
Concise Large-Scale Synthesis of Tomatidine, A Potent Antibiotic Natural Product


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
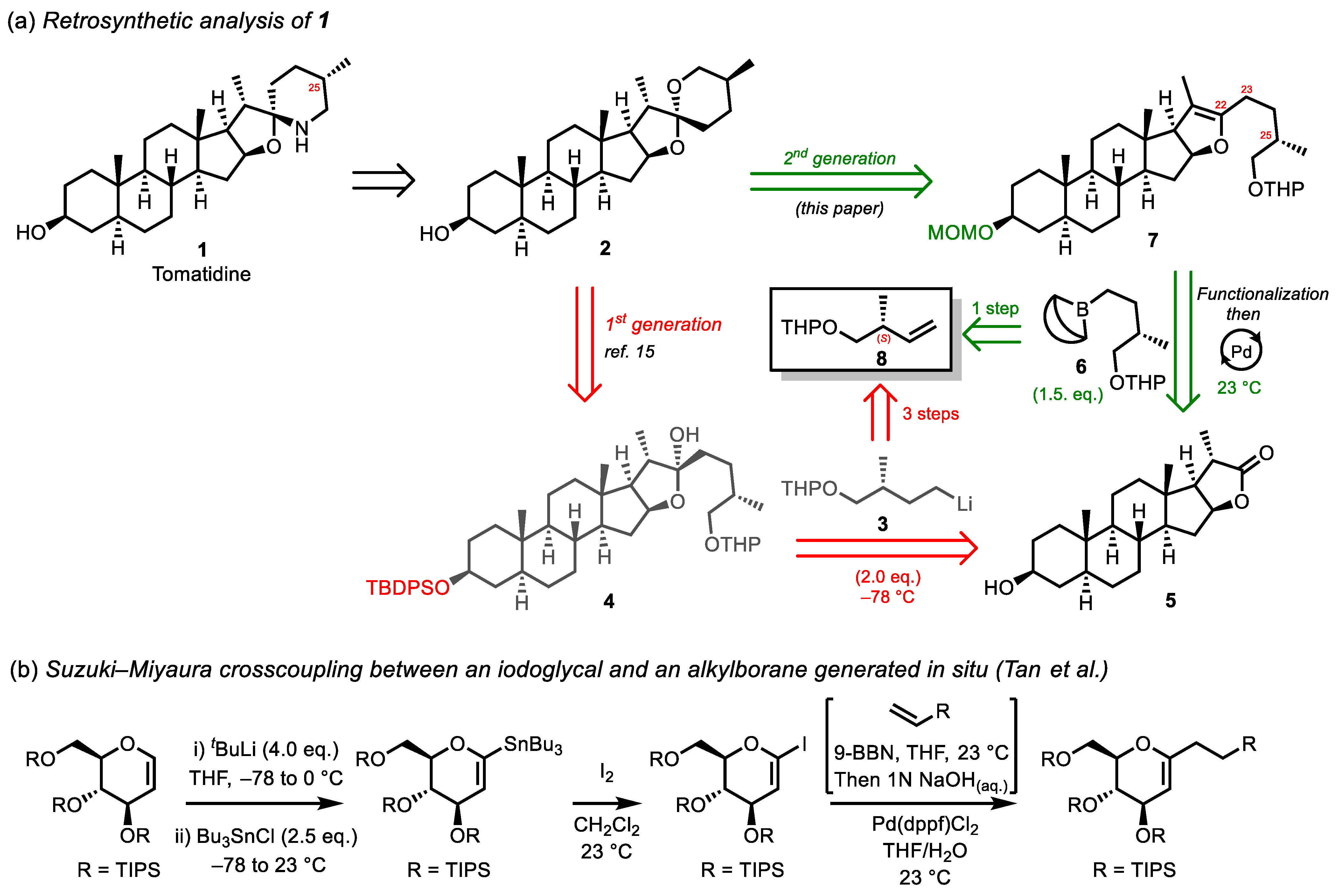
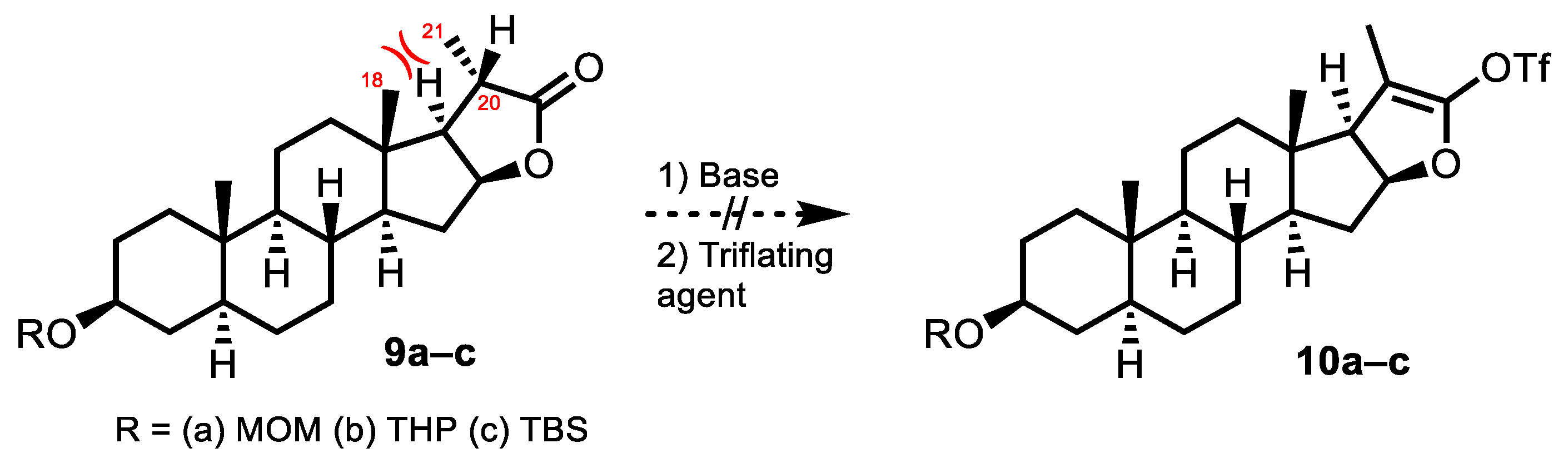
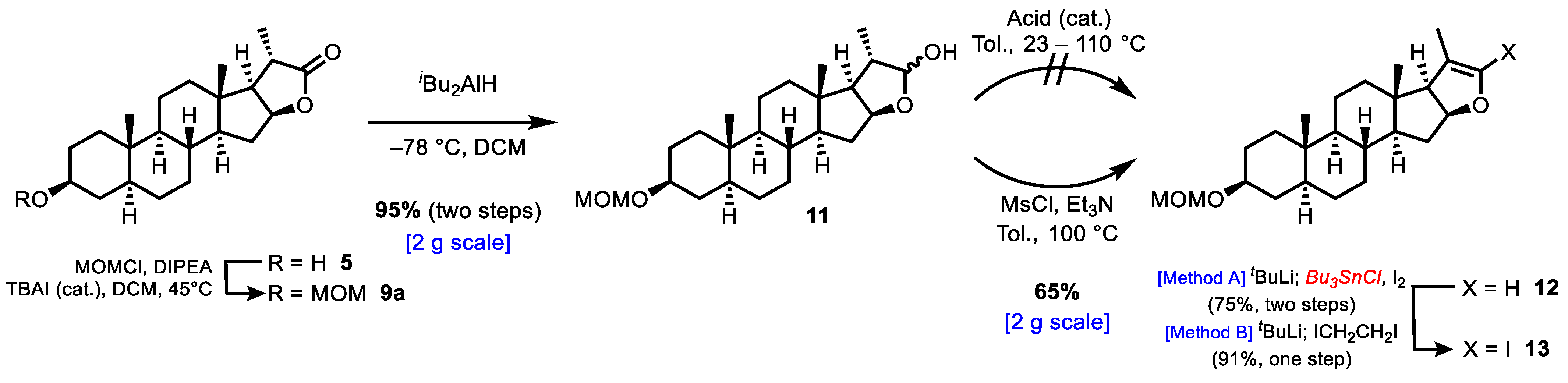
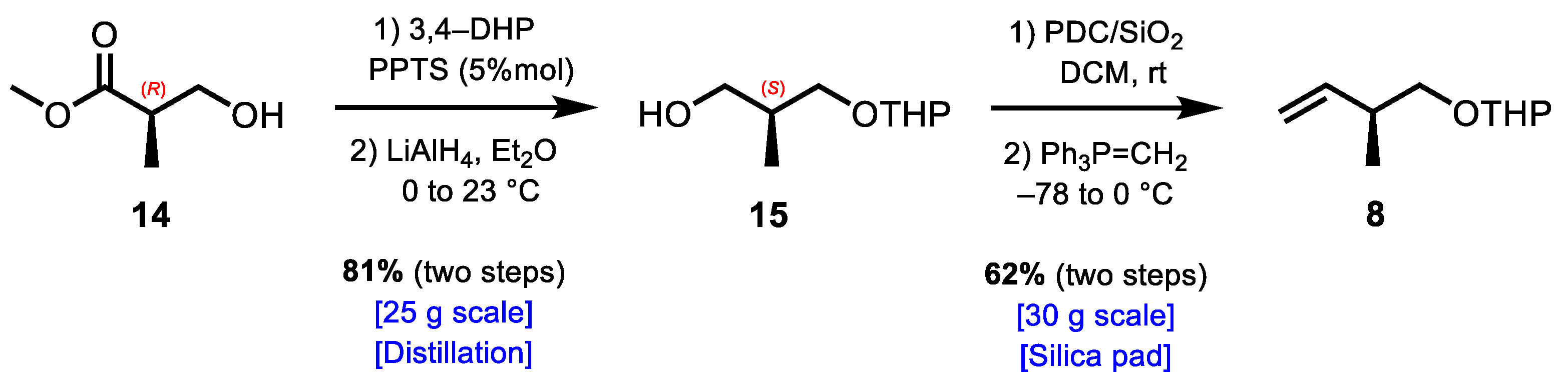
2. Results and Discussion
3. Experimental Section
3.1. General Remarks
3.2. Methods
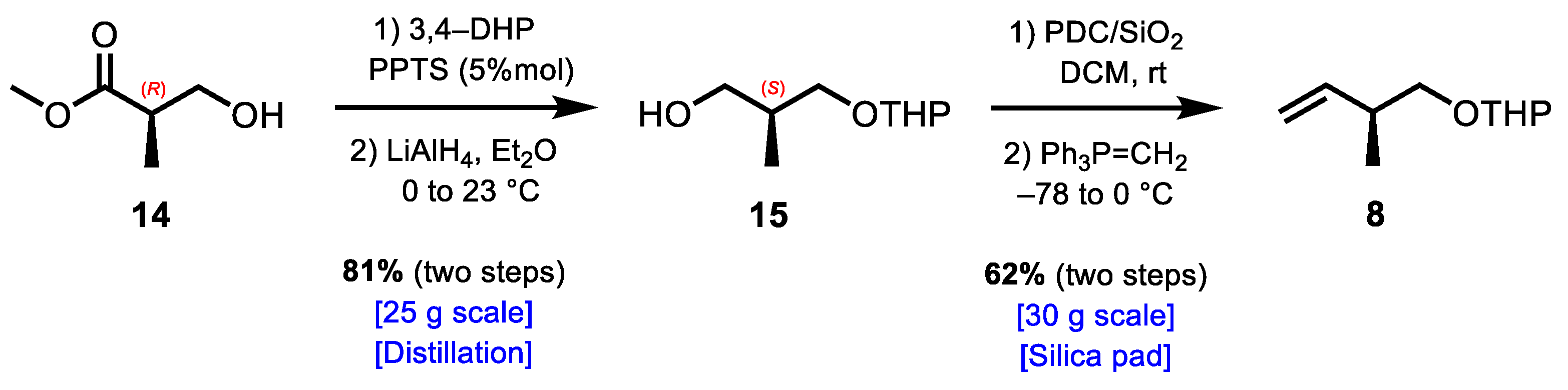
3.2.1. Synthesis of (2S)-2-Methyl-3-((tetrahydro-2H-pyran-2-yl)oxy)propan-1-ol (15)
3.2.2. Synthesis of 2-(((S)-2-Methylbut-3-en-1-yl)oxy)tetrahydro-2H-pyran (8)
3.2.3. Synthesis of (2aS,4S,6aS,6bS,8aS,8bR,9S,11aS,12aS,12bR)-4-(Methoxymethoxy)-6a,8a,9-trimethyloctadecahydro-10H-naphtho[2′,1′:4,5]indeno[2,1-b]furan-10-one (9a)
3.2.4. Synthesis of (2aS,4S,6aS,6bS,8aS,8bR,9S,11aS,12aS,12bR)-4-(Methoxymethoxy)-6a,8a,9-trimethyloctadecahydro-1H-naphtho[2′,1′:4,5]indeno[2,1-b]furan-10-ol (11)
3.2.5. Synthesis of (2aS,4S,6aS,6bS,8aS,8bS,11aS,12aS,12bR)-4-(Methoxymethoxy)-6a,8a,9-trimethyl 2,2a,3,4,5,6,6a,6b,7,8,8a,8b,11a,12,12a, 12b-hexadecahydro-1H-naphtho[2′,1′:4,5]indeno[2,1-b]furan (12)
3.2.6. Synthesis of (2aS,4S,6aS,6bS,8aS,8bS,11aS,12aS,12bR)-10-Iodo-4-(methoxymethoxy)-6a,8a,9-trimethyl-2,2a,3,4,5,6,6a,6b,7,8,8a,8b,11a, 12,12a,12b-hexadecahydro-1H-naphtho[2′,1′:4,5]indeno[2,1-b]furan (13)
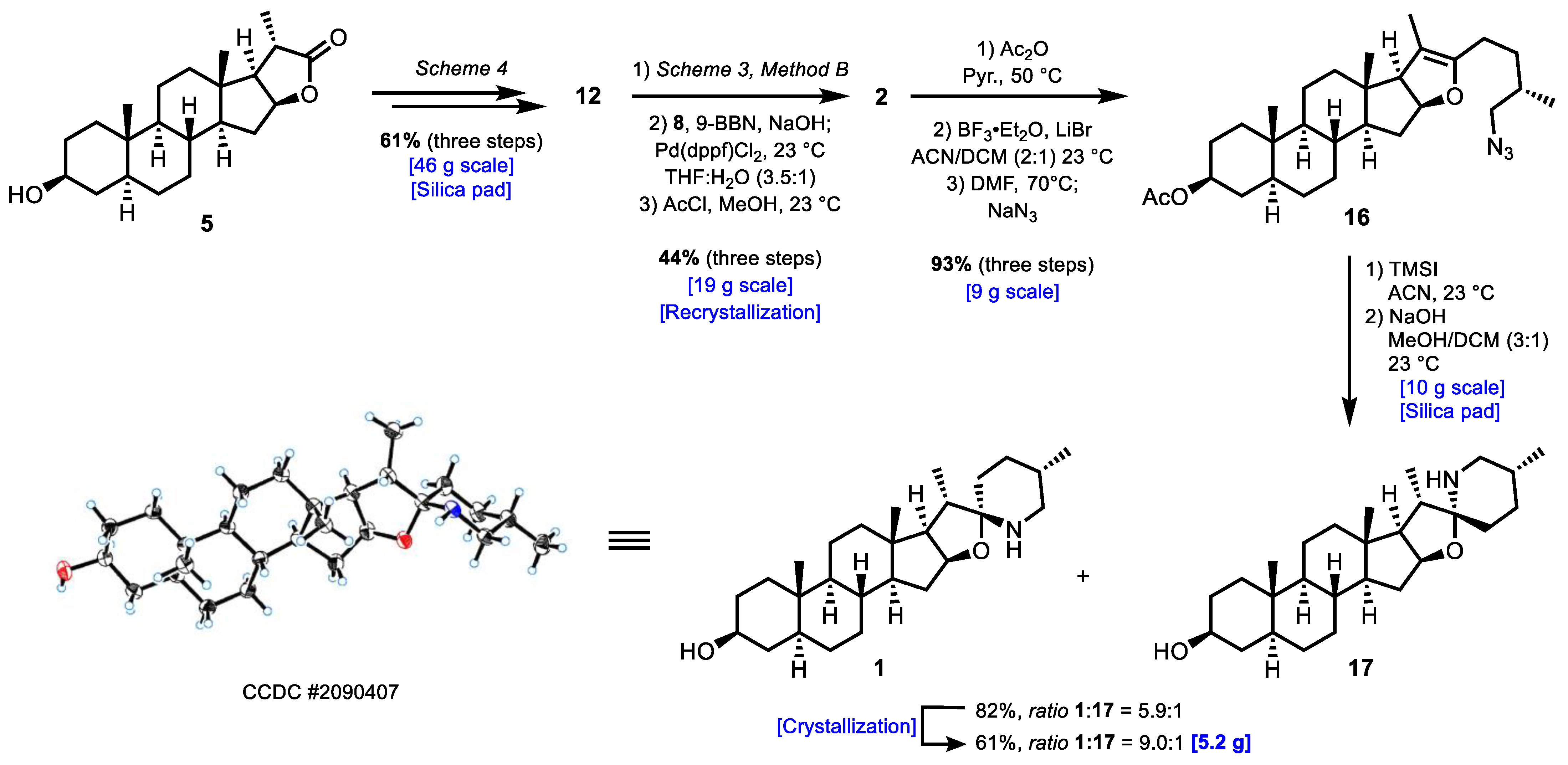
3.2.7. Synthesis of (2aS,4S,6aS,6bS,8aS,8bS,11aS,12aS,12bR)-4-(Methoxymethoxy)-6a,8a,9-trimethyl-10-((3S)-3-methyl-4-((tetrahydro-2H-pyran-2-yl)oxy)butyl)-2,2a,3,4,5,6,6a,6b,7,8,8a,8b,11a,12,12a,12b-hexadecahydro-1H-naphtho[2′,1′:4,5]indeno[2,1-b]furan (7)
3.2.8. Synthesis of (3β,5α,25S)-Spirostan-3-ol (2)
3.2.9. Synthesis of (2aS,4S,6aS,6bS,8aS,8bS,11aS,12aS,12bR)-10-((S)-4-Azido-3-methylbutyl)-6a,8a,9-trimethyl-2,2a,3,4,5,6,6a,6b,7,8,8a,8b,11a,12,12a,12b-hexadecahydro-1H-naphtho[2′,1′:4,5]indeno[2,1-b]furan-4-yl acetate (16)
3.2.10. Synthesis of Tomatidine (1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Dedication
Sample Availability
References and Note
- Kaunda, J.S.; Zhang, Y.-J. The Genus Solanum: An Ethnopharmacological, Phytochemical and Biological Properties Review. Nat. Prod. Bioprospect. 2019, 9, 77–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heretsch, P.; Giannis, A. The Vetratrum and Solanum Alkaloids. Alkaloids Chem Biol. 2015, 74, 201–232. [Google Scholar]
- Dyle, M.C.; Ebert, S.M.; Cook, D.P.; Kunkel, S.D.; Fox, D.K.; Bongers, K.S.; Bullard, S.A.; Dierdorff, J.M.; Adams, C.M. Systems-based Discovery of Tomatidine as a Natural Small Molecule Inhibitor of Skeletal Muscle Atrophy. J. Biol. Chem. 2014, 289, 14913–14924. [Google Scholar] [CrossRef] [Green Version]
- Koduru, S.; Jimoh, F.O.; Grierson, D.S.; Afolayan, A.J. Antioxidant Activity of Two Steroid Alkaloids Extracted from Solanum aculeastrum. J. Pharm. Toxicol. 2007, 2, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Chiu, F.-L.; Lin, J.-K. Tomatidine inhibits iNOS and COX-2 through suppression of NF-κB and JNK pathways in LPS-stimulated mouse macrophages. FEBS Lett. 2008, 582, 2407–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troost, B.; Mulder, L.M.; Diosa-Toro, M.; van de Pol, D.; Rodenhuis-Zybert, I.A.; Smit, J.M. Tomatidine, a natural steroidal alkaloid shows antiviral activity towards chikungunya virus in vitro. Sci. Rep. 2020, 10, 6364–6375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diosa-Toro, M.; Troost, B.; van de Pol, D.; Heberle, A.M.; Urcuqui-Inchima, S.; Thedieck, K.; Smit, J.M. Tomatidine, a Novel Antiviral Compounds Towards Dengue Virus. Antiviral Res. 2019, 161, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.-H.; Lee, L.-M.; Yan, S.-H.; Huang, H.-C.; Li, C.-C.; Lin, H.-T.; Chen, P.-S. Tomatidine Inhibits Invasion of Human Lung Adenocarcinoma Cells A549 by Reducing Matrix Metalloproteinases Expression. Chem.-Biol. Interact. 2013, 203, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Guay, I.; Boulanger, S.; Isabelle, C.; Brouillette, E.; Chagnon, F.; Bouarab, K.; Marsault, E.; Malouin, F. Tomatidine and Analog FC04-100 Possess Bactericidal Activities Against Listeria, acillus and Staphylococcus spp. BMC Pharmacol. Toxicol. 2018, 19, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, G.; Gattuso, M.; Grondin, G.; Marsault, É.; Bouarab, K.; Malouin, F. Tomatidine Inhibits Replication of Staphylococcus aureus Small-Colony Variants in Cystic Fibrosis Airway Epithelial Cells. Antimicrob. Agents Chemother. 2011, 55, 1937–1945. [Google Scholar] [CrossRef] [Green Version]
- Boulet, M.L.; Isabelle, C.; Guay, I.; Brouillette, E.; Langlois, J.-P.; Jacques, P.-E.; Rodrigue, S.; Brzezinski, R.; Beauregard, P.B.; Bouarab, K.; et al. Tomatidine Is a Lead Molecule That Targets Staphylococcus aureus ATP Synthase Subunic C. Antimicrob. Agents Chemother. 2018, 62, e02197-17. [Google Scholar]
- Chagnon, F.; Guay, I.; Bonin, M.-A.; Mitchell, G.; Bouarab, K.; Malouin, F.; Marsault, E. Unraveling the Structure-Activity Relationship of Tomatidine, A Steroid Alkaloid With Unique Antibiotic Properties Against Persistent Forms of Staphylococcus aureus. Eur. J. Med. Chem. 2014, 80, 605–620. [Google Scholar] [CrossRef]
- Mitchell, G.; Lafrance, M.; Boulanger, S.; Seguin, D.L.; Guay, I.; Gattuso, M.; Marsault, E.; Bouarab, K.; Malouin, F. Tomatidine Acts in Synergy with Aminoglycoside Antibiotics against Multiresistant Staphylococcus aureus and Prevents Virulence Gene Expression. J. Antimicrob. Chemother. 2012, 67, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Kuttruff, C.A.; Eastgate, M.D.; Baran, P.S. Natural Product Synthesis in The Age of Scalability. Nat. Prod. Rep. 2014, 31, 419–432. [Google Scholar] [CrossRef]
- Normandin, C.; Malouin, F.; Marsault, E. Gram-Scale Synthesis of Tomatidine, a Steroid Alkaloid with Antibiotic Properties Against Persistent Forms of Staphylococcus aureus. Eur. J. Org. Chem. 2020, 2693–2698. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, G.; Shi, Y.; Tian, W.-S.; Zhuang, C.; Chen, F.-E. Asymmetric Synthesis of (−)-Solanidine and (−)-Tomatidenol. Org. Biomol. Chem. 2020, 18, 3169–3176. [Google Scholar] [CrossRef] [PubMed]
- Gaich, T.; Baran, P. Aiming for the Ideal Synthesis. J. Org. Chem. 2010, 75, 4657–4673. [Google Scholar] [CrossRef] [PubMed]
- Budarin, V.L.; Shuttleworth, P.S.; Clark, J.H.; Luque, R. Industrial Application of C-C Coupling Reactions. Curr. Org. Synth. 2011, 7, 614–627. [Google Scholar] [CrossRef]
- Potuzak, J.S.; Tan, D.S. Synthesis of C1-Alkyl and Acylglycals from Glycals Using a B-alkyl Suzuki-Miyaura Cross Coupling Approach. Tetrahedron Lett. 2004, 45, 1797–1801. [Google Scholar] [CrossRef]
- Wang, S.-S.; Shi, Y.; Tian, W.-S. Highly Efficient and Scalable Synthesis of Clionamide D. Org. Lett. 2014, 16, 2177–2179. [Google Scholar] [CrossRef]
- Shah, H.J.; Lele, S.S. Extraction of Diosgenin, a Bioactive Compound from Natural Source Dioscorea alata Var purpurae. J. Anal. Bioanal. Tech. 2012, 3, 141–143. [Google Scholar] [CrossRef] [Green Version]
- Rafaniello, A.A.; Rizzacasa, M.A. Total Synthesis of (+)-Trachyspic Acid 19-n-Butyl Ester. Org. Lett. 2020, 22, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, K.; Araki, K.; Murai, A. Conversion of Lactones into Substituted Cyclic Ethers. Chem. Lett. 1989, 18, 1313–1316. [Google Scholar] [CrossRef]
- Boeckman, R.K.; Bruza, K.J. Cyclic Vinyl Ether Carbanions I: Synthetic Equivalents of β-acylvinyl and Substituted Acyl Anions. Tetrahedron Lett. 1977, 18, 4187–4190. [Google Scholar] [CrossRef]
- Boeckman, R.K.; Bruza, K.J. Cyclic Vinyl Ether Carbanions—II: Preparation and Applications to The Synthesis of Carbonyl Compounds. Tetrahedron 1981, 37, 3997–4006. [Google Scholar] [CrossRef]
- Torisawa, Y.; Shibasaki, M.; and Ikegami, S. Novel Reactivities on tert-Butyldimethylsilyl and tert-Butyldiphenylsilyl Ethers; Application to the Synthesis of 11-epi-PGF2a. Chem. Pharm. Bull. 1983, 31, 2607–2615. [Google Scholar] [CrossRef] [Green Version]
- Franke, F.; Guthrie, R.D. 6,6'-Di-O-t-Butyldimethylsilylsucrose: Studies on the Rearrangements Accompanying Deblocking of Such Silyl Ethers. Aust. J. Chem. 1978, 31, 1285–1290. [Google Scholar] [CrossRef]
- Shi, Y.; Xiao, Q.; Lan, Q.; Wang, D.-H.; Jia, L.-Q.; Tang, X.-H.; Zhou, T.; Li, M.; Tian, W.-S. A Synthesis of Cephalostatin 1. Tetrahedron 2018, 75, 1722–1738. [Google Scholar] [CrossRef]
- Zenk, P.C.; Wiley, R.A. A General Synthesis for 4-Alkyl-2,3-dihydrofurans. Synthesis 1984, 8, 695–697. [Google Scholar] [CrossRef]
- Garnsey, M.R.; Slutskyy, Y.; Jamison, C.R.; Zhao, P.; Lee, J.; Rhee, Y.H.; Overman, L.E. Short Enantioselective Total Syntheses of Cheloviolenes A and B and Dendrillolide C via Convergent Fragment Coupling Using a Tertiary Carbon Radical. J. Org. Chem. 2017, 83, 6958–6976. [Google Scholar] [CrossRef]
- Friesen, R.W.; Loo, R.W. Preparation of C-aryl Glucals Via the Palladium-Catalyzed Coupling of Metalated Aromatics With 1-iodo-3,4,6-tri-O-(triisopropylsilyl)-D-glucal. J. Org. Chem. 1991, 56, 4821–4823. [Google Scholar] [CrossRef]
- Paquette, L.A.; Lanter, J.C.; Johnston, J.N. Single Stereodifferentiation Associated with Carbon Atom Insertion during the Oxonium Ion-Initiated Pinacol Rearrangement of Dihydrofuranyl and Dihydropyranyl Carbinols. J. Org. Chem. 1997, 62, 1702–1712. [Google Scholar] [CrossRef]
- Genicot, C.; Ley, S.V. Dispiroketals in Synthesis (Part 15): Simultaneous Protection and Enantioselective Desymmetrisation of Glycerol by Reaction with a C2-Symmetric Dimethyl bis-Dihydropyran. Synthesis 1994, 12, 1275–1277. [Google Scholar] [CrossRef]
- Denmark, S.E.; Kobayashi, T.; Regens, C.S. Total Synthesis of (+)-Papulacandin D. Tetrahedron 2010, 66, 4745–4759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, S.V.; Tackett, M.N.; Maddess, M.L.; Anderson, J.C.; Brennan, P.E.; Cappi, M.W.; Heer, J.P.; Helgen, C.; Kori, M.; Kouklovsky, C.; et al. Total Synthesis of Rapamycin. Chem. Eur. J. 2009, 15, 2874–2914. [Google Scholar] [CrossRef]
- Luzzio, F.A.; Fitch, R.W.; Moore, W.J.; Mudd, K.J. A Facile Oxidation of Alcohols Using Pyridinium Chlorochromate/Silica Gel. J. Chem. Educ. 1999, 76, 974–975. [Google Scholar] [CrossRef]
- Smith, A.B.; Lin, Q.; Doughty, V.A.; Zhuang, L.; McBriar, M.D.; Kerns, J.K.; Boldi, A.M.; Murase, N.; Moser, W.H.; Brook, C.S.; et al. Spongipyran synthetic studies. Total Synthesis of (+)-Spongistatin 2. Tetrahedron 2009, 65, 6470–6488. [Google Scholar] [CrossRef] [Green Version]
- Tobari, A.; Teshima, M.; Koyanagi, J.; Kawase, M.; Miyamae, H.; Yoza, K.; Takasaki, A.; Nagamura, A.; Saito, S. Spirostanols Obtained by Cyclization of Pseudosaponin Derivatives and Comparison of Anti-Platelet Agglutination Activities of Spirostanol Glycosides. Eur. J. Med. Chem. 2000, 35, 511–527. [Google Scholar] [CrossRef]
- Kemsley, J.N. Learning from UCLA. Chem. Eng. News 2009, 87, 29–34. [Google Scholar] [CrossRef]
- Wu, J.-J.; Shi, Y.; Tian, W.-S. Facile synthesis of solasodine based on a mild halogenation-ring opening reaction of spiroketals in steroidal sapogenins. Tetrahedron Lett. 2015, 56, 1215–1217. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Guo, T.; Guan, H.; Hao, Y.; Yu, B. Synthesis of the Cytotoxic Gitogenin 3β-O-[2-O-(α-l-Rhamnopyranosyl)-β-d-galactopyranoside] and its Congeners. Synthesis 2006, 5, 775–782. [Google Scholar] [CrossRef]
- Watson, S.C.; Eastham, J.F. Colored Indicators for Simple Direct Titration of Magnesium and Lithium Reagents. J. Organomet. Chem. 1967, 9, 165–168. [Google Scholar] [CrossRef]
- Molander, G.A.; Etter, J.B.; Zinka, P.W. Stereocontrolled Cyclization Reactions Mediated by Samarium Diiodide. J. Am. Chem. Soc. 1987, 109, 453–463. [Google Scholar] [CrossRef]
- The conversion was calculated by integration of the H22 shift at δ = 6.00 ppm.
- Callow, R.K.; James, V.H.T. Epimerisation at C(25) of Steroid Sapogenins: Sarsasapogenin, Neotigogenin, and Sisalagenin. J. Chem. Soc. 1955, 1671–1674. [Google Scholar] [CrossRef]
- Tschesche, R.; Wulff, G.; Balle, G. Über saponine der spirostanolreihe—VIII: Über das gemeinsame vorkommen von 25α- und 25β-sapogeninen in den saponinen von digitalis purpurea L. und digitalis lanata ehrh. Tetrahedron 1962, 18, 959–967. [Google Scholar] [CrossRef]
- Sato, Y.; Latham Jr, H.G. Chemistry of Dihydrotomatidines. J. Am. Chem. Soc. 1956, 78, 3146–3150. [Google Scholar] [CrossRef]
- Matsushita, S.; Yanai, Y.; Fusyuku, A.; Fujiwara, Y.; Ikeda, T.; Ono, M.; Han, C.; Ojika, M.; Nohara, T. Efficient Conversion of Tomatidine into Neuritogenic Pregnane Derivative. Chem. Pharm. Bull. 2007, 55, 1077–1078. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Normandin, C.; Boudreault, P.-L. Concise Large-Scale Synthesis of Tomatidine, A Potent Antibiotic Natural Product. Molecules 2021, 26, 6008. https://doi.org/10.3390/molecules26196008
Normandin C, Boudreault P-L. Concise Large-Scale Synthesis of Tomatidine, A Potent Antibiotic Natural Product. Molecules. 2021; 26(19):6008. https://doi.org/10.3390/molecules26196008
Chicago/Turabian StyleNormandin, Chad, and Pierre-Luc Boudreault. 2021. "Concise Large-Scale Synthesis of Tomatidine, A Potent Antibiotic Natural Product" Molecules 26, no. 19: 6008. https://doi.org/10.3390/molecules26196008
APA StyleNormandin, C., & Boudreault, P.-L. (2021). Concise Large-Scale Synthesis of Tomatidine, A Potent Antibiotic Natural Product. Molecules, 26(19), 6008. https://doi.org/10.3390/molecules26196008