
Protein–Protein Interactions in Translesion Synthesis



Abstract
:1. Introduction
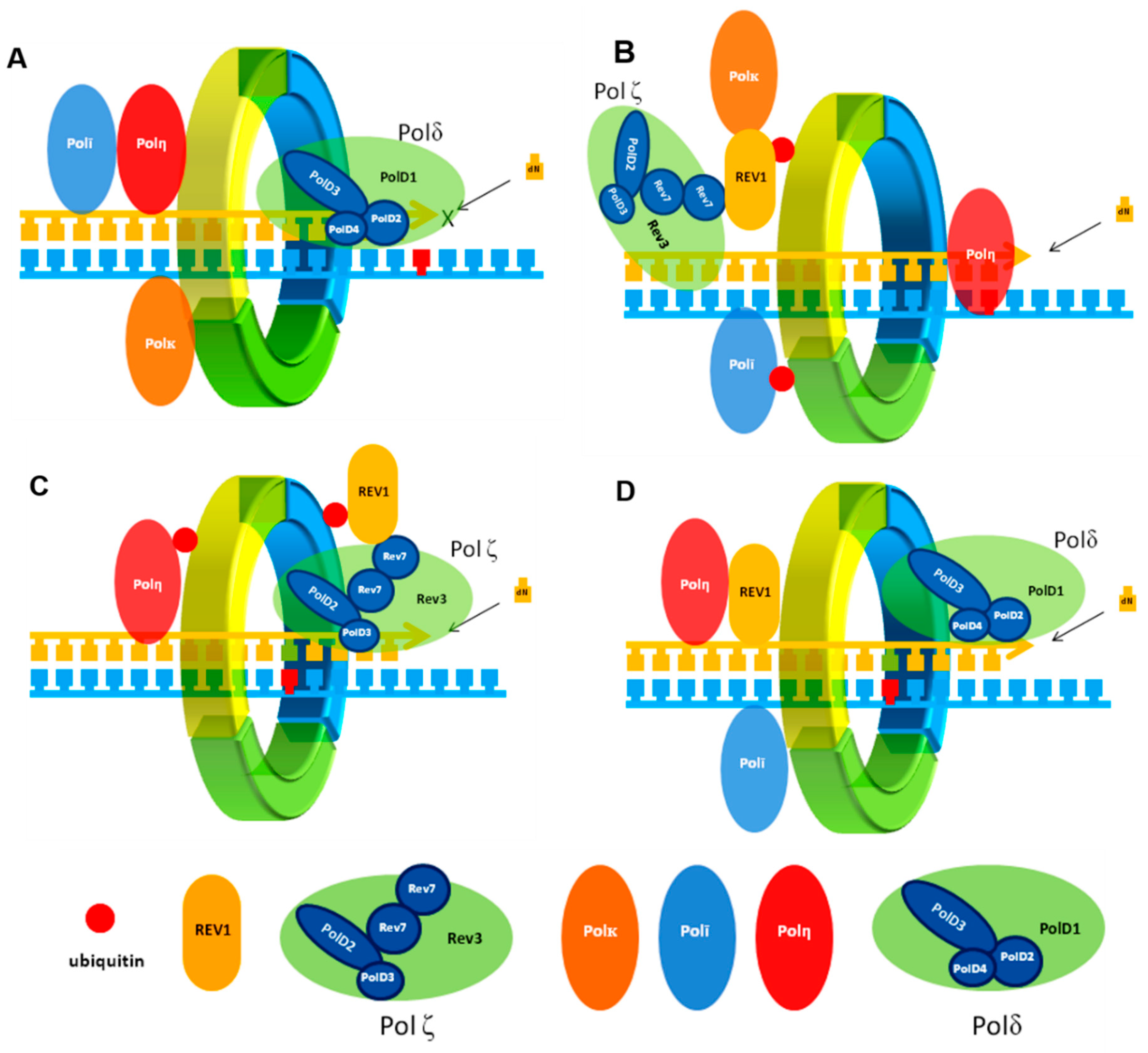
2. Protein–Protein Interactions (PPIs) in TLS
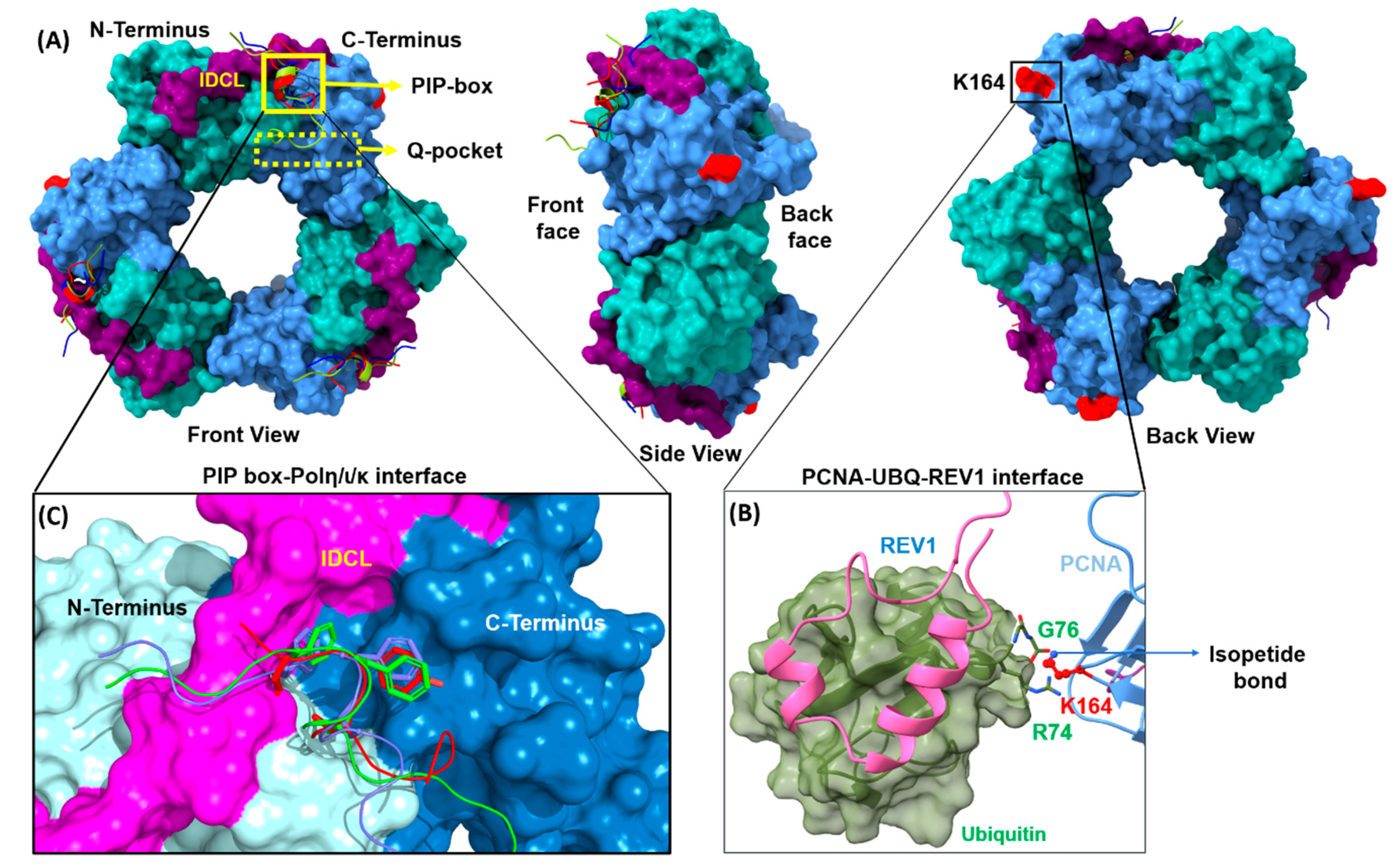
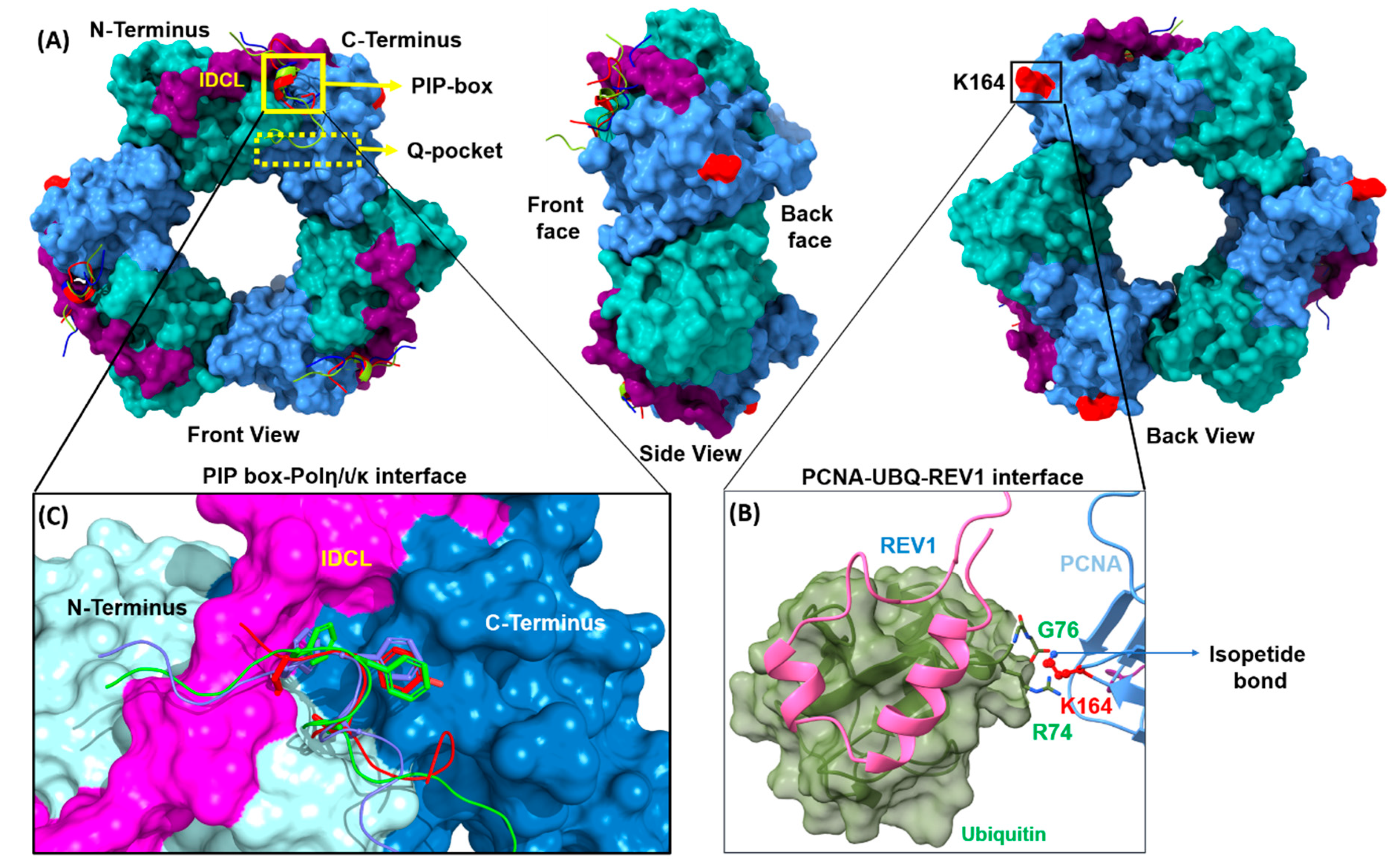
2.1. PCNA PPIs
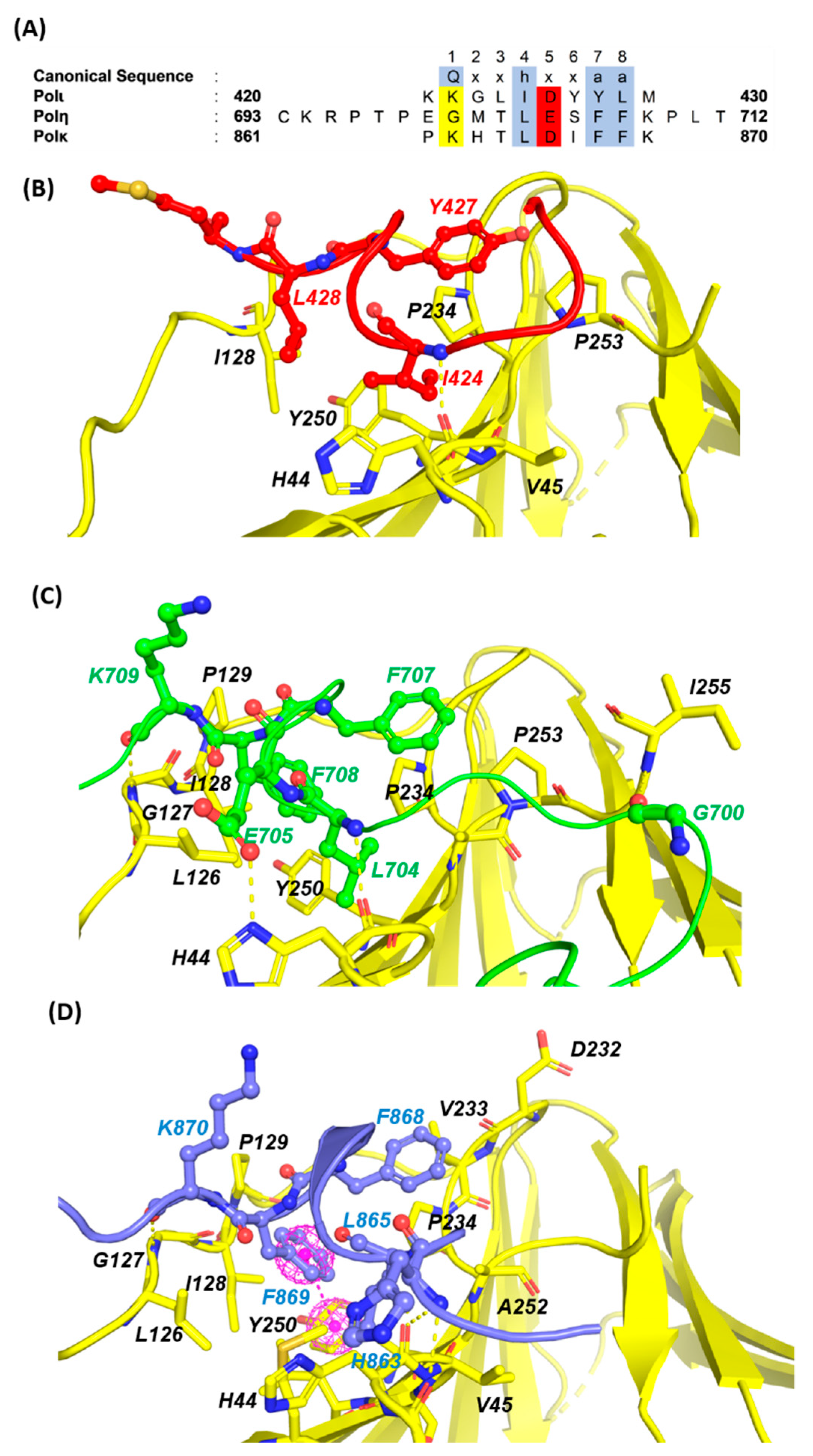
2.1.1. PCNA/UBM and PCNA/UBZ PPIs
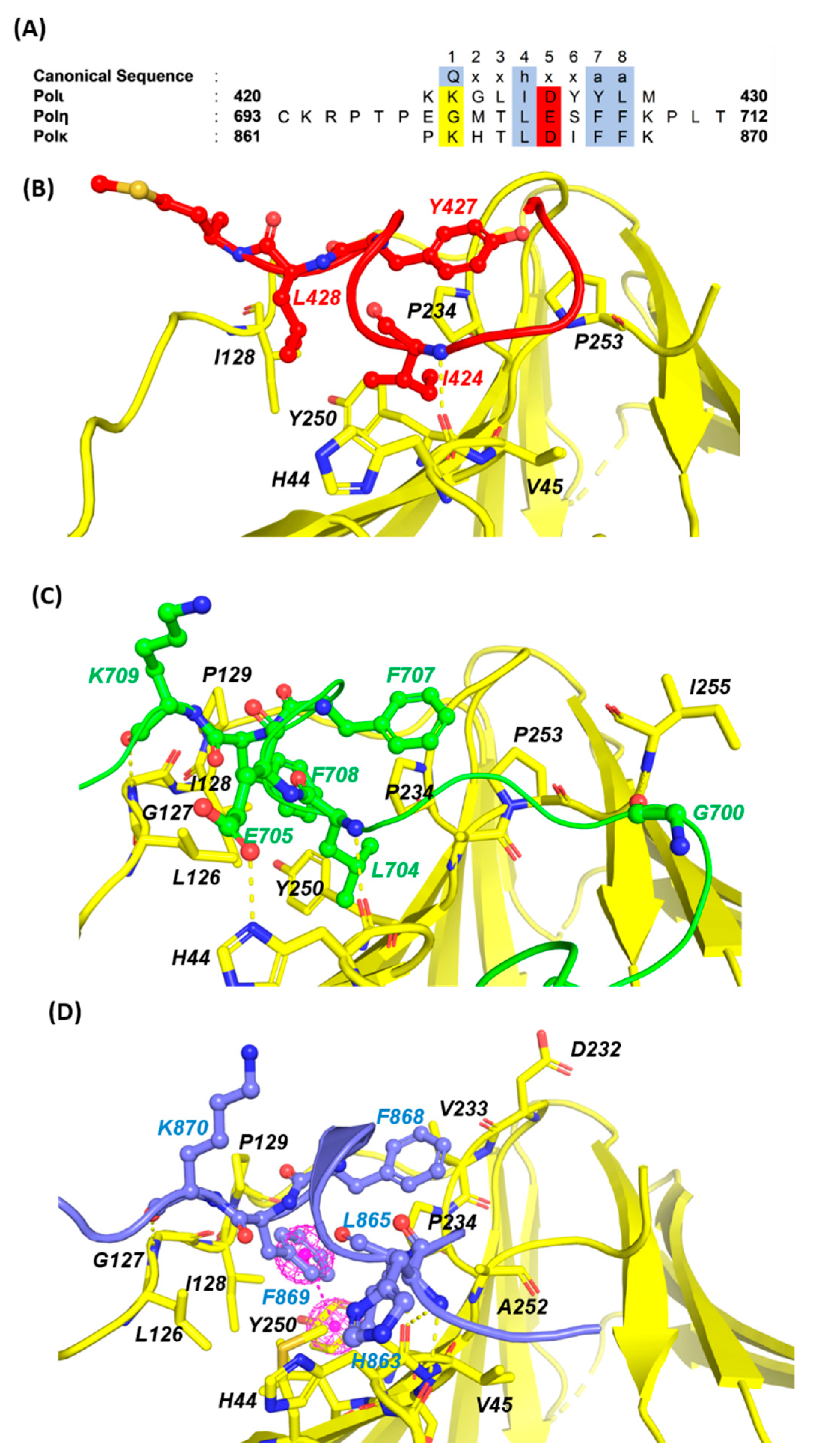
2.1.2. PCNA/PIP-Box PPIs
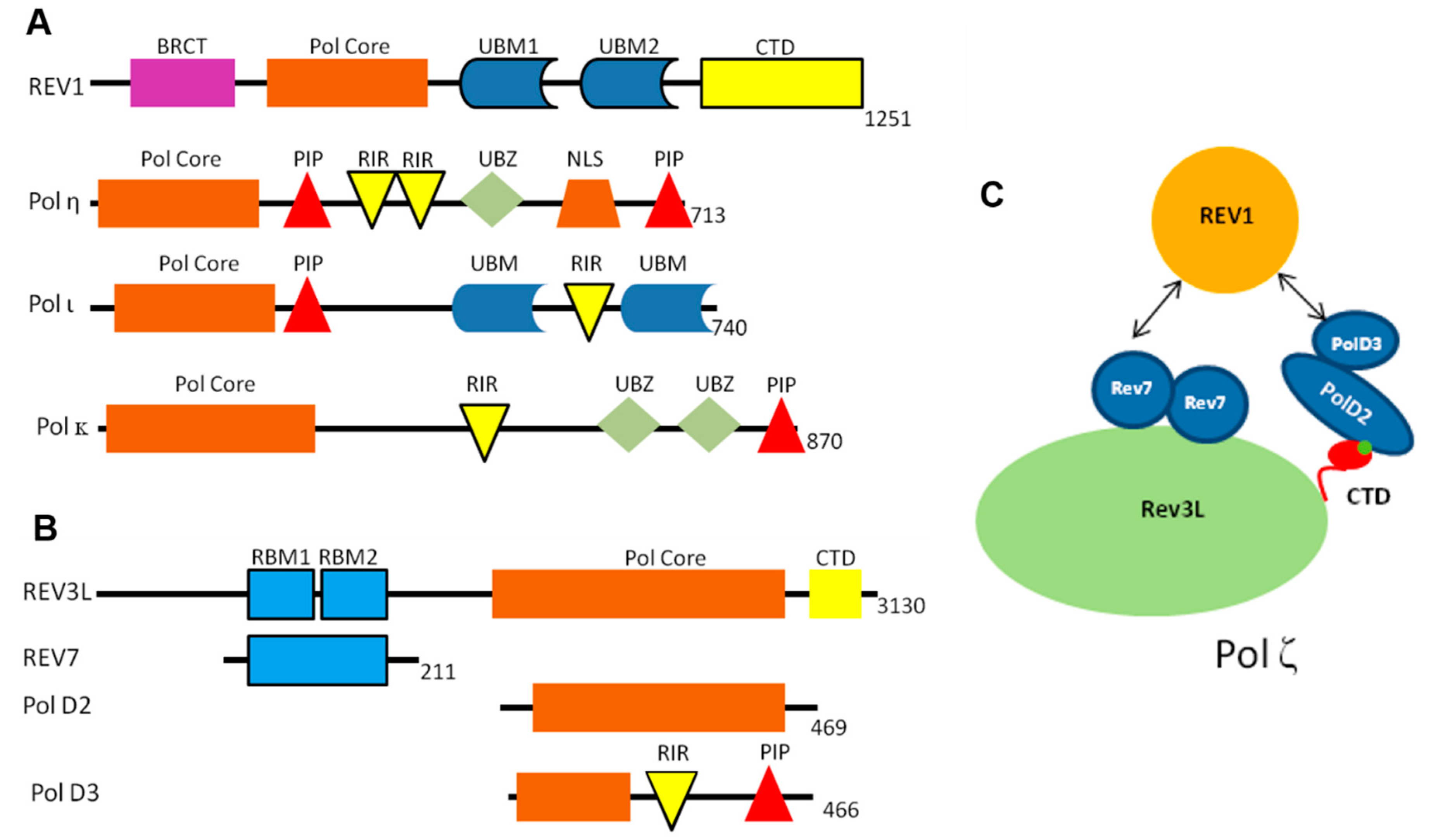
2.2. REV1 PPIs
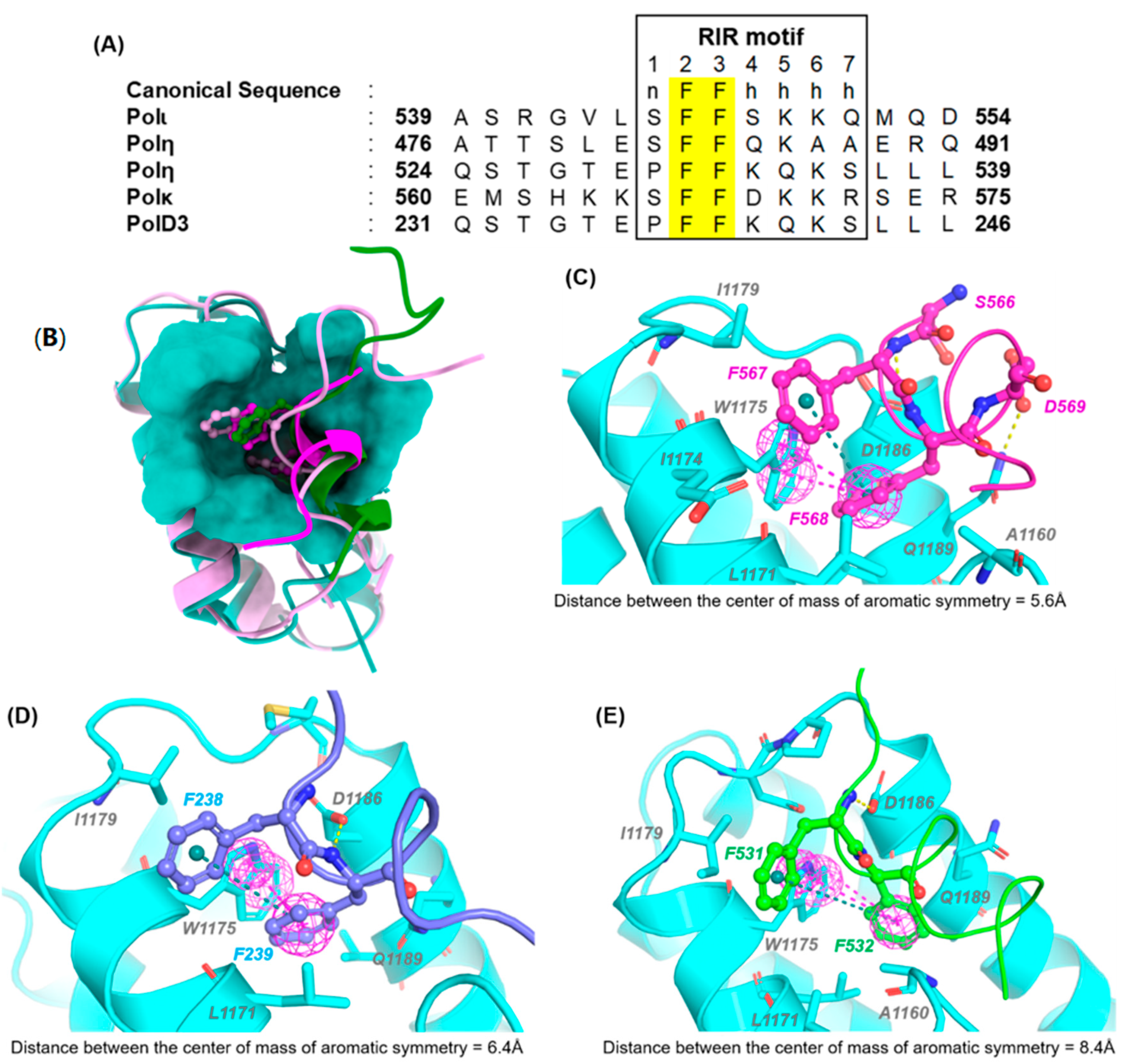
REV1-CT/RIR PPI
2.3. POLζ PPIs
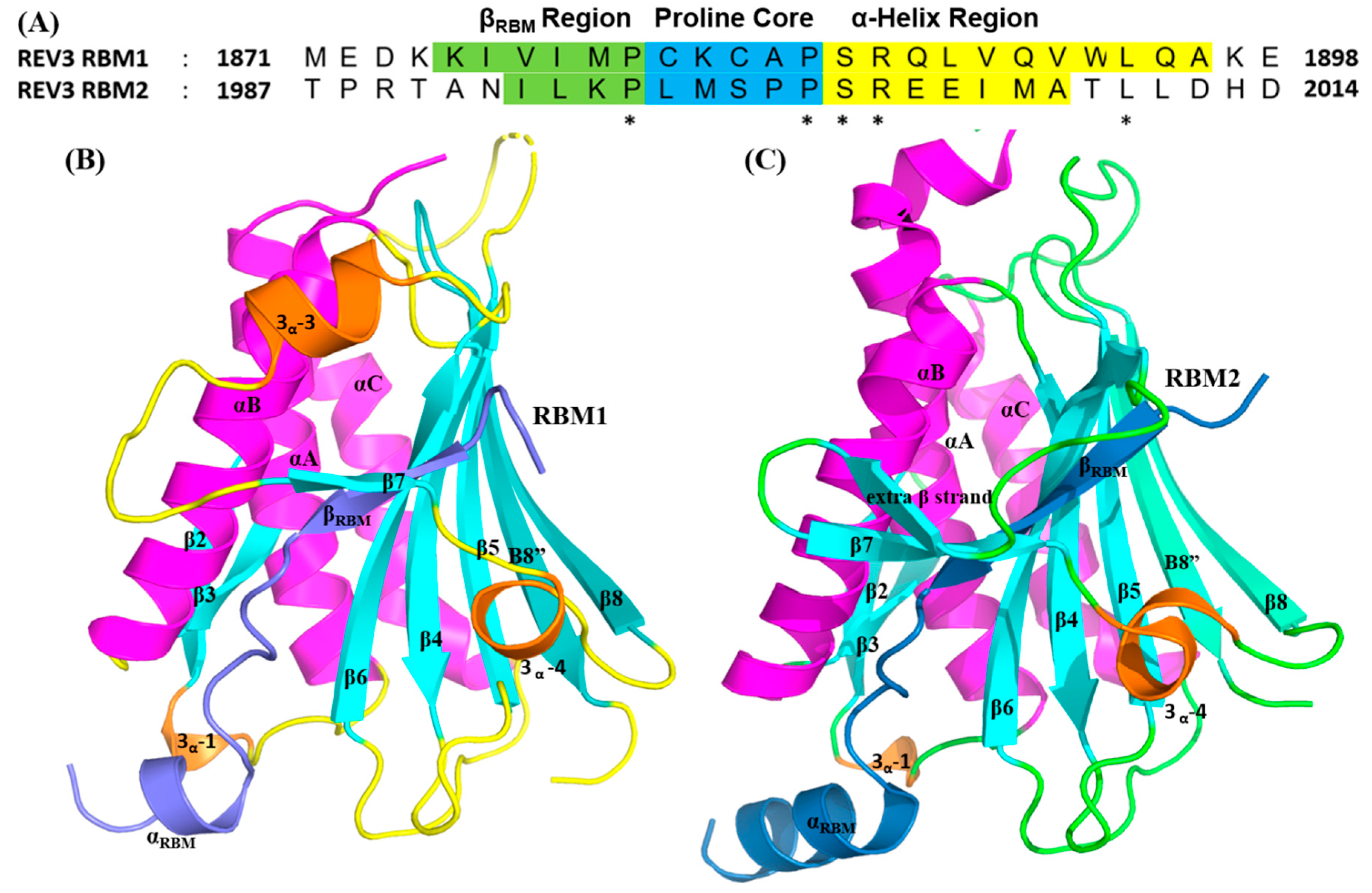
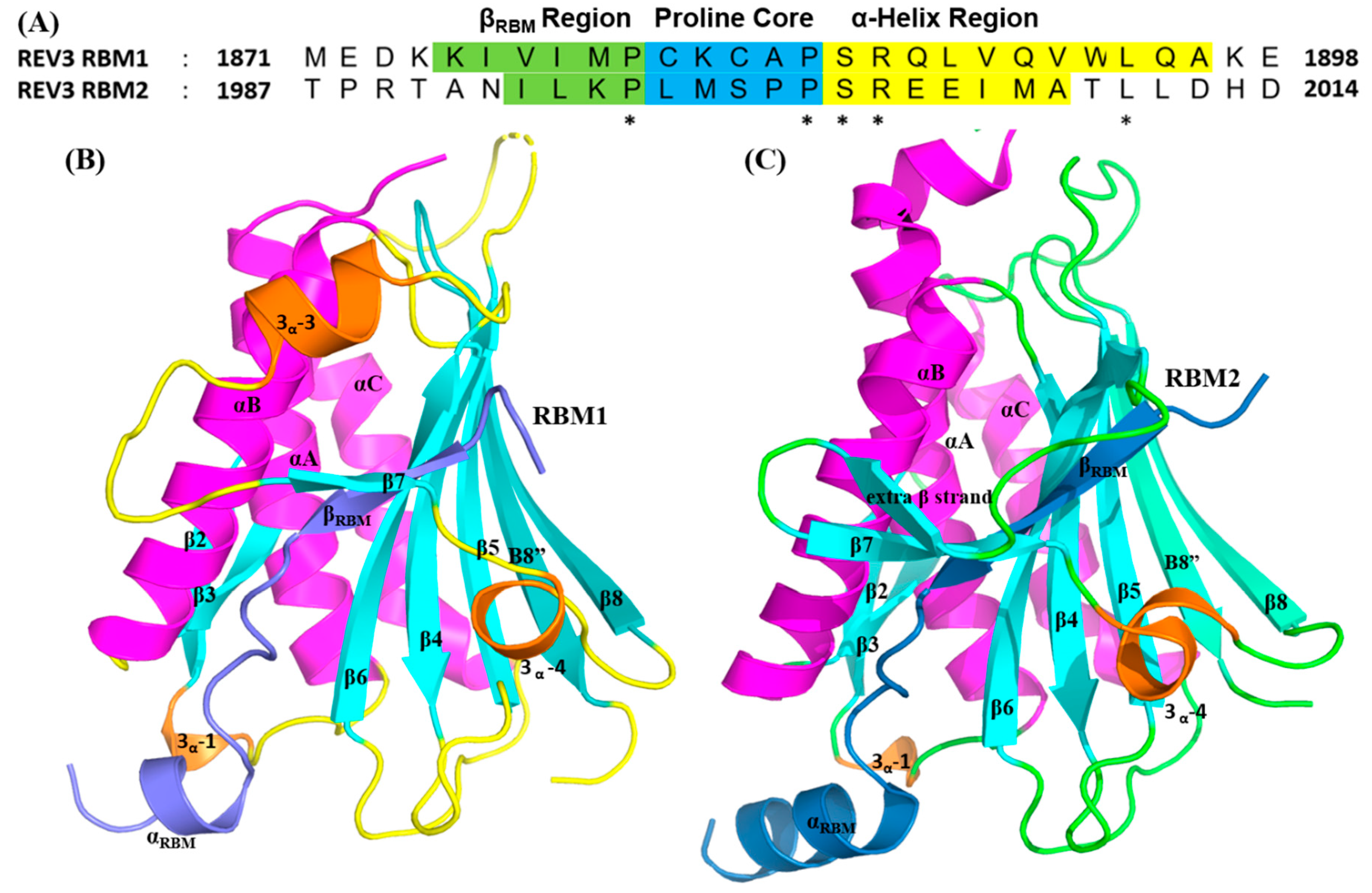
2.3.1. REV7/REV3 PPI
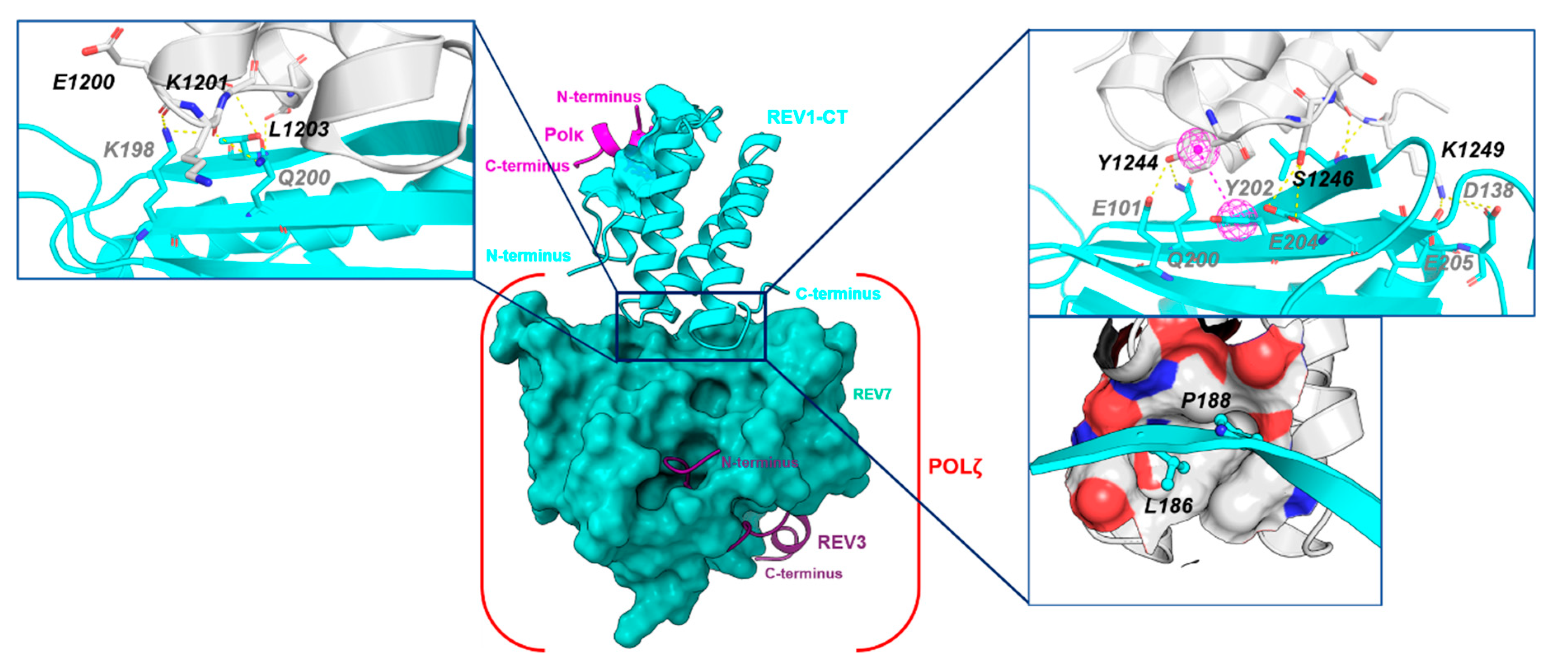
2.3.2. REV1-CT/ REV7 PPI
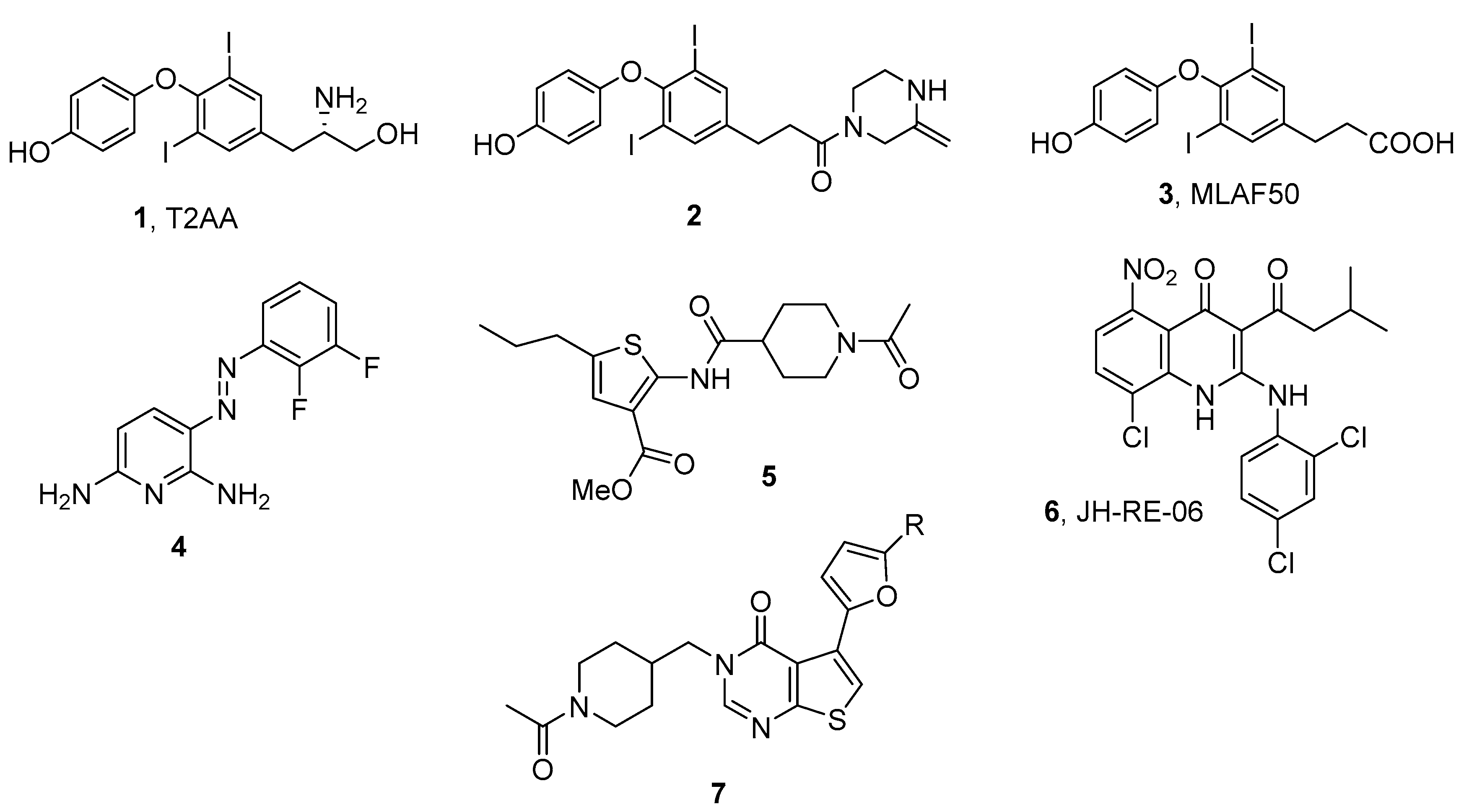
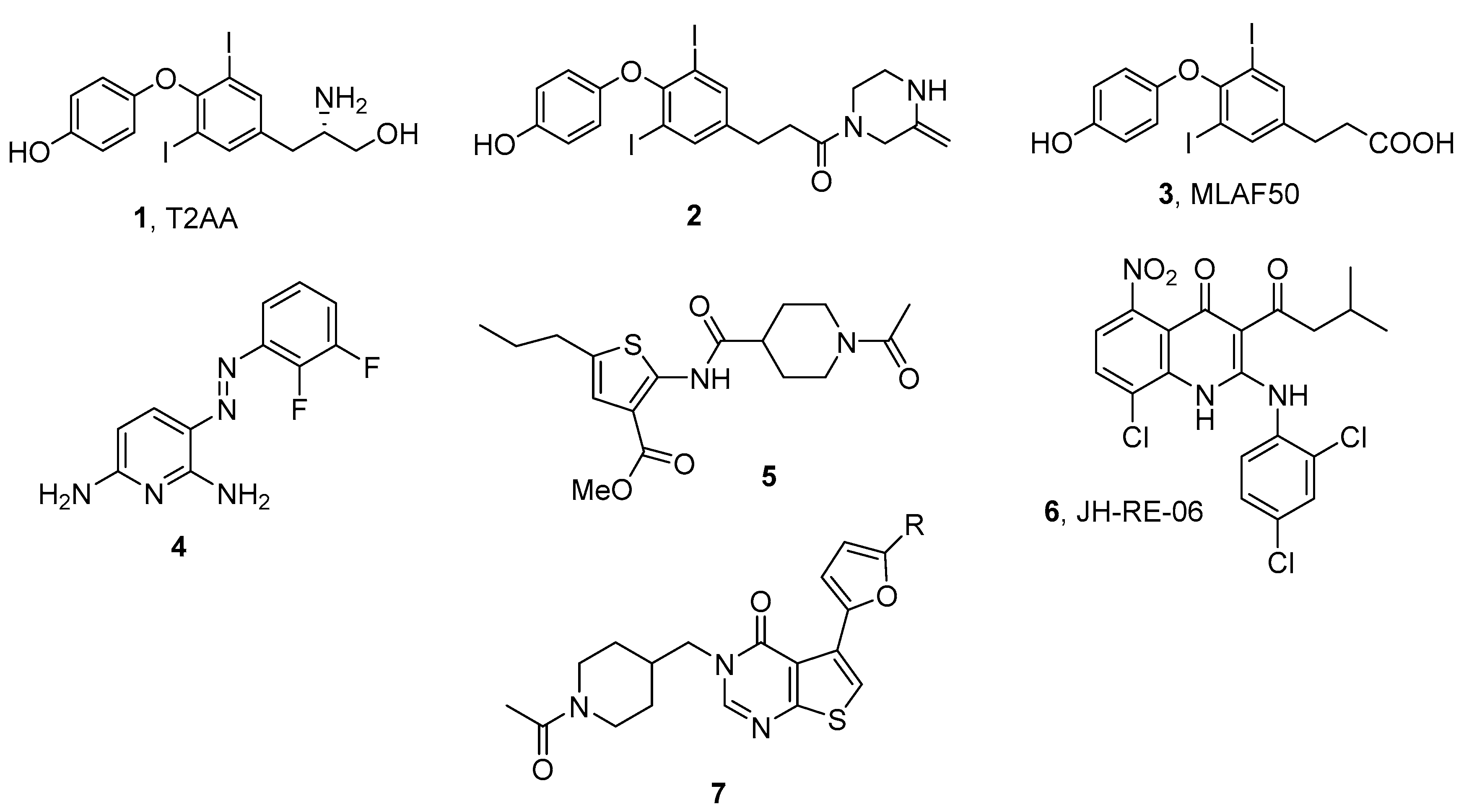
3. Targeting TLS PPIs with Small Molecules
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Crosby, M.E. Cell Cycle: Principles of Control. Yale J. Biol. Med. 2007, 80, 141–142. [Google Scholar]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef]
- Sale, J.E. Competition, collaboration and coordination–Determining how cells bypass DNA damage. J. Cell Sci. 2012, 125, 1633–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedberg, E.C. Suffering in silence: The tolerance of DNA damage. Nat. Rev. Mol. Cell Biol. 2005, 6, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R.; Fuchs, R.P. Gaps and forks in DNA replication: Rediscovering old models. DNA Repair 2006, 5, 1495–1498. [Google Scholar] [CrossRef] [Green Version]
- Shcherbakova, P.V.; Fijalkowska, I.J. Translesion synthesis DNA polymerases and control of genome stability. Front. Biosci. 2006, 11, 2496–2517. [Google Scholar] [CrossRef] [Green Version]
- Waters, L.S.; Walker, G.C. The critical mutagenic translesion DNA polymerase REV1 is highly expressed during G2/M phase rather than S phase. Proc. Nat. Acad. Sci. USA 2006, 103, 8971–8976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Bi, X. Mechanism of DNA damage tolerance. World J. Biol. Chem. 2015, 6, 48–56. [Google Scholar] [CrossRef]
- Shilkin, E.S.; Boldinova, E.O.; Stolyarenko, A.D.; Goncharova, R.I.; Chuprov-Netochin, R.N.; Khairullin, R.F.; Smal, M.P.; Makarova, A.V. Translesion DNA synthesis and carcinogenesis. Biochemistry 2020, 85, 425–435. [Google Scholar]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef] [Green Version]
- Sutton, M.D.; Walker, G.C. Managing DNA polymerases: Coordinating DNA replication, DNA repair, and DNA recombination. Proc. Nat. Acad. Sci. USA 2001, 98, 8342–8349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.-C.; Drac, M.; Schwob, E.; Baldacci, G.; Cazaux, C.; Hoffmann, J.-S. A role for DNA polymerase θ in the timing of DNA replication. Nat. Commun. 2014, 5, 4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, A.; Garg, P.; Burgers, P.M. A ubiquitin-binding motif in the translesion DNA polymerase REV1 mediates its essential functional interaction with ubiquitinated proliferating cell nuclear antigen in response to DNA damage. J. Biol. Chem. 2007, 282, 20256–20263. [Google Scholar] [CrossRef] [Green Version]
- Garg, P.; Burgers, P.M. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases eta and REV1. Proc. Nat. Acad. Sci. USA 2005, 102, 18361–18366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmunds, C.E.; Simpson, L.J.; Sale, J.E. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol. Cell 2008, 30, 519–529. [Google Scholar] [CrossRef]
- Guo, C.; Tang, T.-S.; Bienko, M.; Parker, J.L.; Bielen, A.B.; Sonoda, E.; Takeda, S.; Ulrich, H.D.; Dikic, I.; Friedberg, E.C. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol. Cell. Biol. 2006, 26, 8892–8900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomar, M.G.; D’Souza, S.; Bienko, M.; Dikic, I.; Walker, G.C.; Zhou, P. Unconventional ubiquitin recognition by the ubiquitin-binding motif within the Y family DNA polymerases iota and REV1. Mol. Cell 2010, 37, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanarotti, M.; Grace, C.R.; Miller, D.J.; Actis, M.L.; Inoue, A.; Evison, B.J.; Vaithiyalingam, S.; Singh, A.P.; McDonald, E.T.; Fujii, N. Structures of REV1 UBM2 domain complex with ubiquitin and with a small-molecule that inhibits the REV1 UBM2-ubiquitin interaction. J. Mol. Biol. 2018, 430, 2857–2872. [Google Scholar] [CrossRef]
- Vidal, A.E.; Kannouche, P.; Podust, V.N.; Yang, W.; Lehmann, A.R.; Woodgate, R. Proliferating cell nuclear antigen-dependent coordination of the biological functions of human DNA polymerase iota. J. Biol. Chem. 2004, 279, 48360–48368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Sharma, S.; Helchowski, C.M.; Canman, C.E. The roles of DNA polymerase ζ and the Y family DNA polymerases in promoting or preventing genome instability. Mutat. Res. 2013, 743–744, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tissier, A.; Kannouche, P.; Reck, M.P.; Lehmann, A.R.; Fuchs, R.P.; Cordonnier, A. Co-localization in replication foci and interaction of human Y-family members, DNA polymerase polη and REVl protein. DNA Repair 2004, 3, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [Green Version]
- Zafar, M.K.; Eoff, R.L. Translesion DNA synthesis in cancer: Molecular mechanisms and therapeutic opportunities. Chem. Res. Toxicol. 2017, 30, 1942–1955. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Calvo, J.A. Targeting translesion synthesis (TLS) to expose replication gaps, a unique cancer vulnerability. Sci. Adv. 2021, 25, 27–36. [Google Scholar]
- Yamanaka, K.; Chatterjee, N.; Hemann, M.T.; Walker, G.C. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet. 2017, 13, e1006842. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Gao, Y. Translesion and repair DNA polymerases: Diverse structure and mechanism. Ann. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef]
- Lawrence, C.W. Cellular functions of DNA polymerase zeta and REV1 protein. Adv. Protein Chem. 2004, 69, 167–203. [Google Scholar] [PubMed]
- Prakash, S.; Prakash, L. Translesion DNA synthesis in eukaryotes: A one- or two-polymerase affair. Genes Dev. 2002, 16, 1872–1883. [Google Scholar] [CrossRef] [Green Version]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Halling, P.J. Proteins: Structures and Molecular Properties, 2nd ed.; Thomas, E., Creighton, W.H., Eds.; Freeman and Company: New York, NY, USA, 1995; Volume 62, p. 105. ISBN 0-7167-7030-X. [Google Scholar]
- Glaser, F.; Steinberg, D.M.; Vakser, I.A.; Ben-Tal, N. Residue frequencies and pairing preferences at protein-protein interfaces. Proteins 2001, 43, 89–102. [Google Scholar] [CrossRef]
- Vakser, I.A.; Aflalo, C. Hydrophobic docking: A proposed enhancement to molecular recognition techniques. Proteins 1994, 20, 320–329. [Google Scholar] [CrossRef]
- Lawrence, M.C.; Colman, P.M. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 1993, 234, 946–950. [Google Scholar] [CrossRef]
- Park, J.M.; Yang, S.W.; Yu, K.R.; Ka, S.H.; Lee, S.W.; Seol, J.H.; Jeon, Y.J.; Chung, C.H. Modification of PCNA by ISG15 plays a crucial role in termination of error-prone translesion DNA synthesis. Mol. Cell 2014, 54, 626–638. [Google Scholar] [CrossRef] [Green Version]
- Burschowsky, D.; Rudolf, F.; Rabut, G.; Herrmann, T.; Peter, M.; Wider, G. Structural analysis of the conserved ubiquitin-binding motifs (UBMs) of the translesion polymerase iota in complex with ubiquitin. J. Biol. Chem. 2011, 286, 1364–1373. [Google Scholar] [CrossRef] [Green Version]
- Bomar, M.G.; Pai, M.-T.; Tzeng, S.-R.; Li, S.S.-C.; Zhou, P. Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase eta. EMBO Rep. 2007, 8, 247–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillaire, M.-J.; Bétous, R.; Hoffmann, J.-S. Role of DNA polymerase κ in the maintenance of genomic stability. Mol. Cell Oncol. 2014, 1, e29902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.; Botuyan, M.V.; Mer, G. Structural Basis for the Interaction of Mutasome Assembly Factor REV1 with Ubiquitin. J. Mol. Biol. 2018, 430, 2042–2050. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Benirschke, R.C.; Tuan, H.F.; Juranić, N.; Macura, S.; Botuyan, M.V.; Mer, G. Structural basis of ubiquitin recognition by translesion synthesis DNA polymerase ι. Biochemistry 2010, 49, 10198–10207. [Google Scholar] [CrossRef] [Green Version]
- Hishiki, A.; Hashimoto, H.; Hanafusa, T.; Kamei, K.; Ohashi, E.; Shimizu, T.; Ohmori, H.; Sato, M. Structural basis for novel interactions between human translesion synthesis polymerases and proliferating cell nuclear antigen. J. Biol. Chem. 2009, 284, 10552–10560. [Google Scholar] [CrossRef] [Green Version]
- De Biasio, A.; Blanco, F.J. Proliferating cell nuclear antigen structure and interactions: Too many partners for one dancer? Adv. Protein Chem. Struct. Biol. 2013, 91, 1–36. [Google Scholar]
- Pustovalova, Y.; Magalhães, M.T.; D’Souza, S.; Rizzo, A.A.; Korza, G.; Walker, G.C.; Korzhnev, D.M. Interaction between the REV1 C-Terminal domain and the PolD3 subunit of polζ suggests a mechanism of polymerase exchange upon REV1/Polζ-dependent translesion synthesis. Biochemistry 2016, 55, 2043–2053. [Google Scholar] [CrossRef] [Green Version]
- Pozhidaeva, A.; Pustovalova, Y.; D’Souza, S.; Bezsonova, I.; Walker, G.C.; Korzhnev, D.M. NMR structure and dynamics of the C-terminal domain from human REV1 and its complex with REV1 interacting region of DNA polymerase η. Biochemistry 2012, 51, 5506–5520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Yang, X.; Xu, M.; Jiang, T. Structural insights into the assembly of human translesion polymerase complexes. Protein Cell 2012, 3, 864–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, E.; Hanafusa, T.; Kamei, K.; Song, I.; Tomida, J.; Hashimoto, H.; Vaziri, C.; Ohmori, H. Identification of a novel REV1-interacting motif necessary for DNA polymerase kappa function. Genes Cells 2009, 14, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, G.N.; Wittschieben, J.P.; Wittschieben, B.Ø.; Wood, R.D. DNA polymerase zeta (pol ζ) in higher eukaryotes. Cell Res. 2008, 18, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Gregory, M.T.; Yang, W. Human Pol ζ purified with accessory subunits is active in translesion DNA synthesis and complements Pol η in cisplatin bypass. Proc. Natl. Acad. Sci. USA 2014, 111, 2954–2959. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Hashimoto, H.; Murakumo, Y.; Kobayashi, S.; Kogame, T.; Unzai, S.; Akashi, S.; Takeda, S.; Shimizu, T.; Sato, M. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J. Biol. Chem. 2010, 285, 12299–12307. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, S.; Hara, K.; Shimizu, T.; Sato, M.; Hashimoto, H. Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins. J. Biol. Chem. 2012, 287, 33847–33852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, A.A.; Vassel, F.M.; Chatterjee, N.; D’Souza, S.; Li, Y.; Hao, B. Rev7 dimerization is important for assembly and function of the REV1/Polζ translesion synthesis complex. Proc. Natl. Acad. Sci. USA 2018, 115, E8191–E8200. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, H.; Hishiki, A.; Hara, K.; Kikuchi, S. Structural basis for the molecular interactions in DNA damage tolerances. Biophys. Physicobiol. 2017, 14, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Wells, J.A. A hot spot of binding energy in a hormone-receptor interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Wells, J.A.; Braisted, A.C. Tethering: Fragment-Based Drug Discovery. Ann. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BioLuminate, 2021-3; Schrödinger, LLC: New York, NY, USA, 2021.
- Patel, S.M.; Dash, R.C. Translesion synthesis inhibitors as a new class of cancer chemotherapeutics. Expert Opin. Investig. Drugs 2021, 30, 13–24. [Google Scholar] [CrossRef]
- Inoue, A.; Kikuchi, S.; Hoshiki, A.; Shao, Y.; Heath, R.; Evison, B.J.; Actis, M.; Canman, C.E.; Hashimoto, H.; Fujii, N. A small molecule inhibitor of monoubiquitinated proliferating cell nuclear antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J. Biol. Chem. 2014, 289, 7109–7120. [Google Scholar] [CrossRef] [Green Version]
- McPherson, K.S.; Zaino, A.M.; Dash, R.C.; Rizzo, A.A.; Li, Y.; Hao, B.; Bezsonova, I.; Hadden, M.K.; Korzhnev, D.M. Structure-based drug design of phenazopyridine derivatives as inhibitors of REV1 interactions in translesion synthesis. Chem. Med. Chem. 2021, 16, 1126–1132. [Google Scholar] [CrossRef]
- Wojtaszek, J.L.; Chatterjee, N.; Najeeb, J.; Ramos, A.; Lee, M.; Bian, K.; Xue, J.Y.; Fenton, B.A.; Park, H.; Li, D.; et al. A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 2019, 178, 152–159.e11. [Google Scholar] [CrossRef] [PubMed]
- Actis, M.; Inoue, A.; Evison, B.; Perry, S.; Punchihewa, C.; Fujii, N. Small molecule inhibitors of PCNA/PIP-box interaction suppress translesion DNA synthesis. Bioorg. Med. Chem. 2013, 21, 1972–1977. [Google Scholar] [CrossRef]
- Vanarotti, M.; Evison, B.J.; Actis, M.L.; Inoue, A.; McDonald, E.T.; Shao, Y.; Heath, R.J.; Fujii, N. Small-molecules that bind to the ubiquitin-binding motif of REV1 inhibit REV1 interaction with K164-monoubiquitinated PCNA and suppress DNA damage tolerance. Bioorg. Med. Chem. 2018, 26, 2345–2353. [Google Scholar] [CrossRef]
- Dash, R.C.; Ozen, Z.; Rizzo, A.A.; Lim, S.; Korzhnev, D.M.; Hadden, M.K. Structural approach to identify a lead scaffold that targets the translesion synthesis polymerase REV1. J. Chem. Inf. Model. 2018, 58, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Sail, V.; Rizzo, A.A.; Chatterjee, N.; Dash, R.C.; Ozen, Z.; Walker, G.C.; Korzhnev, D.M.; Hadden, M.K. Identification of small molecule translesion synthesis inhibitors that target the REV1-CT/RIR protein-protein interaction. ACS Chem. Biol. 2017, 12, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Ozen, Z.; Dash, R.C.; McCarthy, K.R.; Chow, S.A.; Rizzo, A.A.; Korzhnev, D.M.; Hadden, M.K. Small molecule scaffolds that disrupt the REV1-CT/RIR protein-protein interaction. Bioorg. Med. Chem. 2018, 26, 4301–4309. [Google Scholar] [CrossRef] [PubMed]
- Actis, M.L.; Ambaye, N.D.; Evison, B.J.; Shao, Y.; Vanarotti, M.; Inoue, A.; McDonald, E.T.; Kikuchi, S.; Heath, R.; Hara, K.; et al. Identification of the first small-molecule inhibitor of the REV7 DNA repair protein interaction. Bioorg. Med. Chem. 2016, 24, 4339–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, E.M.; Washington, M.T. R.I.P. to the PIP: PCNA-binding motif no longer considered specific: PIP motifs and other related sequences are not distinct entities and can bind multiple proteins involved in genome maintenance. Bioessays 2016, 38, 1117–1122. [Google Scholar] [CrossRef]
- Gestwicki, J.E.; Crabtree, G.R.; Graef, I.A. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science 2004, 306, 865–869. [Google Scholar] [CrossRef] [Green Version]
- Gestwicki, J.E.; Marinec, P.S. Chemical control over protein-protein interactions: Beyond inhibitors. Comb. Chem. High Throughput Screen. 2007, 10, 667–675. [Google Scholar] [CrossRef]
- DeLano, W.L.; Ultsch, M.H.; de Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprinzak, E.; Altuvia, Y.; Margalit, H. Characterization and prediction of protein-protein interactions within and between complexes. Proc. Natl. Acad. Sci. USA 2006, 103, 14718–14723. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPI a | Protein/Region | Molecular Surface Area (Å2) b | Hydrophobic Surface Area (Å2) b | Hydrophilic Surface Area (Å2) b |
|---|---|---|---|---|
| REV1-CT/RIR (PDB ID: 4GK5) | FF binding pocket | 446.4 | 243.7 | 202.7 |
| REV1-CT/REV7 (PDB ID 3VU7) | C-terminal tail α2-α3 | 265.3 232.7 | 118.0 --- | 147.3 232.7 |
| REV7/REV3RBM1 (PDB ID:3VU7) | βRBM1 proline core α-helix region | 748.6 380.2 551.3 | 365.3 238.2 247.4 | 383.3 142.0 303.9 |
| REV7/REV3RBM2 (PDB ID: 6BC8) | βRBM1 proline core α-helix region | 1044.0 352.8 644.6 | 578.1 184.1 247.6 | 465.9 168.7 397.0 |
| PCNA/PIP-box (PDB ID: 2ZVK) | Hydrophobic pocket Q-pocket | 451.4 385.2 | 212.3 219.9 | 239.1 165.3 |
| PCNA/PIP-box (PDB ID: 2ZVM) | Hydrophobic pocket Q-pocket | 486.0 283.6 | 241.6 183.6 | 244.4 100.0 |
| PCNA/PIP-box (PDB ID: 2ZVL) | Hydrophobic pocket Q-pocket | 531.9 283.6 | 329.7 183.6 | 202.2 100.0 |
| UbPCNA/UBM-POLι (PDB ID: 2KTF) | α1-α2 region | 893.4 | 338.3 | 555.1 |
| UbPCNA/UBM-REV1 (PDB ID: 5VZM) | α1-α2 region | 860.0 | 355.4 | 504.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, R.C.; Hadden, K. Protein–Protein Interactions in Translesion Synthesis. Molecules 2021, 26, 5544. https://doi.org/10.3390/molecules26185544
Dash RC, Hadden K. Protein–Protein Interactions in Translesion Synthesis. Molecules. 2021; 26(18):5544. https://doi.org/10.3390/molecules26185544
Chicago/Turabian StyleDash, Radha Charan, and Kyle Hadden. 2021. "Protein–Protein Interactions in Translesion Synthesis" Molecules 26, no. 18: 5544. https://doi.org/10.3390/molecules26185544
APA StyleDash, R. C., & Hadden, K. (2021). Protein–Protein Interactions in Translesion Synthesis. Molecules, 26(18), 5544. https://doi.org/10.3390/molecules26185544