
Study on the CID Fragmentation Pathways of Deprotonated 4’-Monophosphoryl Lipid A


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. MS2 Mass Spectra of the [M − H]− Lipid A Precursor Ions
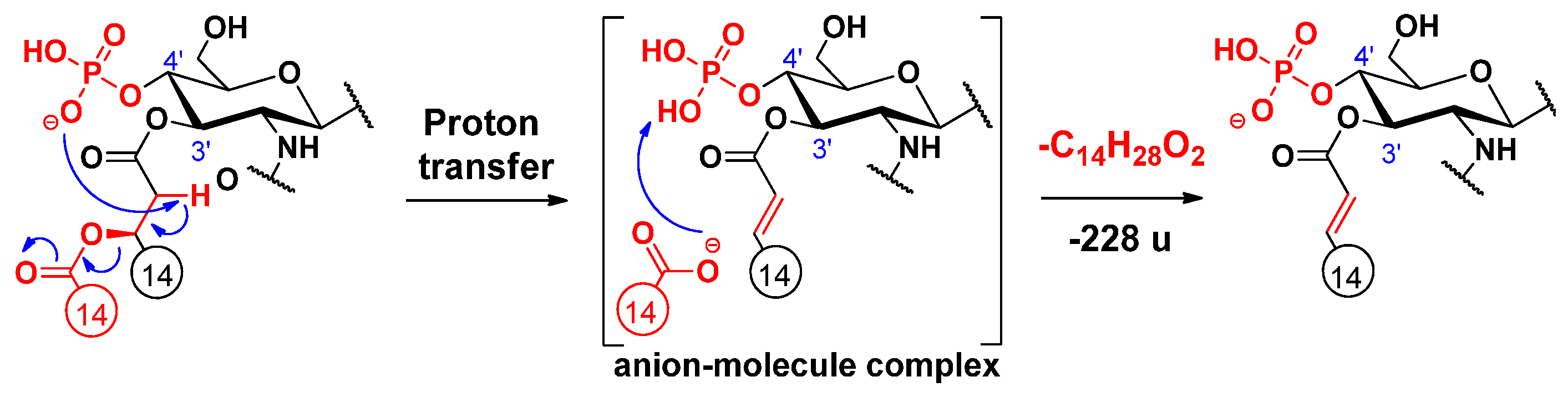
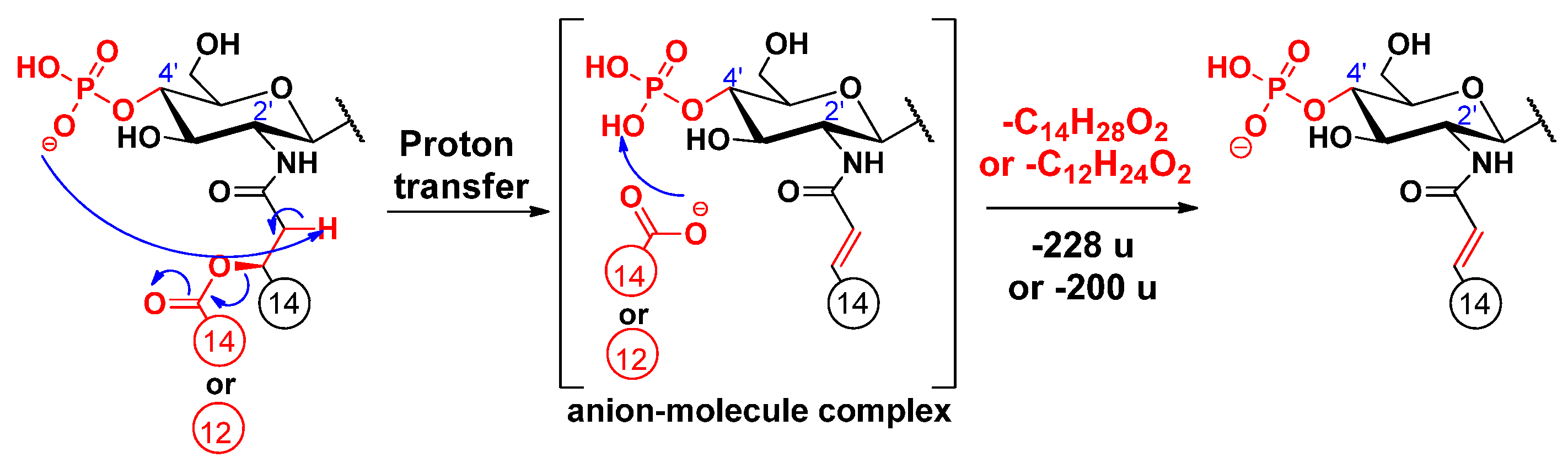
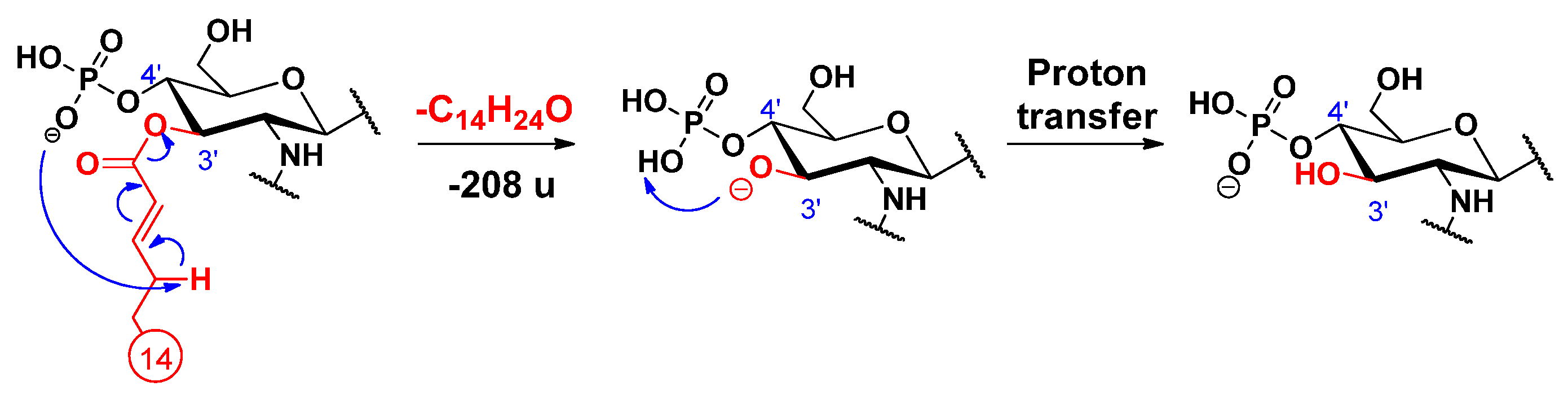
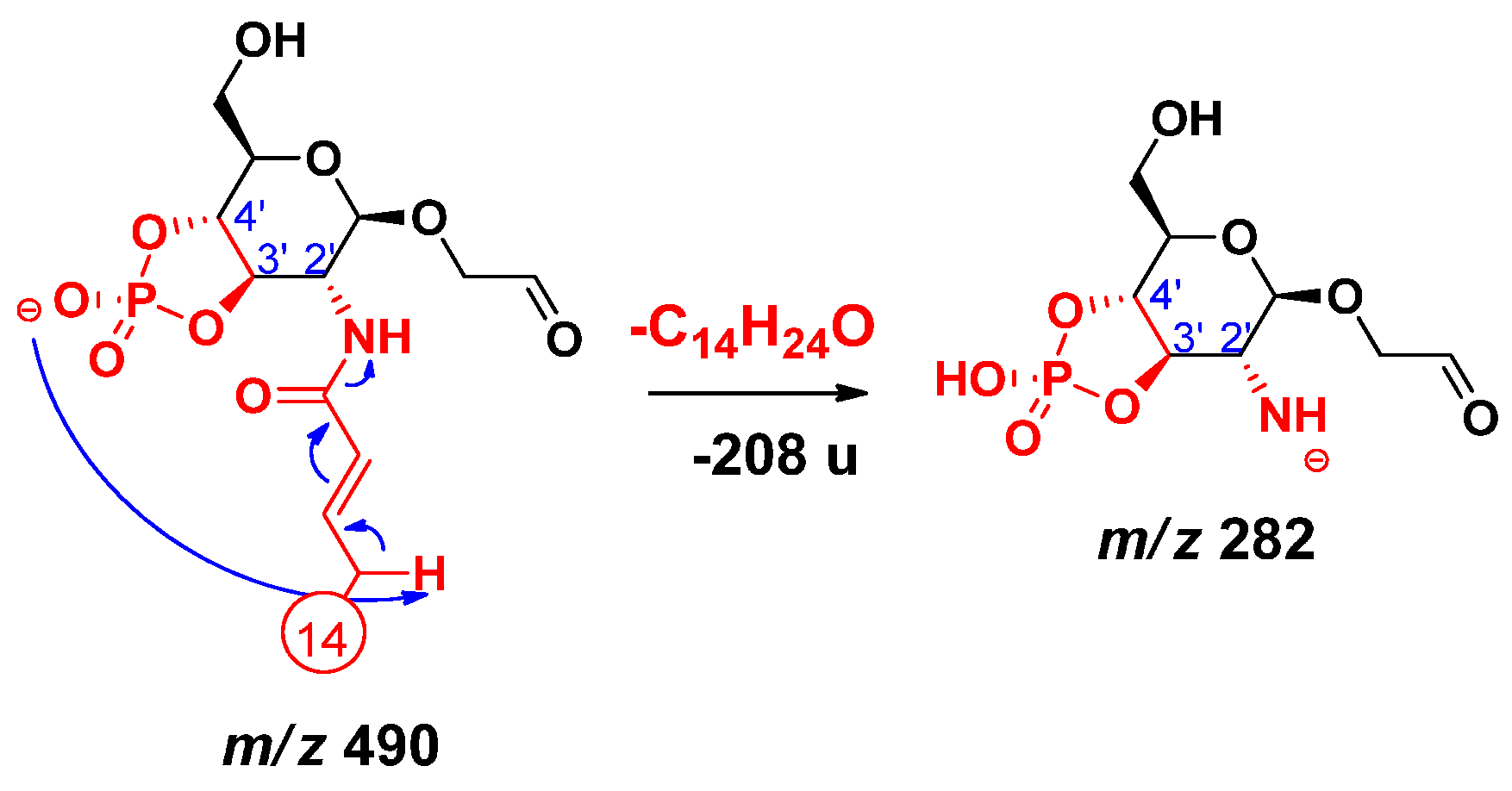
2.2. Proposed Mechanisms for the Release of the Secondary Fatty Acids
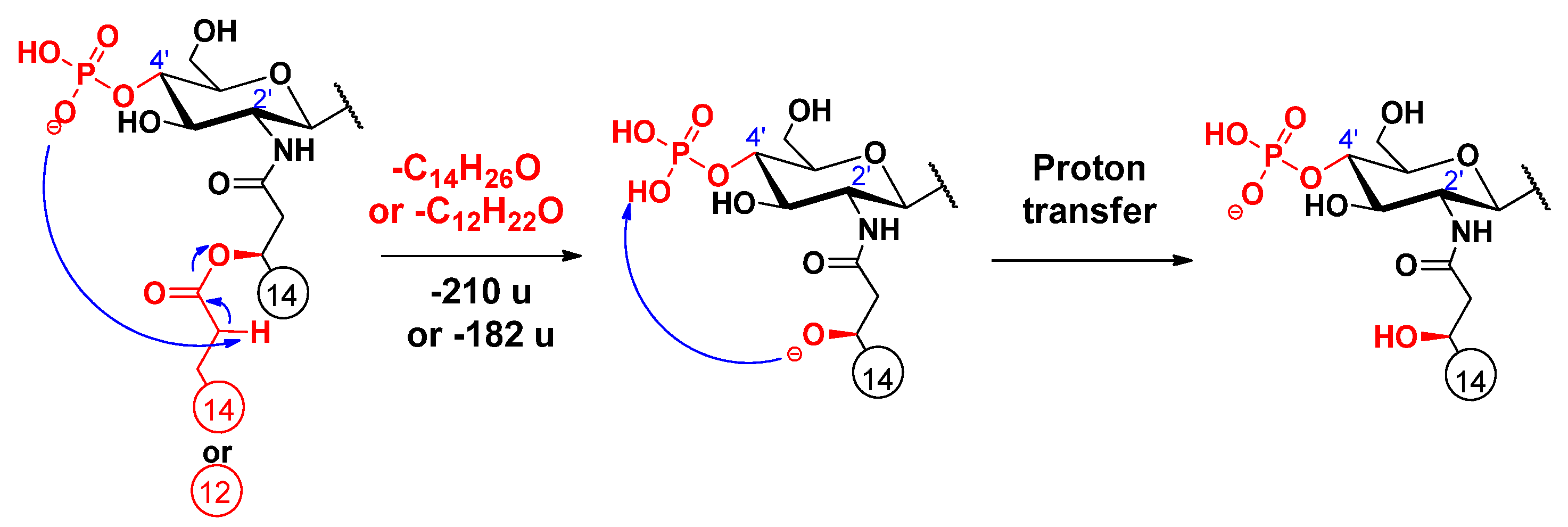
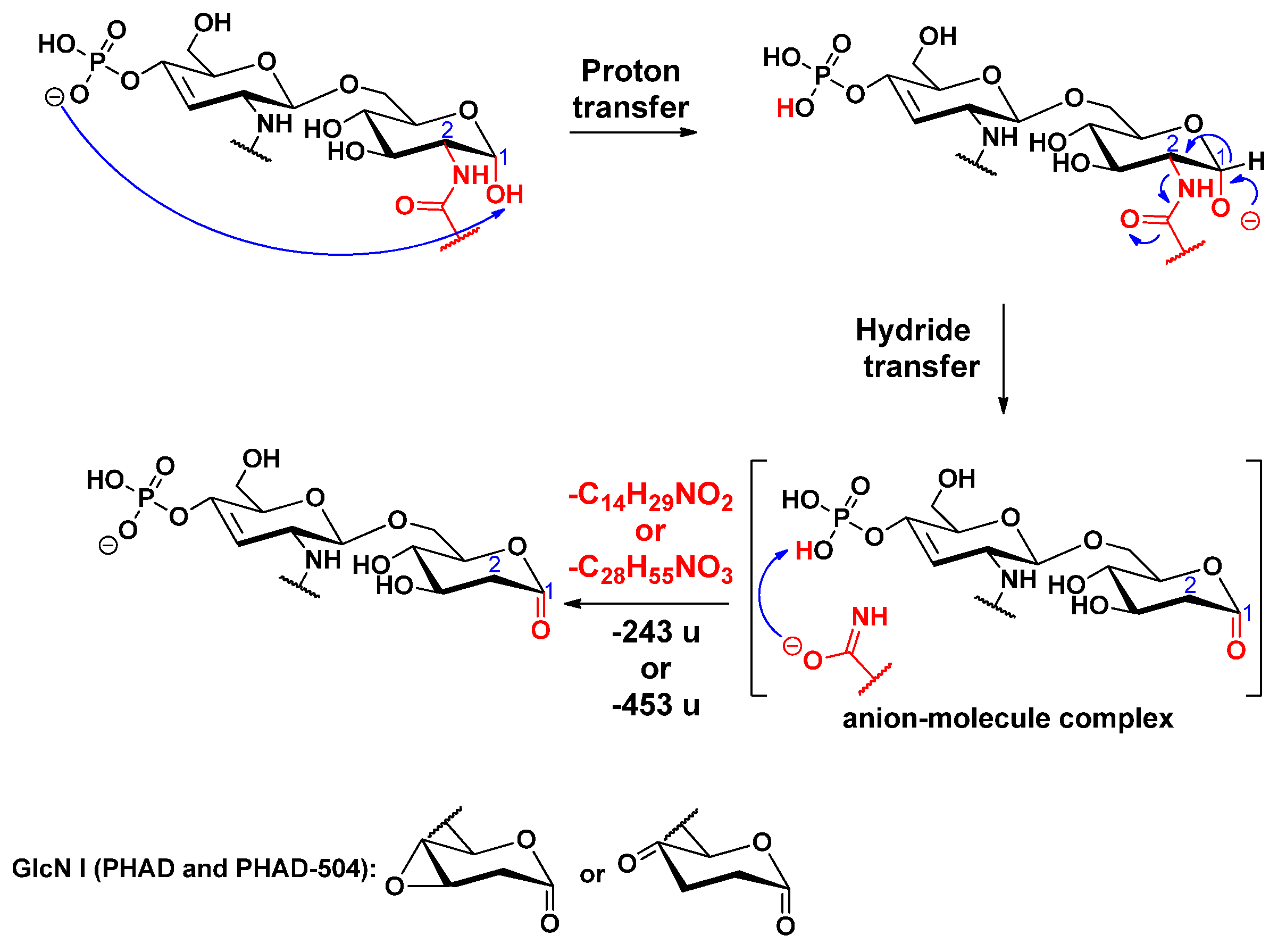
2.3. Proposed Mechanisms for the Release of the Primary Fatty Acids
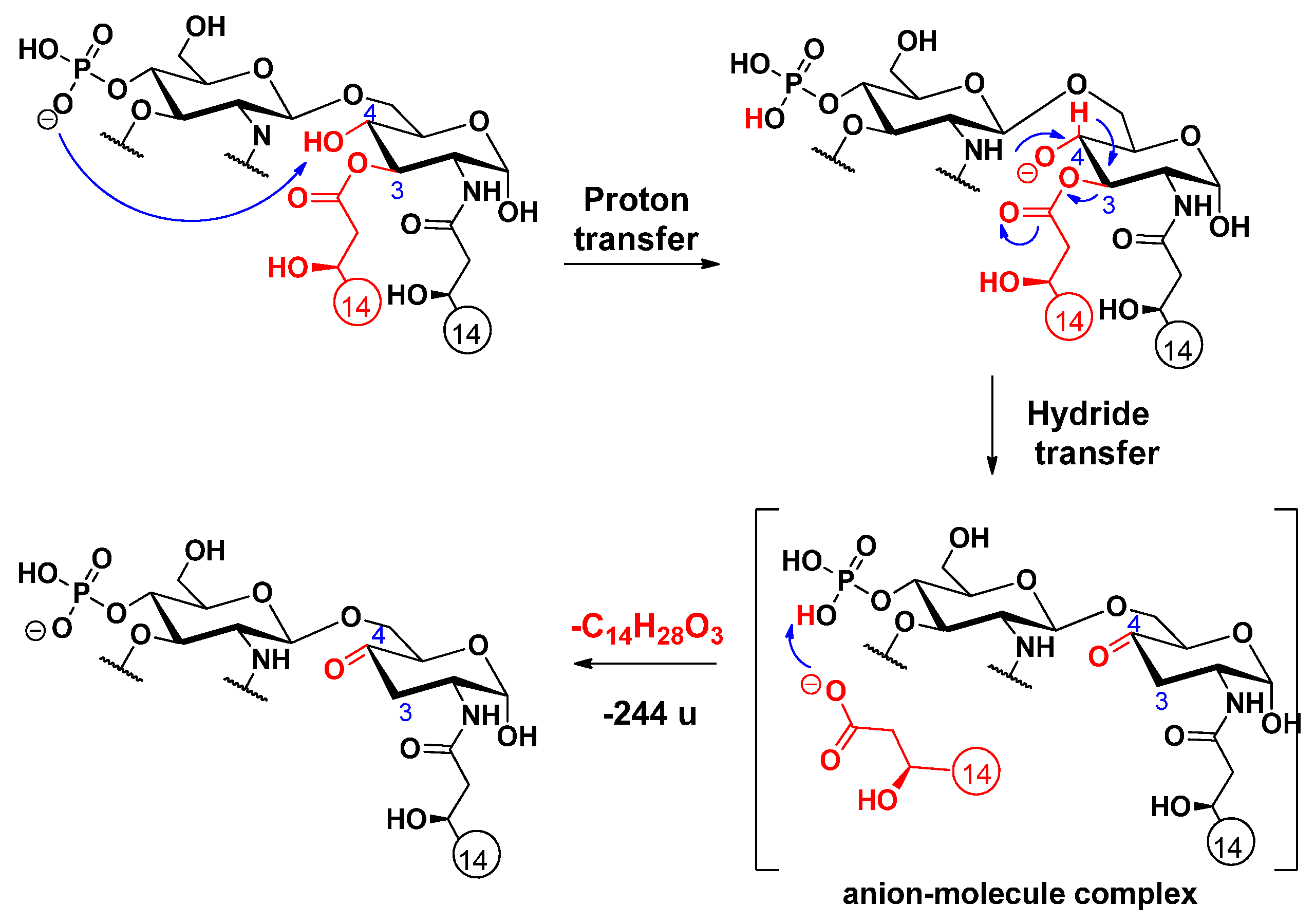
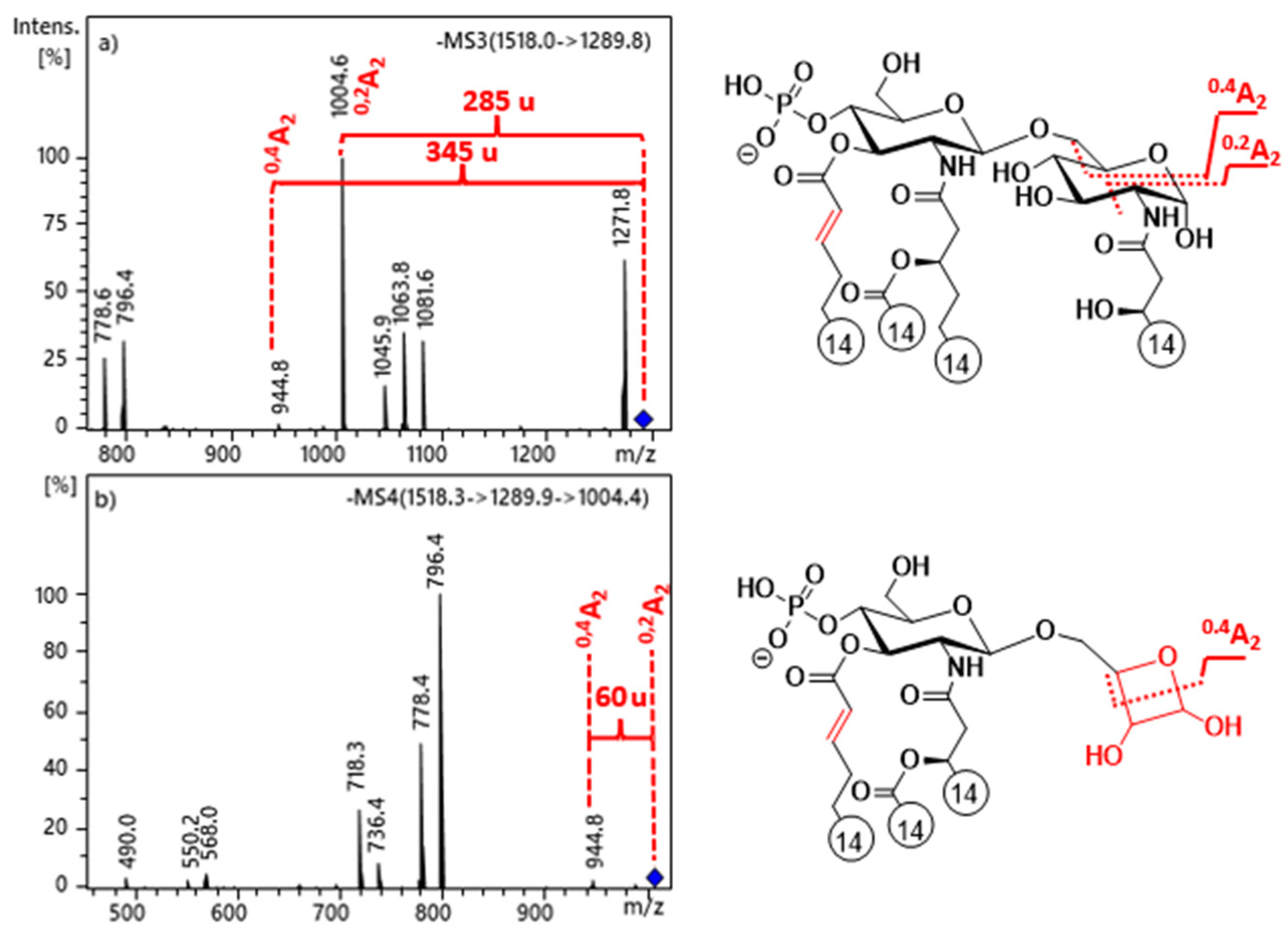
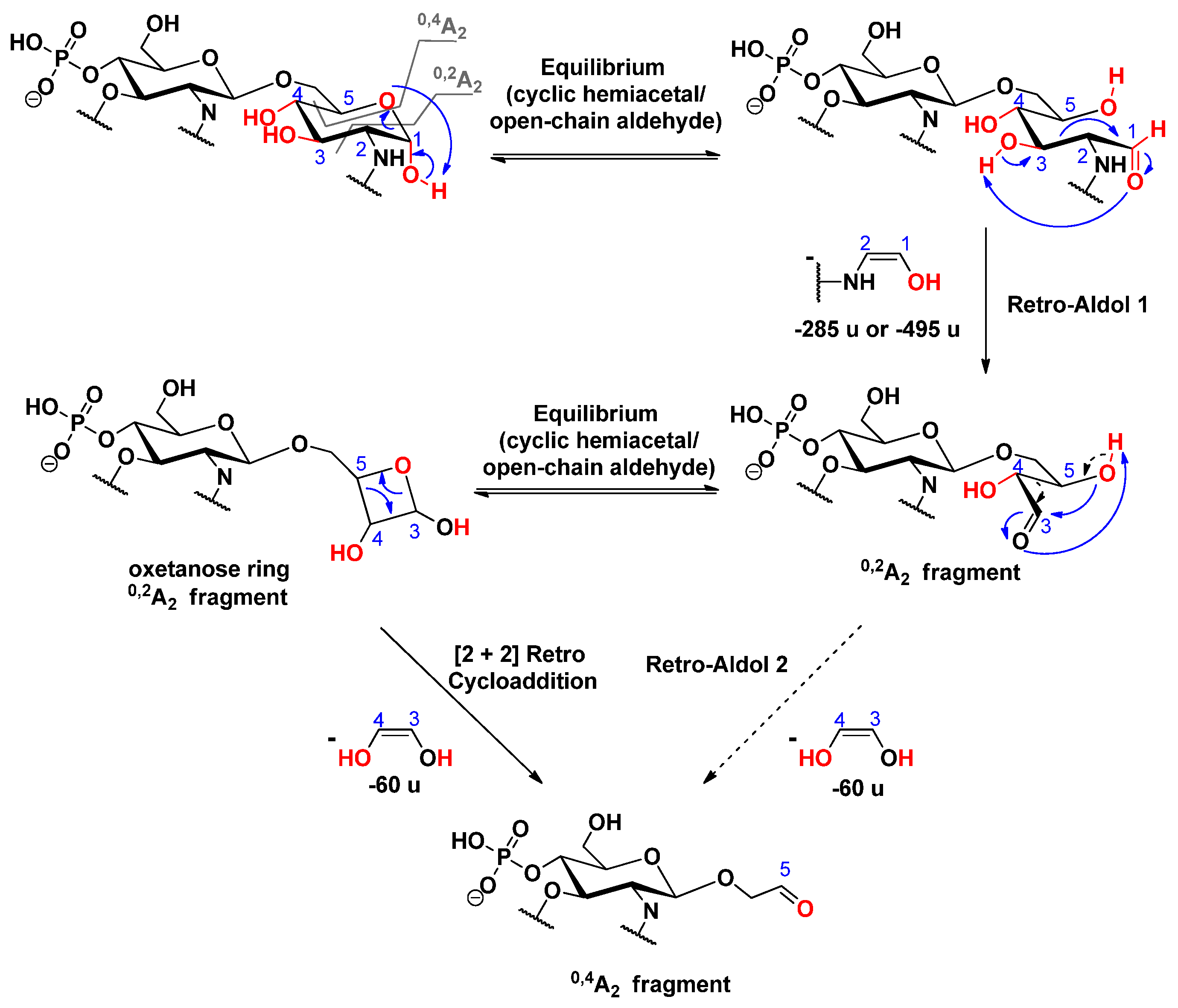
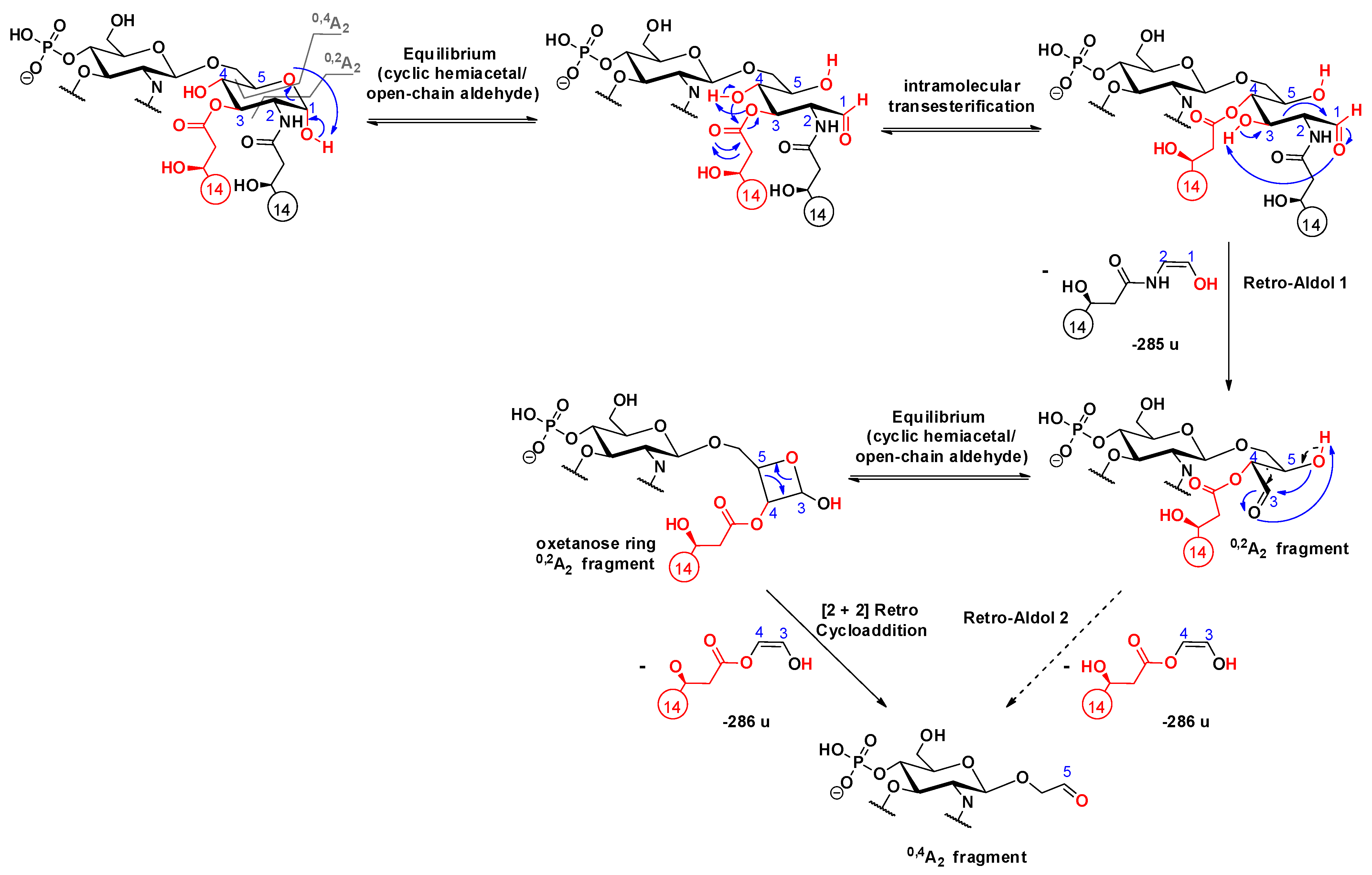
2.4. Cross-ring Fragmentations
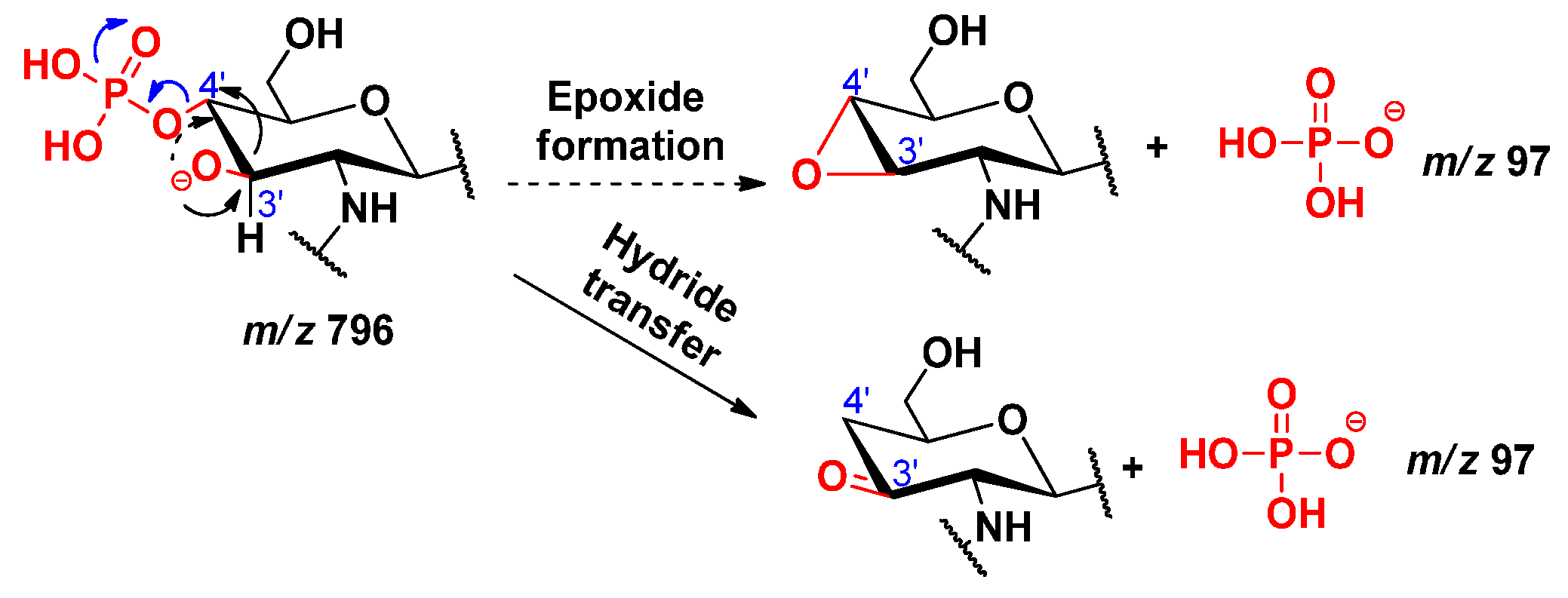
2.5. Dephosphorylation
3. Experimental
3.1. Chemicals
3.2. Lipid A Samples
3.3. Lipid A Sample Preparation
3.4. Mass Spectrometric Analysis
3.5. Data Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Caroff, M.; Novikov, A. Lipopolysaccharides: Structure, Function and Bacterial Identification. OCL: Oilseeds Fats, Crops Lipids 2020, 27, 31. [Google Scholar] [CrossRef]
- Cavaillon, J.M. Exotoxins and Endotoxins: Inducers of Inflammatory Cytokines. Toxicon 2018, 149, 45–53. [Google Scholar] [CrossRef]
- Baldridge, J.R.; Crane, R.T. Monophosphoryl Lipid A (MPL) Formulations for the Next Generation of Vaccines. Methods 1999, 19, 103–107. [Google Scholar] [CrossRef]
- Arenas, J. The Role of Bacterial Lipopolysaccharides as Immune Modulator in Vaccine and Drug Developement. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Zahringer, U.; Lindner, B.; Rietschel, E.T. Molecular-Structure of Lipid-A, the Endotoxic Center of Bacterial Lipopolysaccharides. Adv. Carbohydr. Chem. Biochem. 1994, 50, 211–276. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.; Holst, O.; Di Lorenzo, F.; Callaghan, M.; Nurisso, A.; D’Errico, G.; Zamyatina, A.; Peri, F.; Berisio, R.; Jerala, R.; et al. Chemistry of Lipid A: At the Heart of Innate Immunity. Chem. Eur. J. 2015, 21, 500–519. [Google Scholar] [CrossRef]
- Matsuura, M. Structural Modifications of Bacterial Lipopolysaccharide that Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front. Immunol. 2013, 4, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erridge, C.; Bennett-Guerrero, E.; Poxton, I.R. Structure and Function of Lipopolysaccharides. Microbes Infect. 2002, 4, 837–851. [Google Scholar] [CrossRef]
- Anandan, A.; Vrielink, A. Structure and Function of Lipid A-Modifying Enzymes. Ann. N. Y. Acad. Sci. 2020, 1459, 19–37. [Google Scholar] [CrossRef]
- Mata-Haro, V.; Cekic, C.; Martin, M.; Chilton, P.M.; Casella, C.R.; Mitchell, T.C. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 2007, 316, 1628–1632. [Google Scholar] [CrossRef]
- Casella, C.R.; Mitchell, T.C. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cellular and Molecular Life Sciences 2008, 65, 3231–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilár, A.; Dörnyei, Á.; Kocsis, B. Structural Characterization of Bacterial Lipopolysaccharides With Mass Spectrometry and on- and off-line Separation Techniques. Mass Spectrom. Rev. 2013, 32, 90–117. [Google Scholar] [CrossRef] [PubMed]
- Boue, S.M.; Cole, R.B. Confirmation of the Structure of Lipid A from Enterobacter Agglomerans by Electrospray Ionization Tandem Mass Spectrometry. J. Mass Spectrom. 2000, 35, 361–368. [Google Scholar] [CrossRef]
- Kussak, A.; Weintraub, A. Quadrupole Ion-Trap Mass Spectrometry to Locate Fatty Acids on Lipid A from Gram-Negative Bacteria. Anal. Biochem. 2002, 307, 131–137. [Google Scholar] [CrossRef]
- Bedoux, G.; Vallee-Rehel, K.; Kooistra, O.; Zahringer, U.; Haras, D. Lipid A Components from Pseudomonas Aeruginosa PAO1 (Serotype O5) and Mutant Strains Investigated by Electrospray Ionization Ion-Trap Mass Spectrometry. J. Mass Spectrom. 2004, 39, 505–513. [Google Scholar] [CrossRef]
- Madalinski, G.; Fournier, F.; Wind, F.L.; Afonso, C.; Tabet, J.C. Gram-Negative Bacterial Lipid A Analysis by Negative Electrospray Ion Trap Mass Spectrometry: Stepwise Dissociations of Deprotonated Species under Low Energy CID Conditions. Int. J. Mass Spectrom. 2006, 249, 77–92. [Google Scholar] [CrossRef]
- Schilling, B.; McLendon, M.K.; Phillips, N.J.; Apicella, M.A.; Gibson, B.W. Characterization of Lipid a Acylation Patterns in Francisella Tularensis, Francisella Novicida, and Francisella Philomiragia using Multiple-Stage Mass Spectrometry and Matrix-Assisted Laser Desorption/Ionization on an Intermediate Vacuum Source Linear Ion Trap. Anal. Chem. 2007, 79, 1034–1042. [Google Scholar] [CrossRef] [Green Version]
- Silipo, A.; De Castro, C.; Lanzetta, R.; Molinaro, A.; Parrilli, M.; Vago, G.; Sturiale, L.; Messina, A.; Garozzo, D. Structural Characterizations of Lipids A by MS/MS of Doubly Charged Ions on a Hybrid Linear Ion Trap/Orbitrap Mass Spectrometer. J. Mass Spectrom. 2008, 43, 478–484. [Google Scholar] [CrossRef]
- Jones, J.W.; Cohen, I.E.; Turecek, F.; Goodlett, D.R.; Ernst, R.K. Comprehensive Structure Characterization of Lipid A Extracted from Yersinia Pestis for Determination of its Phosphorylation Configuration. J. Am. Soc. Mass Spectrom. 2010, 21, 785–799. [Google Scholar] [CrossRef] [Green Version]
- Morrison, L.J.; Parker, W.R.; Holden, D.D.; Henderson, J.C.; Boll, J.M.; Trent, M.S.; Brodbelt, J.S. UVliPiD: A UVPD-Based Hierarchical Approach for De Novo Characterization of Lipid A Structures. Anal. Chem. 2016, 88, 1812–1820. [Google Scholar] [CrossRef] [Green Version]
- Tawab, A.; Akbar, N.; Hasssan, M.; Habib, F.; Ali, A.; Rahman, M.; Jabbar, A.; Rauf, W.; Iqbal, M. Mass Spectrometric Analysis of Lipid A Obtained from the Lipopolysaccharide of Pasteurella Multocida. RSC Adv. 2020, 10, 30917–30933. [Google Scholar] [CrossRef]
- Sándor, V.; Kilár, A.; Kilár, F.; Kocsis, B.; Dörnyei, Á. Characterization of Complex, Heterogeneous Lipid A Samples using HPLC-MS/MS Technique II. Structural Elucidation of non-Phosphorylated Lipid A by Negative-Ion Mode Tandem Mass Spectrometry. J. Mass Spectrom. 2016, 51, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Sándor, V.; Dörnyei, Á.; Makszin, L.; Kilár, F.; Péterfi, Z.; Kocsis, B.; Kilár, A. Characterization of Complex, Heterogeneous Lipid A Samples using HPLC-MS/MS Technique I. Overall Analysis with Respect to Acylation, Phosphorylation and Isobaric Distribution. J. Mass Spectrom. 2016, 51, 1043–1063. [Google Scholar] [CrossRef] [PubMed]
- Sándor, V.; Kilár, A.; Kilár, F.; Kocsis, B.; Dörnyei, Á. Characterization of Complex, Heterogeneous Lipid A Samples using HPLC-MS/MS Technique III. Positive-Ion Mode Tandem Mass Spectrometry to Reveal Phosphorylation and Acylation Patterns of Lipid A. J. Mass Spectrom. 2018, 53, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Kilár, A.; Dörnyei, Á.; Sándor, V.; Kilár, F.; Kocsis, B. Phosphoglycolipid Profiling of Bacterial Endotoxins. Hung. J. Ind. Chem. 2018, 46, 7–11. [Google Scholar] [CrossRef]
- Crittenden, C.M.; Akin, L.D.; Morrison, L.J.; Trent, M.S.; Brodbelt, J.S. Characterization of Lipid A Variants by Energy-Resolved Mass Spectrometry: Impact of Acyl Chains. J. Am. Soc. Mass Spectrom. 2017, 28, 1118–1126. [Google Scholar] [CrossRef]
- El-Aneed, A.; Banoub, J. Elucidation of the Molecular Structure of Lipid A Isolated from Both a Rough Mutant and a Wild Strain of Aeromonas Salmonicida Lipopolysaccharides using Electrospray Ionization Quadrupole Time-Of-Flight Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 1683–1695. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q.; Cheng, S.H.; Wu, Y.F.; Guo, D.J.; Fan, Q.H.; Wang, X.F.; Zhang, L.H.; Ye, X.S. Base-Promoted Rearrangement of Sugar Epoxides to Unsaturated Sugars. Org. Lett. 2005, 7, 5577–5579. [Google Scholar] [CrossRef]
- Hsu, F.F.; Turk, J. Charge-Remote and Charge-Driven Fragmentation Processes in Diacyl Glycerophosphoethanolamine upon Low-Energy Collisional Activation: A Mechanistic Proposal. J. Am. Soc. Mass Spectrom. 2000, 11, 892–899. [Google Scholar] [CrossRef]
- Spengler, B.; Dolce, J.W.; Cotter, R.J. Infrared-Laser Desorption Mass-spectrometry of Oligosaccharides-Fragmentation Mechanisms and Isomer Analysis. Anal. Chem. 1990, 62, 1731–1737. [Google Scholar] [CrossRef]
- Chiu, C.C.; Huynh, H.T.; Tsai, S.T.; Lin, H.Y.; Hsu, P.J.; Phan, H.T.; Karumanthra, A.; Thompson, H.; Lee, Y.C.; Kuo, J.L.; et al. Toward Closing the Gap between Hexoses and N-Acetlyhexosamines: Experimental and Computational Studies on the Collision-Induced Dissociation of Hexosamines. J. Phys. Chem. A 2019, 123, 6683–6700. [Google Scholar] [CrossRef] [PubMed]
- Hazelard, D.; Compain, P. Square sugars: Challenges and synthetic strategies. Org. Biomol. Chem. 2017, 15, 3806–3827. [Google Scholar] [CrossRef]
- Popsavin, V.; Radic, L.; Popsavin, M.; Cirin-Novta, V. Unexpected Cycloreversion of a Tosylated Tugar Oxetane Under E2 Conditions. The Facile Formation of 2-(2-Furanyl)-1,3-Dioxolane from a Novel 2,5:4,6-Dianhydro-L-Idose Derivative. J. Serb. Chem. Soc. 2004, 69, 117–122. [Google Scholar] [CrossRef]
- Lee, J.W.; Lewin, N.E.; Blumberg, P.M.; Marquez, V.E. Conformationally Constrained Analogs of Diacylglycerol.9. The Effect of Side-chain Orientation on the Protein-kinase-c (Pk-c) Binding-affinity of Delta-lactones. Bioorg. Med. Chem. Lett. 1994, 4, 2405–2410. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.J.; Altman, E. Structural Characterization of the Lipid A Region of Aeromonas Salmonicida Subsp Salmonicida Lipopolysaccharide. Carbohydr. Res. 2006, 341, 2816–2825. [Google Scholar] [CrossRef]
- Domon, B.; Costello, C.E. A Systematic Nomenclature for Carbohydrate Fragmentations in FAB-MS/MS Spectra of Glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aissa, I.; Kilár, A.; Dörnyei, Á. Study on the CID Fragmentation Pathways of Deprotonated 4’-Monophosphoryl Lipid A. Molecules 2021, 26, 5961. https://doi.org/10.3390/molecules26195961
Aissa I, Kilár A, Dörnyei Á. Study on the CID Fragmentation Pathways of Deprotonated 4’-Monophosphoryl Lipid A. Molecules. 2021; 26(19):5961. https://doi.org/10.3390/molecules26195961
Chicago/Turabian StyleAissa, Ibrahim, Anikó Kilár, and Ágnes Dörnyei. 2021. "Study on the CID Fragmentation Pathways of Deprotonated 4’-Monophosphoryl Lipid A" Molecules 26, no. 19: 5961. https://doi.org/10.3390/molecules26195961
APA StyleAissa, I., Kilár, A., & Dörnyei, Á. (2021). Study on the CID Fragmentation Pathways of Deprotonated 4’-Monophosphoryl Lipid A. Molecules, 26(19), 5961. https://doi.org/10.3390/molecules26195961