
STL1, a New AKT Inhibitor, Synergizes with Flavonoid Quercetin in Enhancing Cell Death in A Chronic Lymphocytic Leukemia Cell Line




Abstract
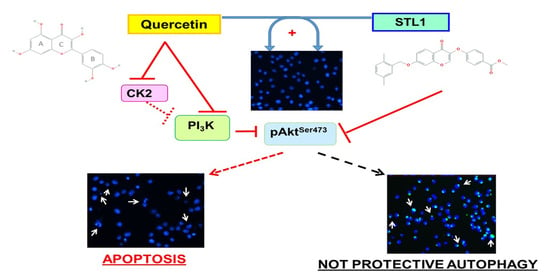
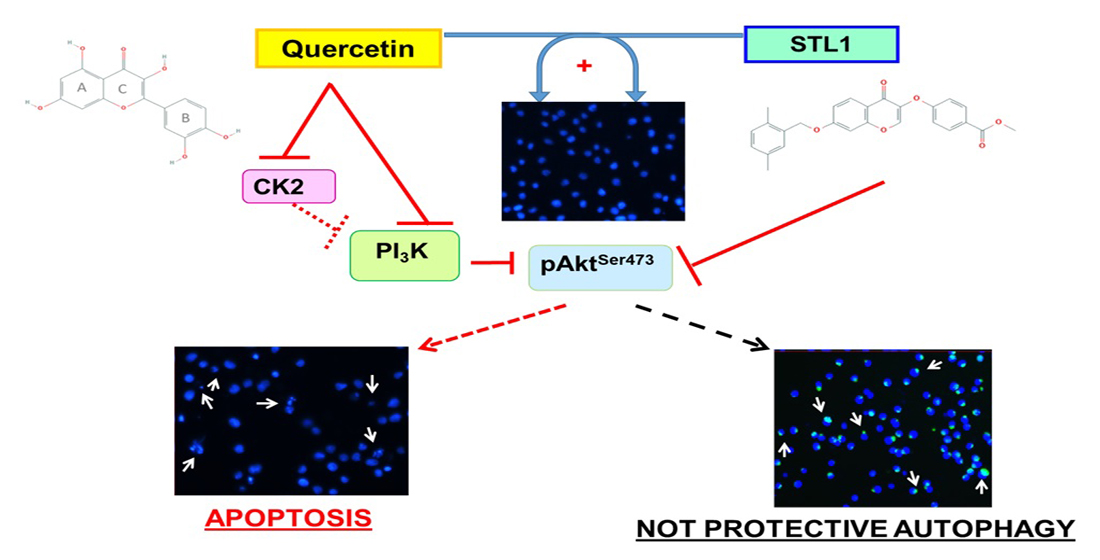
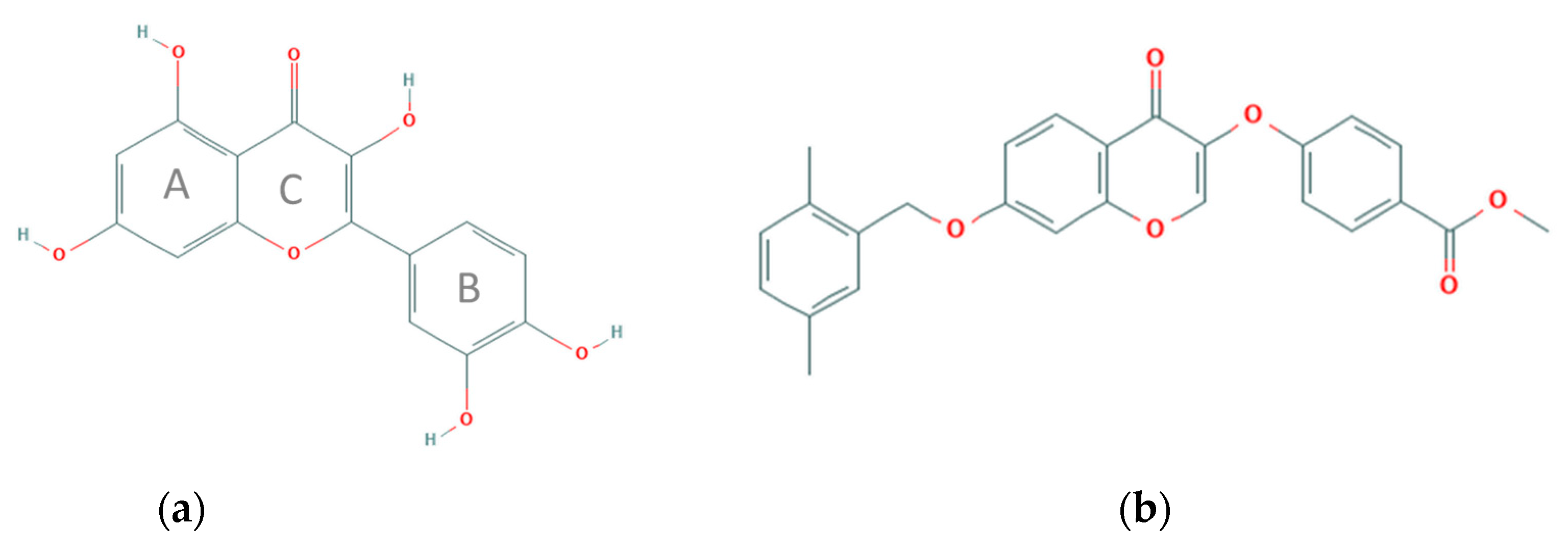
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
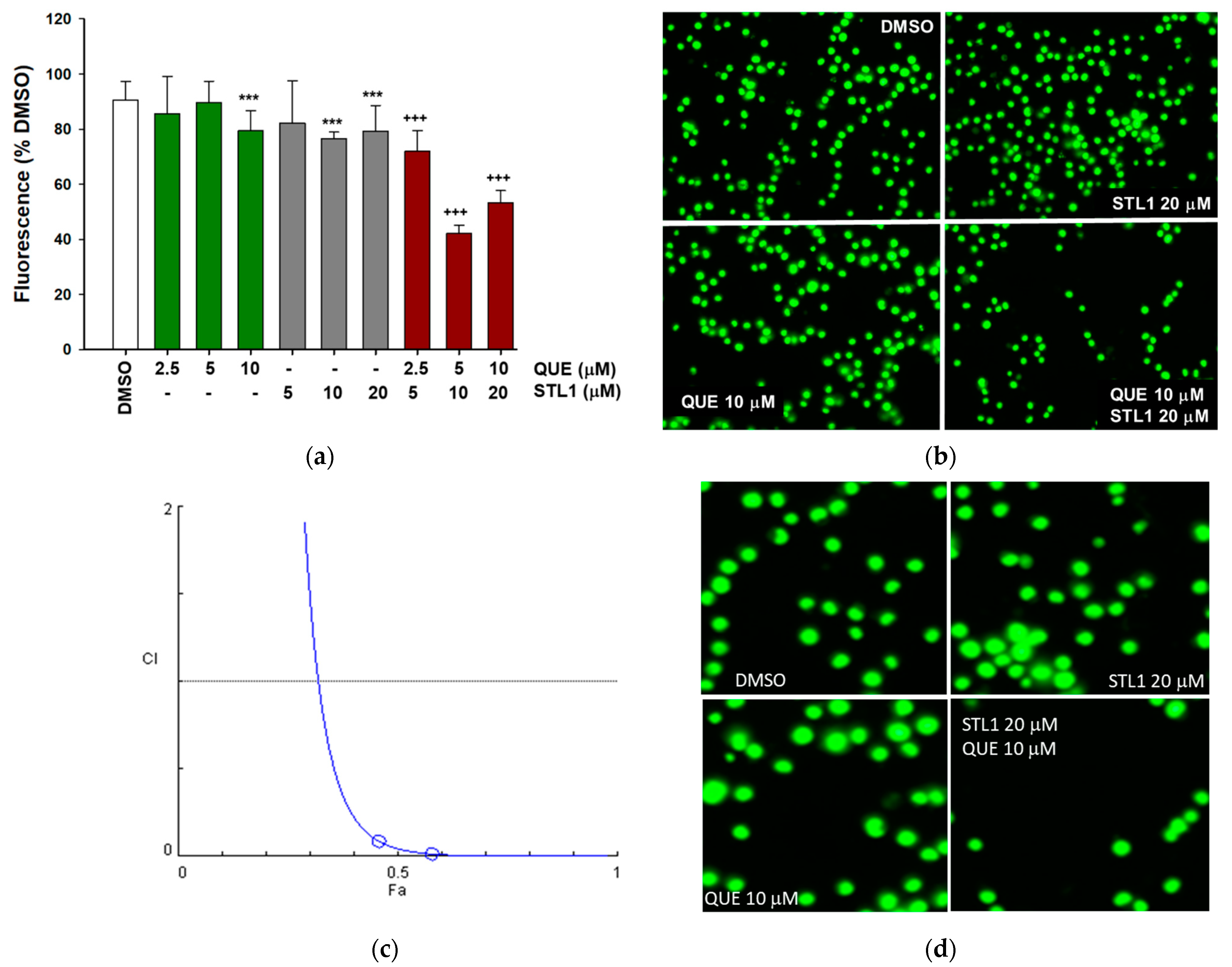
2.1. Quercetin Enhances Cell Death Induced by STL1 in HG-3 Cells
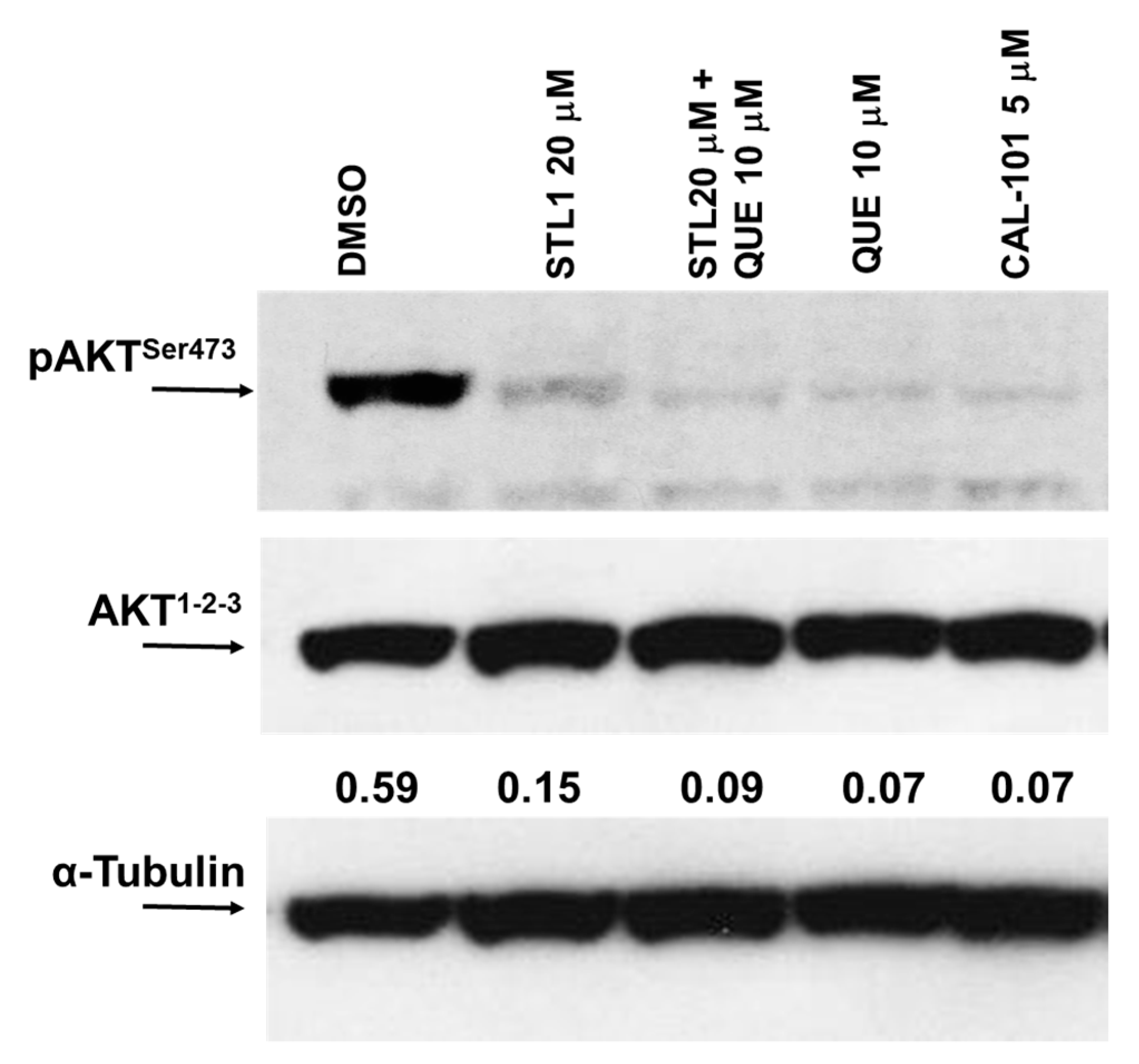
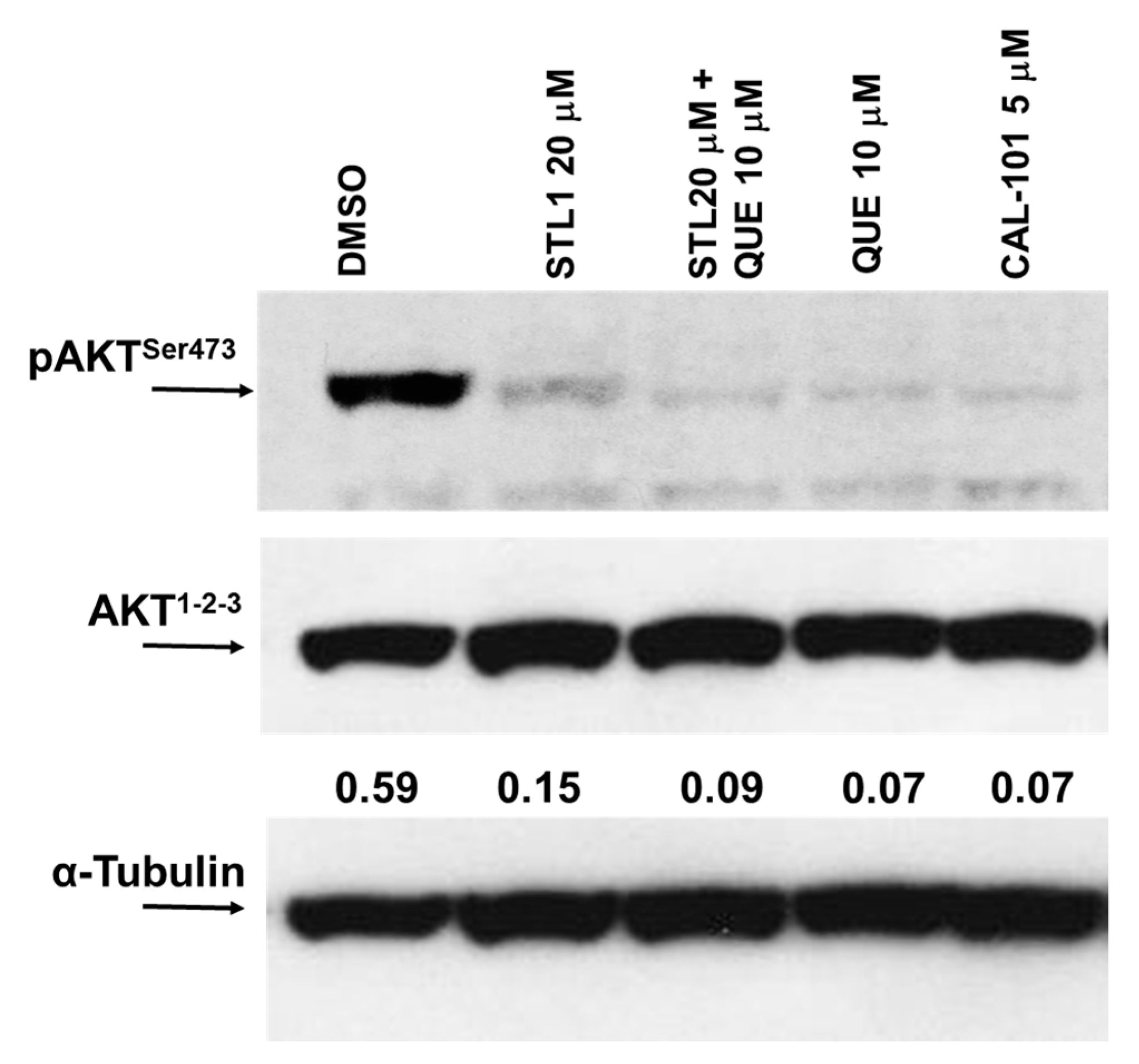
2.2. Quercetin Enhances STL1 Sensitivity in HG-3 through AKT Inhibition
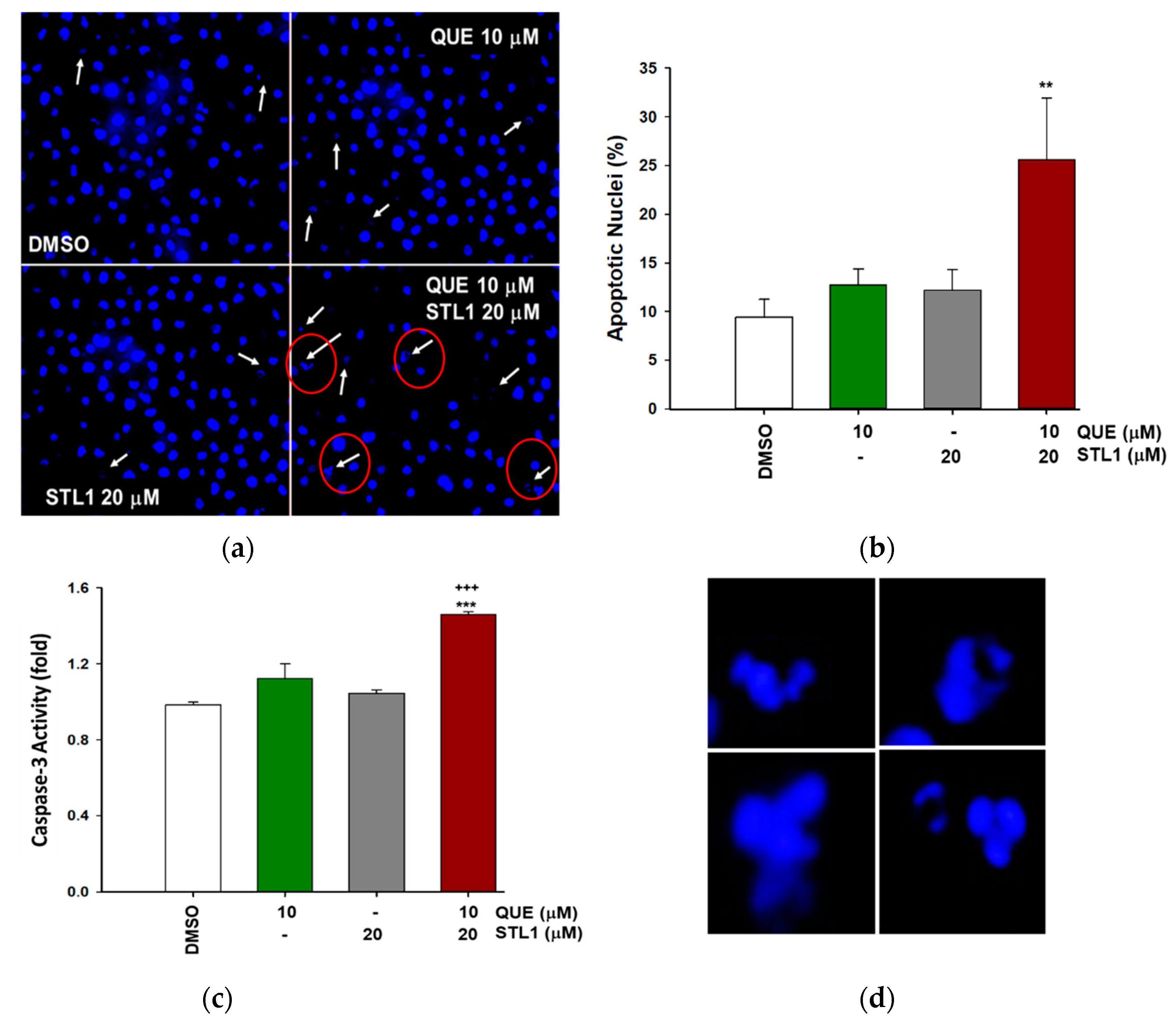
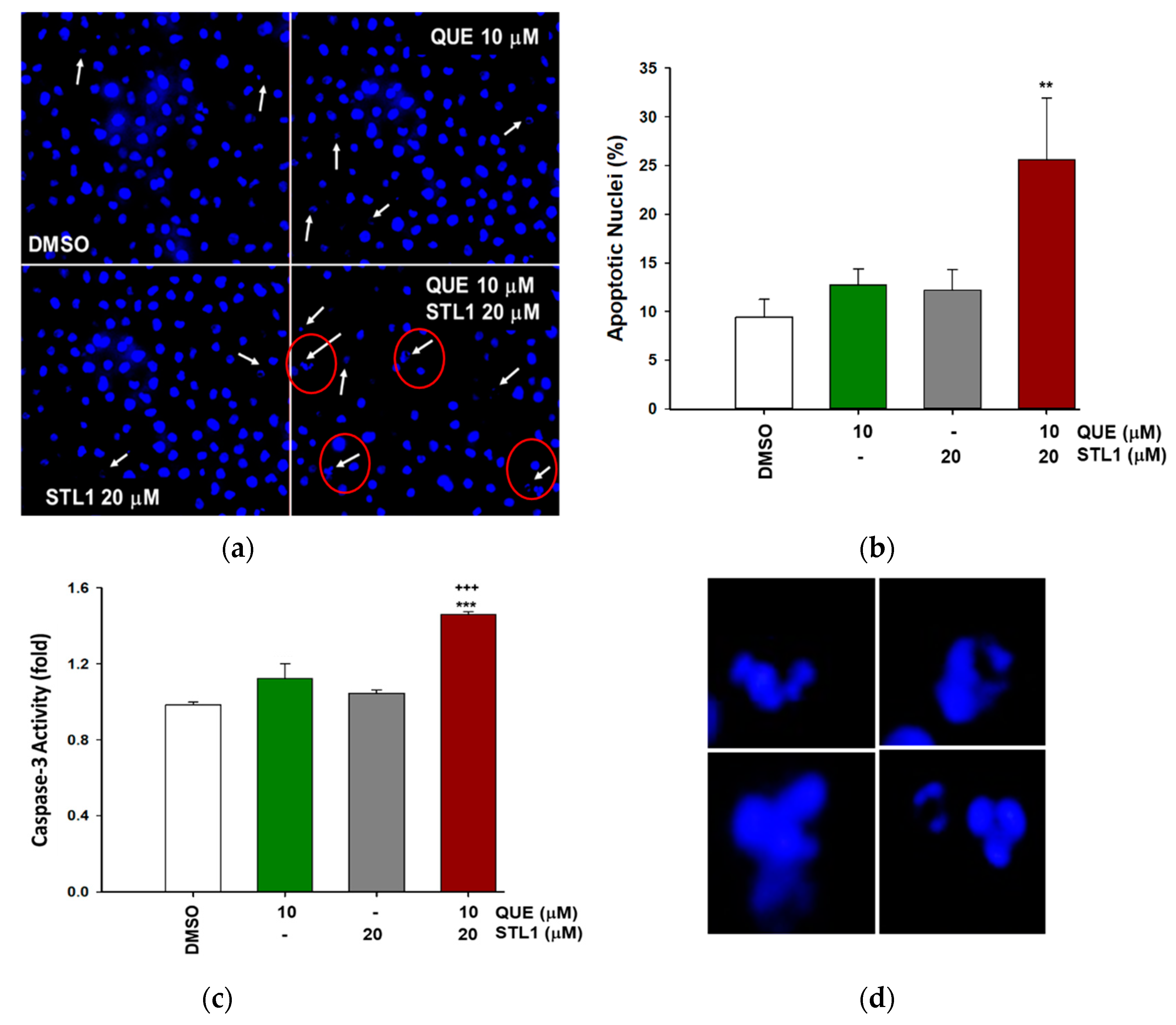
2.3. Quercetin and STL1 Co-Treatment Induce Apoptosis in HG-3 Cells
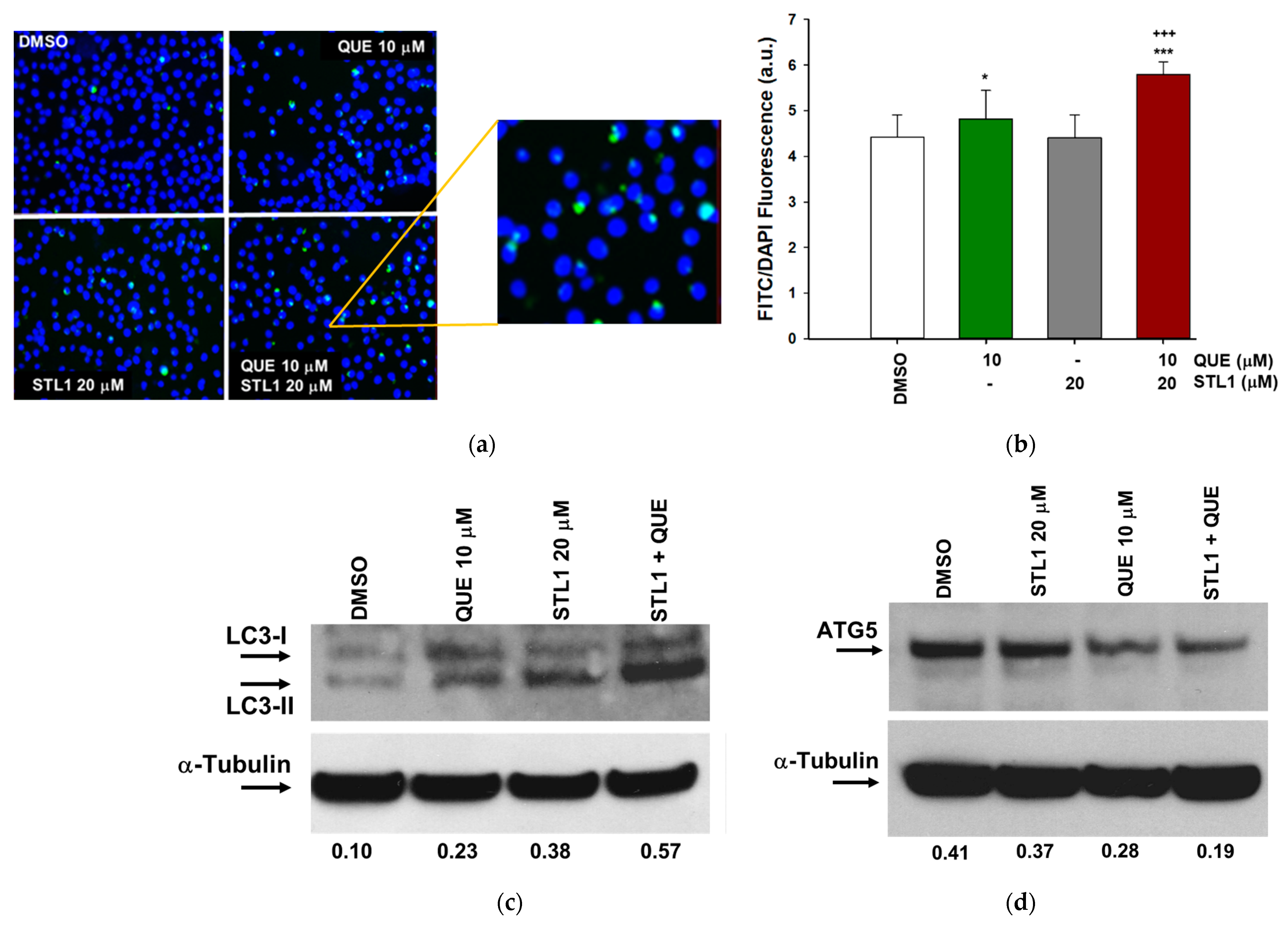
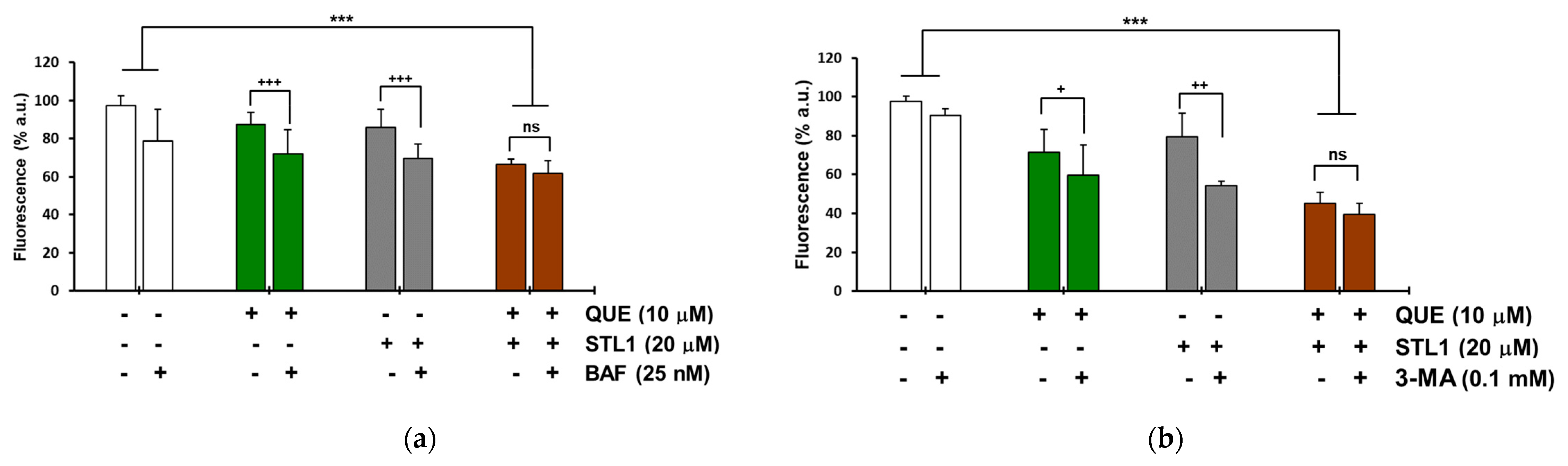
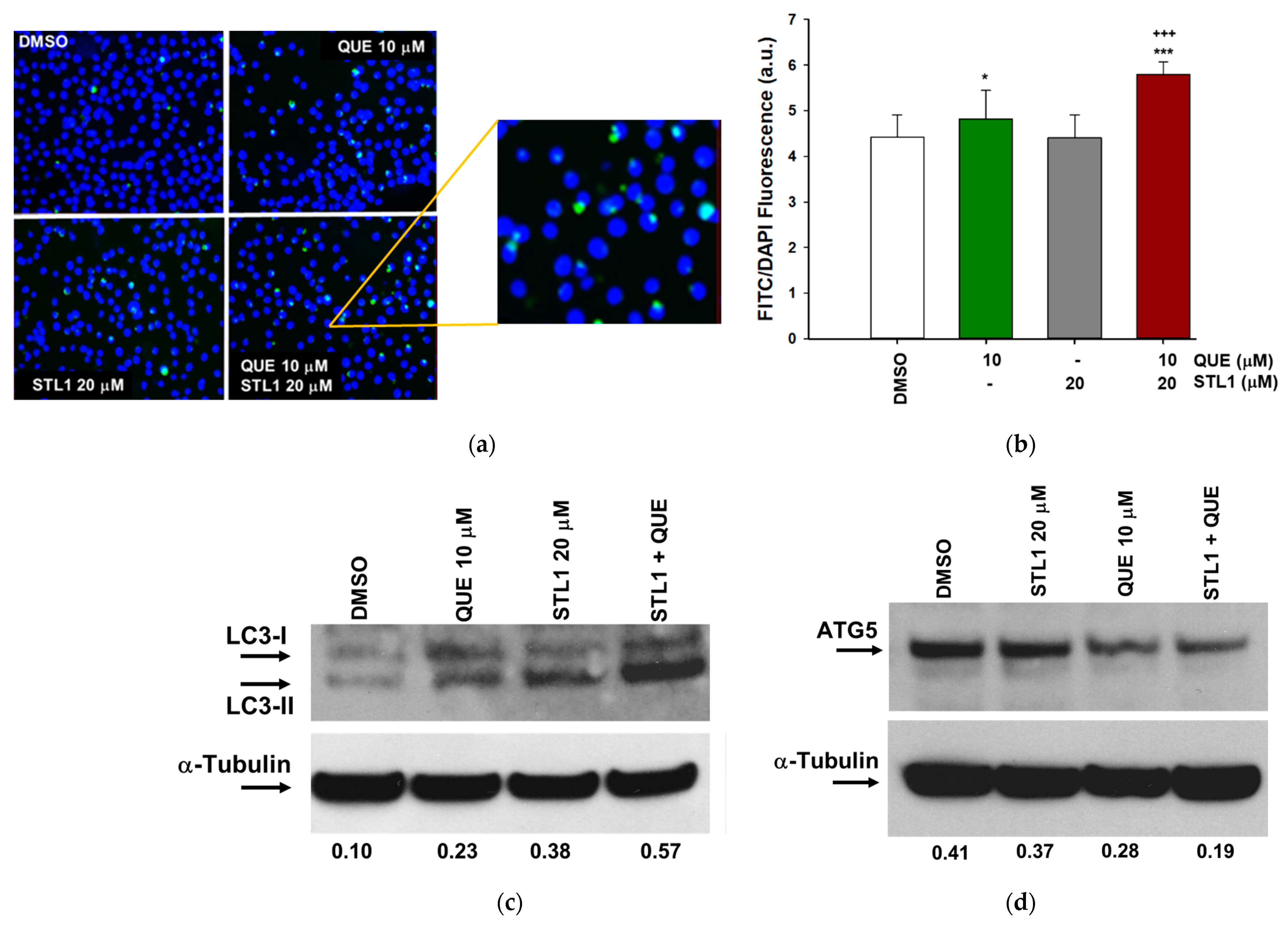
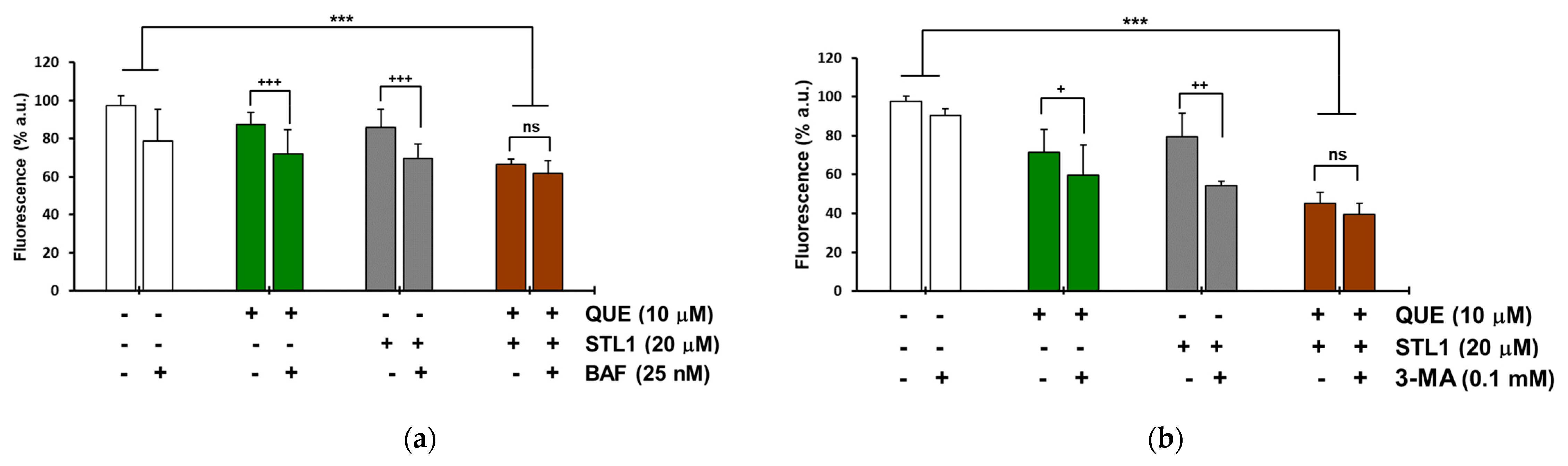
2.4. Quercetin and STL1 Co-Treatment Induces Autophagy in HG-3 Cells
3. Discussion
4. Materials and Methods
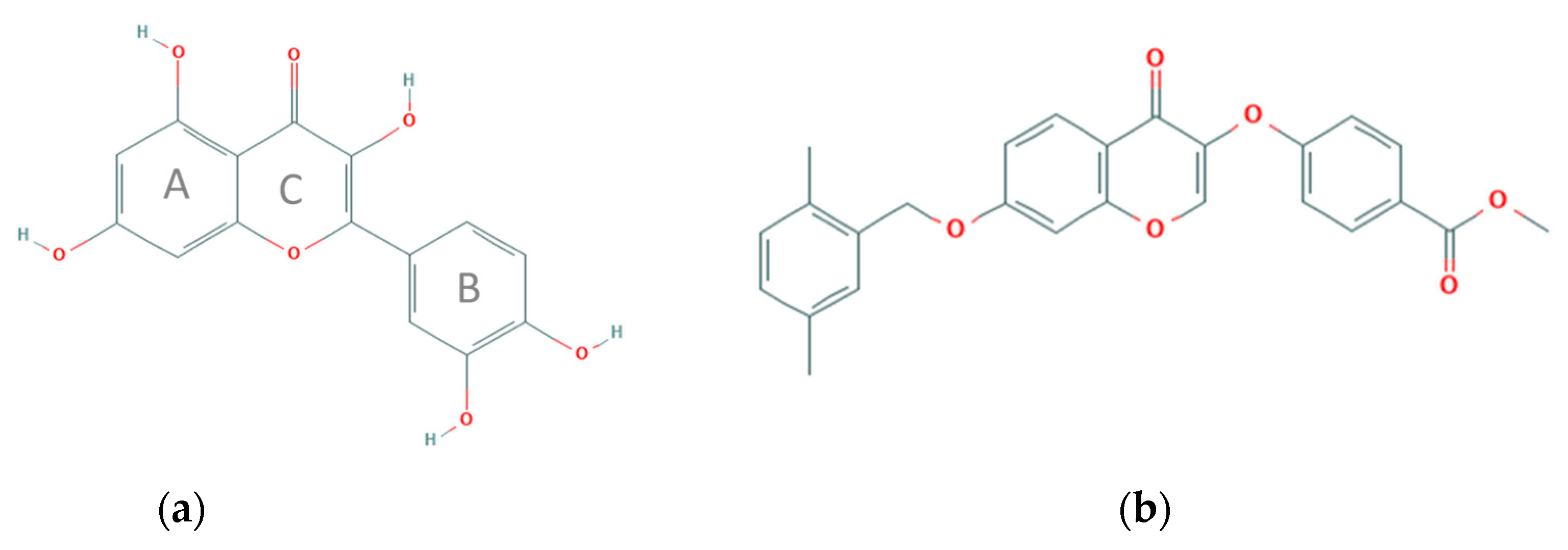
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. Cell Viability Assay and Combination Index Determination
4.4. Apoptosis Assays
4.5. Autophagy Assays
4.6. Immunoblotting
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Crozier, A.; Clifford, M.N.; Ashihara, H. Plant. Secondary Metabolites, Occurrence, Structure and Role in the Human Diet; Blackwell Publishing Ltd.: Oxford, UK, 2006. [Google Scholar]
- Burbulis, I.E.; Winkel-Shirley, B. Interactions among enzymes of the Arabidopsis flavonoid biosynthetic pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 12929–12934. [Google Scholar] [CrossRef] [Green Version]
- Bors, W.; Michel, C.; Saran, M. Flavonoid antioxidants: Rate constants for reactions with oxygen radicals. Methods Enzymol. 1994, 234, 420–429. [Google Scholar]
- Russo, M.; Milito, A.; Spagnuolo, C.; Carbone, V.; Rosen, A.; Minasi, P.; Lauria, F.; Russo, G.L. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget 2017, 8, 42571–42587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, M.; Nigro, P.; Rosiello, R.; D'Arienzo, R.; Russo, G.L. Quercetin enhances CD95- and TRAIL-induced apoptosis in leukemia cell lines. Leukemia 2007, 21, 1130–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, M.; Spagnuolo, C.; Volpe, S.; Tedesco, I.; Bilotto, S.; Russo, G.L. ABT-737 resistance in B-cells isolated from chronic lymphocytic leukemia patients and leukemia cell lines is overcome by the pleiotropic kinase inhibitor quercetin through Mcl-1 down-regulation. Biochem. Pharmacol. 2013, 85, 927–936. [Google Scholar] [CrossRef]
- Dotolo, S.; Cervellera, C.; Russo, M.; Russo, G.L.; Facchiano, A. Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation. Molecules 2021, 26, 492. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Spagnuolo, C.; Tedesco, I.; Bilotto, S.; Russo, G.L. The flavonoid quercetin in disease prevention and therapy: Facts and fancies. Biochem. pharmacol. 2012, 83, 6–15. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Russo, G.L.; Russo, M.; Spagnuolo, C.; Tedesco, I.; Bilotto, S.; Iannitti, R.; Palumbo, R. Quercetin: A pleiotropic kinase inhibitor against cancer. Cancer Treat. Res. 2014, 159, 185–205. [Google Scholar] [CrossRef]
- Patel, K.; Pagel, J.M. Current and future treatment strategies in chronic lymphocytic leukemia. J. Hematol. Oncol. 2021, 14, 69. [Google Scholar] [CrossRef]
- Russo, G.L.; Russo, M.; Spagnuolo, C. The pleiotropic flavonoid quercetin: From its metabolism to the inhibition of protein kinases in chronic lymphocytic leukemia. Food Funct. 2014, 5, 2393–2401. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Liu, X.; Liu, K.; Zhao, R.; Huang, H.; Shi, Y.; Zhang, M.; Zhou, S.; Xie, H.; Chen, H.; et al. Targeting AKT with Oridonin Inhibits Growth of Esophageal Squamous Cell Carcinoma In Vitro and Patient-Derived Xenografts In Vivo. Mol. Cancer Ther. 2018, 17, 1540–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toker, A.; Marmiroli, S. Signaling specificity in the Akt pathway in biology and disease. Adv. Biol. Regul. 2014, 55, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Chen, X.; Hay, N. Akt as a target for cancer therapy: More is not always better (lessons from studies in mice). Br. J. Cancer 2017, 117, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Herman, S.E.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Zirlik, K.; Veelken, H. Idelalisib. Recent Results Cancer Res. 2018, 212, 243–264. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Barrientos, J.C. Oral PI3K-delta, gamma Inhibitor for the Management of People with Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma: A Narrative Review on Duvelisib. Onco Targets Ther. 2021, 14, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Rosen, A.; Bergh, A.C.; Gogok, P.; Evaldsson, C.; Myhrinder, A.L.; Hellqvist, E.; Rasul, A.; Bjorkholm, M.; Jansson, M.; Mansouri, L.; et al. Lymphoblastoid cell line with B1 cell characteristics established from a chronic lymphocytic leukemia clone by in vitro EBV infection. Oncoimmunology 2012, 1, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Liu, R.; Li, J.; Mao, J.; Lei, Y.; Wu, J.; Zeng, J.; Zhang, T.; Wu, H.; Chen, L.; et al. Quercetin induces protective autophagy in gastric cancer cells: Involvement of Akt-mTOR- and hypoxia-induced factor 1alpha-mediated signaling. Autophagy 2011, 7, 966–978. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Moon, J.Y.; Ahn, K.S.; Cho, S.K. Quercetin induces mitochondrial mediated apoptosis and protective autophagy in human glioblastoma U373MG cells. Oxid. Med. Cell. Longev. 2013, 2013, 596496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhou, X.J.; Zhang, H. Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A molecular switch node in the crosstalk between autophagy and apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef]
- Shacka, J.J.; Klocke, B.J.; Roth, K.A. Autophagy, bafilomycin and cell death: The “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy 2006, 2, 228–230. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewirtz, D.A. The Switch between Protective and Nonprotective Autophagy; Implications for Autophagy Inhibition as a Therapeutic Strategy in Cancer. Biology 2020, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. An autophagic switch in the response of tumor cells to radiation and chemotherapy. Biochem. Pharmacol. 2014, 90, 208–211. [Google Scholar] [CrossRef]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [Green Version]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Rubinsztein, D.C. Atg5 and Bcl-2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ. 2007, 14, 1247–1250. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S. Biological Roles of Alternative Autophagy. Mol. Cells 2018, 41, 50–54. [Google Scholar] [CrossRef]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Russo, M.; Russo, G.L. Autophagy inducers in cancer. Biochem. Pharmacol. 2018, 153, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Primikyri, A.; Chatziathanasiadou, M.V.; Karali, E.; Kostaras, E.; Mantzaris, M.D.; Hatzimichael, E.; Shin, J.S.; Chi, S.W.; Briasoulis, E.; Kolettas, E.; et al. Direct binding of Bcl-2 family proteins by quercetin triggers its pro-apoptotic activity. ACS Chem. Biol. 2014, 9, 2737–2741. [Google Scholar] [CrossRef]
- Hirano, I.; Nakamura, S.; Yokota, D.; Ono, T.; Shigeno, K.; Fujisawa, S.; Shinjo, K.; Ohnishi, K. Depletion of Pleckstrin homology domain leucine-rich repeat protein phosphatases 1 and 2 by Bcr-Abl promotes chronic myelogenous leukemia cell proliferation through continuous phosphorylation of Akt isoforms. J. Biol. Chem. 2009, 284, 22155–22165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tromp, J.M.; Geest, C.R.; Breij, E.C.; Elias, J.A.; van Laar, J.; Luijks, D.M.; Kater, A.P.; Beaumont, T.; van Oers, M.H.; Eldering, E. Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin. Cancer Res. 2012, 18, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Moccia, S.; Bilotto, S.; Spagnuolo, C.; Durante, M.; Lenucci, M.S.; Mita, G.; Volpe, M.G.; Aquino, R.P.; Russo, G.L. A Carotenoid Extract from a Southern Italian Cultivar of Pumpkin Triggers Nonprotective Autophagy in Malignant Cells. Oxid. Med. Cell Longev. 2017, 2017, 7468538. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Waterhouse, N.J. Analyzing Cell Death by Nuclear Staining with Hoechst 33342. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cervellera, C.; Russo, M.; Dotolo, S.; Facchiano, A.; Russo, G.L. STL1, a New AKT Inhibitor, Synergizes with Flavonoid Quercetin in Enhancing Cell Death in A Chronic Lymphocytic Leukemia Cell Line. Molecules 2021, 26, 5810. https://doi.org/10.3390/molecules26195810
Cervellera C, Russo M, Dotolo S, Facchiano A, Russo GL. STL1, a New AKT Inhibitor, Synergizes with Flavonoid Quercetin in Enhancing Cell Death in A Chronic Lymphocytic Leukemia Cell Line. Molecules. 2021; 26(19):5810. https://doi.org/10.3390/molecules26195810
Chicago/Turabian StyleCervellera, Carmen, Maria Russo, Serena Dotolo, Angelo Facchiano, and Gian Luigi Russo. 2021. "STL1, a New AKT Inhibitor, Synergizes with Flavonoid Quercetin in Enhancing Cell Death in A Chronic Lymphocytic Leukemia Cell Line" Molecules 26, no. 19: 5810. https://doi.org/10.3390/molecules26195810
APA StyleCervellera, C., Russo, M., Dotolo, S., Facchiano, A., & Russo, G. L. (2021). STL1, a New AKT Inhibitor, Synergizes with Flavonoid Quercetin in Enhancing Cell Death in A Chronic Lymphocytic Leukemia Cell Line. Molecules, 26(19), 5810. https://doi.org/10.3390/molecules26195810