Commercially available reagents were purchased from Fluka (Sydney, NSW, Australia), Aldrich (Sydney, NSW, Australia), Acros Organics (Morris Plains, NJ, USA), Alfa Aesar (Lancashire, UK) and Lancaster (Lancashire, UK) and purified if necessary. The synthetic procedures have been reported for all compounds as general methods and appropriate references have been given for known compounds. 1H (300 MHz) and 13C-NMR (75 MHz) spectra were obtained in the designated solvents on a DPX 300 spectrometer (Bruker, Sydney, NSW, Australia). Melting points were measured using a Mel-Temp melting point apparatus and are uncorrected. Infrared spectra were recorded on Avatar Series FT-IR spectrophotometer as KBr disks (Thermo Nicolet, Waltham, MA, USA). Ultraviolet spectra were measured using a Cary 100 spectrophotometer (Varian, Santa Clara, CA, USA) in the designated solvents and data reported as wavelength (λ) in nm and adsorption coefficient (ε) in cm−1M−1. High-resolution [ESI] mass spectra were recorded by the UNSW Bioanalytical Mass Spectrometry Facility, on an Orbitrap LTQ XL (Thermo Scientific, Waltham, MA, USA) ion trap mass spectrometer using a nanospray (nano-electrospray) ionization source.
3.1.2. GP-2: General Procedure for the Synthesis of Furoindoles
A solution of indole ether (0.26 mmol) in DCM (10 mL) was treated with trifluoroacetic acid (10 drops) and heated at reflux for 24 h. The reaction mixture was poured into crushed ice (100 g), and the precipitate collected via filtration and washed with water. The crude product was purified using gravity column chromatography, eluted with DCM/n-hexane (2:10), to give the furoindole.
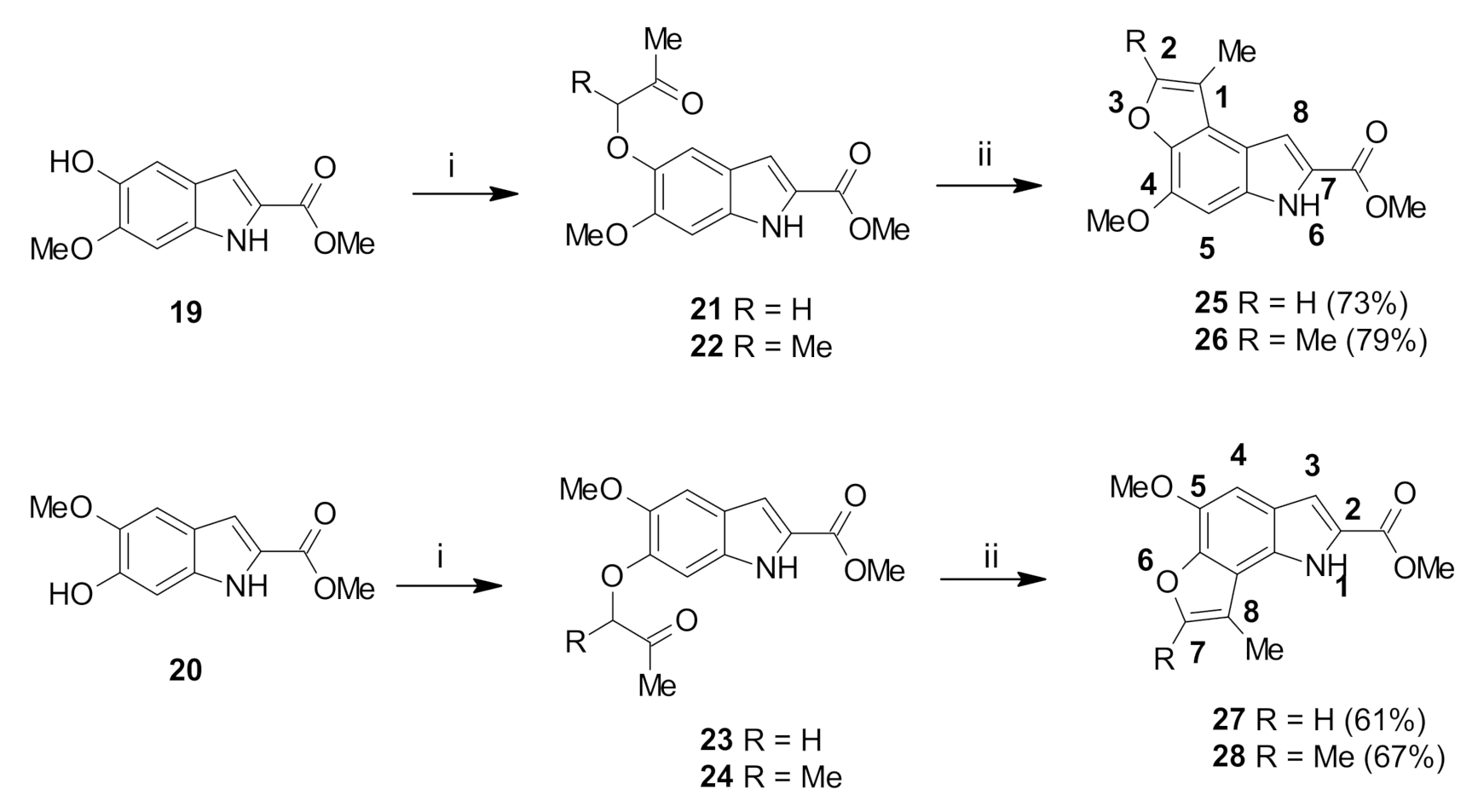
Methyl 6-methoxy-5-(2-oxopropoxy)-1H-indole-2-carboxylate (21): The title compound was prepared as described in GP-1 from methyl 5-hydroxy-6-methoxyindole-2-carboxylate (19) (107.2 mg, 0.485 mmol), chloroacetone (44.2 mg, 0.485 mmol) and potassium carbonate (66.9 mg, 0.485 mmol) to give the product (108.8 mg, 81%) as a white solid; m.p. 164–166 °C; IR (KBr): νmax 3314, 2879, 2828, 2318, 2117, 1925, 1688, 1632, 1521, 1445, 1356, 1250, 1151, 1065, 1019, 998, 893, 759, 671 cm−1; UV-vis (CH3CN): λmax 208 nm (ε 30,600 cm−1 M−1), 318 (22,100); 1H-NMR (300 MHz, CDCl3): δ 2.34 (s, 3H, COMe), 3.95 (s, 3H, CO2Me), 3.97 (s, 3H, OMe), 4.64 (s, 2H, CH2), 6.91 (d, J = 0.9 Hz, 1H, H7), 7.02 (s, 1H, H3), 7.11 (d, J = 0.9 Hz, 1H, H4), 8.90 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 26.5 (COMe), 51.9 (CO2Me), 56.1 (Ome), 75.0 (CH2), 94.4 (C7), 106.0 (C4), 108.9 (C3), 120.3 (aryl C), 126.9 (C2), 132.9 (aryl C), 144.5 (C5), 150.4 (C6), 162.2 (CO2Me), 206.5 (COMe); HRMS (+ESI): Found m/z 300.0832, [M + Na]+, C14H15NO5Na requires 300.0842.
Methyl 5-methoxy-6-(2-oxopropoxy)-1H-indole-2-carboxylate (23): The title compound was prepared as described in GP-1 from methyl 5-methoxy-6-hydroxyindole-2-carboxylate (20) (107.2 mg, 0.485 mmol), chloroacetone (44.2 mg, 0.485 mmol) and potassium carbonate (66.9 mg, 0.485 mmol) to give the product (104.8 mg, 78%) as a white solid; m.p. 170–172 °C; IR (KBr): νmax 3316, 2918, 2845, 2339, 2109, 1910, 1680, 1642, 1520, 1428, 1356, 1285, 1209, 1160, 1017, 890, 817, 761, 666 cm−1; UV-vis (CH3CN): λmax 209 nm (ε 30,700 cm−1 M−1), 309 (18,500); 1H-NMR (300 MHz, CDCl3): δ 2.33 (s, 3H, COMe), 3.94 (s, 3H, CO2Me), 3.95 (s, 3H, OMe), 4.65 (s, 2H, CH2), 6.80 (d, J = 0.7 Hz, 1H, H4), 7.12 (s, 1H, H3), 7.13 (d, J = 0.7 Hz, 1H, H7), 9.05 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ26.5 (COMe), 51.8 (CO2Me), 56.2 (OMe), 74.5 (CH2), 95.5 (C7), 103.4 (C4), 108.6 (C3), 121.6 (aryl C), 126.3 (C2), 131.6 (aryl C), 146.4 (C6), 148.1 (C5), 162.2 (CO2Me), 206.3 (COMe); HRMS (+ESI): Found m/z 300.0842, [M + Na]+, C14H15NO5Na requires 300.0842.
Methyl 6-methoxy-5-((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (22): The title compound was prepared as described in GP-1 from methyl 5-hydroxy-6-methoxyindole-2-carboxylate (19) (107.2 mg, 0.485 mmol), 3-chloro-2-butanone (51.4 mg, 0.485 mmol), potassium carbonate (66.9 mg, 0.485 mmol) to give the product (120 mg, 85%) as a white solid; m.p. 134–136 °C; IR (KBr): νmax 3308, 2928, 2802, 2287, 2116, 1965, 1676, 1622, 1519, 1440, 1358, 1274, 1191, 1142, 1092, 1013, 937, 820, 766, 688 cm−1; UV-vis (CH3CN): λmax 207 nm (ε 31,400 cm−1 M−1), 319 (21,700); 1H-NMR (300 MHz, CDCl3): δ 1.56 (d, J = 6.9 Hz, 3H, CHMe), 2.29 (s, 3H, COMe), 3.94 (s, 3H, CO2Me), 3.95 (s, 3H, OMe), 4.57 (q, J = 6.9 Hz, 1H, CHMe), 6.90 (d, J = 0.8 Hz, 1H, H7), 7.04 (s, 1H, H3), 7.10 (d, J = 0.9 Hz, 1H, H4), 8.92 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 17.6 (CHMe), 24.7 (COMe), 51.8 (CO2Me), 56.0 (Ome), 81.4 (CHMe), 94.4 (C7), 107.8 (C4), 108.9 (C3), 120.3 (aryl C), 126.1 (C2), 133.0 (aryl C), 143.9 (C6), 150.8 (C5), 162.2 (CO2Me), 211.0 (COMe); HRMS (+ESI): Found m/z 314.0988, [M + Na]+, C15H17NO5Na requires 314.0999.
Methyl 5-methoxy-6-((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (24): The title compound was prepared as described in GP-1 from methyl 5-methoxy-6-hydroxyindole-2-carboxylate (20) (107.2 mg, 0.485 mmol), 3-chloro-2-butanone (51.4 mg, 0.485 mmol), potassium carbonate (66.9 mg, 0.485 mmol) to give the product (114 mg, 81%) as a white solid; m.p. 136–138 °C; IR (KBr): νmax 3321, 2915, 2842, 2327, 2108, 1928, 1678, 1640, 1520, 1438, 1354, 1288, 1221, 1101, 1022, 937, 848, 830, 766, 672 cm−1; UV-vis (CH3CN): λmax 210 nm (ε 34,200 cm−1 M−1), 289 (20,300); 1H-NMR (300 MHz, CDCl3): δ 1.58 (d, J = 6.9 Hz, 3H, CHMe), 2.26 (s, 3H, COMe), 3.92 (s, 3H, CO2Me), 3.95 (s, 3H, OMe) 4.63 (q, J = 6.9 Hz, 1H, CHMe), 6.80 (d, J = 0.9 Hz, 1H, H4), 7.12 (s, 1H, H3), 7.14 (d, J = 0.9 Hz, 1H, H7), 8.87 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3):δ 17.6 (CHMe), 24.5 (COMe), 51.8 (CO2Me), 56.2 (OMe), 81.0 (CHMe), 97.8 (C7), 103.6 (C4), 108.6 (C3), 121.7 (aryl C), 126.4 (C2), 131.6 (aryl C), 146.7 (C6), 147.8 (C5), 162.1 (CO2Me), 210.8 (COMe); HRMS (+ESI): Found m/z 314.0991, [M + Na]+, C15H17NO5Na requires 314.0999.
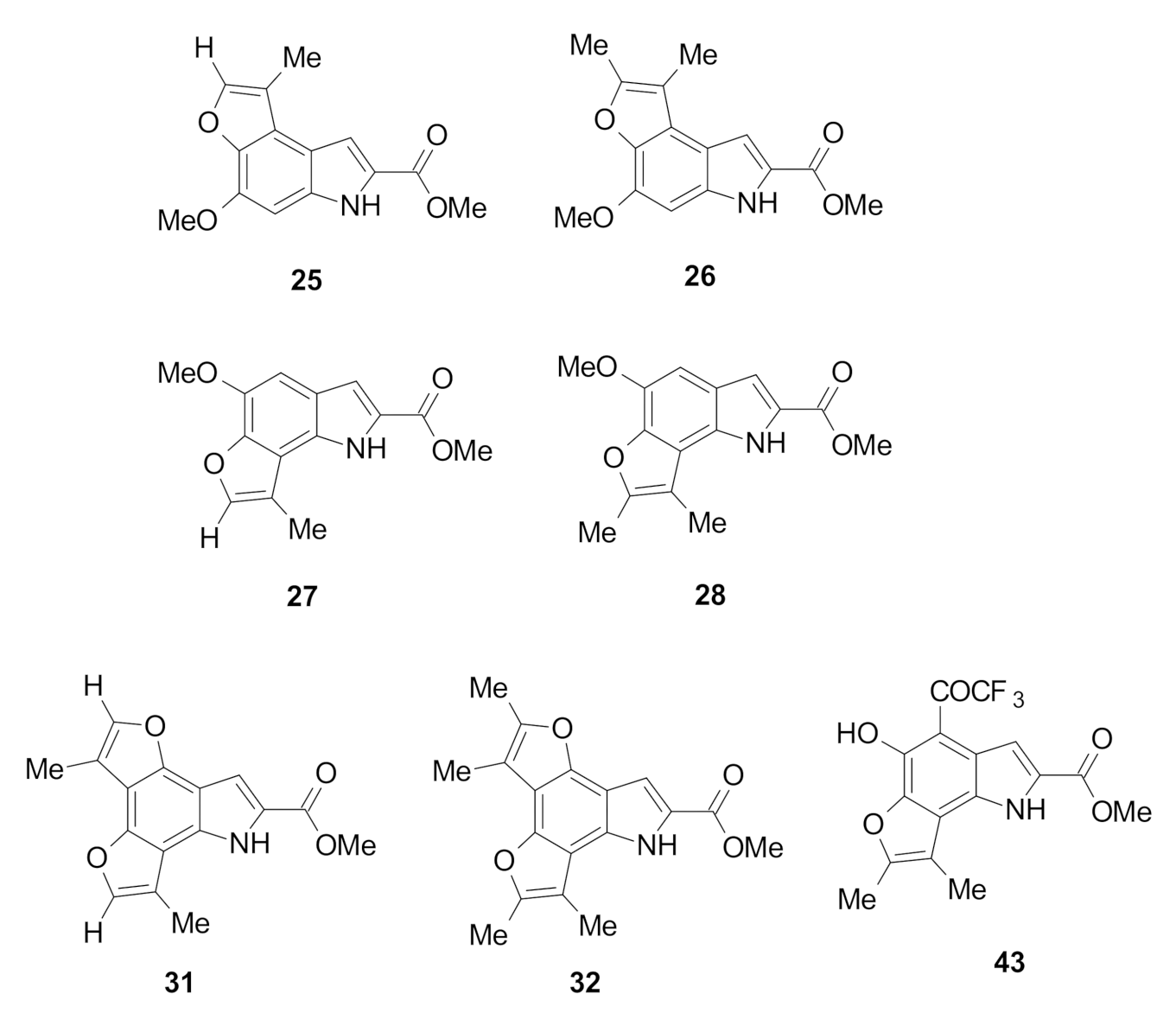
Methyl 4-methoxy-1-methyl-6H-furo[3,2-e]indole-7-carboxylate (25): The title compound was prepared as described in GP-2 from methyl 6-methoxy-5-(2-oxopropoxy)-1H-indole-2-carboxylate (21) (72.0 mg, 0.26 mmol) with trifluoroacetic acid (10 drops) in dichloromethane (10 mL) to give the product (49.1 mg, 73%) as a white solid; m.p. 202–204 °C; IR (KBr): νmax 3289, 2919, 2857, 2342, 2113, 1933, 1673, 1621, 1521, 1439, 1391, 1280, 1186, 1144, 1083, 1030, 997, 916, 761, 693 cm−1; UV-vis (CH3CN): λmax 209 nm (ε 33,800 cm−1 M−1), 312 (37.900), 324 (37,500); 1H-NMR (300 MHz, CDCl3): δ 2.48 (d, J = 1.1 Hz, 3H, Me), 3.97 (s, 3H, CO2Me), 4.05 (s, 3H, OMe), 6.77 (s, 1H, H5), 7.46 (d, J = 1.4 Hz, 1H, H8), 7.51 (d, J = 1.1 Hz, 1H, H2), 9.03 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 9.3 (C-Me), 51.8 (CO2Me), 56.1 (Ome), 107.1 (C8), 114.4 (aryl C), 116.9 (C5), 122.5 (C1), 125.1 (aryl C), 134.3 (C7), 140.8 (aryl C), 141.4 (aryl C), 142.9 (C2), 146.1 (C4), 162.3 (CO2Me); HRMS (+ESI): Found m/z 282.0731, [M + Na]+, C14H13NO4Na requires 282.0737.
Methyl 5-methoxy-8-methyl-1H-furo[2,3-g]indole-2-carboxylate (27): The title compound was prepared as described in GP-2 from methyl 5-methoxy-6-(2-oxopropoxy)-1H-indole-2-carboxylate (23) (72.0 mg, 0.26 mmol) with trifluoroacetic acid (10 drops) in dichloromethane (10 mL) to give the product (41.1 mg, 61%) as a white solid; m.p. 182–184 °C; IR (KBr): νmax 3294, 2919, 2849, 2370, 2119, 1917, 1683, 1625, 1532, 1433, 1380, 1290, 1196, 1146, 1041, 989, 920, 823, 746, 674 cm−1; UV-vis (CH3CN): λmax 213 nm (ε 20,200 cm−1 M−1), 250 (21,900), 294 (16,500); 1H-NMR (300 MHz, CDCl3): δ 2.53 (d, J = 1.3 Hz, 3H, Me), 3.98 (s, 3H, CO2Me), 4.05 (s, 3H, OMe), 6.97 (s, 1H, H4), 7.28 (d, J = 3.0 Hz, 1H, H3), 7.49 (d, J = 1.3 Hz, 1H, H7), 9.12 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): δ 9.7 (C-Me), 51.8 (CO2Me), 56.1 (OMe), 109.7 (C3), 114.8 (C4), 115.1 (C8), 122.8 (C2), 125.4 (aryl C), 125.7 (aryl C), 140.8 (2×aryl C), 142.9 (C7), 145.5 (C5), 162.3 (CO2Me); HRMS (+ESI): Found m/z 282.0730, [M + Na]+, C14H13NO4Na requires 282.0737.
Methyl 4-methoxy-1,2-dimethyl-6H-furo[3,2-e]indole-7-carboxylate (26): The title compound was prepared as described in GP-2 from methyl 6-methoxy-5-((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (22) (75.7 mg, 0.26 mmol) with the catalytic amount of trifluoroacetic acid (10 drops) in dichloromethane (10 mL) to give the product (56.1 mg, 79%) as a white solid; m.p. 182–184 °C; IR (KBr): νmax 3308, 2939, 2855, 2342, 2116, 1933, 1671, 1625, 1520, 1438, 1371, 1280, 1211, 1133, 1035, 1012, 927, 807, 684 cm−1; UV-vis (CH3CN): λmax 211 nm (ε 31,400 cm−1 M−1), 314 (38,400); 1H-NMR (300 MHz, CDCl3): δ 2.41 (d, J = 0.8 Hz, 3H, Me), 2.48 (d, J = 0.8 Hz, 3H, Me), 3.98 (s, 3H, CO2Me), 4.06 (s, 3H, OMe), 6.72 (s, 1H, H5), 7.46 (d, J = 0.9 Hz, 1H, H8), 8.96 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 9.4 (Me), 11.8 (Me), 51.7 (CO2Me), 56.0 (Ome), 107.1 (C1and aryl C), 111.2 (C8), 114.2 (C5), 123.7 (aryl C), 124.9 (C7), 134.4 (aryl C), 139.6 (C4), 145.7 (aryl C), 150.6 (C2), 162.3 (CO2Me); HRMS (+ESI): Found m/z 296.0889, [M + Na]+, C15H15NO4Na requires 296.0893.
Methyl 5-methoxy-7,8-dimethyl-1H-furo[2,3-g]indole-2-carboxylate (28): The title compound was prepared as described in GP-2 from methyl 5-methoxy-6-((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (24) (75.7 mg, 0.26 mmol) with the catalytic amount of trifluoroacetic acid (10 drops) in dichloromethane (10 mL) to give the product (47.6 mg, 67%) as a white solid; m.p. 176–178 °C; IR (KBr): νmax 3297, 2917, 2845, 2342, 2092, 1942, 1684, 1593, 1529, 1431, 1364, 1293, 1236, 1192, 1048, 995, 928, 829, 766, 670 cm−1; UV-vis (CH3CN): λmax 215 nm (ε 29,500 cm−1 M−1), 251 (33,800), 299 (22.300); 1H-NMR (300 MHz, CDCl3): δ 2.45 (d, J = 0.8 Hz, 3H, Me), 2.47 (d, J = 0.8 Hz, 3H, Me), 3.97 (s, 3H, CO2Me), 4.05 (s, 3H, OMe), 6.90 (s, 1H, H4), 7.26 (d, J = 2.2 Hz, 1H, H3), 9.06 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 9.4 (Me), 11.8 (Me), 51.7 (CO2Me), 56.0 (OMe), 107.1 (C3), 111.2 (C4), 114.2 (C8), 123.7 (aryl C), 124.9 (aryl C), 127.0 (C2), 129.1 (aryl C), 134.4 (aryl C), 145.7 (C5), 150.6 (C7), 162.3 (CO2Me); HRMS (+ESI): Found m/z 274.1071, [M + H]+, C15H16NO4 requires 274.1074.
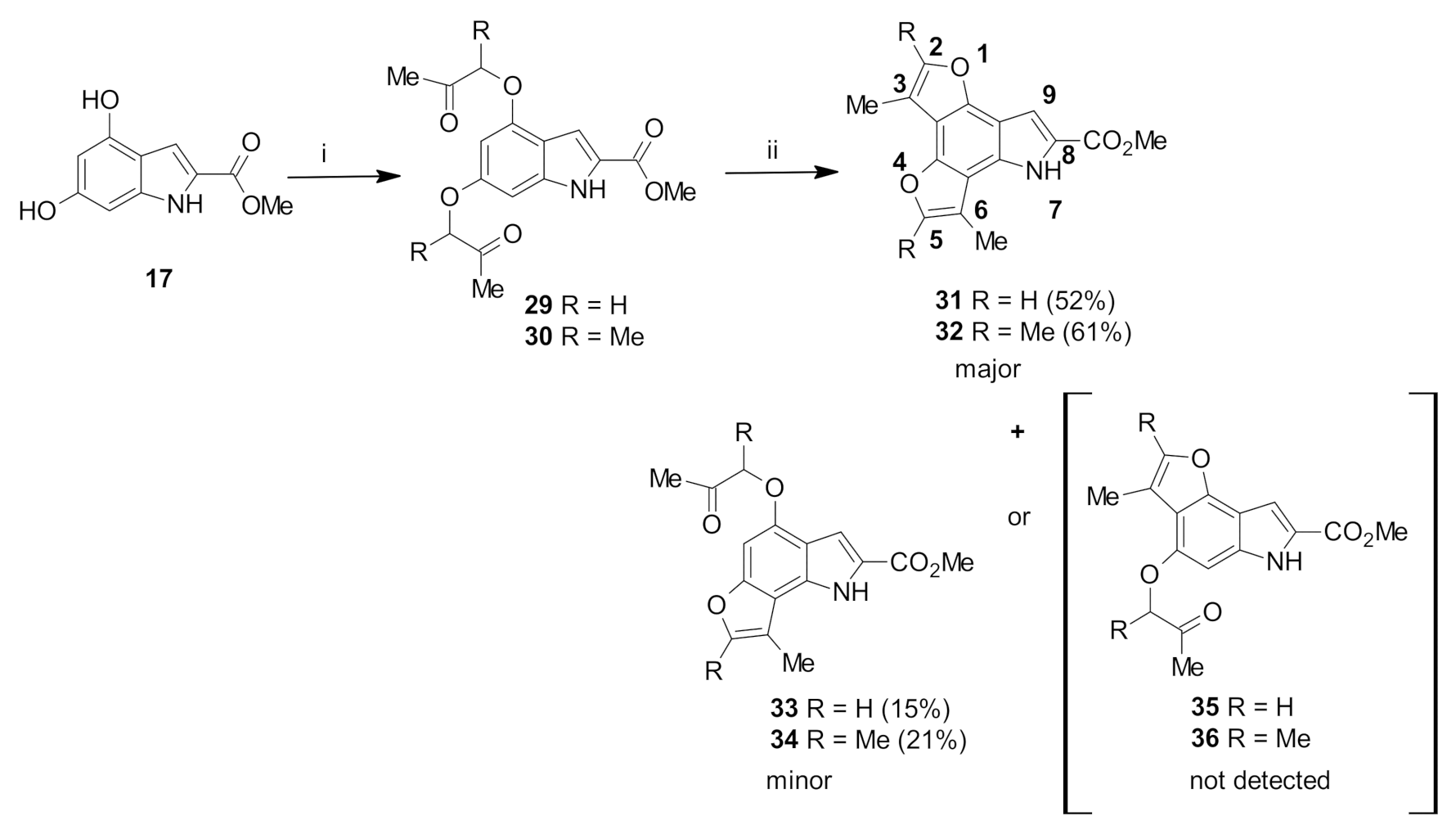
Methyl 4,6-bis(2-oxopropoxy)-1H-indole-2-carboxylate (29): The title compound was prepared as described in GP-1 from methyl 4,6-dihdroxyindole-2-carboxylate (17) (100.4 mg, 0.485 mmol), chloroacetone (88.4 mg, 0.97 mmol), potassium carbonate (123.8 mg, 0.97 mmol) to give the product (126.9 mg, 82%) as a white solid; m.p. 184–186 °C; IR (KBr): νmax 3305, 2919, 2822, 2341, 2107, 1898, 1688, 1626, 1584, 1520, 1435, 1354, 1275, 1200, 1144, 985, 951, 803, 767 cm−1; UV-vis (CH3CN): λmax 203 nm (ε 30,400 cm−1 M−1), 245 (23,000), 305 (23,200); 1H-NMR (300 MHz, CDCl3): δ 2.33 (s, 3H, Me), 2.39 (s, 3H, Me), 3.95 (s, 3H, CO2Me), 4.59 (d, J = 0.8 Hz, 2H, CH2), 4.67 (d, J = 0.8 Hz, 2H, CH2), 6.28 (d, J = 1.7 Hz, 1H, H5), 6.37 (d, J = 1.7 Hz, 1H, H7), 7.26 (d, J = 2.2 Hz, 1H, H3), 8.96 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ30.9 (2 × COMe), 51.9 (CO2Me), 73.0 (O-CH2), 73.4 (O-CH2), 88.1 (C5), 93.8 (C7), 106.6 (C3), 114.3 (aryl C), 125.4 (C2), 138.0 (aryl C), 153.2 (C4), 157.9 (C6), 162.0 (CO2Me), 205.5 (COMe), 207.0 (COMe); HRMS (+ESI): Found m/z 342.0948, [M + Na]+, C16H17NO6Na requires 342.0936.
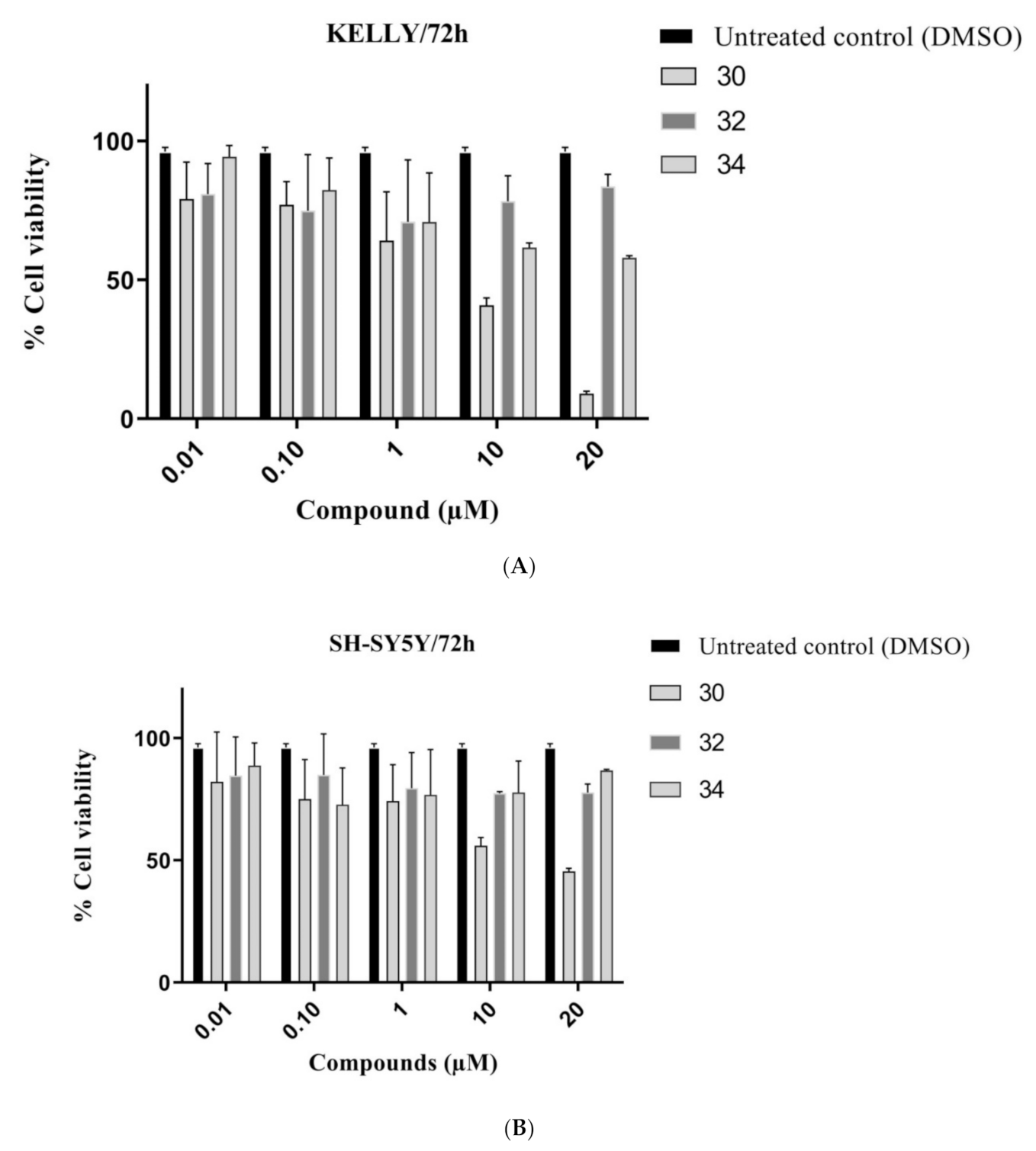
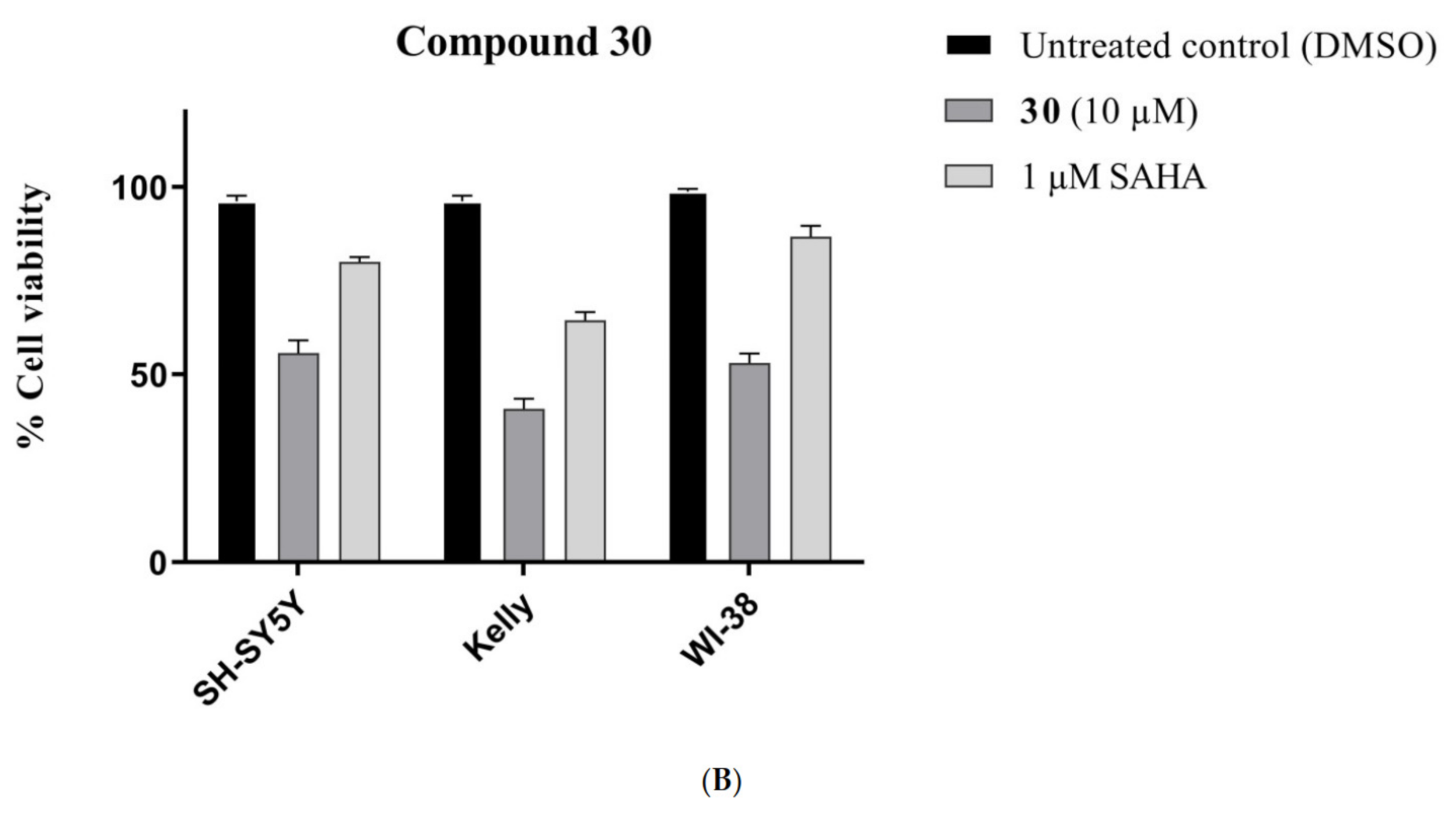
Methyl 4,6-bis((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (30): The title compound was prepared as described in GP-1 from methyl 4,6-dihdroxyindole-2-carboxylate (17) (100.4 mg, 0.485 mmol), 3-chloro-2-butanone (102.8 mg, 0.97 mmol), potassium carbonate (123.8 mg, 0.97 mmol) to give the product (148.1 mg, 88%) as a white solid; m.p. 166–168 °C; IR (KBr): νmax 3309, 2918, 2846, 2315, 2112, 1917, 1683, 1621, 1584, 1520, 1438, 1380, 1280, 1205, 1160, 1088, 991, 934, 811, 769, 685 cm−1; UV-vis (CH3CN): λmax 207 nm (ε 28,500 cm−1 M−1), 245 (21,600), 305 (22,800); 1H-NMR (300 MHz, CDCl3): δ 1.52 (d, J = 6.9 Hz, 3H, CH-Me), 1.59 (d, J = 6.9 Hz, 3H, CH-Me), 2.16 (s, 3H, COMe), 2.24 (s, 3H, COMe), 3.95 (s, 3H, CO2Me), 4.55 (q, J = 6.9 Hz, 1H, CH-Me), 4.78 (q, J = 6.9 Hz, 1H, CH-Me), 6.09 (d, J = 1.8 Hz, 1H, H5) 6.32 (d, J = 1.8 Hz, 1H, H7), 7.32 (t, J = 1.1 Hz, 1H, H3), 8.85 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ17.4 (CH-Me), 17.6 (CH-Me), 24.4 (COMe), 24.6 (COMe), 51.6 (CO2Me), 79.9 (C5), 88.5 (C7), 94.5 (CH-Me), 94.6 (CH-Me), 109.2 (C3), 112.9 (aryl C), 125.3 (C2), 141.1 (aryl C), 153.2 (C4), 158.0 (C6), 162.3 (CO2Me), 202.6 (COMe), 210.6 (COMe); HRMS (+ESI): Found m/z 370.1260, [M + Na]+, C18H21NO6Na requires 370.1267.
Methyl 8-methyl-4-(2-oxopropoxy)-1H-furo[2,3-g]indole-2-carboxylate (33): The title compound was prepared as described in GP-2 from methyl 4,6-bis(2-oxopropoxy)-1H-indole-2-carboxylate (29) (82.9 mg, 0.26 mmol) with trifluoroacetic acid (10 drops) in dichloromethane (10 mL) to give the product (11.7 mg, 15%) as a white solid; m.p. 142–144 °C; IR (KBr): νmax 3357, 2916, 2848, 2340, 2120, 1927, 1700, 1635, 1560, 1506, 1435, 1355, 1280, 1149, 1093, 1050, 994, 940, 801, 770, 704 cm−1; UV-vis (CH3CN): λmax 208 nm (ε 16,600 cm−1 M−1), 261 (40,200), 298 (15,100); 1H-NMR (300 MHz, CDCl3): δ 2.40, (s, 3H, COMe), 2.52 (d, J = 1.3 Hz, 3H, C-Me), 4.00 (s, 3H, CO2Me), 4.70 (s, 2H, CH2), 6.63 (s, 1H, H5), 7.39 (q, J = 1.3 Hz, 1H, H7), 7.52 (d, J = 2.2 Hz, 1H, H3), 9.14 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3):δ 9.8 (C-Me), 26.7 (COMe), 51.9 (CO2Me), 73.3 (CH2), 88.7 (C5), 107.7 (C3), 108.4 (aryl C), 114.3 (C8), 115.5 (aryl C), 124.9 (C2), 130.8 (aryl C), 139.6 (C7), 150.3 (C4), 155.3 (aryl C), 162.2 (CO2Me), 207.9 (COMe); HRMS (+ESI): Found m/z 324.0846, [M + Na]+, C16H15NO5Na requires 324.0848.
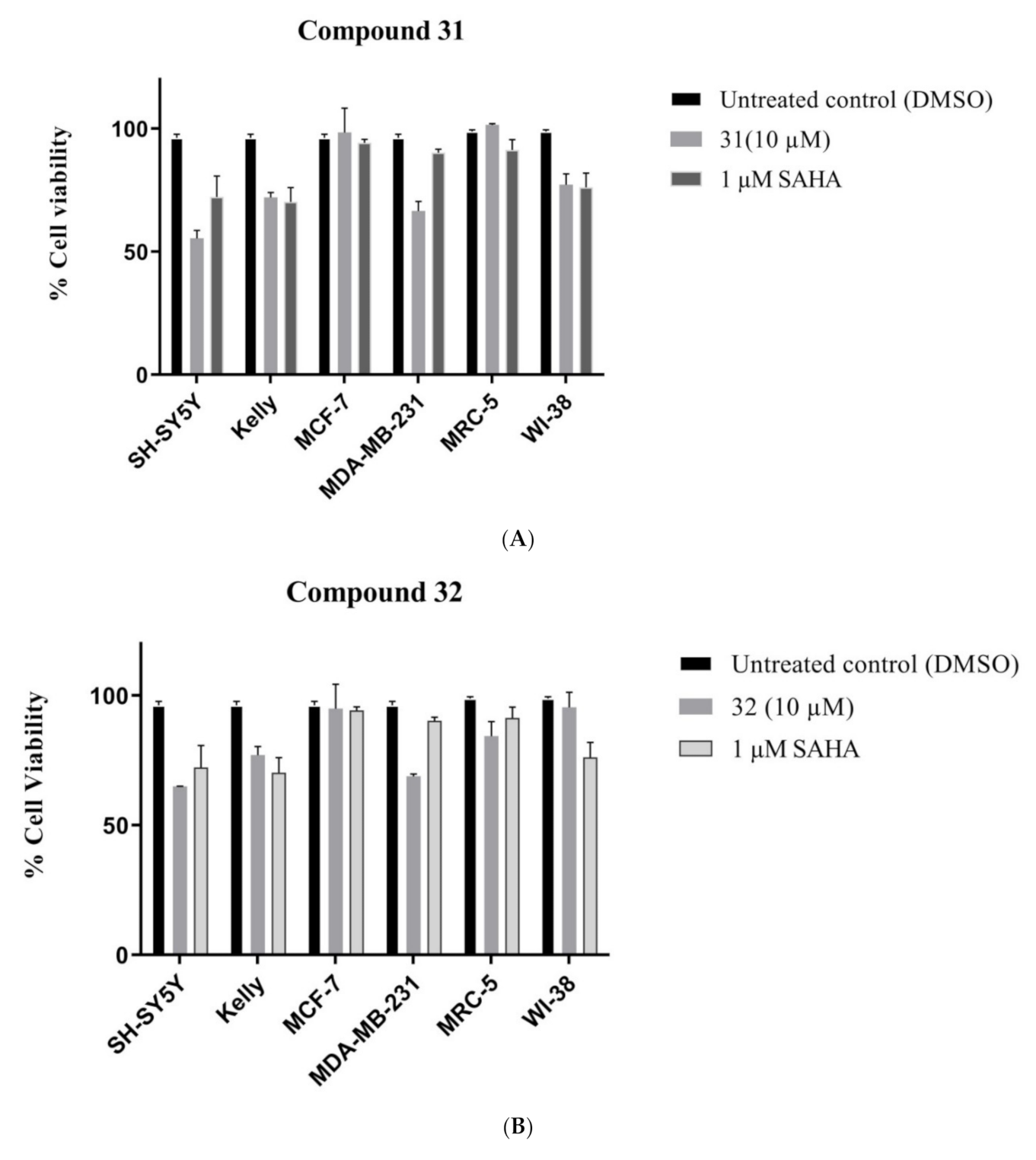
Methyl 3,6-dimethyl-7H-difuro[2,3-e:2′,3′-g]indole-8-carboxylate (31): The title compound was prepared as described in GP-2 from methyl 4,6-bis(2-oxopropoxy)-1H-indole-2-carboxylate (29) (82.9 mg, 0.26 mmol) with trifluoroacetic acid (10 drops) in dichloromethane (10 mL) to give the product (38.3 mg, 52%) as a white solid; m.p. 182–184 °C; IR (KBr): νmax 3292, 2918, 2849, 2318, 2116, 1917, 1676, 1641, 1524, 1432, 1400, 1343, 1281, 1298, 1103, 990, 924, 875, 800, 688 cm−1; UV-vis (CH3CN): λmax 238 nm (ε 33,400 cm−1 M−1), 255 (31,200), 299 (15,400); 1H-NMR (300 MHz, CDCl3): δ 2.55 (d, J = 1.3 Hz, 3H, Me), 2.57 (d, J = 1.3 Hz, 3H, Me), 3.99 (s, 3H, CO2Me), 7.43 (d, J = 1.3 Hz, 1H, H5), 7.48 (d, J = 1.3 Hz, 1H, H2), 7.55 (d, J = 2.1 Hz, 1H, H9), 9.20 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): 9.4 (CH-Me), 9.9 (CH-Me), 51.9 (CO2Me), 105.8 (C3 and C6), 109.5 (C9), 110.0 (aryl C), 111.1 (aryl C), 114.3 (aryl C), 115.2 (aryl C), 124.9 (C8), 129.0 (aryl C), 139.1 (C5), 139.4 (C2), 147.8 (aryl C), 162.2 (CO2Me); HRMS (+ESI): Found m/z 306.0733, [M + Na]+, C16H13NO4Na requires 306.0737.
Methyl 7,8-dimethyl-4-((3-oxobutan-2-yl)oxy)-1H-furo[2,3-g]indole-2-carboxylate (34): The title compound was prepared as described in GP-2 from methyl 4,6-bis((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (30) (90.2 mg, 0.26 mmol) with the catalytic amount of trifluoroacetic acid (10 drops) in dichloromethane (10 mL) to give the product (18 mg, 21%) as a white solid; m.p. 150–152 °C; IR (KBr): νmax 3326, 2921, 2831, 2338, 2110, 1907, 1686, 1628, 1590, 1528, 1501, 1437, 1354, 1271, 1161, 1115, 994, 941, 795, 770, 747 cm−1; UV-vis (CH3CN): λmax 263 nm (ε 58,400 cm−1 M−1), 301 (21,000); 1H-NMR (300 MHz, CDCl3): δ 1.62 (d, J = 7.0 Hz, 3H, CH-Me), 1.64 (s, 3H, COMe), 2.40 (s, 3H, C-Me), 2.42 (s, 3H, C-Me), 3.99 (s, 3H, CO2Me), 4.77 (q, J = 7.0 Hz, 1H, CH-Me), 6.65 (s, 1H, H5), 7.49 (d, J = 2.2 Hz, 1H, H3), 9.02 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ9.9 (C-Me), 11.5 (C-Me), 17.6 (CH-Me), 29.7 (CO-Me), 51.9 (CO2Me), 79.5 (CH-Me), 89.1 (C5), 107.8 (C3), 108.2 (aryl C), 115.7 (C8), 122.7 (aryl C), 124.6 (C2), 131.3 (aryl C), 148.4 (C4), 149.0 (C7), 153.2 (aryl C), 162.8 (CO2Me), 210.6 (CO-Me); HRMS (+ESI): Found m/z 352.1154, [M + Na]+, C18H19NO5Na requires 352.1161.
Methyl 2,3,5,6-tetramethyl-7H-difuro[2,3-e:2′,3′-g]indole-8-carboxylate (32): The title compound was prepared as described in GP-2 from methyl 4,6-bis((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (30) (90.2 mg, 0.26 mmol) with trifluoroacetic acid (10 drops) in dichloromethane (10 mL) to give the product (49.3 mg, 61%) as a white solid; m.p. 226–228 °C; IR (KBr): νmax 3329, 2918, 2864, 2318, 2105, 1921, 1680, 1639, 1528, 1437, 1368, 1275, 1298, 1180, 1080, 998, 945, 800, 749, 651 cm−1; UV-vis (CH3CN): λmax 238 nm (ε 33,600 cm−1 M−1), 259 (30,600), 304 (14,200); 1H-NMR (300 MHz, CDCl3): δ 2.44 (s, 3H, Me), 2.45 (s, 3H, Me), 2.46 (s, 3H, Me), 2.47 (s, 3H, Me), 3.97 (s, 3H, CO2Me), 7.49 (d, J = 2.1 Hz, 1H, H9), 9.08 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ9.5 (C-Me), 10.0 (C-Me), 11.6 (2×C-Me), 51.8 (CO2Me), 105.4 (C3 and C6), 108.6 (C9), 108.9 (aryl C), 110.2 (aryl C), 110.7 (aryl C), 110.7 (aryl C), 124.6 (C8), 128.3 (aryl C), 146.5 (aryl C),147.7 (C2), 147.9 (C5), 162.4 (CO2Me); HRMS (+ESI): Found m/z 312.1232, [M + H]+, C18H18NO4 requires 312.1230.
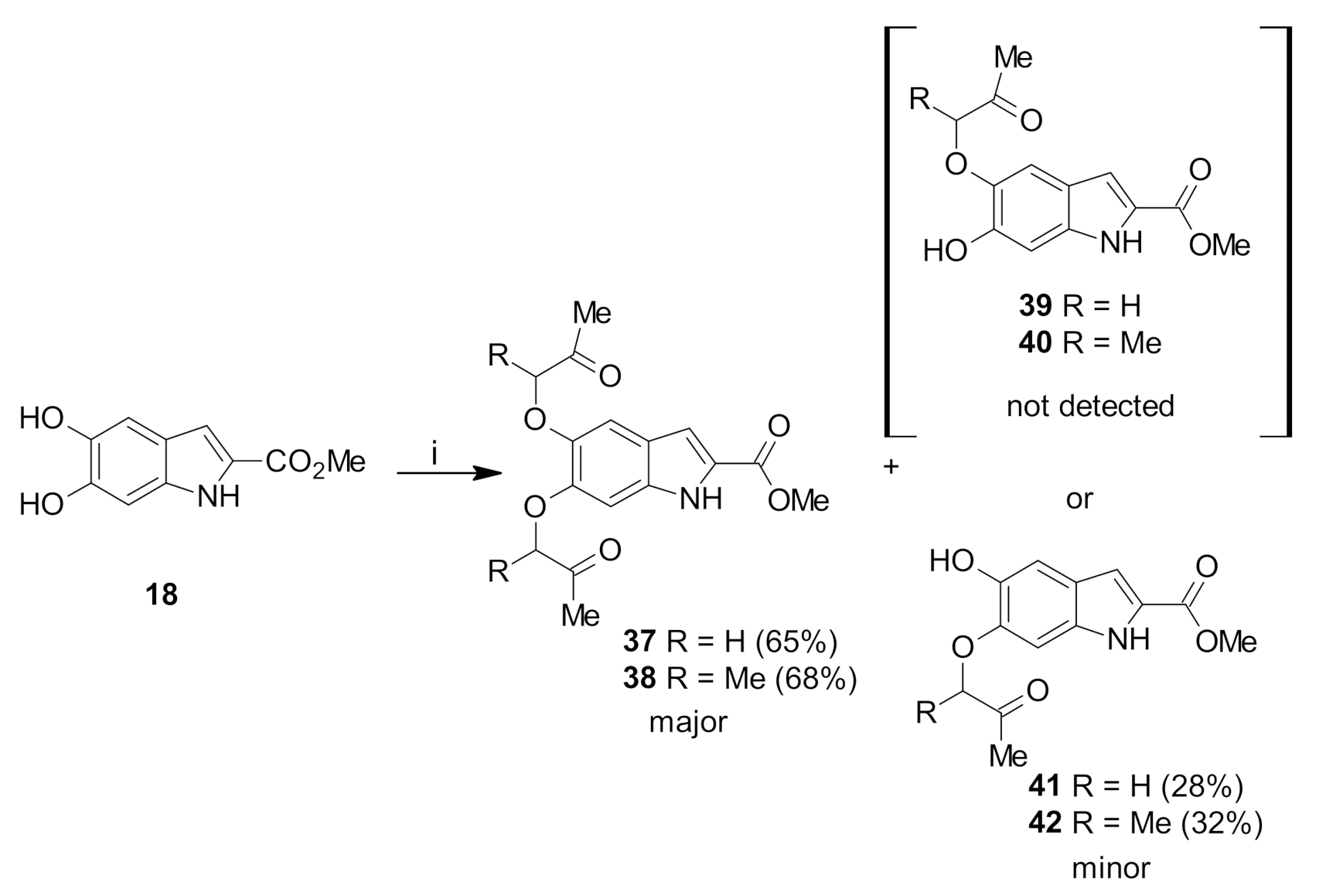
Methyl 5,6-bis(2-oxopropoxy)-1H-indole-2-carboxylate (37): The title compound was prepared as described in GP-1 from methyl 5,6-dihdroxyindole-2-carboxylate (18) (100.4 mg, 0.485 mmol), chloroacetone (88.4 mg, 0.97 mmol), potassium carbonate (123.8 mg, 0.97 mmol) to give the product (100.6 mg, 65%) as a white solid; m.p. 138–140 °C; IR (KBr): νmax 3326, 2915, 2849, 2362, 2137, 1966, 1677, 1633, 1522, 1480, 1434, 1357, 1244, 1207, 1145, 1041, 991, 897, 842, 761, 676 cm−1; UV-vis (CH3CN): λmax 209 nm (ε 35,500 cm−1 M−1), 310 (23,900); 1H-NMR (300 MHz, CDCl3): δ 2.36 (s, 6H, 2 × Me), 3.95 (s, 3H, CO2Me), 4.59 (s, 2H, CH2), 4.67 (s, 2H, CH2), 6.84 (d, J = 0.6 Hz, 1H, H4), 7.08 (s, 1H, H3), 7.12 (dd, J = 2.1, 0.9 Hz, 1H, H7), 8.89 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): δ 26.6 (CO-Me), 29.7 (CO-Me), 51.9 (CO2Me), 74.3 (CH2), 74.8 (CH2), 98.3 (C7), 106.5 (C4), 108.5 (C3), 121.5 (aryl C), 126.8 (C2), 132.3 (aryl C), 144.5 (C6), 148.3 (C5), 162.0 (CO2Me), 205.5 (CO-Me), 205.8 (CO-Me); HRMS (+ESI): Found m/z 342.0940, [M + Na]+, C16H17NO6Na requires 342.0948.
Methyl 5,6-bis((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (38): The title compound was prepared as described in GP-1 from methyl 5,6-dihydroxyindole-2-carboxylate (18) (100.4 mg, 0.485 mmol), 3-chloro-2-butanone (102.8 mg, 0.97 mmol), potassium carbonate (123.8 mg, 0.97 mmol) to give the product (114.5 mg, 68%) as a white solid; m.p. 120–122 °C; IR (KBr): νmax 3335, 2917, 2832, 2359, 2100, 1895, 1687, 1597, 1519, 1468, 1434, 1350, 1252, 1208, 1162, 1082, 977, 919, 838, 755 cm−1; UV-vis (CH3CN): λmax 212 nm (ε 34,100 cm−1 M−1), 305 (22,400); 1H-NMR (300 MHz, CDCl3): δ 1.51–1.57 (m, 6H, 2×CH-Me), 2.27 (d, J = 1.8 Hz, 3H, CO-Me), 2.31 (d, J = 1.8 Hz, 3H, CO-Me), 3.88 (s, 3H, CO2Me), 4.60–4.69 (m, 2H, 2×CH-Me), 6.59 (s, 1H, H3), 7.09 (q, J = 1.8 Hz, 1H, H7), 7.24 (q, J = 1.8 Hz, 1H, H4), 8.81 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): δ 17.5 (CH-Me), 17.6 (CH-Me), 24.6 (CO-Me), 24.8 (CO-Me), 51.6 (CO2Me), 81.4 (C7), 96.5 (C4), 108.7 (CH-Me), 108.9 (CH-Me), 111.0 (C3), 120.2 (aryl C), 126.6 (C2), 135.2 (aryl C), 144.6 (C6), 148.5 (C5), 162.3 (CO2Me), 202.7 (2 × CO-Me); HRMS (+ESI): Found m/z 370.1254, [M + Na]+, C18H21NO6Na requires 370.1261.
Methyl 5-hydroxy-6-(2-oxopropoxy)-1H-indole-2-carboxylate (41): The title compound was prepared as described in GP-1 from methyl 5,6-dihydroxyindole-2-carboxylate (18) (100.4 mg, 0.485 mmol), chloroacetone (88.4 mg, 0.97 mmol), potassium carbonate (123.8 mg, 0.97 mmol) to give the product (35.7 mg, 28%) as a white solid; m.p. 182–184 °C; IR (KBr): νmax 3334, 2918, 2855, 2318, 2106, 1948, 1682, 1622, 1519, 1486, 1430, 1351, 1248, 1203, 995, 920, 826, 799, 749, 694 cm−1; UV-vis (CH3CN): λmax 238 nm (ε 37,500 cm−1 M−1), 259 (23,200);1H-NMR (300 MHz, CDCl3): δ 2.37 (s, 3H, Me), 3.90 (s, 3H, CO2Me), 4.67 (s, 2H, CH2), 5.26 (bs, 1H, OH), 6.62 (s, 1H, H7), 7.09 (s, 1H, H4), 7.27 (d, J = 1.8 Hz. 1H, H3), 8.85 (bs, 1H, NH); 13C-NMR (75.6 MHz, CDCl3): δ 23.4 (CO-Me), 50.8 (CO2Me), 70.1 (CH2), 92.8 (aryl C), 98.1 (C7), 107.5 (C4), 107.6 (C3), 122.1 (aryl C), 126.9 (C2), 138.7 (C5), 142.9 (C6), 161.6 (CO2Me), 204.9 (CO-Me); HRMS (+ESI): Found m/z 286.0684, [M + Na]+, C13H13NO5Na requires 286.0691.
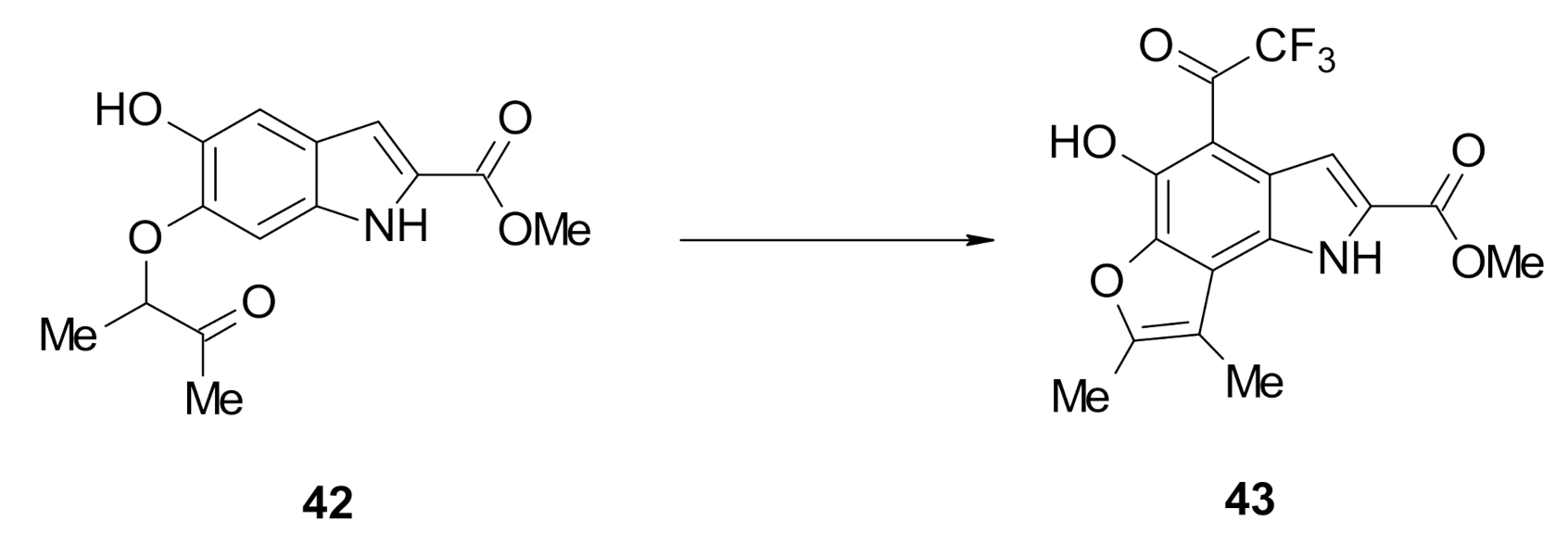
Methyl 5-hydroxy-6-((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (42): The title compound was prepared as described in GP-1 from methyl 5,6-dihydroxyindole-2-carboxylate (18) (100.4 mg, 0.485 mmol), 3-chloro-2-butanone (102.8 mg, 0.97 mmol), potassium carbonate (123.8 mg, 0.97 mmol) to give the product (43.0 mg, 32%) as a white solid; m.p. 198–200 °C; IR (KBr): νmax 3379, 2987, 2943, 2343, 2107, 1946, 1694, 1618, 1523, 1439, 1379, 1294, 1238, 1144, 1069, 928, 857, 760, 726 cm−1; UV-vis (CH3CN): λmax 209 nm (ε 40,300 cm−1 M−1), 319 (27,500); 1H-NMR (300 MHz, CDCl3): δ 1.45 (d, J = 2.1 Hz, 3H, CH-Me), 2.89 (s, 3H, C-Me), 3.87 (s, 3H, CO2Me), 4.02–4.09 (m, 1H, CH-Me), 5.61 (bs, 1H, OH), 6.97 (dd, J = 2.1, 0.9 Hz, 1H, H7), 7.03 (q, J = 1.3 Hz, 1H, H3), 7.04 (s, 1H, H4), 10.57 (bs, 1H, NH), 13C-NMR (75.6 MHz, CDCl3): δ 15.2 (CH-Me), 23.6 (CO-Me), 50.8 (CO2Me), 94.6 (CH-Me), 97.8 (C7), 107.2 (C4), 107.5 (C3), 121.9 (aryl C), 126.7 (C2), 133.5 (aryl C), 138.9 (C6), 144.0 (C5), 161.6 (CO2Me), 205.3 (CO-Me); HRMS (+ESI): Found m/z 300.0846, [M + Na]+, C14H15NO5Na requires 300.0842.
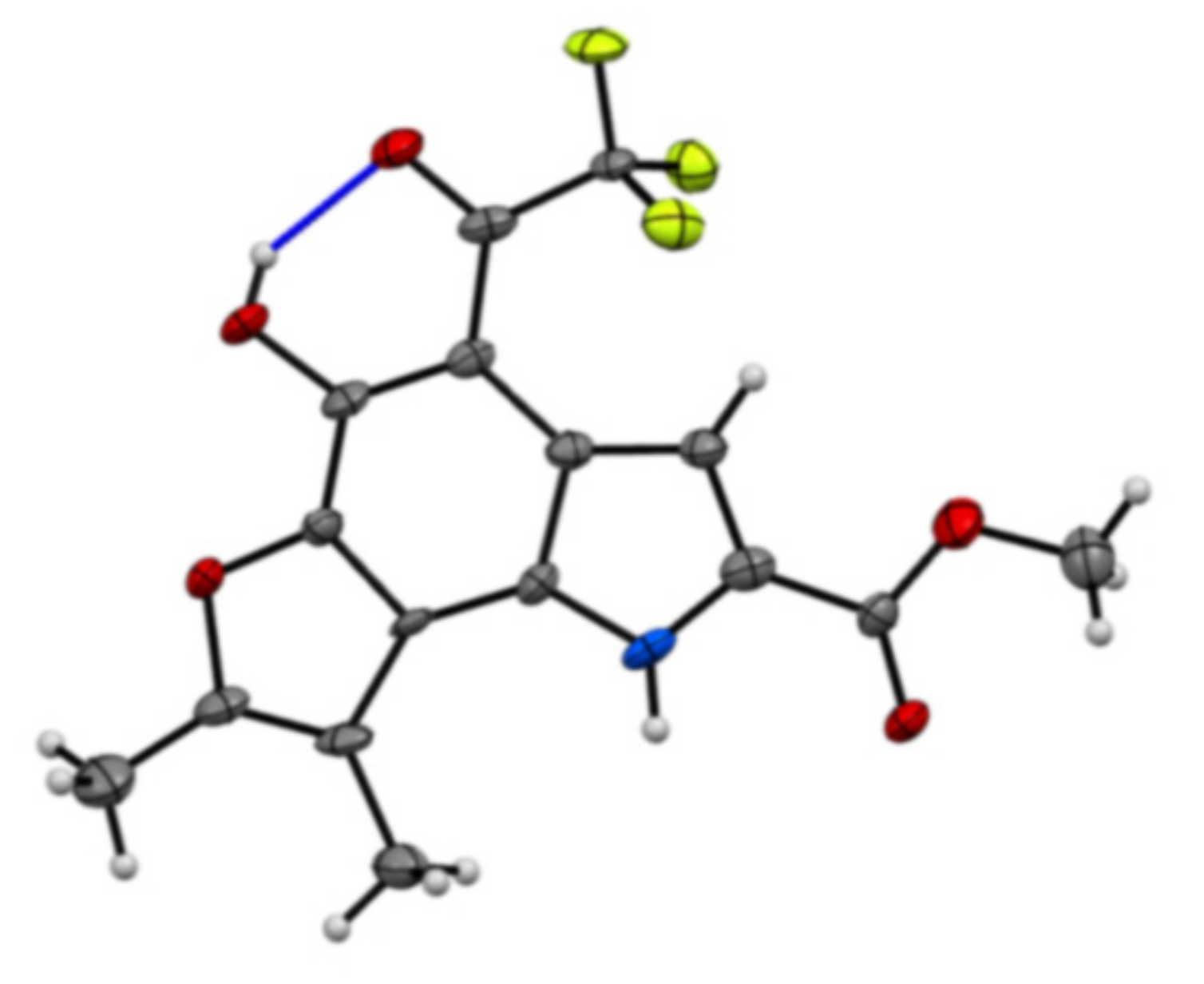
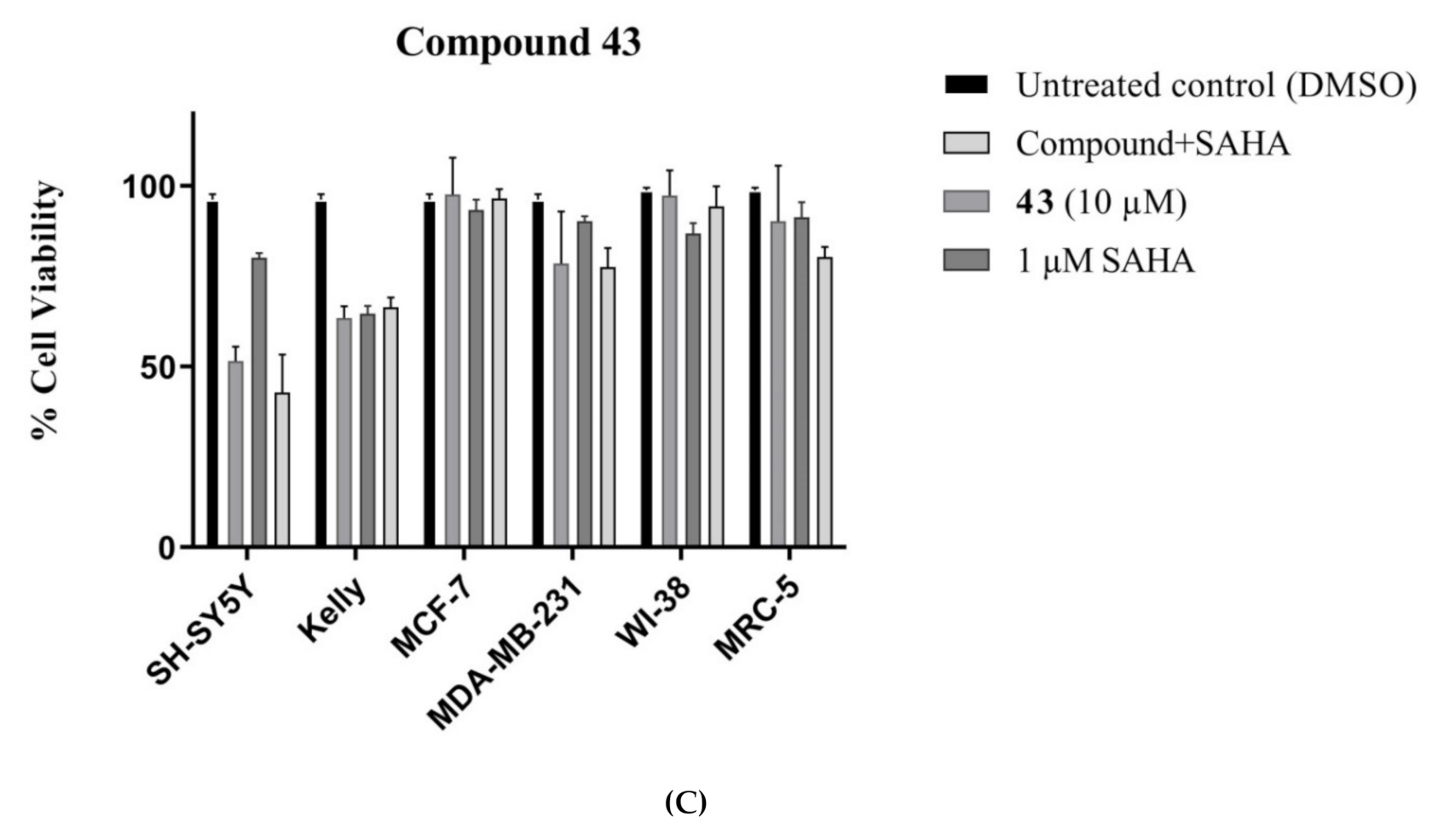
Methyl 5-hydroxy-7,8-dimethyl-4-(2,2,2-trifluoroacetyl)-1H-furo[2,3-g]indole-2-carboxylate (43): The title compound was prepared as described in GP-2 from methyl 5-hydroxy-6-((3-oxobutan-2-yl)oxy)-1H-indole-2-carboxylate (42) (72.6 mg, 0.26 mmol) with 1:1 DCM:TFA (10 mL) to give the product (59.1 mg, 64%) as a yellow solid; m.p. 224–226 °C; IR (KBr): νmax 3369, 2972, 2845, 2330, 2122, 1902, 1700, 1635, 1601, 1521, 1429, 1288, 1238, 1135, 1075, 927, 874, 790, 760 cm−1; UV-vis (CH3CN): λmax 235 nm (ε 26,600 cm−1 M−1), 272 (23,100), 381 (31,000); 1H-NMR (300 MHz, CDCl3): δ 2.55 (d, J = 1.8 Hz, 3H, Me), 2.47 (d, J = 1.8 Hz, 3H, Me), 4.01 (s, 3H, CO2Me), 7.59 (t, J = 1.8 Hz, 1H, H3), 9.27 (bs, 1H, NH), 13.44 (bs, 1H, OH), 13C-NMR (75.6 MHz, CDCl3): δ 9.6 (C-Me), 12.3 (C-Me), 52.2 (CO2Me), 102.7 (C4), 109.7 (C3), 109.8, (C8), 110.7 (CF3) 118.6 (aryl C), 124.1 (C2), 126.3 (aryl C), 126.6 (aryl C), 140.8 (C5), 153.5 (aryl C), 157.7 (C7), 161.9 (CO2Me), 181.9 (CO-CF3); HRMS (+ESI): Found m/z 378.0568, [M + Na]+, C16H12F3NO5Na requires 378.0565.


 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}