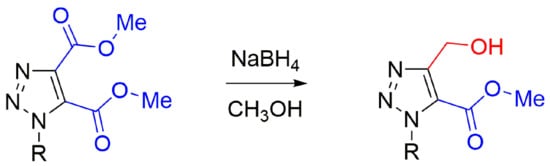
2. Results
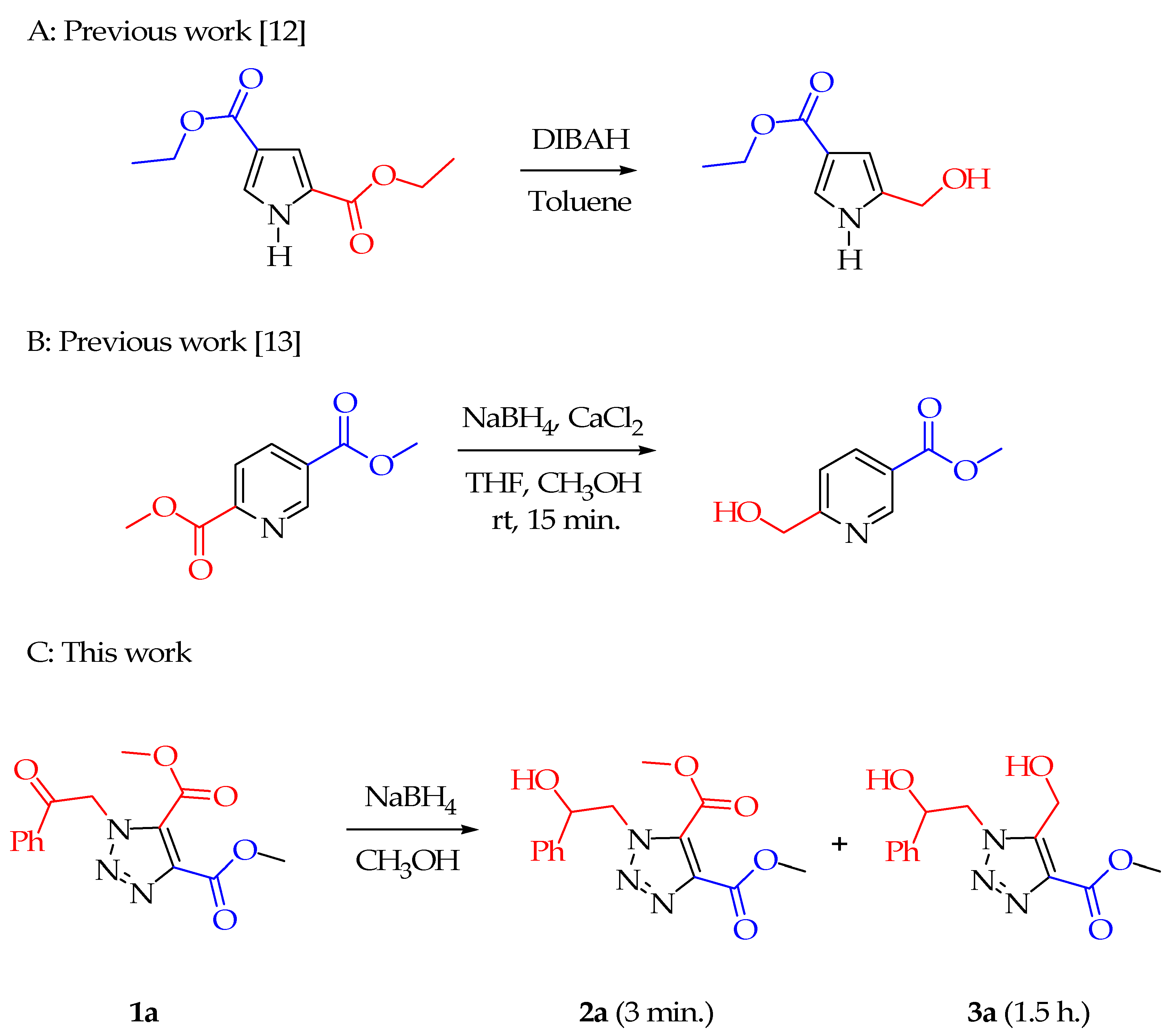
We elected to pursue work directed towards possible selective reduction of five-membered ring heteroaromatic diesters. As representatives of these, neither of the ester groups of dimethyl imidazole-4,5-dicarboxylate were reduced by NaBH
4 in MeOH/THF, but chemoselective reduction [
25] was achieved for the N(1) ketone of dimethyl 1-(2-oxo-2-phenylethyl)-1
H-1,2,3-triazole-4,5-dicarboxylate (
1a) (C of
Scheme 1) to form dimethyl 1-(2-hydroxy-2-phenylethyl)-1
H-1,2,3-triazole-4,5-dicarboxylate (
2a), followed later by regioselective reduction to form methyl 5-(hydroxymethyl)-1-(2-hydroxy-2-phenylethyl)-1
H-1,2,3-triazole-4-carboxylate (
3a). In our earliest experiment for reaction of
1a, we anticipated the reduction with NaBH
4 to reduce the ketone, but not the ester. The ketone was reduced, but some ester reduction was also observed. Adding more NaBH
4 and waiting for the reaction to finish gave
3a in good yield, with the ester group at C(4) being unreactive, although initially we did not know which of the ester substituents had been reduced. This turned out to be an excellent example of a regioselective process. We chose to investigate the scope of the selectivity of diester reduction of
1a, along with related compounds. Ester reduction was understandably more likely for
1a than for the more electron-rich imidazole 4,5-diesters. An important issue was how a hydroxy group as part of the N(1) substituent might influence the outcome of the ester reduction or the reduction rate of
1a. We posited that a hydroxy group might be required in order to observe selective reduction of
1a, which turned out not to be the case.
A detailed study of a series of the 1
H-1,2,3-triazole diesters revealed that a carbonyl function at N(1) was not necessary for reduction of the C(5) ester, but reduction might be enhanced via six-membered lactone formation between an OH group on an N(1) substituent and the C(5) ester prior to reduction. Five-membered lactones are known to be reduced by NaBH
4 [
26]. We have not investigated the possibility of lactone formation. However, evidence was not seen for formation of a reactive borate complex [
27]. The C(5) ester group was found to be much more reactive with NaBH
4 than a C(4) ester in the presence or absence of a carbonyl group (or a hydroxy group) at N(1). The scope of the selective ester reduction of triazole diesters at C(5) and the factors responsible for the selectivity were examined by reduction of a number of triazole esters and diesters.
Several dimethyl
1H-1,2,3-triazole-4,5-dicarboxylates (
1a–
f) were prepared by thermal cycloaddition [
4,
5] of organic azides with dimethyl acetylenedicarboxylate (DMAD). Reduction of
1a–
f with NaBH
4 in methanol afforded
2a and
2b in a chemoselective process after just 3 min (
Table 1). The N(1) ester group of dimethyl 1-(2-ethoxy-2-oxoethyl)-1
H-1,2,3-triazole-4,5-dicarboxylate (
1c) was reduced faster than the C(5) ester, but not fast enough to be synthetically useful.
Additional reaction time allowed all C(5) esters to undergo reduction. The products
3a–
f were formed in the times indicated, along with the number of equivalents of NaBH
4 needed for the starting diesters to be fully converted to the C(4) esters
3a–
f [
28,
29]. Notably, diesters
1d–
f, containing only alkyl or aralkyl functionality at N(1), required longer times for reduction. The structures of the products were elucidated using 1D and 2D-NMR spectroscopy. As it was not clear from 1D spectroscopy alone which ester group had been reduced, 2D-NMR HMBC spectroscopy was necessary to help elucidate the product structures of
3a–
f [
29]. The structure of the reduction product
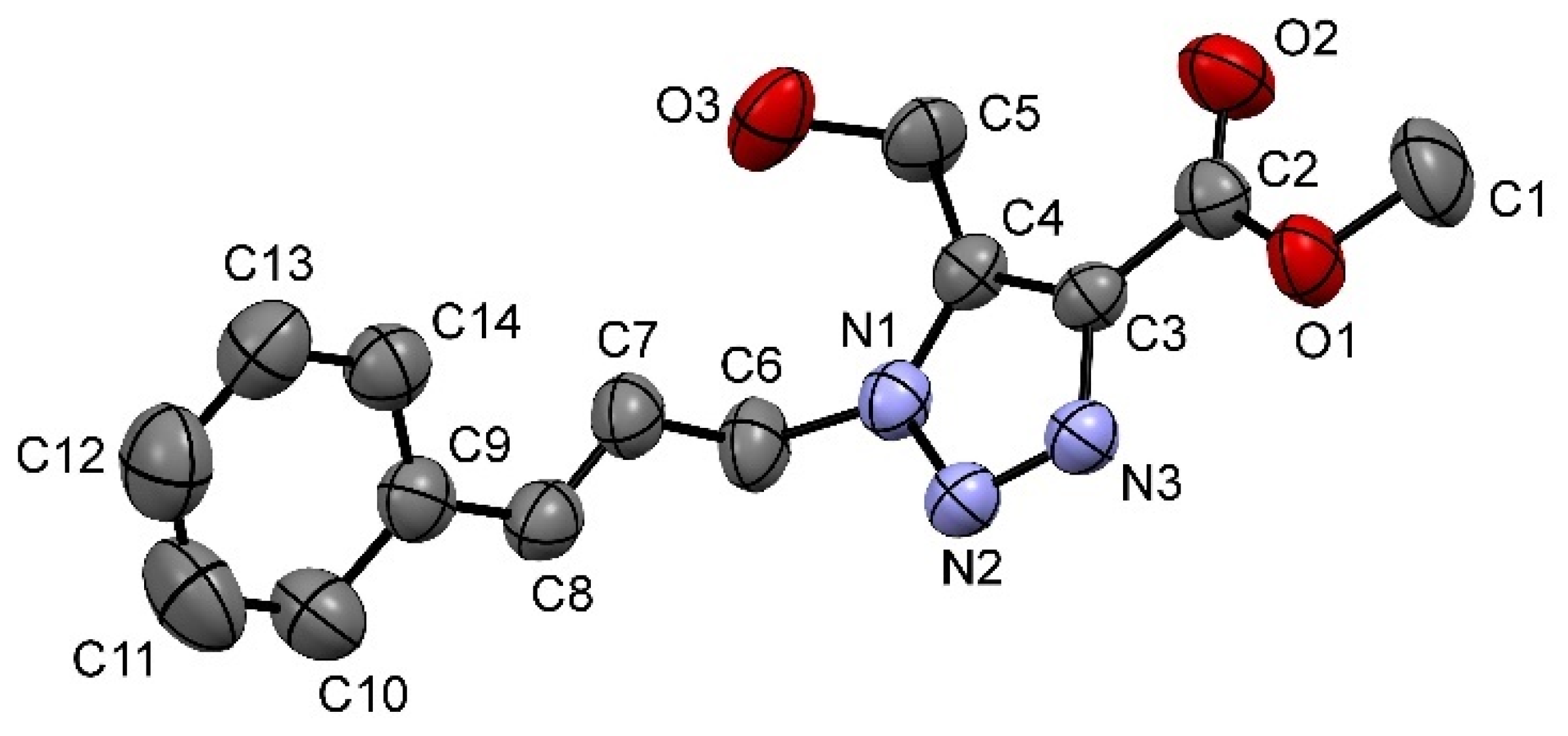
3f was further confirmed by X-ray crystallography, as shown in
Figure 1.
Triazoles with substituents at the C(5) position were prepared using ruthenium catalysis [
10] by dichloro(pentamethylcyclopentadienyl)ruthenium polymer (RuCl
2CP*)
x producing 1-substituted 1
H-1,2,3-triazole-5-esters
4a–
d, as shown in
Table 2. The reduction of
4a–
d with NaBH
4 in methanol afforded
5a–
d. The reduction of
4d required more NaBH
4 and more time than
4a–
c, similar to the conditions needed for the reduction of
1f. The absence of an electron-withdrawing ester group at C4 for
4a–
d also slowed the reduction rate of the C5 ester group compared to the rates of
1a–
f.
Triazoles with substituents at the C(4) position were prepared using copper catalysis [
6,
7] producing methyl 1-substituted 1
H-1,2,3-triazole-4-carboxylates
6a–
d, as shown in
Table 3. The reduction of
6a–
d with NaBH
4 in methanol afforded
7a–
b and
8c–
d. It was observed that the reduction of the N(1) carbonyl for
6a–
b occurred at an acceptable rate, but the reduction of the C(4) position ester group of
8c–
d was slow and required multiple days and a large excess of NaBH
4, as shown in
Table 3.
Observations based on the data in
Table 1,
Table 2 and
Table 3 are: 1. β-Ketone substituents at N(1) undergo rapid reduction as expected. 2. β-Ester substituents at N(1) undergo reduction more rapidly than C(5) ester substituents. 3. Reduction of C(5) ester substituents is facilitated by a hydroxy group at the β carbon of N(1) substituents, whereas C(5) esters having only aralkyl substituents at N(1) react slower with NaBH
4. 4. A ketone substituent at N(1) facilitates reduction of a C(5) ester more than an ester at N(1) because the ketone is reduced more rapidly, exposing a facilitating hydroxy group for reduction of a C(5) ester. 5. Regardless of the substituents at N(1), C(5) ester substituents are reduced more rapidly than C(4) ester substituents. 6. The presence of a C(4) ester substituent enhances the rate of reduction of a C(5) ester substituent. Stronger reducing agents such as LiAlH
4 should be chosen when complete reduction of triazole and other heteroaromatic diesters is desired.
Compounds 1a–f and 4a–d each gave reduction of the C(5) ester. The relative rates of reduction varied considerably with the nature of the N(1) substituents. An N(1) substituent containing a ketone was reduced by NaBH4 to the corresponding alcohol within a few minutes, as observed for products 2a–b. The resulting hydroxy substituent situated at the δ-position relative to the ester carbonyl undergoing reduction showed a rate-enhancing effect on the ester at C(5), such as in the reduction of 1a–b to 3a–b as compared to the reduction rates of 1d–f to 3d–f. When N(1) contained a keto or ester group, as for 1a–c and 4a–c, reduction of that keto or ester group to an alcohol was observed. The rate-enhancement was lower for the reduction of 1c because the N(1) ester group was reduced more slowly than the N(1) ketones of 1a and 1b, also observed for the reduction rate of 6b compared to that of 6a. The reduction rate of the C(5) ester group was significantly slower when the N(1) substituent contained only alkyl, alkenyl, or aralkyl groups as in 1d–f and 4d. The times necessary to complete the reactions in this study provide a clear distinction between triazole C(5) esters bearing an N(1) substituent containing a β-carbonyl group (1a–c and 4a–c) and those having N(1) alkyl or alkenyl substituents (1d–f and 4d). All ketone and ester groups of 1a–c and 4a–c were reduced to alcohols. The resulting alcohols 2a–c and 5a–c gave reduction of the C(5) ester groups more readily than C(5) esters 1d–f and 4d.
Our hypothesis to explain the difference in reactivity of the C(4) and C(5) diesters is that there are consistent differences in electron density at the ester carbonyl carbons, leading to preferred reaction of the C(5) carbonyl group of the C(5) esters. Molecular modeling studies [
30] furnished insight into the preferential reduction of the C(5) esters, which indicated that the calculated electron density at the C(5) carbonyl carbon was lower than at the C(4) carbonyl carbon for
1a–
f and
4a–
d, supporting the observation that NaBH
4 gave reduction of the C(5) ester group in preference to the C(4) ester group. Plots of electron densities of the C(5) carbonyl and C(5) carbons of
1a–
f and separately
4a–
d showed linear correlations with R-squared values of >0.9. When intramolecular hydrogen bonding was included in the calculations and compared to the data for no hydrogen bonding, a lower electron density was observed at C(5) and a much lower electron density at the C(5) carbonyl carbon. However, the R-squared values were only 0.87 and 0.49, respectively (See
Supplementary Material).
Electron density differences and, where relevant, intramolecular hydrogen bonding, are postulated to explain the faster rates of reduction of the C(5) esters. Other possible explanations of a higher rate of reactivity at C(5) such as for
2a and
2b are the formation of a lactone intermediate [
26] or a borate complex intermediate [
27]. These explanations are not possible for
1d–
f, which lack reducible carbonyl groups in substituents at N(1) such as in
1a–
c. Similar observations were made for the modeling studies of
4a–
d. The overall rates of reduction of
4a–
d showed a decrease compared to those for
1a–
f, indicating a favorable rate enhancement for reduction of a C(5) ester when an ester group is also present at C(4).
4. Materials and Methods
All starting materials were purchased from commercially available sources and used as obtained. All synthesized organic azides were stored at 0 °C until needed. All azides should be considered as hazardous and potentially explosive. Plastic or ceramic spoons were used when weighing solid azides. All synthesized organic azides were stored at 2–8 °C to ensure safety of potentially explosive material and to reduce degradation of the azide moiety. All reactions were performed in a ventilated hood. Thin layer chromatography (TLC) was performed on Agela Technologies aluminum-backed silica dioxide plates and products observed under 254 nm UV light. Flash column and radial chromatography (T-Squared Technology, Inc., 206 Lassen Dr., San Bruno CA 94066 USA) were performed with SiliCycle silica gel 60, 0.040−0.063 mm (230−400 mesh) using distilled ethyl acetate and distilled hexanes. Microwave-assisted synthesis was performed using a CEM Discover SP Microwave Synthesizer. NMR spectra (400 or 500 MHz for 1H and 100 MHz for 13C) were measured in CDCl3 or DMSO-d6. Chemical shifts (δ) are given in ppm relative to the resonance of their respective residual solvent peak, CHCl3 (7.27 ppm, 1H; 77.16 ppm, the middle peak, 13C). Multiplicities were described using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, dt = doublet of triplets, m = multiplet. Regioselectivity of the reduction was determined by HSQC and HMBC correlation 2D-NMR analysis. FTIR experiments were performed on a Perkin Elmer Spectrum 1 instrument (Perkin Elmer Inc., 732 E. Utah Valley Dr., Suite 120, American Fork, UT 84003, USA) or Fisher Scientific Nicolet iS5 instrument (Thomas Scientific, PO Box 99, Swedesboro, NJ 08085, USA). Melting points were performed on a Stanford Research Systems Digimelt MPA160 SRS instrument (Stanford Research Systems, 1290-D Reamwood Ave., Sunnyvale, CA 94089, USA) and all melting points are uncorrected. All elemental analyses were performed by Atlantic Microlabs Inc., Norcross, GA. High-resolution MS experiments were recorded at 70 eV by electrospray ionization using a Voyager DE-STR MALDI-TOF (ABI) instrument (AB Sciex, LLC, 500 Old Connecticut Path, Framingham, MA 10701, USA). Single crystals of 3f suitable for X-ray crystallographic analysis were obtained by slow recrystallization from a THF solution of 3f at room temperature. X-ray diffraction data were collected using a Bruker SMART APEX CCD diffractometer (Bruker Corp., 40 Manning Rd., Billerica, MA 01821, USA).
4.1. General Procedures for the Synthesis of Organic Azides
Metal azides are shock sensitive and should be handled with smooth edged, non-metallic spoons or spatulas. Purification of initially isolated products is required to remove traces of azides.
Procedure A. To
tert-butyl alcohol/water (1:1, 100 mL) in a 250 mL round-bottom flask were added 2-bromoacetophenone, cinnamyl bromide or benzyl bromide (10 mmol), and sodium azide (0.98 mg, 15 mmol) and the mixture was stirred at rt for 2 h. The mixture was extracted with ethyl acetate (3 × 25 mL), the organic layers were combined, washed with brine, dried over anhydrous sodium sulfate, and evaporated in vacuo to dryness, affording the organic azide in 80–95% yield. The azide was stored at 0 °C and used without further purification [
29].
2-Azidoacetophenone [
31]. Dark yellow oil. (1.435 g, 89.0% yield);
1H-NMR (CDCl
3, 400 MHz) δ 7.92–7.90 (d, 2H), δ 7.65–7.61 (t, 1H), δ 7.52–7.48 (t, 2H), δ 4.57 (s, 2H); FTIR cm
−1 3062, 2096, 1691, 1596, 1448, 1213.
Cinnamyl azide [
32]. Yellow oil. (1.586 g, 99.7% yield);
1H-NMR (CDCl
3, 400 MHz) δ7.41–7.38 (d, 2H), δ 7.35–7.31 (t, 2H), δ 7.29–7.25 (t, 1H), δ 6.66–6.62 (d,
J = 15.6 Hz, 1H), δ 6.27–6.19 (m, 1H), δ 3.94–3.92 (d, 2H); FTIR cm
−1 3027, 2090, 1654, 1598, 1448, 1234.
Benzyl azide [
33]. Yellow oil. (1.187 g, 87.6%);
1H-NMR (CDCl
3, 400 MHz). δ 7.39–7.29 (m, 5H), δ 4.31 (s, 2H); FTIR cm
−1 3031, 2089, 1597, 1453, 1252.
Procedure B. Into a 100 mL round-bottom flask, chloroacetone, ethyl bromoacetate, or 1-bromopentane (10 mmol), DMSO (50 mL), and sodium azide (0.98 mg, 15 mmol) were added. The mixture was stirred at rt for 12 h and extracted with diethyl ether (3 × 25 mL). The combined organic layers were washed with water (2 × 25 mL), dried over anhydrous sodium sulfate, and evaporated to dryness in vacuo, affording the organic azide in 70–85% yield. The azide was stored at 0 °C and used without further purification.
1-Azidopropan-2-one [
34]. Yellow oil. (831 mg, 83.9% yield);
1H-NMR (CDCl
3, 400 MHz) δ 3.99 (s, 2H), δ 2.21 (s, 3H);
13C NMR (CDCl
3, 100 MHz) δ 202.0, 57.8, 27.1; FTIR cm
−1 2097, 1724, 1281.
Ethyl azidoacetate [
35]. Yellow oil. (1.058 g, 82.1% yield);
1H-NMR (CDCl
3, 400 MHz) δ 4.28–4.21 (q, 2H), δ 3.82 (s, 2H), δ 1.34–1.26 (t, 3H); FTIR cm
−1 2102, 1740, 1286, 1190.
1-Azidopentane [
36]. Colorless oil. (1.120 g, 99.0% yield);
1H-NMR (CDCl
3, 400 MHz) δ 3.28–3.22 (t, 2H), δ 1.66–1.56 (m, 2H), δ 1.40–1.29 (m, 4H), δ 0.95–0.88 (t, 3H);
13C-NMR (CDCl
3, 100 MHz) δ 51.35, 28.75, 28.42, 22.13, 13.81; FTIR cm
−1 2089.
4.2. Experimental Procedure for the Synthesis of 1H-1,2,3-Triazole-4,5-Diesters (1a–f)
Into a 100 mL round-bottom flask, the organic azide (4 mmol), tert-butyl alcohol/H2O (1:1) soln. (60 mL), dimethyl acetylenedicarboxylate (582 mg, 4.1 mmol) were added and the mixture heated under reflux for 2 h, cooled and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, and evaporated in vacuo to dryness. Purification was performed by recrystallization or flash column chromatography.
Dimethyl 1-(2-oxo-2-phenylethyl)-1H-[1,2,3]triazole-4,5-dicarboxylate (
1a) [
37].
Dimethyl 1-(2-oxopropyl)-1
H-1,2,3-triazole-4,5-dicarboxylate (
1b) [
34]. Colorless solid, purified by flash column chromatography (1:6, ethyl acetate/hexanes). (689 mg, 71.4% yield); m.p.: 72.4–73.9 °C;
1H-NMR (CDCl
3, 400 MHz) δ 5.54 (s, 2H), δ 3.98 (s, 3H), δ 3.94 (s, 3H), δ 2.32 (s, 3H);
13C-NMR (CDCl
3, 100 MHz) δ 198.6, 160.3, 158.9, 140.1, 129.9, 59.1, 53.4, 52.8, 27.1; FTIR cm
−1 1752, 1725 1244, 1110, 1064; Anal. Calcd. for C
9H
11N
3O
5: C, 44.82; H, 4.60; N, 17.42% Found: C, 45.09; H, 4.63; N, 17.55%.
Dimethyl 1-(2-ethoxy-2-oxoethyl)-1
H-1,2,3-triazole-4,5-dicarboxylate (
1c) [
38]. Colorless solid, purified by recrystallization from ethanol. (881 mg, 81.2% yield); m.p.: 120.8–121.6 °C;
1H-NMR (CDCl
3, 400 MHz) δ 5.44 (s, 2H), δ 4.25 (q,
J = 7.1 Hz, 2H), δ 3.99 (s, 3H), δ 3.96 (s, 3H), δ 1.28 (t,
J = 7.1 Hz, 3H);
13C-NMR (CDCl
3, 100 Hz), δ 165.5, 160.2, 158.7, 140.2, 130.0, 62.6, 53.4, 52.8, 51.6, 14.0; FTIR cm
−1 3024, 1692, 1664, 1585, 1463, 1248, 1106, 1042; Anal. Calcd. for C
10H
13N
3O
6: C, 44.28; H, 4.83; N, 15.49% Found: C, 44.30; H, 4.88; N, 15.43%.
Dimethyl 1-pentyl-1
H-1,2,3-triazole-4,5-dicarboxylate (
1d) [
39]. Colorless oil, purified by flash column chromatography (1:10, ethyl acetate/hexanes). (819 mg, 80.3% yield);
1H NMR (CDCl
3, 400 MHz) δ 4.16–4.50 (t, 2H), δ 3.97 (s, 3H), δ 3.93 (s, 3H), δ 1.93–1.81 (m, 2H), δ 1.41–1.81 (m, 4H), δ 0.85 (t,
J = 7.0 Hz, 3H);
13C-NMR (CDCl
3, 100 MHz) δ 160.5, 159.0, 139.8, 129.8, 53.4, 52.6, 50.6, 29.9, 28.3, 21.9, 13.7; FTIR cm
−1 1729, 1212, 1127, 1060; HRMS (TOF MS ES+)
m/
z [m + H]
+ calc. C
11H
17N
3O
4 256.1292. Found: 256.1299.
Dimethyl 1-benzyl-1
H-1,2,3-triazole-4,5-dicarboxylate (
1e) [
40]. Light yellow solid, purified by recrystallization from ethanol/water. (982 mg, 89.2% yield); m.p.: 45.8–46.4 °C;
1H-NMR (CDCl
3, 400 MHz) δ 7.38–7.25 (m, 5H), δ 5.82 (s, 2H), δ 3.97 (s, 3H), δ 3.88 (s, 3H); FTIR cm
−1 3028, 1724, 1572, 1466, 1227, 1140, 1057.
Dimethyl 1-(3-phenylprop-2-en-1-yl)-1
H-1,2,3-triazole-4,5-dicarboxylate (
1f) [
41]. Yellow oil, purified by flash column chromatography (1:8, ethyl acetate/hexanes). (1.032 g, 85.7% yield);
1H-NMR (CDCl
3, 400 MHz) δ 7.37–7.24 (m, 5H), δ 6.65–6.62 (d,
J = 15.79 Hz, 1H), δ 6.29 (m, 1H), δ 5.38 (dd,
J = 6.73 Hz, 2H), δ 3.97 (s, 3H), δ 3.94 (s, 3H);
13C-NMR (CDCl
3, 100 MHz) δ 160.4, 159.0, 140.0, 136.0, 135.3, 129.9, 128.7, 128.6, 126.7, 121.0, 53.5, 52.8, 52.6; FTIR cm
−1 3028, 1728, 1553, 1450, 1213, 1100, 1059; HRMS (TOF MS ES+)
m/
z [m + H]
+ calc. C
15H
15N
3O
4 302.1136. Found: 302.1135.
4.3. Experimental Procedure for the Synthesis of 1H-1,2,3-Triazole Monoalcohol Esters (2a–2b)
Into a 25 mL round-bottom flask, 1H-1,2,3-triazoloester (1a or 1b, 1 mmol) and methanol (20 mL) were added, followed by NaBH4 (38 mg, 1 mmol) and the solution stirred for three min. The reaction was quenched with 1 M HCl until a pH of 6 was reached (~1.5 mL) and evaporated in vacuo to dryness. The crude product was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated in vacuo to dryness.
Dimethyl 1-(2-hydroxy-2-phenylethyl)-1
H-1,2,3-triazole-4,5-dicarboxylate (
2a) [
42]. Colorless oil, purified by flash column chromatography (1:4, ethyl acetate/hexanes). (300 mg, 98.5% yield);
1H-NMR (CDCl
3, 400 MHz) δ 7.42–7.37 (m, 5H), δ 5.19–5.14 (m, 1H), δ 4.86–4.83 (dd, 2H), δ 3.98 (s, 3H), δ 3.97 (s, 3H) δ 2.91 (s, 1H);
13C-NMR (CDCl
3, 100 MHz) δ 160.3, 159.3, 139.6, 139.3, 131.3, 128.9, 128.7, 125.8, 72.9, 56.5, 53.5, 52.7; FTIR cm
−1 3382, 2954, 1726, 1454, 1220, 1061; HRMS (TOF MS ES+)
m/
z [m + H]
+ calc. for C
14H
15N
3O
5 306.1090 Found: 306.1087.
Dimethyl 1-(2-hydroxypropyl)-1H-1,2,3-triazole-4,5-dicarboxylate (2b). Colorless oil, purified by flash column chromatography (1:4, ethyl acetate/hexanes). (197 mg, 80.8% yield); 1H-NMR (CDCl3, 400 MHz) δ 4.52–4.57 (dd, 2H), δ 4.23–4.29 (m, 1H), δ 4.02 (s, 3H), δ 3.99 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ 160.36, 159.44, 139.46, 131.17, 66.81, 56.48, 53.49, 52.73, 20.62; FTIR cm−1 3407, 2957, 1727, 1459, 1219, 1117, 1064; HRMS (TOF MS ES+) m/z [m + H]+ calc. for C9H13N3O5 244.0928 Found: 244.0932.
4.4. Experimental Procedure for the 1H-1,2,3-Triazole Ester Reduction to Alcohols 3a–f, 5a–d, 7a–b and 8c–d
Into a 25 mL round-bottom flask, the 1
H-1,2,3-triazoloester (
1a–
f,
4a–
d,
6a–
d, 2 mmol) and methanol (10 mL) were added. Sodium borohydride was added to the flask in portions (76 mg, 2 mmol) every 0.5 h until reaching the number of equivalents specified in
Table 1,
Table 2 and
Table 3. TLC analysis was performed on the reaction mixture prior to each addition until no starting material remained in the reaction mixture. Upon completion, the methanol was evaporated in vacuo to dryness.
Methyl 5-(hydroxymethyl)-1-(2-hydroxy-2-phenylethyl)-1
H-1,2,3-triazole-4-carboxylate (
3a) [
29]. Tan solid, purified by flash column chromatography, (1:4, ethyl acetate/hexanes). (404 mg, 72.9% yield); m.p.: 96.8–98.5 °C;
1H-NMR (CDCl
3, 400 MHz) δ 7.46–7.33 (m, 5H), δ 5.27–5.23 (d, 1H), δ 5.02–4.95 (dd, 1H), δ 4.86–4.81 (ds, 1H), δ 4.69–4.64 (dd, 1H), δ 4.51–4.46 (dd, 1H), δ 4.38–4.34 (s, 2H);
13C-NMR (CDCl
3, 100 MHz) δ162.1, 142.0, 139.7, 136.0, 128.9, 128.6, 125.8, 72.8, 55.6, 53.0, 52.5; FTIR cm
−1 3510, 3290, 3083, 1708, 1234, 1203, 1068, 1032; Anal. Calcd. for C
13H
15N
3O
4: C, 56.31; H, 5.45; N, 15.16% Found: C, 56.39; H, 5.44; N, 15.11%.
Methyl 5-(hydroxymethyl)-1-(2-hydroxypropyl)-1H-1,2,3-triazole-4-carboxylate (3b). Tan solid, purified by flash column chromatography, (1:4, ethyl acetate/hexanes). (334 mg, 77.7% yield); m.p.: 72.1–73.8 °C; 1H-NMR (DMSO-d6, 400 MHz) δ 5.49 (t, 1H), δ 5.12 (d, 1H), δ 4.97–4.74 (m, 2H), δ 4.50–4.28 (m, 2H), δ 4.15–3.97 (m, 1H), δ 3.84 (s, 3H), δ 1.12 (d, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 161.9, 141.9, 135.5, 65.9, 55.5, 52.2, 51.3, 21.3; FTIR cm−1 3381, 3210, 1714, 1250, 1179, 1334, 1033; Anal. Calcd. for C8H13N3O4: C, 44.650; H, 6.09; N, 19.53% Found: C, 44.58; H, 5.98; N, 19.27%
Methyl 1-(2-hydroxyethyl)-5-(hydroxymethyl)-1H-1,2,3-triazole-4-carboxylate (3c). Colorless solid, initially purified by flash column chromatography (1:2.5, ethyl acetate/hexanes) and further purified by recrystallization from dichloromethane/hexanes. (205 mg, 50.9% yield); m.p.: 72.0–72.5 °C; 1H-NMR (DMSO-d6, 400 MHz) δ 5.53 (t, 1H), δ 5.09 (t, 1H), δ 4.88 (d, 2H), δ 4.53 (t, 2H), δ 3.84 (s, 3H), δ 3.80 (q, 2H); 13C-NMR (DMSO-d6, 100 MHz) δ 161.9, 141.8, 135.5, 60.4, 52.2, 51.29, 51.26; FTIR cm−1 3342, 3223, 1708, 1241, 1195, 1123, 1052; Anal. Calcd. for C7H11N3O4: C, 41.79; H, 5.51; N, 20.89% Found: C, 41.64; H, 5.36; N, 20.82%.
Methyl 5-(hydroxymethyl)-1-pentyl-1H-1,2,3-triazole-4-carboxylate (3d). Colorless solid, purified by flash column chromatography (1:5, ethyl acetate/hexanes). (234 mg, 51.4% yield); m.p.: 60.5–60.8 °C; 1H-NMR (CDCl3, 400 MHz) δ 4.89 (s, 2H), δ 4.46–4.30 (m, 2H), δ 3.97 (s, 4H), δ 1.99–1.79 (m, 2H), δ 1.42–1.18 (m, 4H), δ 0.88 (t, 3H); 13C-NMR (CDCl3, 100 MHz) δ 163.1, 140.9, 136.9, 53.2, 52.6, 48.8, 30.0, 28.5, 22.1, 13.8; FTIR cm−1 3247, 1708, 1224, 1194, 1047; Anal. Calcd. for C10H17N3O3: C, 52.85; H, 7.54; N, 18.49% Found: C, 53.00; H, 7.38; N, 18.46%
Methyl 1-benzyl-5-(hydroxymethyl)-1H-1,2,3-triazole-4-carboxylate (3e). Colorless solid, purified by recrystallization from diethyl ether/hexanes. (411 mg, 83.1% yield); m.p.: 89.1–89.8 °C; 1H-NMR (CDCl3, 400 MHz) δ 7.35–7.18 (m, 5H), δ 5.62 (s, 2H), δ 4.76 (s, 2H), δ 3.95 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ 162.9, 141.1, 137.3, 134.0, 129.2, 128.8, 127.3, 53.3, 52.7, 52.6; FTIR cm−1 3238, 3032, 1710, 1570, 1453, 1238, 1192, 1048; Anal. Calcd. for C12H13N3O3: C, 58.29; H, 5.30; N, 17.00% Found: C, 58.16; H, 5.36; N, 16.95; HRMS (TOF MS ES+) m/z [m + H]+ calc. for C12H13N3O3 248.1035 Found: 248.1042.
Methyl 5-(hydroxymethyl)-1-(3-phenylprop-2-en-1-yl)-1H-1,2,3-triazole-4-carboxylate (3f). Colorless solid, purified by recrystallization from ethyl acetate/hexanes. (345 mg, 63.1% yield); m.p.: 155.4–155.7 °C; 1H-NMR (CDCl3, 400 MHz) δ 7.38–7.22 (m, 5H), δ 6.59 (d, J = 15.9 Hz, 1H), δ 6.3 (m, 1H), δ 5.23 (d, 2H), δ 4.96 (s, 2H), δ 3.99 (s, 3H), δ 3.55 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 162.9, 141.1, 137.2, 135.4, 135.0, 128.7, 128.5, 126.7, 121.4, 53.3, 52.4, 50.9; FTIR cm−1 3222, 3058, 3026, 1708, 1570, 1470, 1246, 1192, 1045; Anal. Calcd. for C14H15N3O3: C, 61.530; H, 5.53; N, 15.38% Found: C, 61.53; H, 5.59; N, 15.47%.
4.5. Experimental Procedure for the Synthesis of 1H-1,2,3-Triazole-5-Monoesters (4a–d)
Into a 35 mL microwave vessel, the organic azide (4 mmol), THF (15 mL), and dichloro (pentamethylcyclopentadienyl)ruthenium polymer (91 mg, 0.31 mmol) were added, in that order. The vessel was flushed with N2, sealed, and heated at 100 °C in the CEM microwave for 20 min. Upon completion the crude mixture was purified by flash column chromatography.
Methyl 1-(2-oxopropyl)-1
H-1,2,3-triazole-5-carboxylate (
4a) [
39].
Methyl 1-(2-ethoxy-2-oxoethyl)-1H-1,2,3-triazole-5-carboxylate (4b). Yellow oil, further purified by radial chromatography (1:8, ethyl acetate/hexanes). (378 mg, 44.3% yield); 1H-NMR (CDCl3, 400 MHz) δ 8.18 (s, 1H), δ 5.49 (s, 2H), δ 4.30–4.25 (q, 2H), δ 3.93 (s, 3H), δ 1.32–1.29 (t, 3H); 13C-NMR (CDCl3, 100 MHz) δ 166.1, 158.7, 137.3, 128.4, 62.1, 52.5, 51.1, 13.9; FTIR cm−1 3140, 1726, 1272, 1121, 1092; HRMS (TOF MS ES+) m/z [m + H]+ calc. for C8H11N3O4 214.0823 Found: 214.0822.
Methyl 1-(2-oxo-2-phenylethyl)-1H-1,2,3-triazole-5-carboxylate (4c). Tan solid, further purified by an additional flash column chromatography (1:5, ethyl acetate/hexanes). (322 mg, 32.8% yield); m.p.: 122.3–123.5 °C; 1H-NMR (CDCl3, 400 MHz) δ 8.23 (s, 1H), δ 8.04–7.99 (d, 2H), δ 7.72–7.66 (t, 1H), δ 7.60–7.53 (t, 2H), δ 6.22 (s, 2H), δ 3.88 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ 189.9, 159.1, 137.6, 134.4, 134.1, 129.1, 128.11, 128.06, 56.1, 52.6; FTIR cm−1 3140, 3022, 1731, 1699, 1596, 1460, 1226, 1131; Anal. Calcd. for C12H11N3O3: C, 58.772; H, 4.521; N, 17.134% Found: C, 59.02; H, 4.67; N, 17.05%.
Methyl 1-(3-phenylprop-2-en-1-yl)-1H-1,2,3-triazole-5-carboxylate (4d). Yellow oil, further purified by radial chromatography (1:10, ethyl acetate/hexanes). (128 mg, 13.2% yield); 1H-NMR (CDCl3, 400 MHz) δ 8.16 (s, 1H), δ 7.38–7.26 (m, 5H), δ 6.69–6.65 (d, J = 15.6, 1H), δ 6.41–6.34 (m, 1H), δ 5.51–5.49 (d, 2H), δ 3.94 (s, 3H); 13C-NMR (CDCl3, 100MHz) δ 158.8, 138.1, 135.7, 135.1, 128.6, 128.3, 127.4, 126.7, 122.2, 52.5, 52.1; FTIR cm−1 3104, 3026, 1725, 1598, 1448, 1246, 1122; HRMS (TOF MS ES+) m/z [m + Li]+ calc. for C13H13N3O2 250.1168 Found: 250.1168.
1-[5-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl]propan-2-ol (5a). Colorless solid, further purified by radial chromatography (1:3, ethyl acetate/hexanes). (235 mg, 74.8% yield); m.p. 92.2–92.7 °C; 1H-NMR (DMSO-d6, 400 MHz) δ 7.57 (s, 1H), δ 5.40 (t, 1H), δ 5.07 (d, 1H), δ 4.61 (2H), δ 4.38–4.12 (m, 2H), δ 4.01 (ddd, 1H), δ 1.08 (d, 3H); 13C-NMR (DMSO-d6, 100 MHz) δ 138.6, 132.4, 66.1, 54.9, 52.6, 21.4; FTIR cm−1 3326, 3137, 3093, 1245, 1168, 1017; Anal. Calcd. for C6H11N3O2: C, 45.85; H, 7.05; N, 26.74% Found: C, 45.83; H, 6.98; N, 26.49%.
2-[5-(Hydroxymethyl)-1H-1,2,3-triazole-1-yl]ethanol (5b). Yellow oil, purified by an additional flash column chromatography (1:2, ethyl acetate/hexanes). (153 mg, 53.4% yield); 1H-NMR (DMSO-d6, 400 MHz) δ 7.58 (s, 1H), δ 5.46 (s, 1H), δ 5.09 (s, 1H), δ 4.61 (s, 2H), δ 4.40–4.37 (t, 2H), δ 3.76 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz) δ 138.5, 132.5, 60.7, 52.4, 50.5; FTIR cm−1 3260, 3098, 1238, 1111, 1022; HRMS (TOF MS ES+) m/z [m + H]+ calc. C5H9N3O2 144.0773. Found: 144.0771.
2-[5-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl]-1-phenyl-1-ethanol (5c). Tan solid, purified by radial chromatography (1:7, ethyl acetate/hexanes). (304 mg, 69.2% yield); m.p.: 119.1 –122.3 °C; 1H-NMR (DMSO-d6, 400 MHz) δ 7.56 (s, 1H), δ 7.36–7.27 (m, 5H), δ 5.83–5.82 (d, 1H), δ 5.45–5.42 (t, 1H), δ 5.01–4.97 (q, 1H), δ 4.55–4.42 (m, 4H); 13C-NMR (DMSO-d6, 100 MHz) δ 142.7, 138.7, 132.3, 128.7, 128.1, 126.5, 72.4, 55.1, 52.5; FTIR cm−1 3261, 3036, 1241, 1059, 1023; Anal. Calcd. for C11H13N3O2: C, 44.65; H, 6.09; N, 19.53% Found: C, 44.58; H, 5.98; N, 19.27%
1-[(3-Phenylprop-2-en-1-yl)-1H-1,2,3-triazole-5-yl]-methanol (5d). Colorless solid, further purified by recrystallization from diethyl ether/hexanes. (343 mg, 79.8% yield); m.p.: 66.1–66.6 °C; 1H-NMR (CDCl3, 400 MHz) δ 7.51 (s, 1H), δ 7.38–7.21 (m, 5H), δ 6.57 (d, J = 15.8, 1H), δ 6.31 (dt, 1H), δ 5.19 (d, 2H), δ 4.76 (s, 2H), δ 3.71 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 136.3, 135.6, 134.3, 133.1, 128.7, 128.4, 126.6, 122.2, 53.1, 50.7; FTIR cm−1 3229, 1621, 1593, 1470, 1188, 1045; Anal. Calcd. for C12H13N3O: C, 66.96; H, 6.09; N, 19.55% Found: C, 66.66; H, 6.09; N, 19.26%
4.6. Experimental Procedure for the Synthesis of 1H-1,2,3-Triazole-4-Monoesters (6a–d)
Into a 100 mL round-bottom flask, the organic azide (4 mmol), tert-butyl alcohol/H2O (1:1) soln. (60 mL), methyl propiolate (345 mg, 4.1 mmol), 1M copper sulfate pentahydrate solution (200 uL), and sodium ascorbate (40 mg, 0.20 mmol) were added, in that order. The mixture was refluxed for 2 h. Upon completion, the mixture was cooled to rt., 20 mL of a 10% ammonia solution added, and the mixture stirred for 5 min. The mixture was extracted ethyl acetate (20 mL × 3), the combined organic layers washed with brine, dried over anhydrous sodium sulfate, and evaporated in vacuo to dryness.
Methyl 1-(2-oxo-2-phenylethyl)-1
H-1,2,3-triazole-4-carboxylate (
6a) [
43].
Methyl 1-(2-ethoxy-2-oxoethyl)-1
H-1,2,3-triazole-4-carboxylate (
12b) [
44]. Colorless solid, purified by recrystallization from ethanol/hexanes. (477 mg, 55.9% yield); m.p.: 105.2–105.7 °C;
1H-NMR (CDCl
3, 400 MHz) δ 8.26 (s, 1H), δ 5.22 (s, 2H), δ 4.28 (q, 2H), δ 3.95 (s, 3H), δ 1.3 (t, 3H);
13C-NMR (CDCl
3, 100 MHz) δ 165.6, 160.9, 140.4, 129.0, 62.8, 52.3, 51.0, 14.0; FTIR cm
−1 3152, 3005, 1754, 1715, 1241, 1028, 1014.
Methyl 1-benzyl-1
H-1,2,3-triazole-4-carboxylate (
6c) [
44,
45].
Methyl 1-(3-phenylprop-2-en-1-yl)-1H-1,2,3-triazole-4-carboxylate (6d). Colorless solid, purified by recrystallization from ethyl acetate/hexanes. (831 mg, 85.4% yield); m.p.: 131.7 –132.5 °C; 1H-NMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), δ 7.41–7.31 (m, 5H), δ 6.74–6.70 (d, J = 15.6 Hz, 1H), δ 6.38–6.31 (m, 1H), δ 5.21–5.19 (d, 2H), δ 3.95 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ 161.1, 140.2, 136.5, 135.1, 128.83, 128.80, 127.2, 126.8, 120.7, 52.6, 52.2; FTIR cm−1 3124, 3027, 1717, 1658, 1578, 1451, 1228, 1046; Anal. Calcd. for C13H13N3O2: C, 64.19; H, 5.39; N, 17.27% Found: C, 64.03; H, 5.53; N, 17.23%.
Methyl 1-(2-hydroxy-2-phenylethyl)-1
H-1,2,3-triazole-4-carboxylate (
7a) [
29]. Colorless solid, purified by flash column chromatography (1:5, ethyl acetate/hexanes). (389 mg, 78.7% yield) m.p. 84.5–85.3 °C;
1H-NMR (CDCl
3, 500 MHz) δ 8.19 (s, 1H), δ 7.45–7.30 (m, 5H), δ 5.20 (d, 1H), δ 4.69 (d, 1H), δ 4.44 (dd, 1H), δ 3.91 (s, 3H), δ 3.53 (s, 1H);
13C-NMR (CDCl
3, 100 MHz) δ 161.1, 139.8, 139.4, 129.0, 128.9, 128.6, 125.8, 72.5, 57.5, 52.2; FTIR cm
−1 3393, 3143, 3002, 1730, 1257, 1199, 1061; Anal. Calcd. for C
13H
15N
3O
3: C, 58.29; H, 5.30; N, 17.00% Found: C, 58.41; H, 5.18; N, 17.17%; HRMS (TOF MS ES+)
m/
z [m + H]
+ calc. C
13H
15N
3O
3 248.1035. Found: 248.1037.
Methyl 1-(2-hydroxyethyl)-1
H-1,2,3-triazole-4-carboxylate (
7b) [
46]
. Colorless solid, purified by flash column chromatography (1:4, ethyl acetate/hexanes). (293 mg, 85.7% yield) m.p. 81.3–82.7 °C;
1H-NMR (DMSO-
d6, 400 MHz) δ 8.73 (s, 1H), δ 5.05 (t, 1H) δ 4.46 (t, 2H), δ 3.83–3.78 (m, 5H);
13C-NMR (DMSO-
d6, 100 MHz) δ 160.8, 138.3, 129.4, 59.4, 52.4, 51.6; FTIR cm
−1 3372, 3149, 1728; Anal. Calcd. for C
6H
9N
3O
3: C, 42.11; H, 5.30; N, 24.55% Found: C, 42.09; H, 5.30; N, 24.68%.
(1-Benzyl-1
H-1,2,3-triazol-4-yl)methanol (
8c) [
44]. Colorless solid, purified by flash column chromatography (1:4, ethyl acetate/hexanes). (173 mg, 91.2% yield); m.p.: 67.9–68.6 °C;
1H-NMR (CDCl
3, 400 MHz) δ 7.50 (s, 1H), δ 7.39–7.278 (m, 5H), δ 5.51 (s, 2H), δ 4.76 (s, 2H); FTIR cm
−1 3237, 3136, 1221, 1014.
1-[(3-Phenylprop-2-en-1-yl)-1H-1,2,3-triazol-4-yl]methanol (8d). Colorless solid, purified initially by flash column chromatography (1:5, ethyl acetate/hexanes) and then radial chromatography (1:8, ethyl acetate/hexanes). (87 mg, 40.4% yield); m.p.: 80.0–81.9 °C; 1H-NMR (CDCl3, 400 MHz) δ 7.60 (s, 1H), δ 7.40–7.30 (m, 5H), δ 6.70–6.66 (d, J = 15.6, 1H), δ 6.38–6.31 (dt, 1H), δ 5.15–5.13 (d, 2H), δ 4.01 (s, 2H), δ 2.03 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 148.2, 135.45, 135.38, 128.7, 128.5, 126.7, 121.8, 121.74, 56.1, 52.34; FTIR cm−1 3279, 3121, 3078, 1220, 1039; Anal. Calcd. for C12H13N3O: C, 66.96; H, 6.09; N, 19.52% Found: C, 66.67; H, 6.07; N, 19.51%
4.7. Experimental Procedure for Reduction Completion Analyses (1a–f, 4a–d, 6a–d)
To each of 1, 4 or 6 (1 mmol) in CH3OH (10 mL), NaBH4 (1 eq) was added with stirring at rt. for 30 min. Additional NaBH4 was added in 1eq increments for 30 min periods until 1, 4 or 6 were no longer detectable by TLC analysis under UV (254 nm) light. Aliquots were spotted at the origin of a TLC plate and developed in ethyl acetate/hexanes (3/7). Towards the end of each analysis, larger aliquots were spotted (x5) and developed. Detection limits were <1% for 1a, e, f; 4c–d; and 6a, c, d and < 2% for 1b–d; 4a–b and 6b. Detection limits were determined by visualizing developed plates spotted with standard samples of 1, 4 and 6 by the same technique used for each of the reduction analyses. Spotting (x5) per standard solution gave detectable spots for 0.1 mmol/mL of 1a, e, f; 4c–d; and 6a, c, d, and 0.2 mmol/mL of 1b–d; 4a–b and 6b.





{kind=link}
{kind=link}
{kind=link}


