, V.A.; Csakai, A.J.; Camara, F.; Paquette, W.D.; Wang, H.-J.; Takahashi, H.; Fleck, R.; et al. 2-Trifluoroacetyl aminoindoles as useful intermediates for the preparation of 2-acylamino indoles. Tetrahedron Lett. 2011, 52, 1292–1295. [Google Scholar] [CrossRef]
- Newhouse, T.; Lewis, C.A.; Eastman, K.J.; Baran, P.S. Scalable Total Syntheses of N-Linked Tryptamine Dimers by Direct Indole-Aniline Coupling: Psychotrimine and Kapakahines B and F. J. Am. Chem. Soc. 2010, 133, 7119–7137. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.; Engqvist, R.; Stalhandske, C.; Wallberg, H. Studies of the reactions between indole-2,3-diones (isatins) and 2-aminobenzylamine. Tetrahedron 2003, 59, 1033–1048. [Google Scholar] [CrossRef]
- Coste, A.; Karthikeyan, G.; Couty, F.; Evano, G. Second-generation, biomimetic total synthesis of chaetominine. Synthesis 2009, 17, 2927–2934. [Google Scholar] [CrossRef]
- Ohno, M.; Spande, T.F.; Witkop, B. Cyclization of tryptophan and tryptamine derivatives to pyrrolo[2,3-b]indoles. J. Am. Chem. Soc. 1968, 90, 6521–6522. [Google Scholar] [CrossRef]
- Li, Y.; Dolphin, D.; Patrick, B.O. Synthesis of a BF2 complex of indol-2-yl-isoindol-1-ylidene-amine: A fully conjugated azadipyrromethene. Tetrahedron Lett. 2010, 51, 811–814. [Google Scholar] [CrossRef]
- Seo, J.H.; Artman, G.D., III; Weinreb, S.M. Synthetic Studies on Perophoramidine and the Communesins: Construction of the Vicinal Quaternary Stereocenters. J. Org. Chem. 2006, 71, 8891–8900. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Voute, N.; Neal, A.R.; Medda, F.; Johnston, C.A.; Slawin, A.M.Z.; Westwood, N.J. From one to two quaternary centers: Ester or nitrile α-alkylation applied to bioactive alkaloids. Tetrahedron 2018, 74, 7399–7407. [Google Scholar] [CrossRef]
- Bowden, K.; Heilbron, I.M.; Jones, E.R.H. 13. Researches on acetylenic compounds. Part I. The preparation of acetylenic ketones by oxidation of acetylenic carbinols and glycols. J. Chem. Soc. 1946, 39–45. [Google Scholar] [CrossRef]
- Johnston, C.A.; Wilkie, R.P.; Krauss, H.; Neal, A.R.; Slawin, A.M.Z.; Lebl, T.; Westwood, N.J. Polycyclic ethers and an unexpected dearomatisation reaction during studies towards the bioactive alkaloid, perophoramidine. Tetrahedron 2018, 74, 3339–3347. [Google Scholar] [CrossRef]
- Freeman-Cook, K.D.; Halcomb, R.L. A Symmetry-Based Formal Synthesis of Zaragozic Acid A. J. Org. Chem. 2000, 65, 6153–6159. [Google Scholar] [CrossRef] [PubMed]
- CrystalClear-SM Expert v2.0rc13; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2009.
- Beurskens, P.T.; Beurskens, G.; de Gelder, R.; Garcia-Granda, S.; Gould, R.O.; Israel, R.; Smits, J.M.M. DIRDIF-99; Crystallography Laboratory, University of Nijmegen: Nijmegen, The Netherlands, 1999. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- CrystalStructure v4.3.0; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2018.











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnston, C.A.; Cordes, D.B.; Lebl, T.; Slawin, A.M.Z.; Westwood, N.J. Synthesis of Indoloquinolines: An Intramolecular Cyclization Leading to Advanced Perophoramidine-Relevant Intermediates. Molecules 2021, 26, 6039. https://doi.org/10.3390/molecules26196039
Johnston CA, Cordes DB, Lebl T, Slawin AMZ, Westwood NJ. Synthesis of Indoloquinolines: An Intramolecular Cyclization Leading to Advanced Perophoramidine-Relevant Intermediates. Molecules. 2021; 26(19):6039. https://doi.org/10.3390/molecules26196039
Chicago/Turabian StyleJohnston, Craig A., David B. Cordes, Tomas Lebl, Alexandra M. Z. Slawin, and Nicholas J. Westwood. 2021. "Synthesis of Indoloquinolines: An Intramolecular Cyclization Leading to Advanced Perophoramidine-Relevant Intermediates" Molecules 26, no. 19: 6039. https://doi.org/10.3390/molecules26196039
APA StyleJohnston, C. A., Cordes, D. B., Lebl, T., Slawin, A. M. Z., & Westwood, N. J. (2021). Synthesis of Indoloquinolines: An Intramolecular Cyclization Leading to Advanced Perophoramidine-Relevant Intermediates. Molecules, 26(19), 6039. https://doi.org/10.3390/molecules26196039