
In Situ Monitored Vortex Fluidic-Mediated Protein Refolding/Unfolding Using an Aggregation-Induced Emission Bioprobe

 ,
, 

Abstract
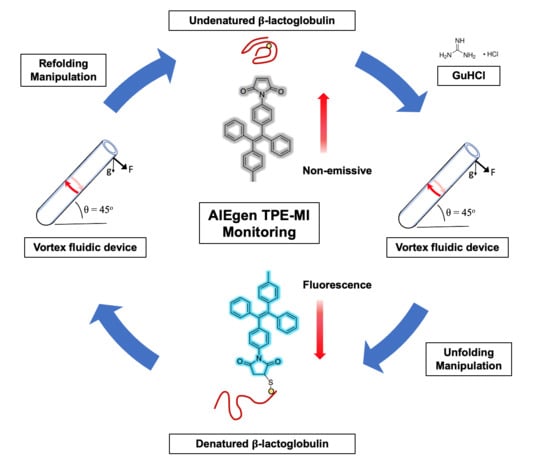
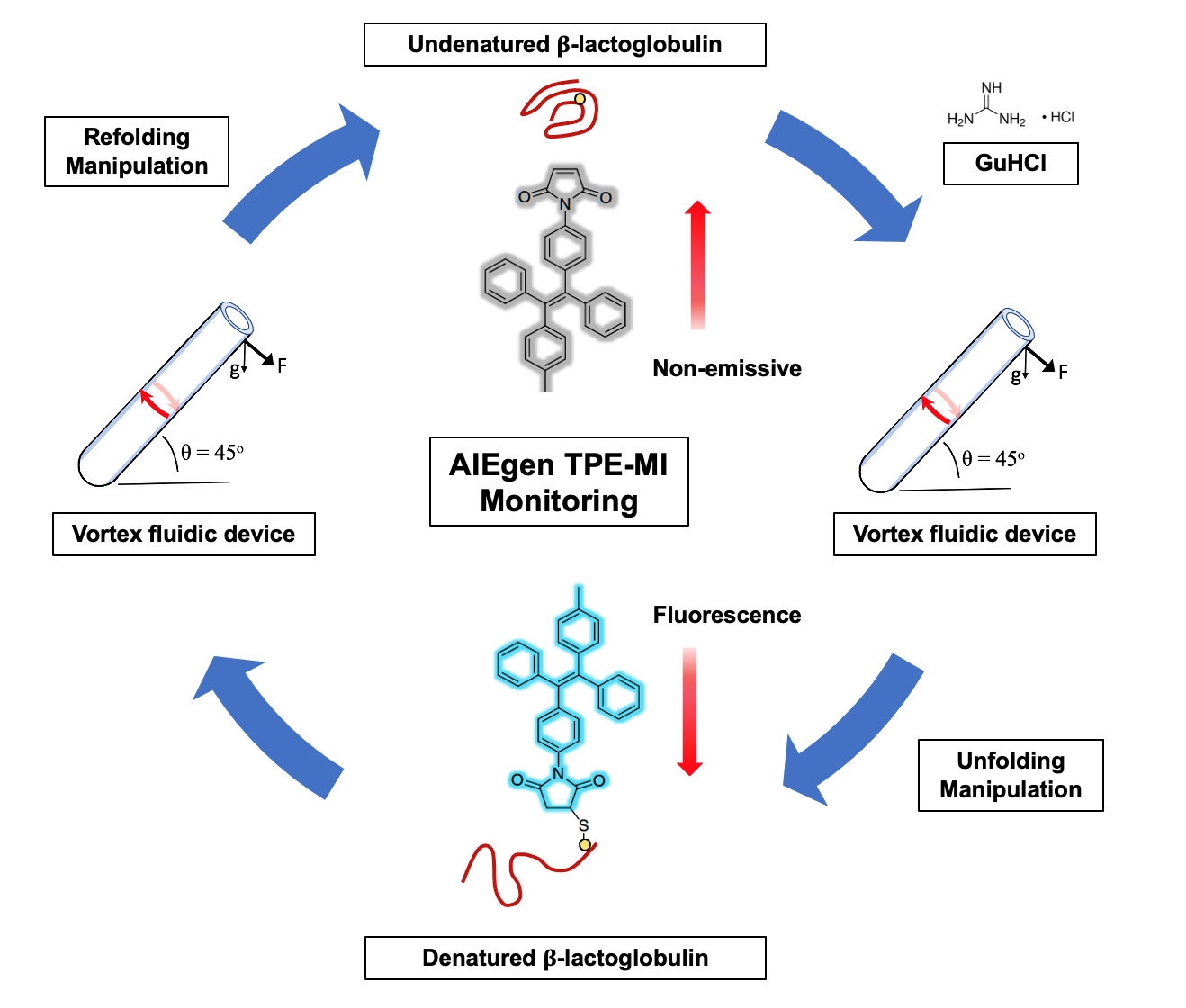
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Characterisations
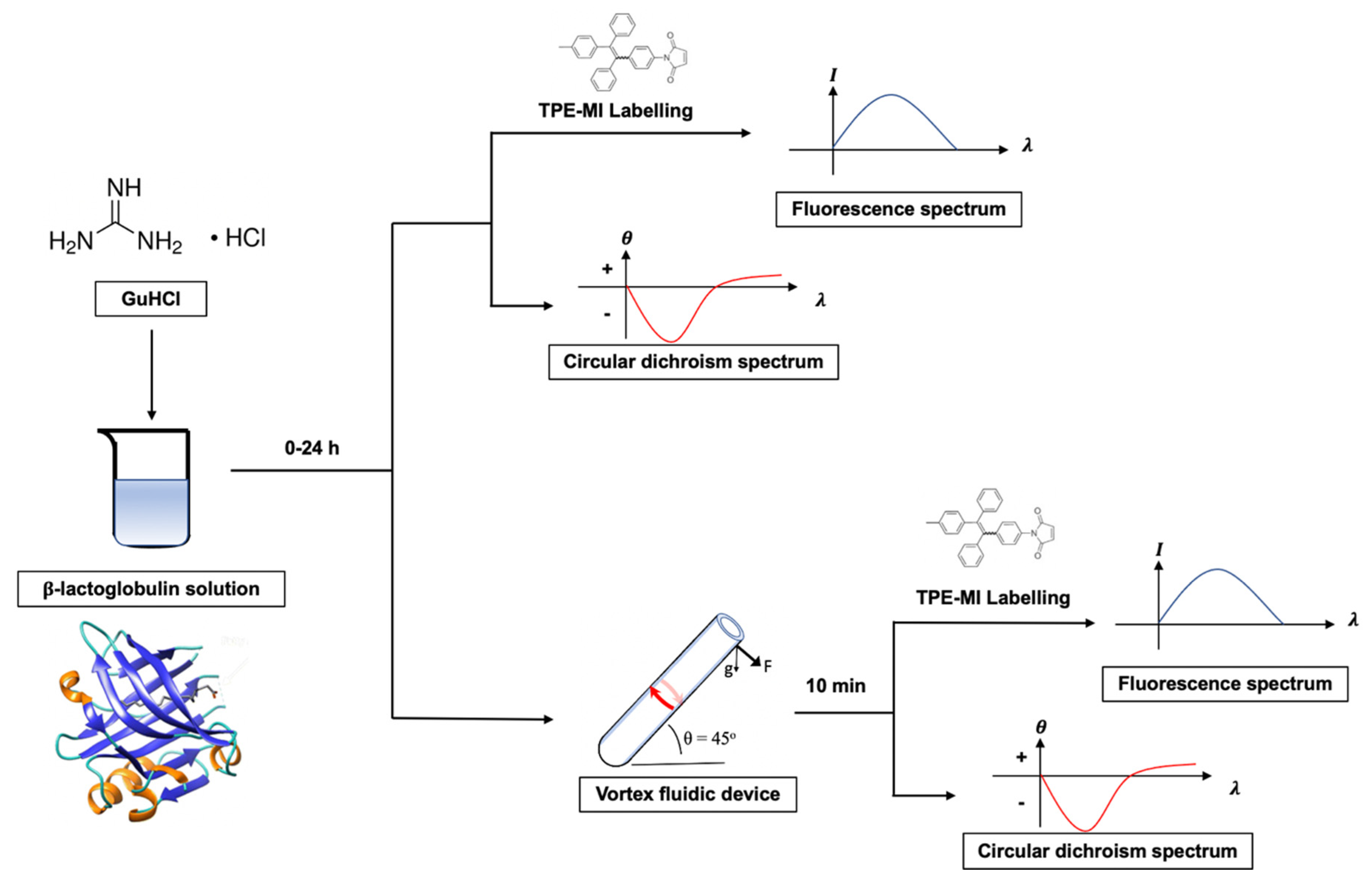
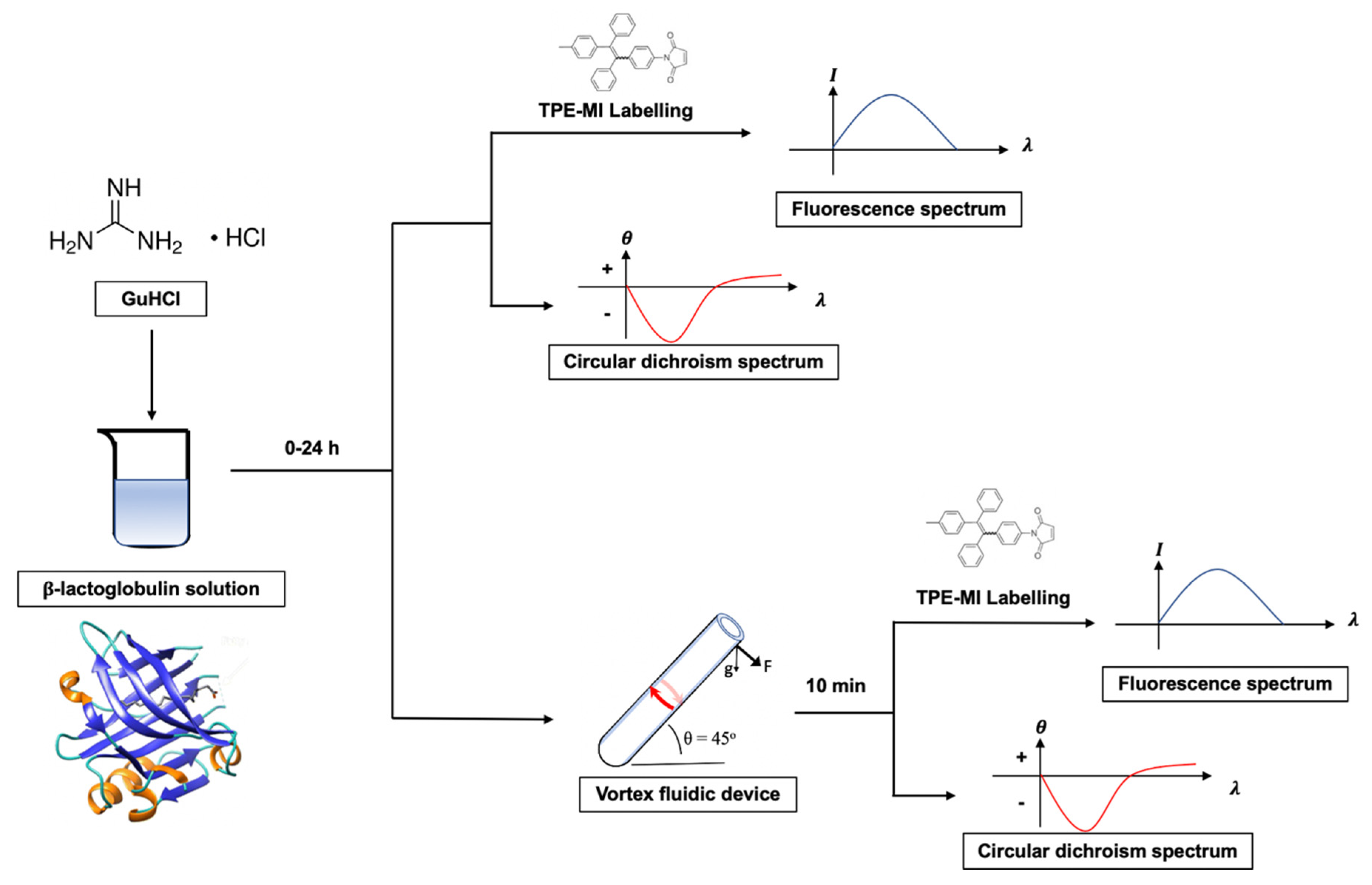
2.3. Protocol Development
3. Results and Discussion
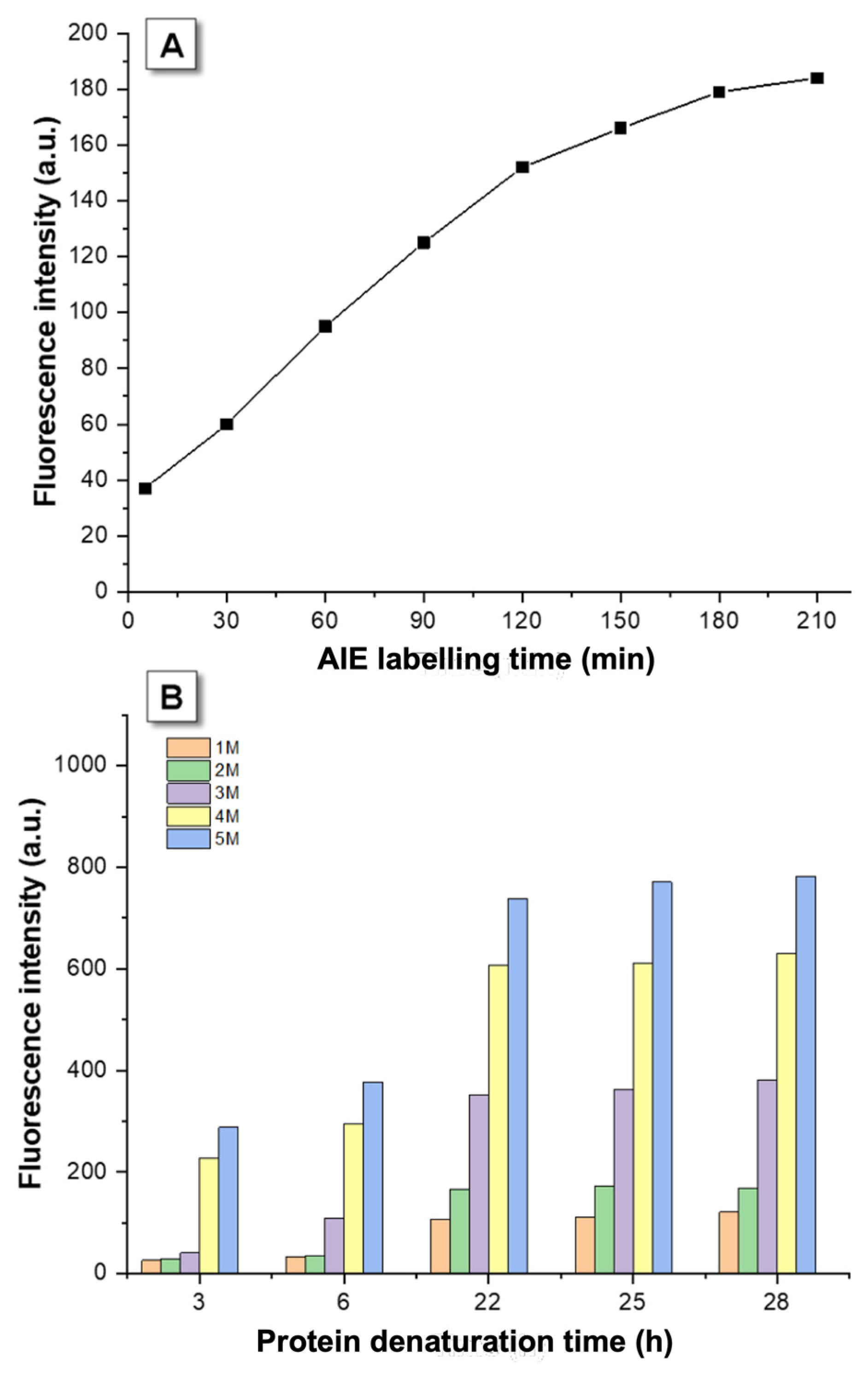
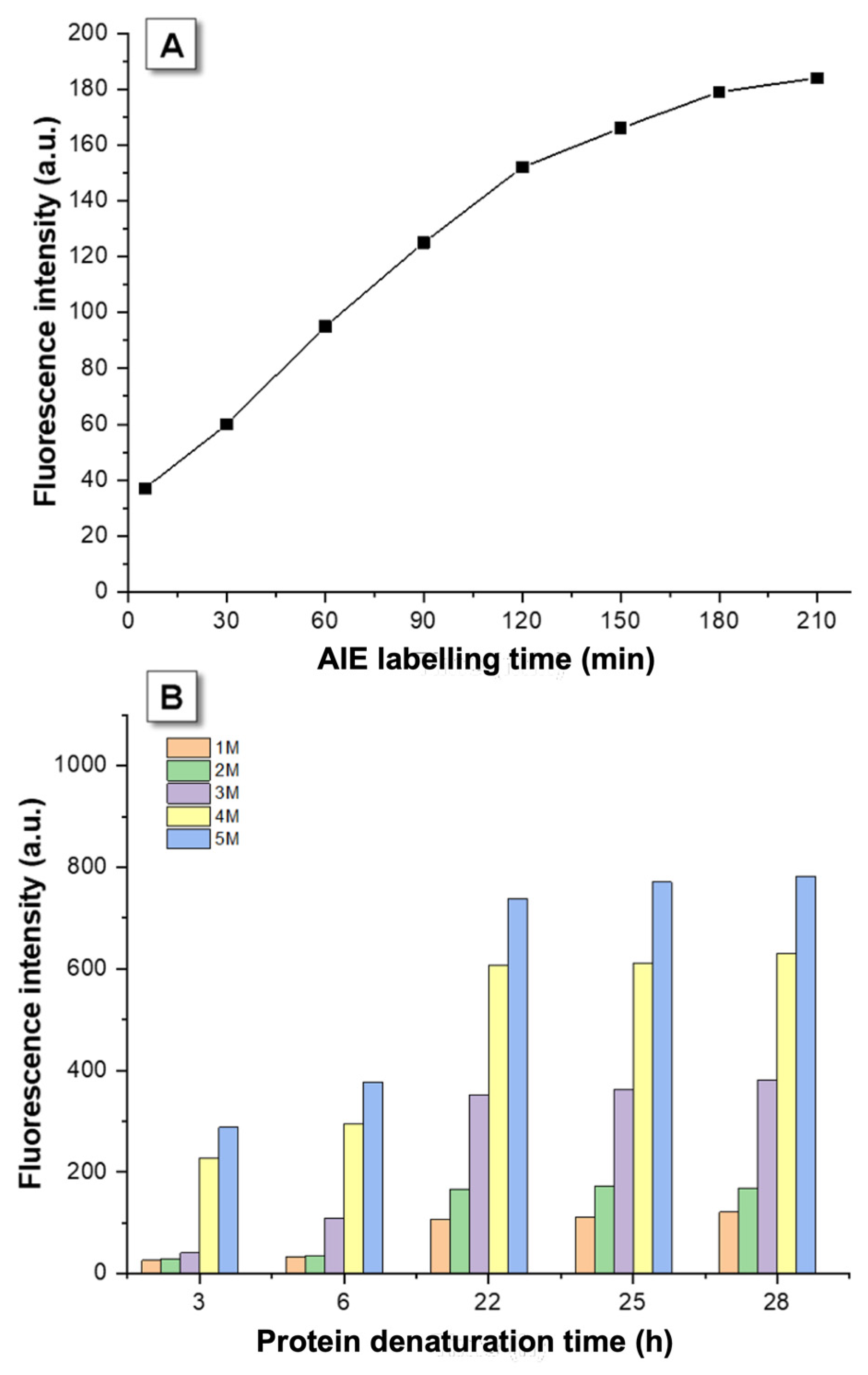
3.1. Kinetic Optimization
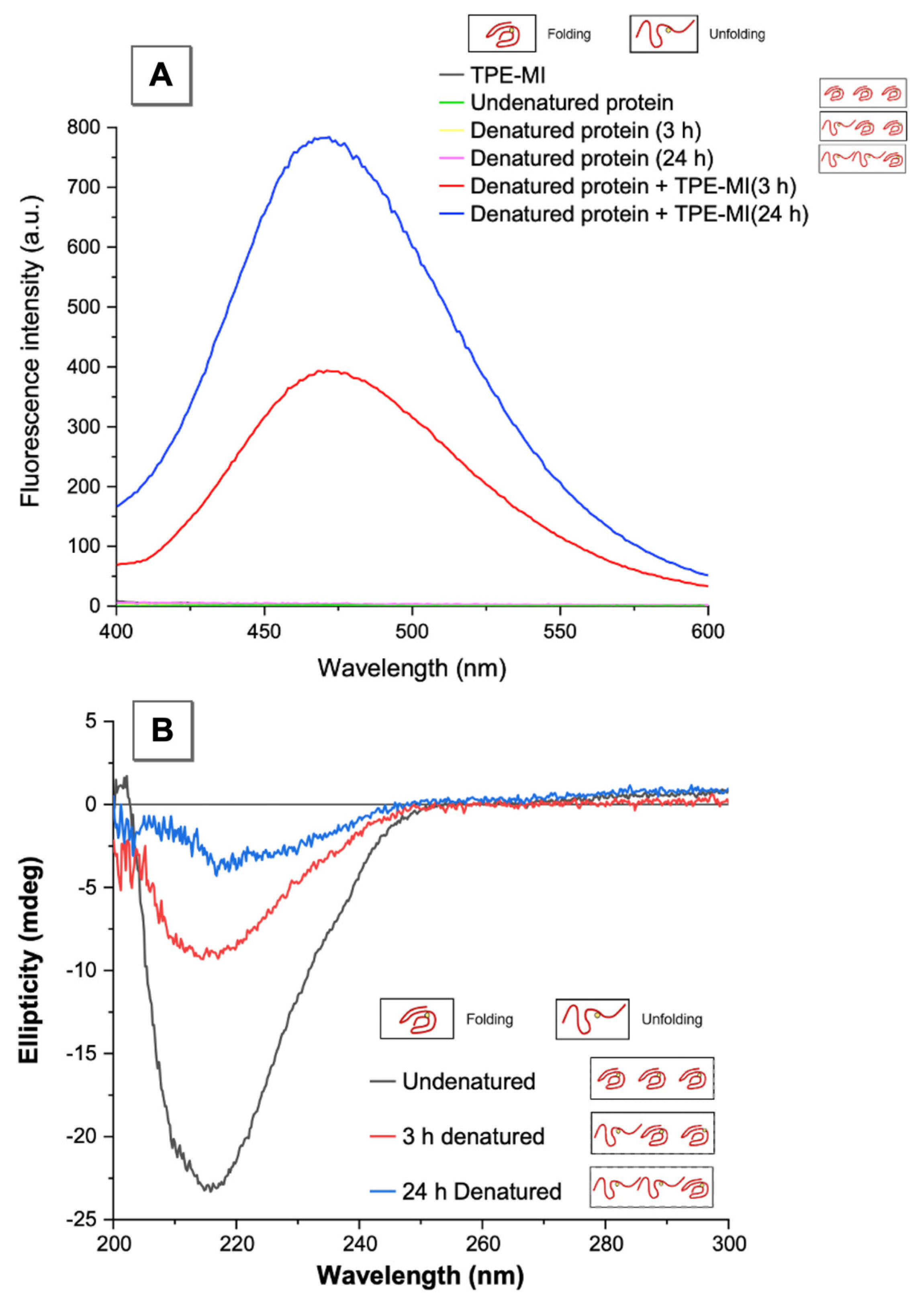
3.2. AIEgen Monitoring Protein Denaturation
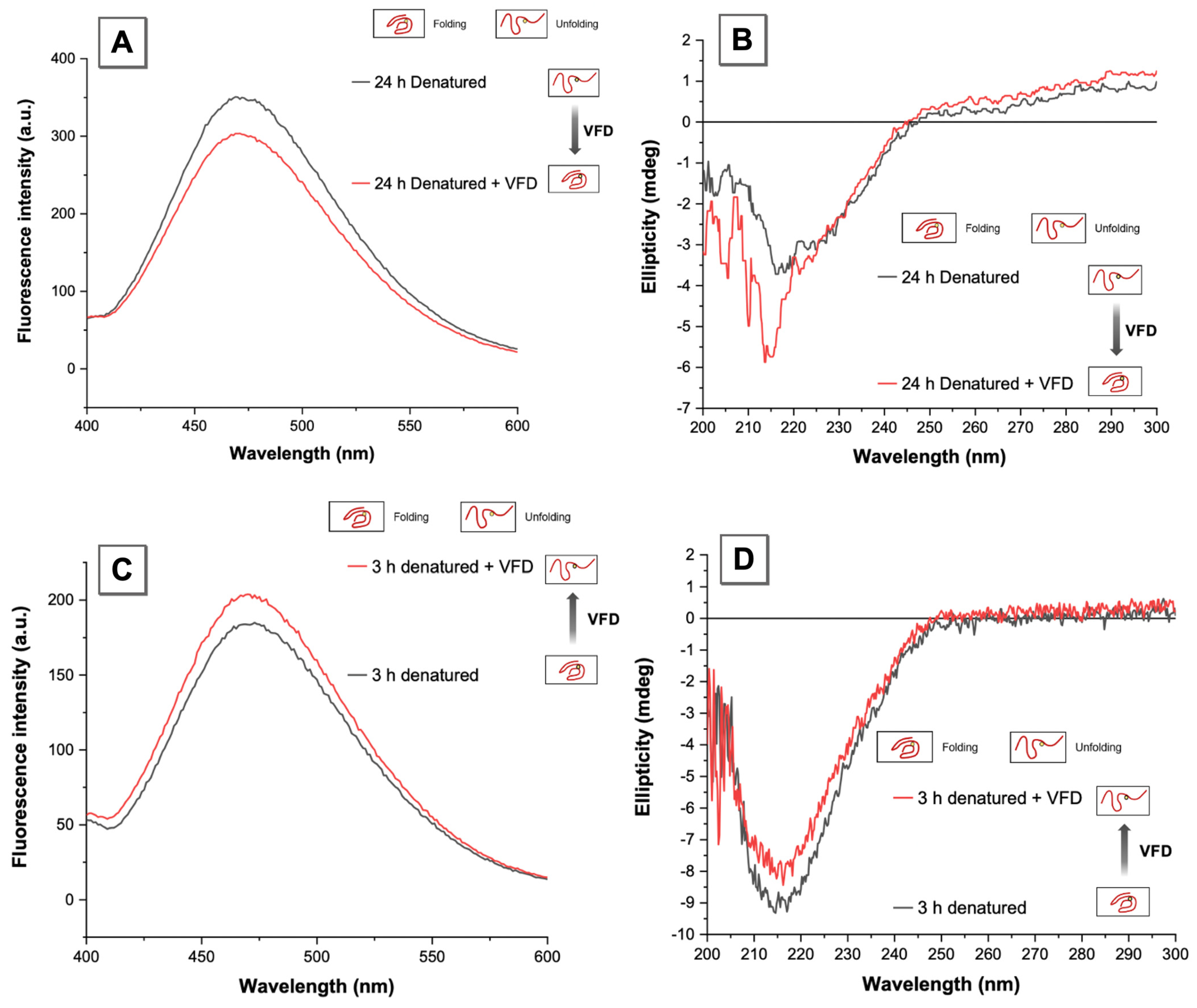
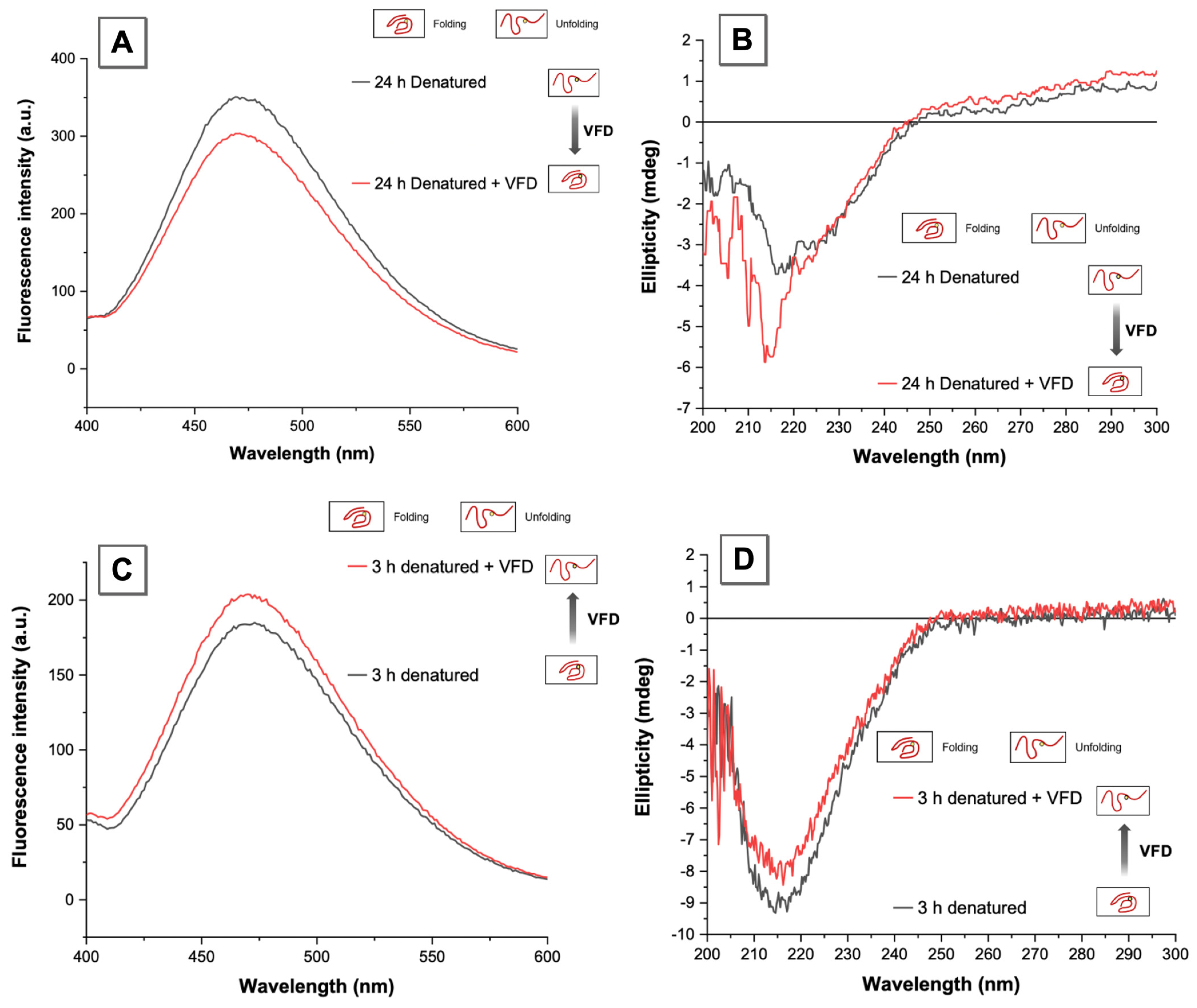
3.3. VFD Manipulating Protein Refolding/Unfolding
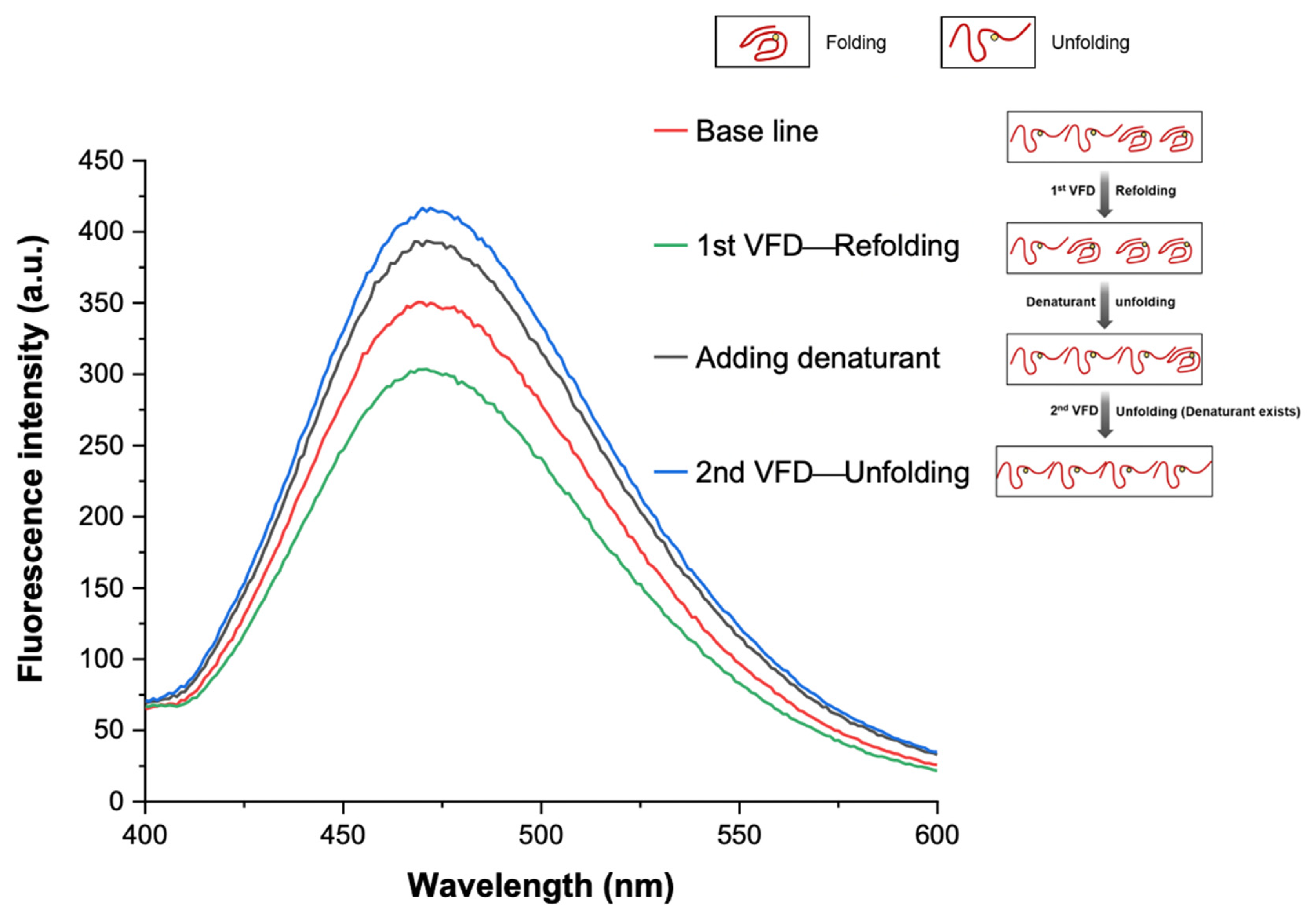
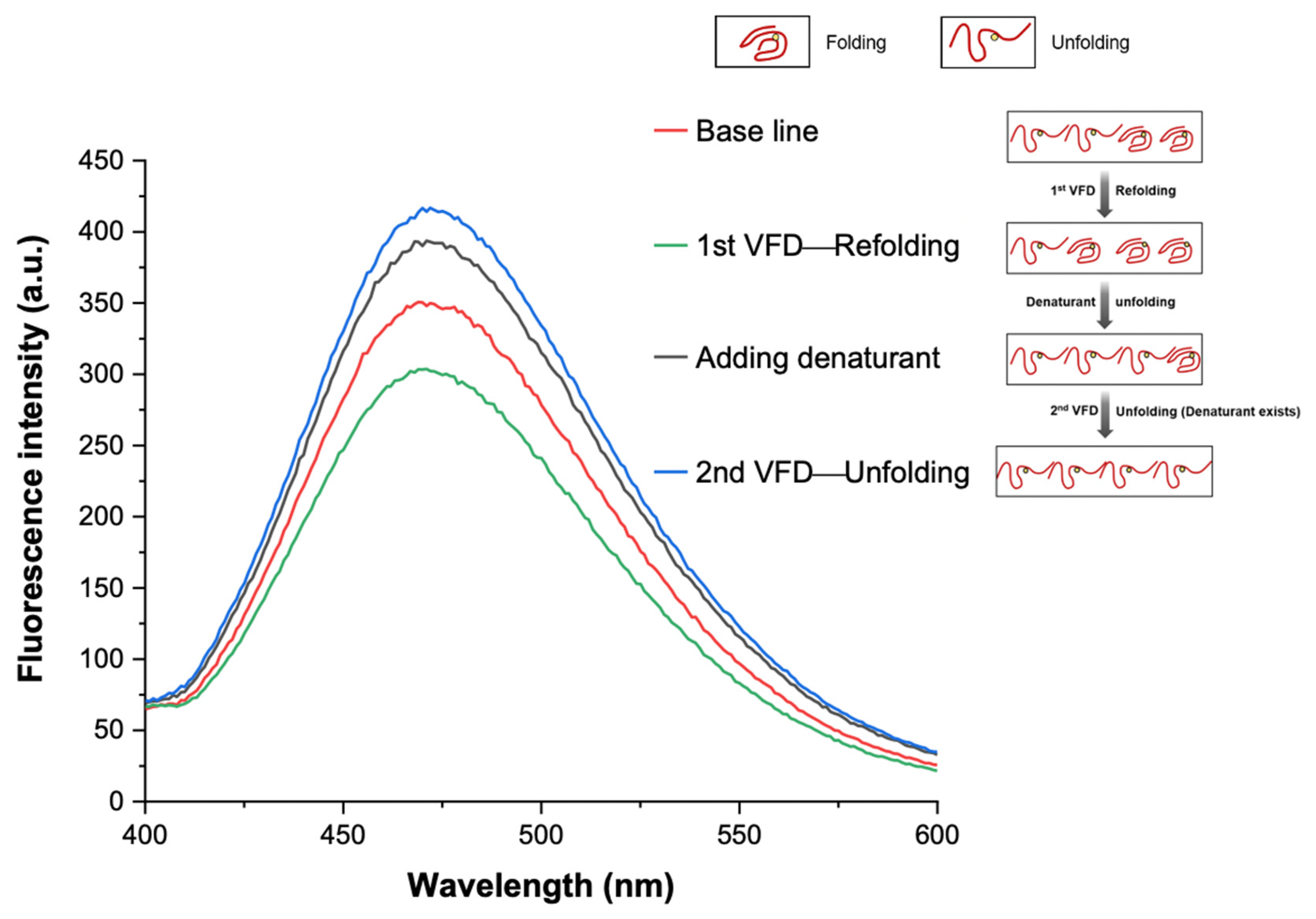
3.4. Interpretation of Equilibrium of Unfolding and Refolding Proteins by VFD and AIEgen
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Gupta, A.; Hong, Y.; Carver, J.A.; Maiti, S. A Spectroscopic marker for structural transitions associated with amyloid-β aggregation. Biochemistry 2020, 59, 1813–1822. [Google Scholar] [CrossRef]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204. [Google Scholar] [CrossRef] [Green Version]
- Igder, A.; Pye, S.; Al-Antaki, A.H.M.; Keshavarz, A.; Raston, C.L.; Nosrati, A. Vortex fluidic mediated synthesis of polysulfone. RSC Adv. 2020, 10, 14761–14767. [Google Scholar] [CrossRef]
- Alharbi, T.M.; Jellicoe, M.; Luo, X.; Vimalanathan, K.; Alsulami, I.K.; AL Harbie, B.S.; Igder, A.; Alrashaidi, F.A.J.; Chen, X.; Stubbs, K.A.; et al. Sub-micron moulding topological mass transport regimes in angled vortex fluidic flow. Nanoscale Adv. 2021, 3, 3064. [Google Scholar] [CrossRef]
- Yuan, T.Z.; Ormonde, C.F.; Kudlacek, S.T.; Kunche, S.; Smith, J.N.; Brown, W.A.; Pugliese, K.M.; Olsen, T.J.; Iftikhar, M.; Raston, C.L. Shear stress mediated refolding of proteins from aggregates and inclusion bodies. ChemBioChem 2015, 16, 393–396. [Google Scholar] [CrossRef]
- Yasmin, L.; Chen, X.; Stubbs, K.A.; Raston, C.L. Optimising a vortex fluidic device for controlling chemical reactivity and selectivity. Sci. Rep. 2013, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Yasmin, L.; Eggers, P.K.; Skelton, B.W.; Stubbs, K.A.; Raston, C.L. Thin film microfluidic synthesis of fluorescent highly substituted pyridines. Green Chem. 2014, 16, 3450–3453. [Google Scholar] [CrossRef]
- Britton, J.; Dalziel, S.B.; Raston, C.L. Continuous flow Fischer esterifications harnessing vibrational-coupled thin film fluidics. RSC Adv. 2015, 5, 1655–1660. [Google Scholar] [CrossRef]
- Britton, J.; Dalziel, S.B.; Raston, C.L. The synthesis of di-carboxylate esters using continuous flow vortex fluidics. Green Chem. 2016, 18, 2193–2200. [Google Scholar] [CrossRef] [Green Version]
- Vimalanathan, K.; Raston, C.L. Dynamic thin films in controlling the fabrication of nanocarbon and its composites. Adv. Mater. Technol. 2017, 2, 1600298. [Google Scholar] [CrossRef]
- Britton, J.; Stubbs, K.A.; Weiss, G.A.; Raston, C.L. Vortex fluidic chemical transformations. Chemistry 2017, 23, 13270. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Al-Antaki, A.H.M.; Harvey, D.P.; Ruan, Y.; He, S.; Zhang, W.; Raston, C.L. Vortex fluidic mediated synthesis of macroporous bovine serum albumin-based microspheres. ACS Appl. Mater. Interfaces 2018, 10, 27224–27232. [Google Scholar] [CrossRef]
- Britton, J.; Smith, J.N.; Raston, C.L.; Weiss, G.A. Protein folding using a vortex fluidic device. In Heterologous Gene Expression in E. coli; Springer: Berlin/Heidelberg, Germany, 2017; pp. 211–220. [Google Scholar]
- Berova, N.; Nakanishi, K.; Woody, R.W. Circular Dichroism: Principles and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- Lobley, A.; Whitmore, L.; Wallace, B. DICHROWEB: An interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 2002, 18, 211–212. [Google Scholar] [CrossRef] [Green Version]
- Mao, D.; Wachter, E.; Wallace, B. Folding of the mitochondrial proton adenosine triphosphatase proteolipid channel in phospholipid vesicles. Biochemistry 1982, 21, 4960–4968. [Google Scholar] [CrossRef]
- Sreeranna, N.; Woody, R. Estimation of protein secondary structure from CD spectra: Comparison of CONTIN, SELCON and CDSSTR niethods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef]
- Kelly, S.M.; Price, N.C. The application of circular dichroism to studies of protein folding and unfolding. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1997, 1338, 161–185. [Google Scholar] [CrossRef]
- Sreerama, N.; Venyaminov, S.Y.; Woody, R.W. Estimation of the number of α-helical and β-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 1999, 8, 370–380. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolym. Orig. Res. Biomol. 2008, 89, 392–400. [Google Scholar] [CrossRef]
- Chen, T.; Xie, N.; Viglianti, L.; Zhou, Y.; Tan, H.; Tang, B.Z.; Tang, Y. Quantitative urinalysis using aggregation-induced emission bioprobes for monitoring chronic kidney disease. Faraday Discuss. 2017, 196, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Eftink, M.R. Fluorescence techniques for studying protein structure. Methods Biochem. Anal. 1991, 35, 127–205. [Google Scholar]
- Hong, Y.; Lam, J.W.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Feng, C.; Yu, Y.; Liu, J.; Lam, J.W.Y.; Luo, K.Q.; Tang, B.Z. Quantitation, visualization, and monitoring of conformational transitions of human serum albumin by a tetraphenylethene derivative with aggregation-induced emission characteristics. Anal. Chem. 2010, 82, 7035–7043. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Hong, Y.; Dong, Y.; Häussler, M.; Li, Z.; Lam, J.W.; Dong, Y.; Sung, H.H.-Y.; Williams, I.D.; Tang, B.Z. Protein detection and quantitation by tetraphenylethene-based fluorescent probes with aggregation-induced emission characteristics. J. Phys. Chem. B 2007, 111, 11817–11823. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Liu, M.; Hong, Y. Biothiol-specific fluorescent probes with aggregation-induced emission characteristics. Sci. China Chem. 2018, 61, 882–891. [Google Scholar] [CrossRef]
- Chen, M.Z.; Moily, N.S.; Bridgford, J.L.; Wood, R.J.; Radwan, M.; Smith, T.A.; Song, Z.; Tang, B.Z.; Tilley, L.; Xu, X. A thiol probe for measuring unfolded protein load and proteostasis in cells. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yu, Y.; Lam, J.W.; Hong, Y.; Faisal, M.; Yuan, W.Z.; Tang, B.Z. Simple biosensor with high selectivity and sensitivity thiol-specific biomolecular probing and intracellular imaging by AIE fluorogen on a TLC plate through a thiolene click mechanism. Chem. A Eur. J. 2010, 16, 8433–8438. [Google Scholar] [CrossRef]
- Luo, J.; Xie, Z.; Lam, J.W.; Cheng, L.; Chen, H.; Qiu, C.; Kwok, H.S.; Zhan, X.; Liu, Y.; Zhu, D. Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun. 2001, 1740–1741. [Google Scholar] [CrossRef]
- Gao, M.; Tang, B.Z. Fluorescent sensors based on aggregation-induced emission: Recent advances and perspectives. ACS Sens. 2017, 2, 1382–1399. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.; Milam, S.L.; Clark, A.C. Practical approaches to protein folding and assembly: Spectroscopic strategies in thermodynamics and kinetics. Methods Enzymol. 2009, 455, 1–39. [Google Scholar] [PubMed] [Green Version]
- Matsuura, J.E.; Manning, M.C. Heat-induced gel formation of β-lactoglobulin: A study on the secondary and tertiary structure as followed by circular dichroism spectroscopy. J. Agric. Food Chem. 1994, 42, 1650–1656. [Google Scholar] [CrossRef]
- Woody, R.W. Circular dichroism. Methods Enzymol. 1995, 246, 34–71. [Google Scholar] [PubMed]
- Dong, A.; Matsuura, J.; Allison, S.D.; Chrisman, E.; Manning, M.C.; Carpenter, J.F. Infrared and circular dichroism spectroscopic characterization of structural differences between β-lactoglobulin A and B. Biochemistry 1996, 35, 1450–1457. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Q.; Hu, H.; Zhang, X.; Fan, K.; Hong, Y.; Raston, C.L.; Tang, Y. In Situ Monitored Vortex Fluidic-Mediated Protein Refolding/Unfolding Using an Aggregation-Induced Emission Bioprobe. Molecules 2021, 26, 4273. https://doi.org/10.3390/molecules26144273
Hu Q, Hu H, Zhang X, Fan K, Hong Y, Raston CL, Tang Y. In Situ Monitored Vortex Fluidic-Mediated Protein Refolding/Unfolding Using an Aggregation-Induced Emission Bioprobe. Molecules. 2021; 26(14):4273. https://doi.org/10.3390/molecules26144273
Chicago/Turabian StyleHu, Qi, Haozhen Hu, Xinyi Zhang, Kyle Fan, Yuning Hong, Colin L. Raston, and Youhong Tang. 2021. "In Situ Monitored Vortex Fluidic-Mediated Protein Refolding/Unfolding Using an Aggregation-Induced Emission Bioprobe" Molecules 26, no. 14: 4273. https://doi.org/10.3390/molecules26144273
APA StyleHu, Q., Hu, H., Zhang, X., Fan, K., Hong, Y., Raston, C. L., & Tang, Y. (2021). In Situ Monitored Vortex Fluidic-Mediated Protein Refolding/Unfolding Using an Aggregation-Induced Emission Bioprobe. Molecules, 26(14), 4273. https://doi.org/10.3390/molecules26144273