
Application of Polyamines and Amino Acid Derivatives Based on 2-Azabicycloalkane Backbone in Enantioselective Aldol Reaction


Abstract
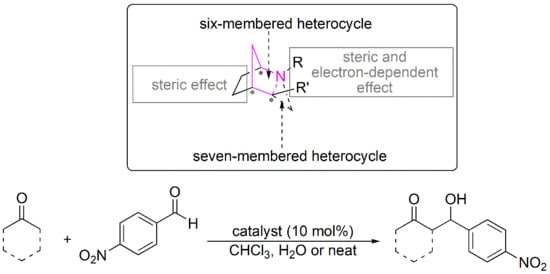
:
1. Introduction
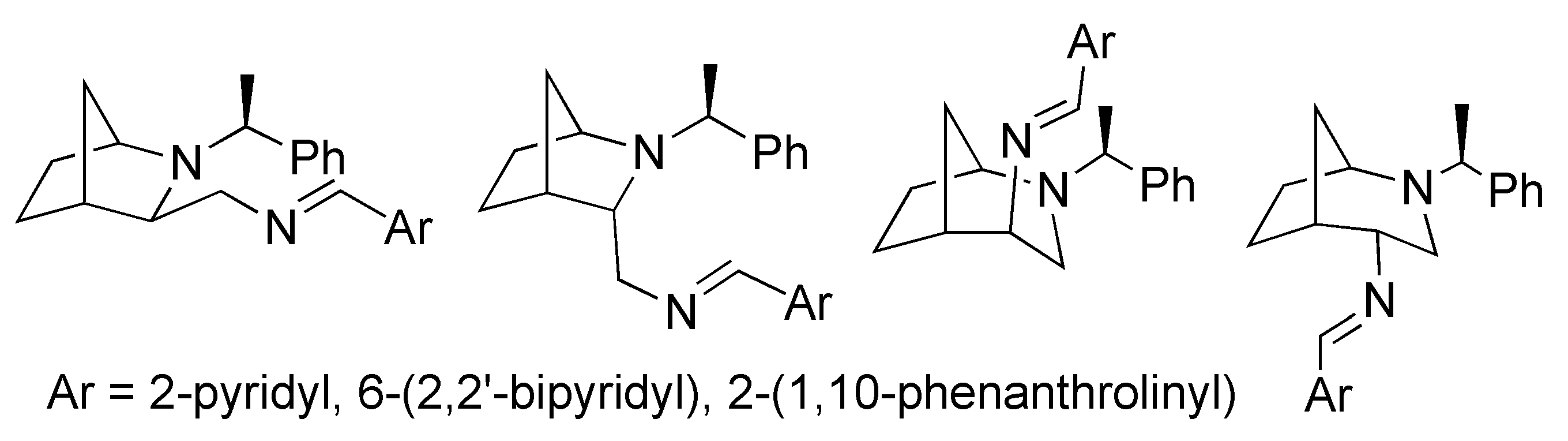
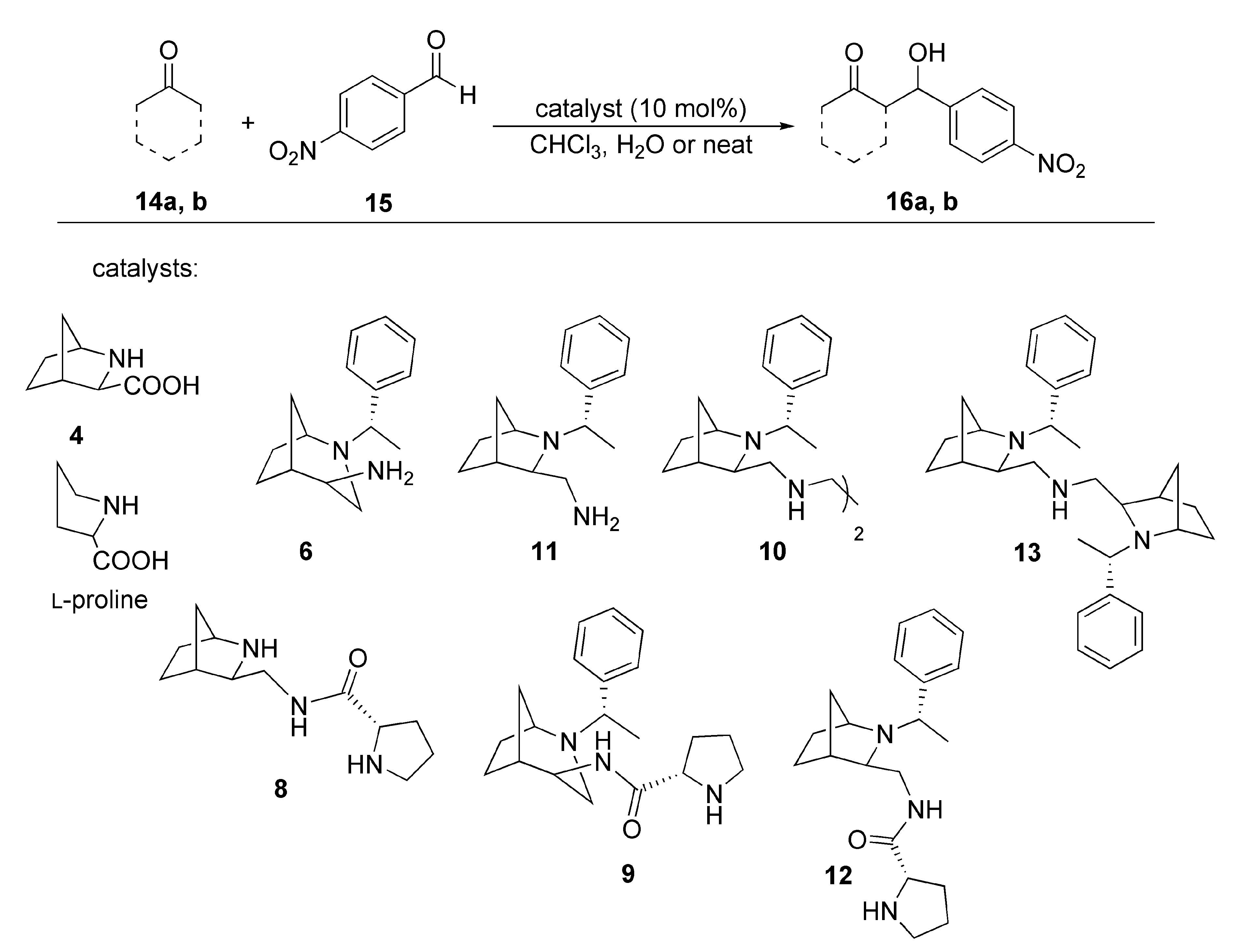
2. Results and Discussion
3. Materials and Methods
3.1. Measurements
3.2. Preparation of Compounds
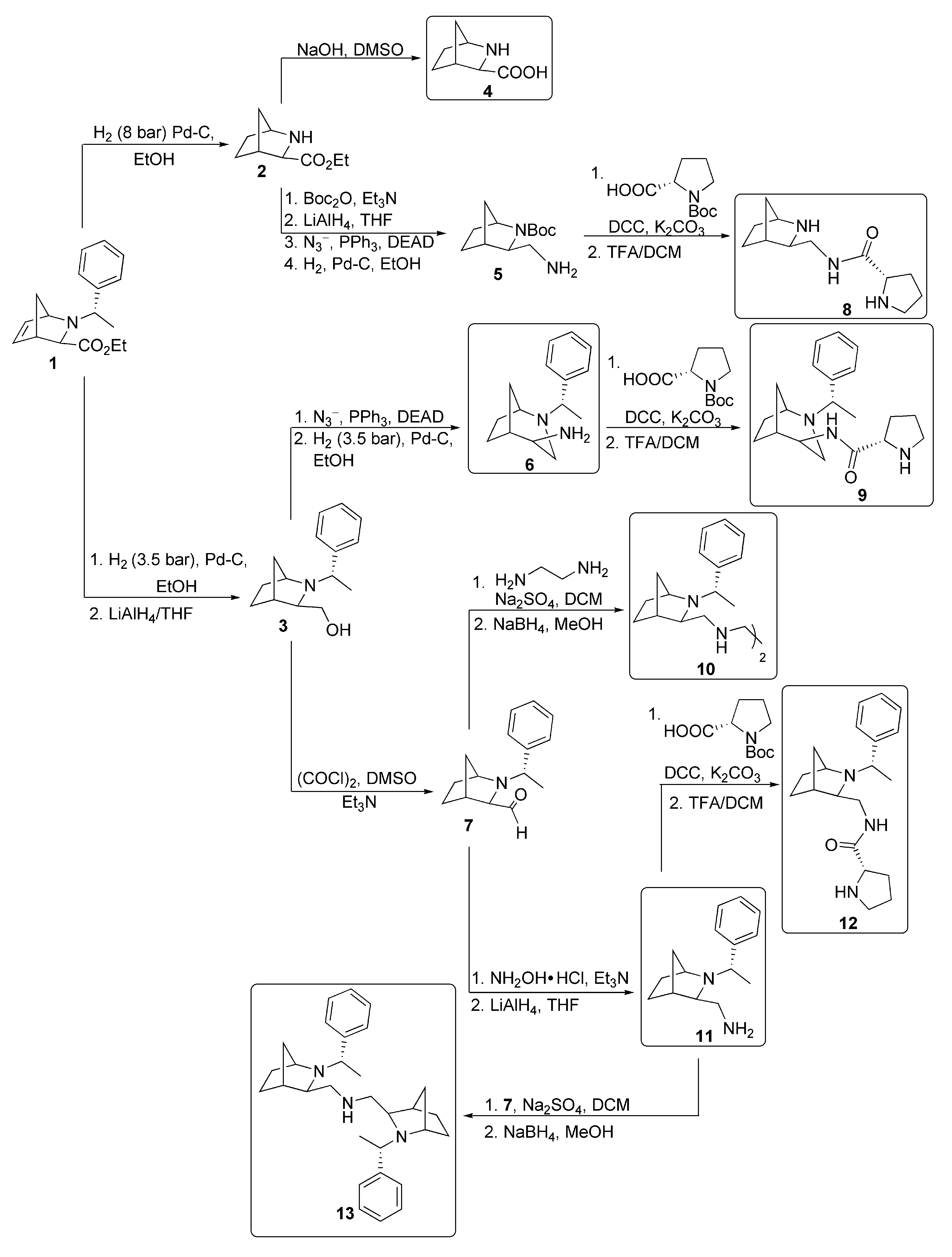
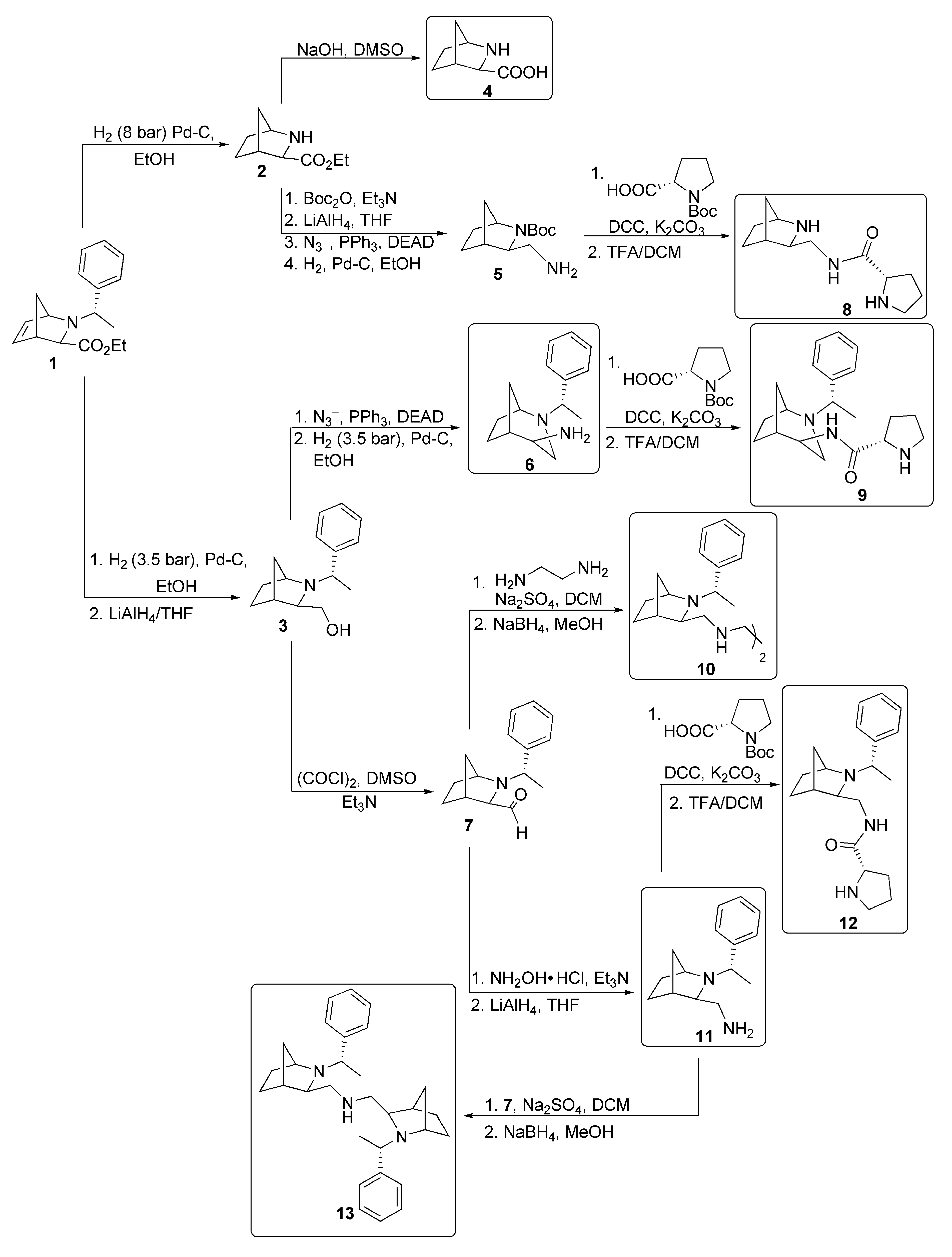
3.2.1. Synthesis of Starting Compounds
3.2.2. Preparation of Polyamines (5, 6, 11, 10, 13)
3.2.3. General Procedure for Amide Synthesis
3.2.4. Catalytic Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mandal, S.; Mandal, S.; Ghosh, S.K.; Ghosh, A.; Saha, R.; Banerjee, S.; Saha, B. Review of the aldol reaction. Synth. Commun. 2015, 46, 1327–1342. [Google Scholar] [CrossRef]
- Braun, M. Fundamentals and transition-state models. Aldol additions of Group 1 and 2 enolates. In Modern Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2004; pp. 1–62. [Google Scholar] [CrossRef]
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kotanosono, T.; Kobayashi, S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. [Google Scholar] [CrossRef]
- Trost, B.M.; Brindle, C.S. The direct catalytic asymmetric aldol reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajos, Z.G.; Parrish, D.R. Synthesis and conversion of 2-methyl-2-(3-oxobutyl)-1,3-cyclopentanedione to the isomeric racemic ketols of the [3.2.1]bicyclooctane and of the perhydroindane series. J. Org. Chem. 1974, 39, 1612–1615. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Eder, U.; Sauer, G.; Wiechert, R. New type of asymmetric cyclization to optically active steroid CD partial structures. Angew. Chem. Int. Ed. Engl. 1971, 10, 496–497. [Google Scholar] [CrossRef]
- Schneider, J.F.; Ladd, C.L.; Bräse, S. Proline as an asymmetric organocatalyst. In Sustainable Catalysis: Without Metals or Other Endangered Elements, 1st ed.; North, M., Ed.; RSC Publishing: Cambridge, UK, 2015; pp. 79–119. [Google Scholar] [CrossRef]
- List, B.; Hoang, L.; Martin, H.J. New mechanistic studies on the proline-catalyzed aldol reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 5839–5842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Carter, R.G. Proline sulfonamide-based organocatalysis: Better late than never. Synlett 2010, 19, 2827–2838. [Google Scholar] [CrossRef] [Green Version]
- Vachan, B.S.; Karuppasamy, M.; Vinoth, P.; Kumar, S.V.; Perumal, S.; Sridharan, V.; Menendezd, J.C. Proline and its derivatives as organocatalysts for multi-component reactions in aqueous media: Synergic pathways to the green synthesis of heterocycles. Adv. Synth. Catal. 2020, 362, 87–110. [Google Scholar] [CrossRef]
- Yadav, G.D.; Singh, S. Prolinamide-catalysed asymmetric organic transformations. ChemistrySelect 2019, 4, 5591–5618. [Google Scholar] [CrossRef]
- List, B. Amine-catalyzed aldol reactions. In Modern Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2004; pp. 161–200. [Google Scholar] [CrossRef]
- Wojaczyńska, E.; Wojaczyński, J.; Kleniewska, K.; Dorsz, M.; Olszewski, T.K. 2-Azanorbornane—A versatile chiral aza-Diels–Alder cycloadduct: Preparation, applications in stereoselective synthesis and biological activity. Org. Biomol. Chem. 2015, 13, 6116–6148. [Google Scholar] [CrossRef]
- Ogasawara, A.; Subba Reddy, U.V.; Seki, C.; Okuyama, Y.; Uwai, K.; Tokiwa, M.; Takeshita, M.; Nakano, H. 2-Azanorbornane-based amine organocatalyst for enantioselective aldol reaction of isatins with ketones. Tetrahedron Asymmetry 2016, 27, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Wojaczyńska, E.; Skarżewski, J.; Sidorowicz, Ł.; Wieczorek, R.; Wojaczyński, J. Zinc complexes formed by 2,2′-bipyridine and 1,10-phenanthroline moieties combined with 2-azanorbornane: Modular chiral catalysts for aldol reactions. New J. Chem. 2016, 40, 9795–9805. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.D.; Wilson, R.D.; Brown, G.R. Stereoselective synthesis of pipecolic acid derivatives using aza-Diels-Alder reactions. Tetrahedron Lett. 1989, 30, 6781–6784. [Google Scholar] [CrossRef]
- Stella, H.; Abraham, H.; Feneau-Dupont, J.; Tinant, B.; Declercq, J.P. Asymmetric aza-Diels-Alder reaction using the chiral 1-phenyl ethyl imine of methyl glyoxylate. Tetrahedron Lett. 1990, 31, 2603–2606. [Google Scholar] [CrossRef]
- Waldmann, H.; Braun, M. Asymmetric synthesis of bicyclic amino acid derivatives by aza-Diels-Alder reactions in aqueous solution. Liebigs Ann. Chem. 1991, 1991, 1045–1048. [Google Scholar] [CrossRef]
- Nakano, H.; Kumagai, N.; Kabuto, C.; Matsuzaki, H.; Hongo, H. Synthesis of new chiral catalysts, N-alkyl-2-azanorbornyl-methanols, for the enantioselective addition of diethylzinc to arylaldehydes. Tetrahedron Asymmetry 1995, 6, 1233–1236. [Google Scholar] [CrossRef]
- Ekegren, J.K.; Modin, S.A.; Alonso, D.A.; Andersson, P.G. Multigram scale synthesis of a useful aza-Diels–Alder adduct in a one-step procedure. Tetrahedron Asymmetry 2002, 13, 447–449. [Google Scholar] [CrossRef]
- Iwan, D.; Kamińska, K.; Wojaczyńska, E.; Psurski, M.; Wietrzyk, J.; Daszkiewicz, M. Biaryl sulfonamides based on the 2-azabicycloalkane skeleton—Synthesis and antiproliferative activity. Materials 2020, 13, 5010. [Google Scholar] [CrossRef] [PubMed]
- Wojaczyńska, E.; Turowska-Tyrk, I.; Skarżewski, J. Novel chiral bridged azepanes: Stereoselective ring expansion of 2-azanorbornan-3-yl methanols. Tetrahedron 2012, 68, 7848–7854. [Google Scholar] [CrossRef]
- Kamińska, K.; Wojaczyńska, E.; Wietrzyk, J.; Turlej, E.; Błażejczyk, A.; Wieczorek, R. Synthesis, structure and antiproliferative activity of chiral polyamines based on a 2-azanorbornane skeleton. Tetrahedron Asymmetry 2016, 27, 753–758. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, Y.; Zhang, X.; Huang, Y.; Li, B.; Zhang, G. Rationally designed organocatalyst for direct asymmetric aldol reaction in the presence of water. Tetrahedron Lett. 2007, 48, 4281–4285. [Google Scholar] [CrossRef]
- Maya, V.; Raj, M.; Singh, V.K. Highly enantioselective organocatalytic direct aldol reaction in an aqueous medium. Org. Lett. 2007, 9, 2593–2595. [Google Scholar] [CrossRef] [PubMed]
- Chimni, S.S.; Singh, S.; Mahajan, D. Protonated (S)-prolinamide derivatives—Water compatible organocatalysts for direct asymmetric aldol reaction. Tetrahedron Asymmetry 2008, 19, 2276–2284. [Google Scholar] [CrossRef]
- Shen, C.; Shen, F.; Zhou, G.; Xia, H.; Chen, X.; Liu, X.; Zhang, P. Novel carbohydrate-derived prolinamide as a highly efficient, recoverable catalyst for direct aldol reactions in water. Catal. Commun. 2012, 26, 6–10. [Google Scholar] [CrossRef]
- Paladhi, S.; Das, J.; Mishra, P.K.; Dash, J. Multifunctional “click” prolinamides: A new platform for asymmetric aldol reactions in the presence of water with catalyst recycling. Adv. Synth. Catal. 2013, 355, 274–280. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ueki, H.; Yasumoto, M.; Mekala, S.; Hirschi, J.S.; Singleton, D.A. Phenomenon of optical self-purification of chiral non-racemic compounds. J. Am. Chem. Soc. 2007, 129, 12112–12113. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Roussel, C.; Kitagawa, O.; Sorochinsky, A.E. Self-disproportionation of enantiomers via achiral chromatography: A warning and extra dimension in optical purifications. Chem. Soc. Rev. 2012, 41, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Wzorek, A.; Klika, K.D. A question of policy: Should tests for the self-disproportionation of enantiomers (SDE) be mandatory for reports involving scalemates? Tetrahedron Asymmetry 2017, 28, 1430–1434. [Google Scholar] [CrossRef]
- Han, J.; Klika, K.D.; Wzorek, A.; Soloshonok, V.A. Recommended Tests for the Self-Disproportionation of Enantiomers (SDE) to Ensure Accurate Reporting of the Stereochemical Outcome of Enantioselective Reactions. Molecules 2021, 26, 2757. [Google Scholar] [CrossRef]
- Han, J.; Dembinski, R.; Soloshonok, V.A.; Klika, K.D. A Call for a Change in Policy Regarding the Necessity for SDE Tests to Validate the Veracity of the Outcome of Enantioselective Syntheses, the Inherent Chiral State of Natural Products, and Other Cases Involving Enantioenriched Samples. Molecules 2021, 26, 3994. [Google Scholar] [CrossRef] [PubMed]
- Södergren, M.J.; Andersson, P.G. Chiral, bicyclic proline derivatives and their application as ligands for copper in the catalytic asymmetric allylic oxidation of olefins. Tetrahedron Lett. 1996, 37, 7577–7580. [Google Scholar] [CrossRef]
- Jin, H.; Lazerwith, S.E.; Martin, T.A.T.; Bacon, E.M.; Cottell, J.J.; Cai, Z.R.; Pyun, H.; Morganelli, P.A.; Ji, M.; Taylor, J.G.; et al. Polycyclic-Carbamoylpyridone Compounds and Their Pharmaceutical Use. U.S. Patent 14/133,858, 26 June 2014. [Google Scholar]
- Illa, O.; Porcar-Tost, O.; Robledillo, C.; Carlos, E.; Nolis, P.; Reiser, O.; Branchadell, V.; Ortuño, R.M. Stereoselectivity of Proline/Cyclobutane Amino Acid-Containing Peptide Organocatalysts for Asymmetric Aldol Additions: A Rationale. J. Org. Chem. 2018, 83, 350–363. [Google Scholar] [CrossRef] [PubMed]
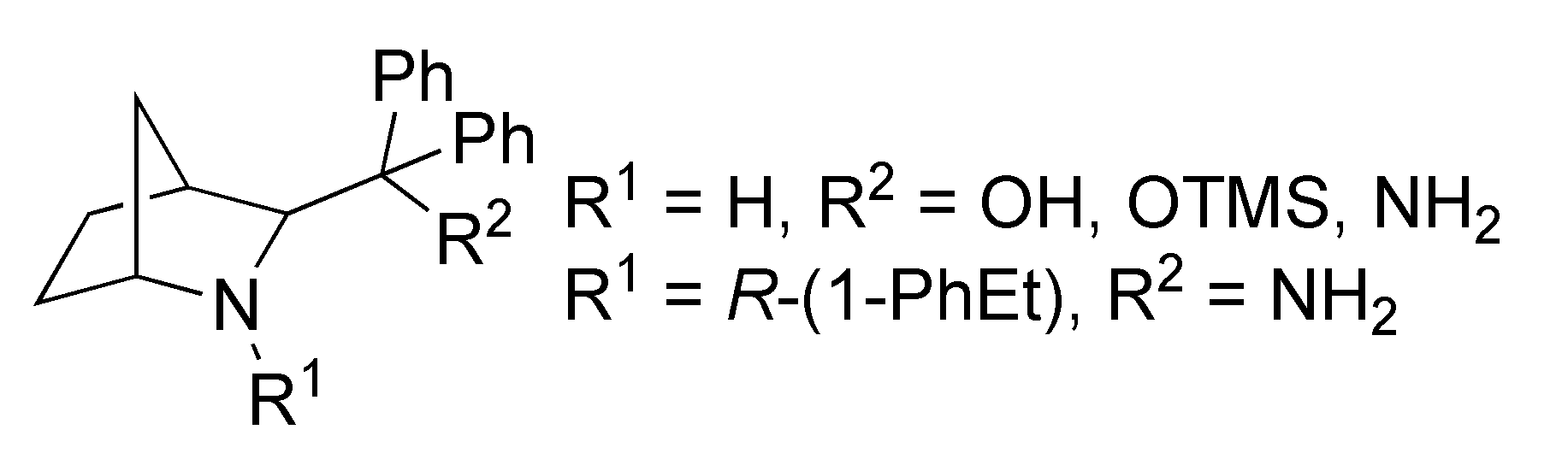
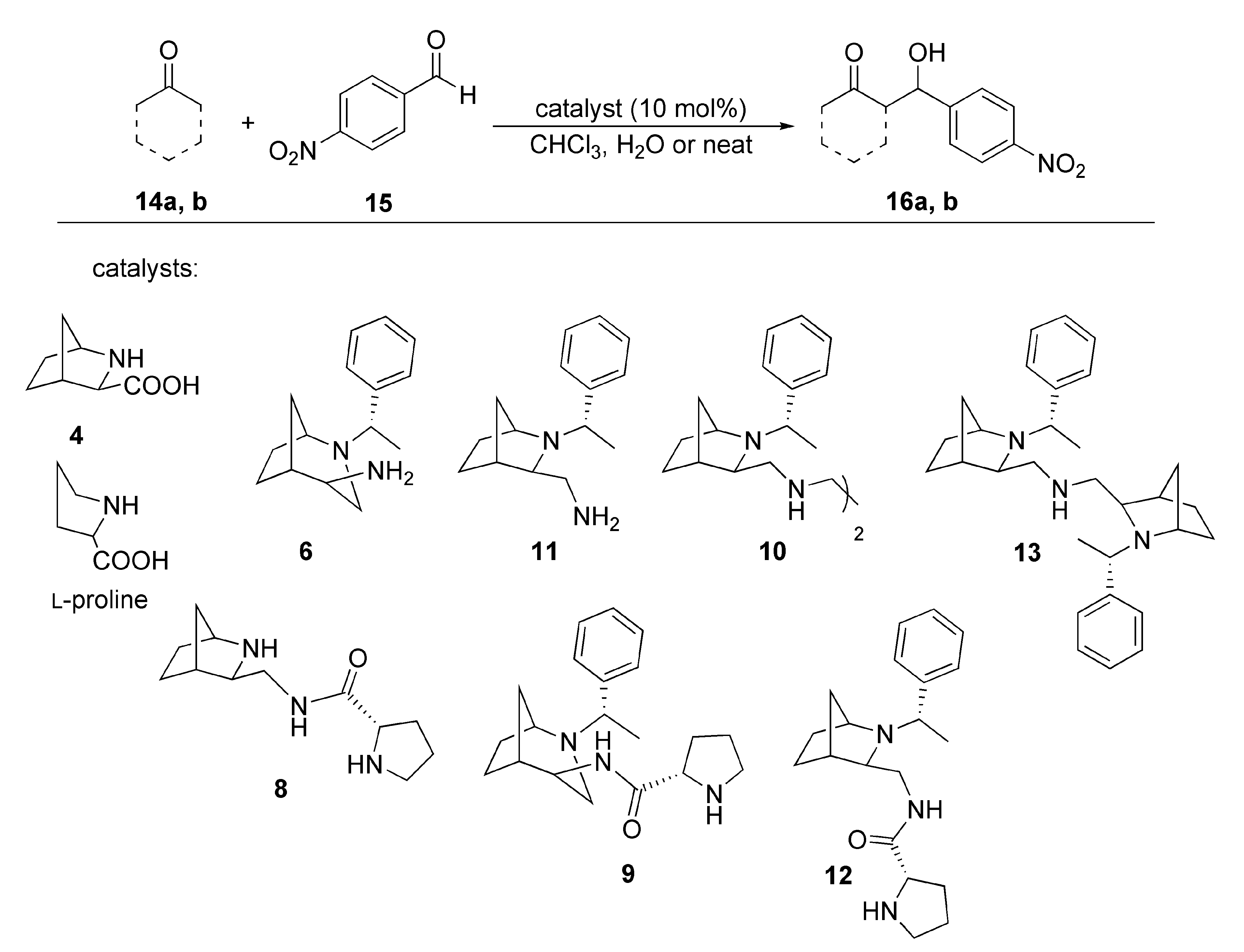

{kind=link}
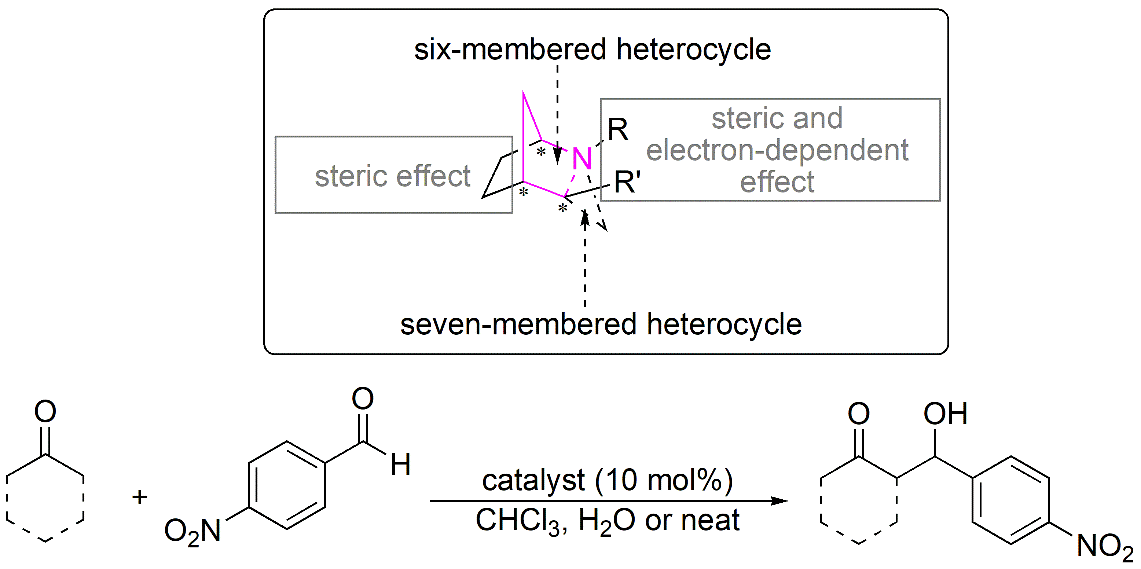
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Ketone | Solvent | Yield [%] d | anti/syne | ee [%] f |
|---|---|---|---|---|---|---|
| 1 a | l-proline | acetone | neat | >95 | - | 66 (R) |
| 2 a | 4 | acetone | neat | 89 | - | 22 (R) |
| 3 a | 8 | acetone | neat | <8 | - | - |
| 4 a | 9 | acetone | neat | 59 g | - | 60 (R) |
| 5 a | 12 | acetone | neat | 15 | - | 52 (R) |
| 6 b | l-proline | cyclohexanone | CHCl3 | 26 | 73:27 | (anti) 63(syn) 56 |
| 7 b | 4 | cyclohexanone | CHCl3 | 13 | 79:21 | (anti) 11(syn) 6 |
| 8 b | 6 | cyclohexanone | CHCl3 | 16 | 26:74 | (anti) 22(syn) 0 |
| 9 b | 8 | cyclohexanone | CHCl3 | traces | - | - |
| 10 b | 9 | cyclohexanone | CHCl3 | >95 h | 78:22 | (anti) 57(syn) 15 |
| 11 b | 10 | cyclohexanone | CHCl3 | traces | - | - |
| 12 b | 11 | cyclohexanone | CHCl3 | 43 | 26:74 | (anti) 20(syn) 21 |
| 13 b | 12 | cyclohexanone | CHCl3 | 63 e | 84:16 | (anti) 38(syn) 40 |
| 14 b | 13 | cyclohexanone | CHCl3 | 0 | - | - |
| 15 c | l-proline | cyclohexanone | brine | traces | - | - |
| 16 c | 4 | cyclohexanone | brine | traces | - | - |
| 17 c | 6 | cyclohexanone | brine | 86 | 65:35 | (anti) 16(syn) 31 |
| 18 c | 8 | cyclohexanone | brine | traces | - | - |
| 19 c | 9 | cyclohexanone | brine | >95 | 73:27 | (anti) 63(syn) 44 |
| 20 c | 10 | cyclohexanone | brine | traces | - | - |
| 21 c | 11 | cyclohexanone | brine | >95 | 81:19 | (anti) 3(syn) 14 |
| 22 c | 12 | cyclohexanone | brine | traces | - | - |
| 23 c | 13 | cyclohexanone | brine | traces | - | - |
| Sample | Fractions | Mass/mg | dr (anti:syn) | ee (anti)/% | ee (syn)/% |
|---|---|---|---|---|---|
| 1 | Raw mixture | 16.0 | 88:12 | 3 | 11 |
| 1 | 1.6 | 85:15 | 5 | 11 | |
| 2 | 5.5 | 95:5 | 2 | 14 | |
| 3 | 1.7 | 97:3 | 2 | 9 | |
| 4 | 5.4 | 97:3 | 2 | 12 | |
| 5 | 1.7 | 97:3 | 2 | 11 | |
| 2 | Raw mixture | 60.0 | 24:76 | 57 | 15 |
| 1 | 8.6 | 33:67 | nd b | ||
| 2 | 27.5 | 19:81 | 61 | 4 | |
| 3 | 18.6 | 1:99 | 43 | 15 | |
| 4 | 2.9 | 1:99 | 52 | 46 | |
| 3 | Raw mixture | 48.0 | 80:20 | 54 | 52 |
| 1 | 16.9 | 57:43 | nd b | ||
| 2 | 17.6 | 88:12 | 48 | 6 | |
| 3 | 13.5 | 98:2 | 61 | 56 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwan, D.; Kamińska, K.; Wojaczyńska, E. Application of Polyamines and Amino Acid Derivatives Based on 2-Azabicycloalkane Backbone in Enantioselective Aldol Reaction. Molecules 2021, 26, 5166. https://doi.org/10.3390/molecules26175166
Iwan D, Kamińska K, Wojaczyńska E. Application of Polyamines and Amino Acid Derivatives Based on 2-Azabicycloalkane Backbone in Enantioselective Aldol Reaction. Molecules. 2021; 26(17):5166. https://doi.org/10.3390/molecules26175166
Chicago/Turabian StyleIwan, Dominika, Karolina Kamińska, and Elżbieta Wojaczyńska. 2021. "Application of Polyamines and Amino Acid Derivatives Based on 2-Azabicycloalkane Backbone in Enantioselective Aldol Reaction" Molecules 26, no. 17: 5166. https://doi.org/10.3390/molecules26175166
APA StyleIwan, D., Kamińska, K., & Wojaczyńska, E. (2021). Application of Polyamines and Amino Acid Derivatives Based on 2-Azabicycloalkane Backbone in Enantioselective Aldol Reaction. Molecules, 26(17), 5166. https://doi.org/10.3390/molecules26175166